
Mycobacterium tuberculosis Deficient in PdtaS Cytosolic Histidine Kinase Displays Attenuated Growth and Affords Protective Efficacy against Aerosol M. tuberculosis Infection in Mice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Bacteria Culture Conditions
2.3. Selection and Development of M. tuberculosis pdtaS Mutant Strains
2.4. In Vitro Infection of Macrophages
2.5. Vaccination and Infection of Mice
2.6. Immunogenicity Studies
2.7. Statistics
3. Results
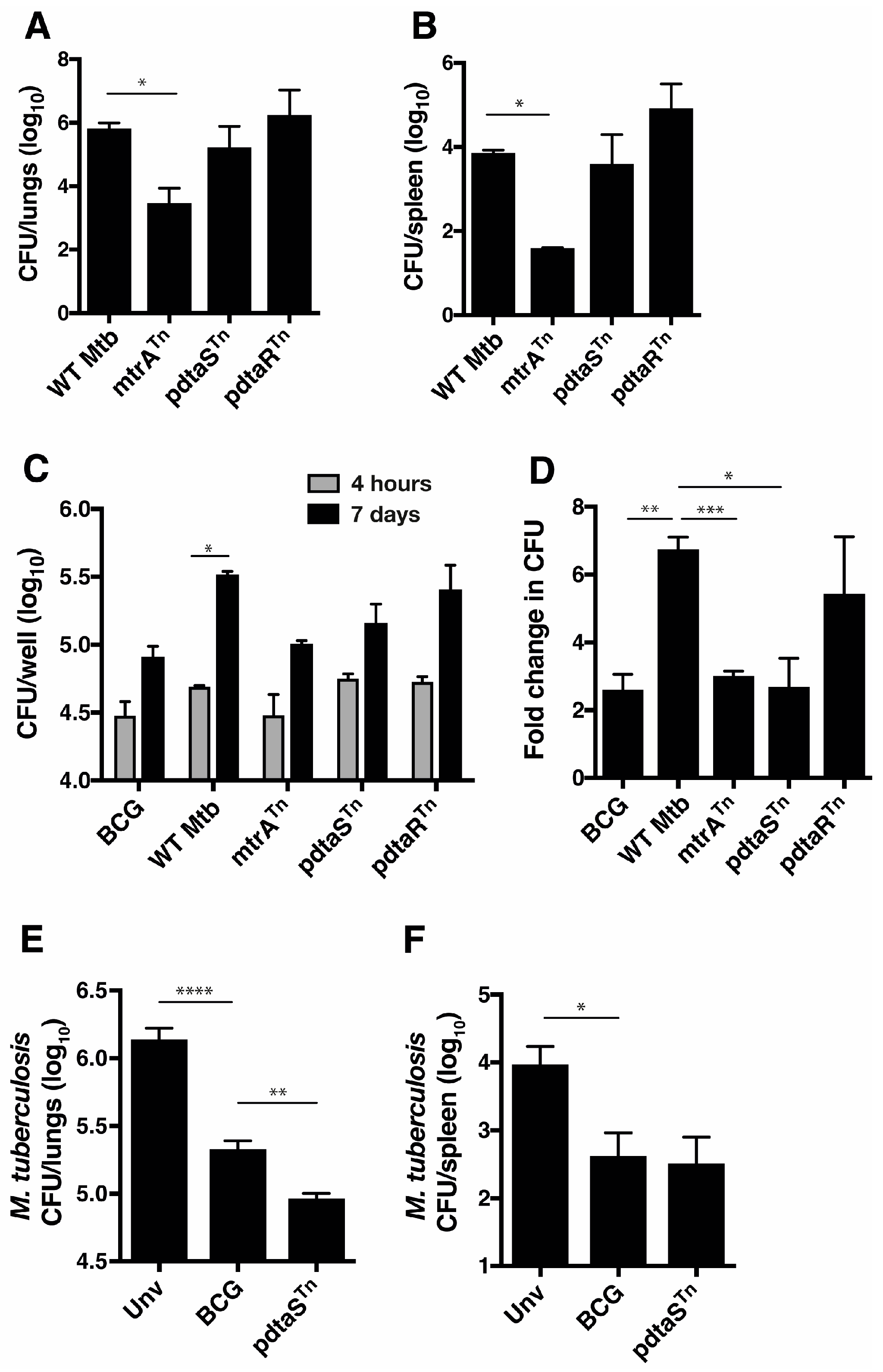
3.1. Identifying In Vivo Attenuated M. tuberculosis Transposon Mutants of the Two-Component System Family
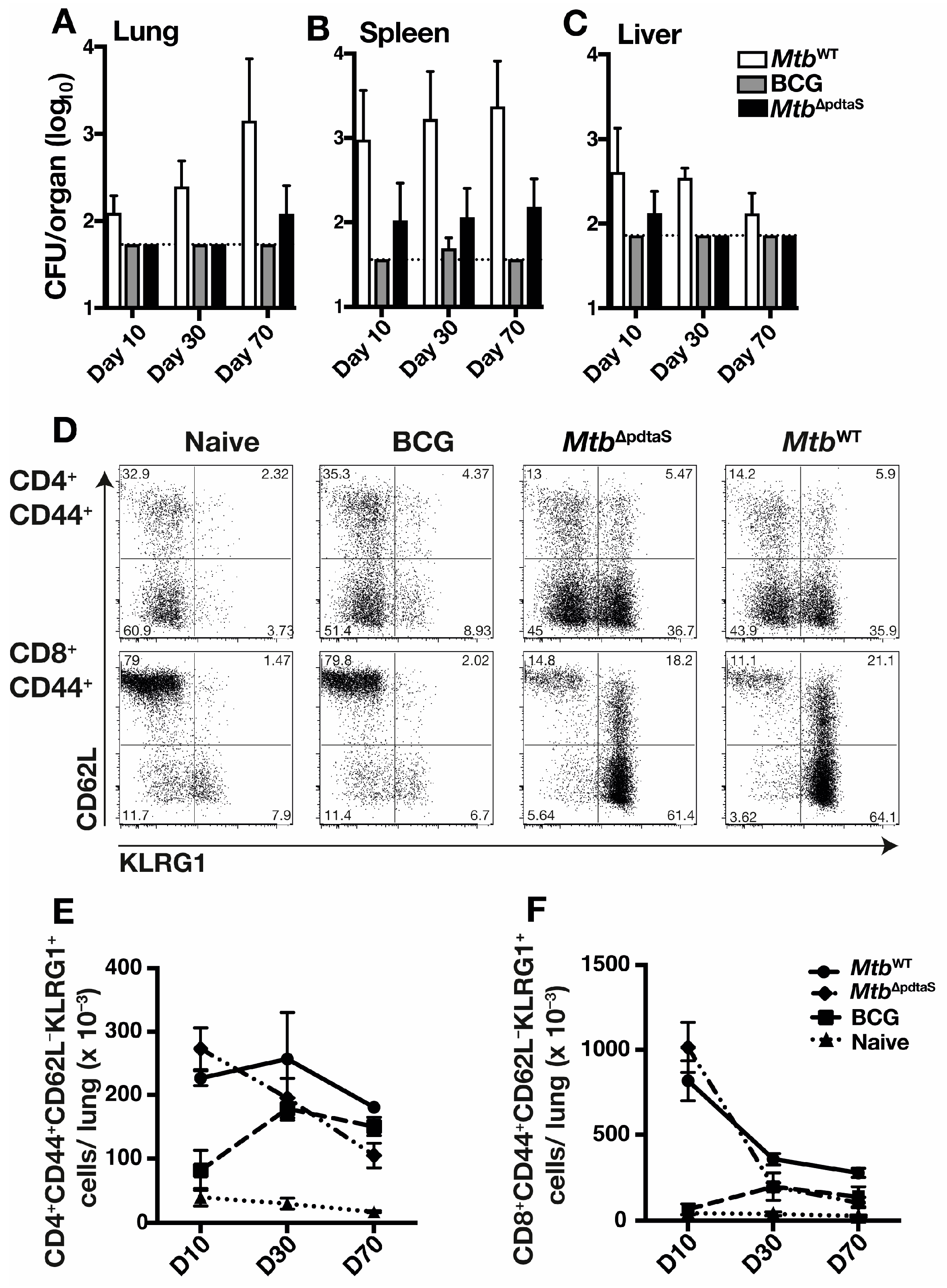
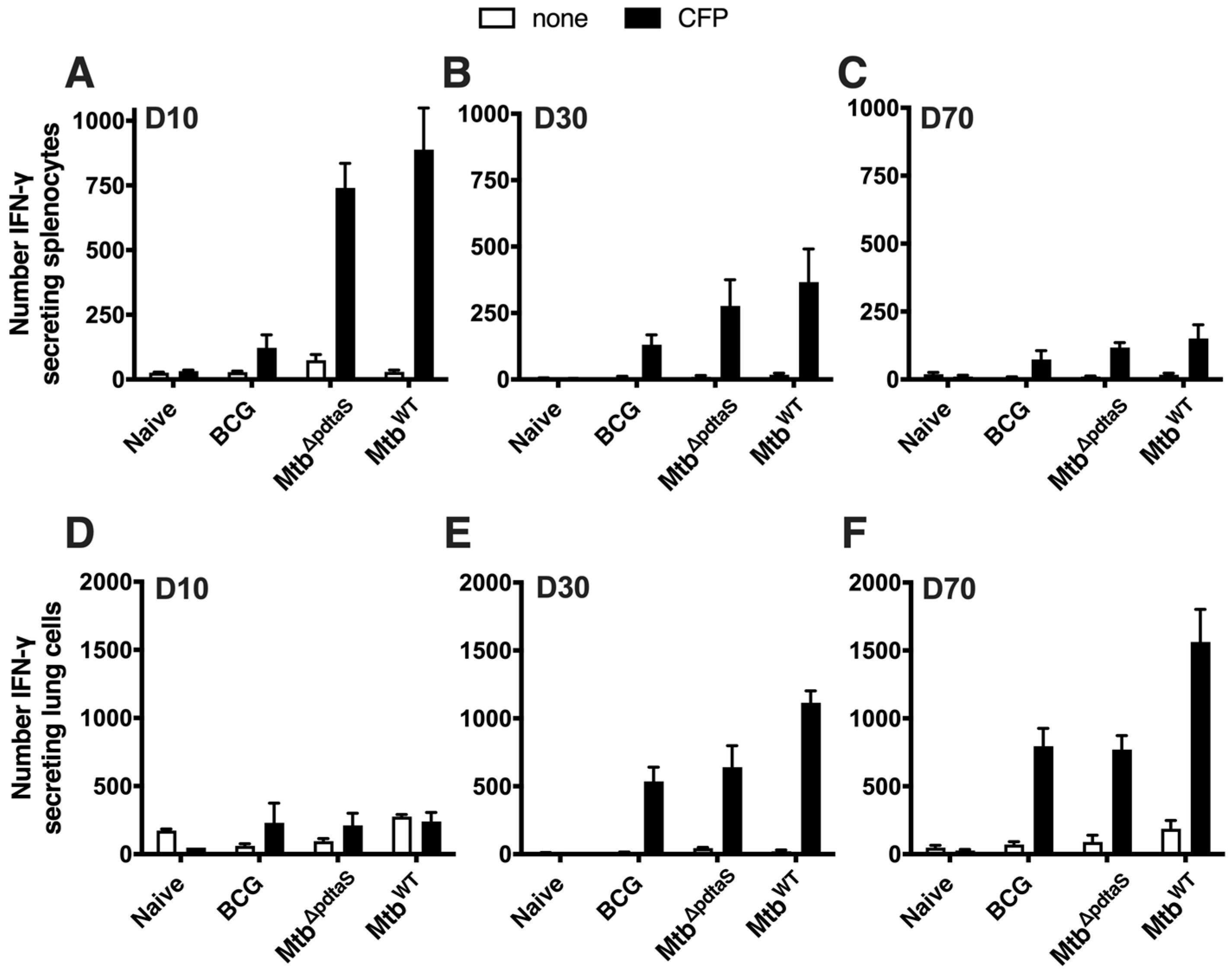
3.2. Persistence and Immunogenicity of a pdtaS Deletion Mutant of M. tuberculosis
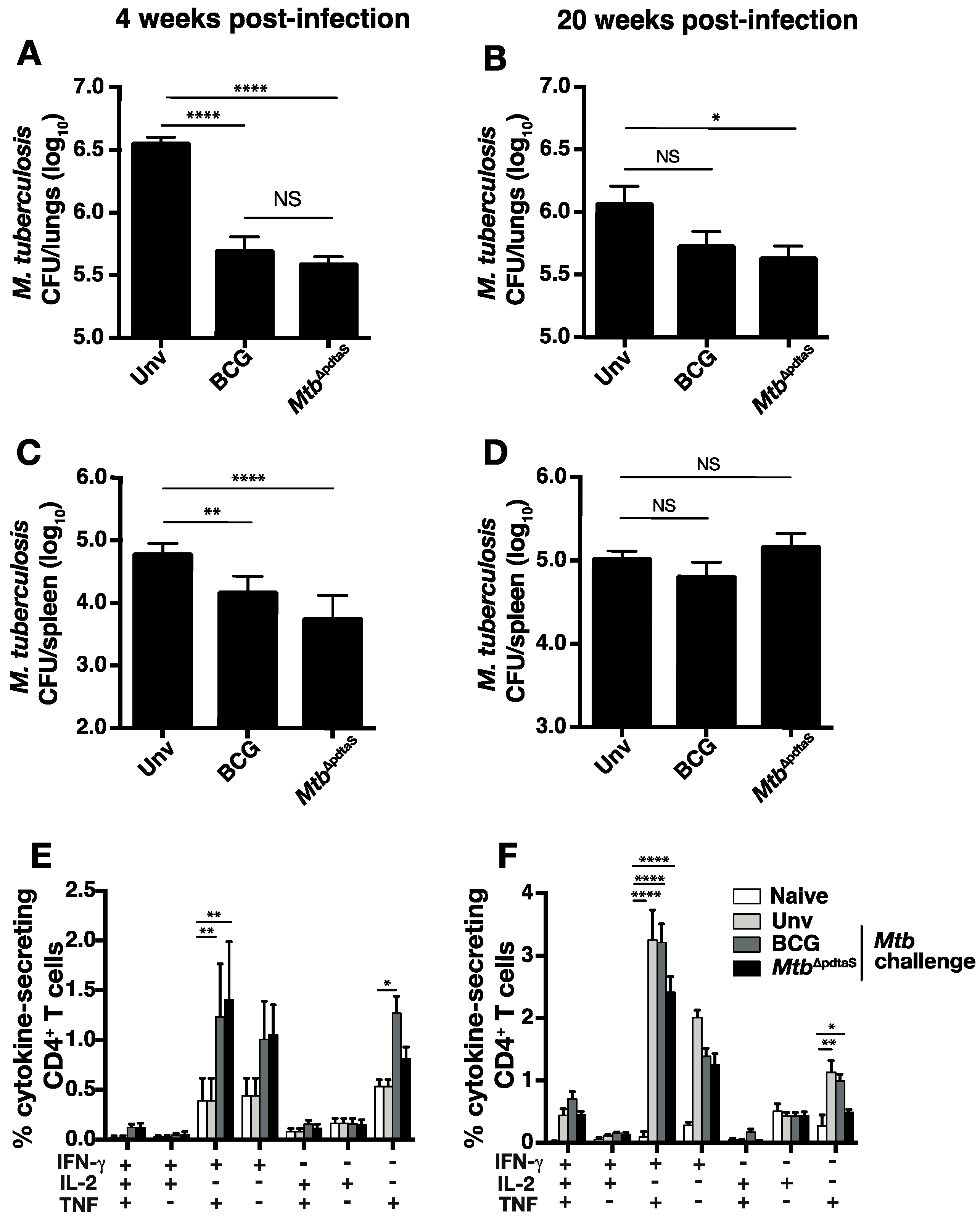
3.3. Protective Efficacy of MtbΔpdtaS against Aerosol M. tuberculosis Infection
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organisation. Global Tuberculosis Report 2023; WHO: Geneva, Switzerland, 2023. [Google Scholar]
- Colditz, G.A.; Brewer, T.F.; Berkey, C.S.; Wilson, M.E.; Burdick, E.; Fineberg, H.V.; Mosteller, F. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of the published literature. JAMA 1994, 271, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Counoupas, C.; Triccas, J.A. The generation of T-cell memory to protect against tuberculosis. Immunol. Cell Biol. 2019, 97, 656–663. [Google Scholar] [CrossRef]
