
Oral Administration of Cancer Vaccines: Challenges and Future Perspectives

 , ,
, ,
Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
| Type of Vaccine | Strategy | Advantages | Disadvantages | References |
|---|---|---|---|---|
| Peptide-based vaccines | They are based on specific epitopes expressed on the surfaces of cancer cells | High specificity | Poorly immunogenic, adjuvants required | [16] |
| Tolerable toxicity profile | ||||
| Bacteria-based vaccines | Bacteria are genetically engineered to express TSA or TAA | Upon oral administration, probiotics embed and colonize within the preexisting microbiome | Toxicity of bacteria toxins and virulence factors | [12,17] |
| Short half-life of bacteria proteins | ||||
| Recombinant plasmid delivered by bacteria might mutate | ||||
| Nucleic acid-based vaccines (DNA, RNA) | DNA and RNA vaccines introduce TAA or TSA in host cells to produce specific antigen | They can activate specific and multiple epitopes simultaneously. Inexpensive | Further studies are needed to evaluate the safety and efficacy | [18,19] |
| Virus-based vaccines | Viral vectors deliver specific antigens to the immune system | Stimulation of both innate and adaptive immune response | Repeated administration might be avoided to disadvantage the induction of antiviral immune responses | [8,19] |
| Long-lasting immune response | ||||
| No adjuvants are required | ||||
| Cell-based vaccines | Cancer cells elicit a targeted immune response through the inclusion of complete tumour antigens inherent to these cells. | Cell-based vaccines just include the epitopes of both T helper and CTLs | Low immunogenicity | [8,20] |
| No selection of a specific tumour antigen is required | The preparation method must maintain the integrity and functionality of antigens |
2. Immunological Basis for Oral Vaccination