- Spertini, F.; Audran, R.; Chakour, R.; Karoui, O.; Steiner-Monard, V.; Thierry, A.C.; Mayor, C.E.; Rettby, N.; Jaton, K.; Vallotton, L.; et al. Safety of human immunisation with a live-attenuated Mycobacterium tuberculosis vaccine: A randomised, double-blind, controlled phase I trial. Lancet Respir. Med. 2015, 3, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Mahairas, G.G.; Sabo, P.J.; Hickey, M.J.; Singh, D.C.; Stover, C.K. Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis. J. Bacteriol. 1996, 178, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Guinn, K.M.; Hickey, M.J.; Mathur, S.K.; Zakel, K.L.; Grotzke, J.E.; Lewinsohn, D.M.; Smith, S.; Sherman, D.R. Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol. Microbiol. 2004, 51, 359–370. [Google Scholar] [CrossRef]
- Parish, T.; Smith, D.A.; Roberts, G.; Betts, J.; Stoker, N.G. The senX3-regX3 two-component regulatory system of Mycobacterium tuberculosis is required for virulence. Microbiology 2003, 149, 1423–1435. [Google Scholar] [CrossRef]
- Perez, E.; Samper, S.; Bordas, Y.; Guilhot, C.; Gicquel, B.; Martin, C. An essential role for phoP in Mycobacterium tuberculosis virulence. Mol. Microbiol. 2001, 41, 179–187. [Google Scholar] [CrossRef]
- Bretl, D.J.; Demetriadou, C.; Zahrt, T.C. Adaptation to environmental stimuli within the host: Two-component signal transduction systems of Mycobacterium tuberculosis. Microbiol. Mol. Biol. Rev. 2011, 75, 566–582. [Google Scholar] [CrossRef]
- Frigui, W.; Bottai, D.; Majlessi, L.; Monot, M.; Josselin, E.; Brodin, P.; Garnier, T.; Gicquel, B.; Martin, C.; Leclerc, C.; et al. Control of M. tuberculosis ESAT-6 secretion and specific T cell recognition by PhoP. PLoS Pathog. 2008, 4, e33. [Google Scholar] [CrossRef]
- Aguilar, D.; Infante, E.; Martin, C.; Gormley, E.; Gicquel, B.; Hernandez Pando, R. Immunological responses and protective immunity against tuberculosis conferred by vaccination of Balb/C mice with the attenuated Mycobacterium tuberculosis (phoP) SO2 strain. Clin. Exp. Immunol. 2007, 147, 330–338. [Google Scholar] [CrossRef]
- Martin, C.; Williams, A.; Hernandez-Pando, R.; Cardona, P.J.; Gormley, E.; Bordat, Y.; Soto, C.Y.; Clark, S.O.; Hatch, G.J.; Aguilar, D.; et al. The live Mycobacterium tuberculosis phoP mutant strain is more attenuated than BCG and confers protective immunity against tuberculosis in mice and guinea pigs. Vaccine 2006, 24, 3408–3419. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, J.K.; Pinto, R.; Aguilo, J.I.; Takatsu, K.; Martin, C.; Britton, W.J.; Triccas, J.A. Protective immunity afforded by attenuated, PhoP-deficient Mycobacterium tuberculosis is associated with sustained generation of CD4+ T-cell memory. Eur. J. Immunol. 2012, 42, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Arbues, A.; Aguilo, J.I.; Gonzalo-Asensio, J.; Marinova, D.; Uranga, S.; Puentes, E.; Fernandez, C.; Parra, A.; Cardona, P.J.; Vilaplana, C.; et al. Construction, characterization and preclinical evaluation of MTBVAC, the first live-attenuated M. tuberculosis-based vaccine to enter clinical trials. Vaccine 2013, 31, 4867–4873. [Google Scholar] [CrossRef] [PubMed]
- Walker, K.B.; Brennan, M.J.; Ho, M.M.; Eskola, J.; Thiry, G.; Sadoff, J.; Dobbelaer, R.; Grode, L.; Liu, M.A.; Fruth, U.; et al. The second Geneva Consensus: Recommendations for novel live TB vaccines. Vaccine 2010, 28, 2259–2270. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Marinova, D.; Aguilo, N.; Gonzalo-Asensio, J. MTBVAC, a live TB vaccine poised to initiate efficacy trials 100 years after BCG. Vaccine 2021, 39, 7277–7285. [Google Scholar] [CrossRef] [PubMed]
- Stupar, M.; Furness, J.; De Voss, C.J.; Tan, L.; West, N.P. Two-component sensor histidine kinases of Mycobacterium tuberculosis: Beacons for niche navigation. Mol. Microbiol. 2022, 117, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Morth, J.P.; Feng, V.; Perry, L.J.; Svergun, D.I.; Tucker, P.A. The crystal and solution structure of a putative transcriptional antiterminator from Mycobacterium tuberculosis. Structure 2004, 12, 1595–1605. [Google Scholar] [CrossRef]
- Morth, J.P.; Gosmann, S.; Nowak, E.; Tucker, P.A. A novel two-component system found in Mycobacterium tuberculosis. FEBS Lett. 2005, 579, 4145–4148. [Google Scholar] [CrossRef]
- Preu, J.; Panjikar, S.; Morth, P.; Jaiswal, R.; Karunakar, P.; Tucker, P.A. The sensor region of the ubiquitous cytosolic sensor kinase, PdtaS, contains PAS and GAF domain sensing modules. J. Struct. Biol. 2012, 177, 498–505. [Google Scholar] [CrossRef]
- Dadura, K.; Plocinska, R.; Rumijowska-Galewicz, A.; Plocinski, P.; Zaczek, A.; Dziadek, B.; Zaborowski, A.; Dziadek, J. PdtaS Deficiency Affects Resistance of Mycobacteria to Ribosome Targeting Antibiotics. Front. Microbiol. 2017, 8, 2145. [Google Scholar] [CrossRef]
- Hariharan, V.N.; Yadav, R.; Thakur, C.; Singh, A.; Gopinathan, R.; Singh, D.P.; Sankhe, G.; Malhotra, V.; Chandra, N.; Bhatt, A.; et al. Cyclic di-GMP sensing histidine kinase PdtaS controls mycobacterial adaptation to carbon sources. FASEB J. 2021, 35, e21475. [Google Scholar] [CrossRef] [PubMed]
- Buglino, J.A.; Sankhe, G.D.; Lazar, N.; Bean, J.M.; Glickman, M.S. Integrated sensing of host stresses by inhibition of a cytoplasmic two-component system controls M. tuberculosis acute lung infection. eLife 2021, 10, e65351. [Google Scholar] [CrossRef] [PubMed]
- Serafini, A.; Tan, L.; Horswell, S.; Howell, S.; Greenwood, D.J.; Hunt, D.M.; Phan, M.D.; Schembri, M.; Monteleone, M.; Montague, C.R.; et al. Mycobacterium tuberculosis requires glyoxylate shunt and reverse methylcitrate cycle for lactate and pyruvate metabolism. Mol. Microbiol. 2019, 112, 1284–1307. [Google Scholar] [CrossRef] [PubMed]
- Bardarov, S.; Bardarov, S., Jr.; Pavelka, M.S., Jr.; Sambandamurthy, V.; Larsen, M.; Tufariello, J.; Chan, J.; Hatfull, G.; Jacobs, W.R., Jr. Specialized transduction: An efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 2002, 148, 3007–3017. [Google Scholar] [CrossRef]
- van Kessel, J.C.; Hatfull, G.F. Mycobacterial recombineering. Methods Mol. Biol. 2008, 435, 203–215. [Google Scholar] [PubMed]
- Zahrt, T.C.; Deretic, V. An essential two-component signal transduction system in Mycobacterium tuberculosis. J. Bacteriol. 2000, 182, 3832–3838. [Google Scholar] [CrossRef]
- Zhuang, L.; Ye, Z.; Li, L.; Yang, L.; Gong, W. Next-Generation TB Vaccines: Progress, Challenges, and Prospects. Vaccines 2023, 11, 1304. [Google Scholar] [CrossRef]
- Neyrolles, O.; Mintz, E.; Catty, P. Zinc and copper toxicity in host defense against pathogens: Mycobacterium tuberculosis as a model example of an emerging paradigm. Front. Cell Infect. Microbiol. 2013, 3, 89. [Google Scholar] [CrossRef]
- Parish, T.; Smith, D.A.; Kendall, S.; Casali, N.; Bancroft, G.J.; Stoker, N.G. Deletion of two-component regulatory systems increases the virulence of Mycobacterium tuberculosis. Infect. Immun. 2003, 71, 1134–1140. [Google Scholar] [CrossRef]
- Counoupas, C.; Pinto, R.; Nagalingam, G.; Hill-Cawthorne, G.A.; Feng, C.G.; Britton, W.J.; Triccas, J.A. Mycobacterium tuberculosis components expressed during chronic infection of the lung contribute to long-term control of pulmonary tuberculosis in mice. NPJ Vaccines 2016, 1, 16012. [Google Scholar] [CrossRef]
- Sterne, J.A.; Rodrigues, L.C.; Guedes, I.N. Does the efficacy of BCG decline with time since vaccination? Int. J. Tuberc. Lung Dis. 1998, 2, 200–207. [Google Scholar] [PubMed]
- Pinto, R.; Saunders, B.M.; Camacho, L.R.; Britton, W.J.; Gicquel, B.; Triccas, J.A. Mycobacterium tuberculosis defective in phthiocerol dimycocerosate translocation provides greater protective immunity against tuberculosis than the existing bacille Calmette-Guerin vaccine. J. Infect. Dis. 2004, 189, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Pym, A.S.; Brodin, P.; Majlessi, L.; Brosch, R.; Demangel, C.; Williams, A.; Griffiths, K.E.; Marchal, G.; Leclerc, C.; Cole, S.T. Recombinant BCG exporting ESAT-6 confers enhanced protection against tuberculosis. Nat. Med. 2003, 9, 533–539. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prendergast, K.A.; Nagalingam, G.; West, N.P.; Triccas, J.A. Mycobacterium tuberculosis Deficient in PdtaS Cytosolic Histidine Kinase Displays Attenuated Growth and Affords Protective Efficacy against Aerosol M. tuberculosis Infection in Mice. Vaccines 2024, 12, 50. https://doi.org/10.3390/vaccines12010050
Prendergast KA, Nagalingam G, West NP, Triccas JA. Mycobacterium tuberculosis Deficient in PdtaS Cytosolic Histidine Kinase Displays Attenuated Growth and Affords Protective Efficacy against Aerosol M. tuberculosis Infection in Mice. Vaccines. 2024; 12(1):50. https://doi.org/10.3390/vaccines12010050
Chicago/Turabian StylePrendergast, Kelly A., Gayathri Nagalingam, Nicholas P. West, and James A. Triccas. 2024. "Mycobacterium tuberculosis Deficient in PdtaS Cytosolic Histidine Kinase Displays Attenuated Growth and Affords Protective Efficacy against Aerosol M. tuberculosis Infection in Mice" Vaccines 12, no. 1: 50. https://doi.org/10.3390/vaccines12010050
APA StylePrendergast, K. A., Nagalingam, G., West, N. P., & Triccas, J. A. (2024). Mycobacterium tuberculosis Deficient in PdtaS Cytosolic Histidine Kinase Displays Attenuated Growth and Affords Protective Efficacy against Aerosol M. tuberculosis Infection in Mice. Vaccines, 12(1), 50. https://doi.org/10.3390/vaccines12010050