2.1. Fisiology and Immunology of the Intestine
2.2. The Intestinal Immune System
Inductive Sites of the Intestinal Immune System
- Mesenteric lymph nodes
- Peyer’s patches
- Isolated lymphoid follicles
2.3. Intestinal T Cells
- T cells in the intestinal epithelium
- T cells in the lamina propria
2.4. Intestinal Dendritic Cells
2.5. Intestinal Monocytes and Macrophages
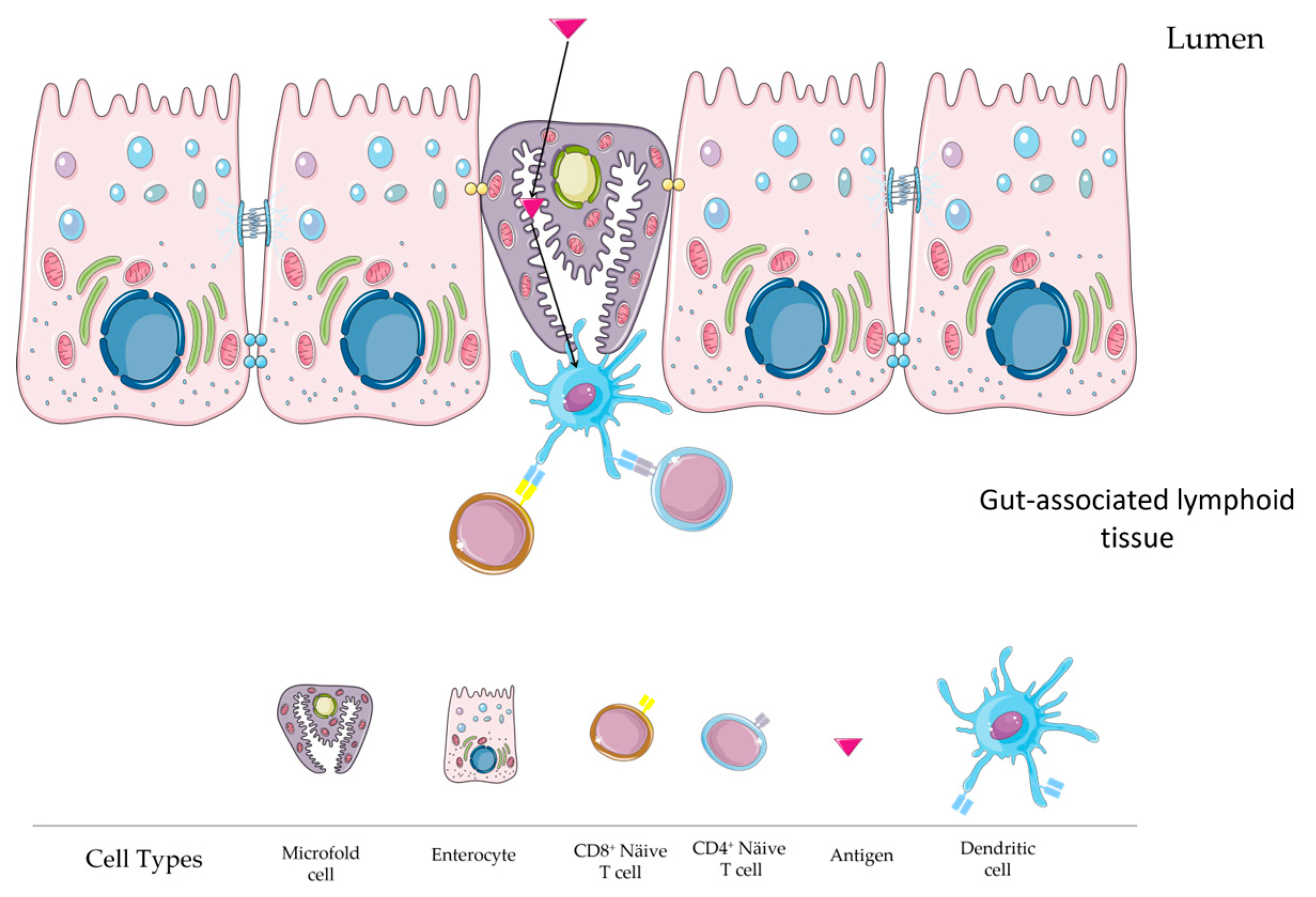
2.6. Microfold (M) Cell Role
2.7. Cellular and Molecular Mechanisms in Mucosal Immunity Induction via Oral Vaccines
2.7.1. The Synthesis and Impact of IgA and IgG in Intestinal Immune Defence
2.7.2. The Role of Cytotoxic T Lymphocytes in Gut Immunity
2.7.3. Molecular Mechanisms of Gut Homing in Mucosally Primed Effector B and T Cells
3. Stimulating Intestinal Immune Responses through Oral Vaccination
4. Commercially Available Oral Vaccines
5. Oral Administration of Cancer Vaccines
| Type of Oral Cancer Vaccine | Subtypes | Cancer | Antigen | Delivery System | Species | Outcome | Ref |
|---|---|---|---|---|---|---|---|
| Peptide-based vaccines | Long peptide | Colorectal cancer | AH1 | Liposome | Mouse | Oral emulsion vaccine reduces colorectal tumour | [103] |
| - | OVA-expressing tumours | Ovalbumin | ovalbumin as the antigen encapsulated in whole glucan particles | Mouse | WGP-OVA: A Promising Cancer Vaccine | [121] | |
| - | B cell lymphoma A20 | MHC class-I restricted peptides derived from survivin | Oral co-administration of β-glucan with peptide vaccination | Mouse | β-Glucan Enhances Peptide Vaccination Efficacy | [122] | |
| Bacteria-based vaccines | Bifidobacterium longum | Renal Carcinoma | WT1 | Oral Bifidobacterium | Mouse | Enhances RCC Immunotherapy | [104,110] |
| Bifidobacterium longum | Bladder cancer | WT1 | Oral Bifidobacterium | Mouse | Enhances Bladder Cancer Immunotherapy | [123] | |
| Bifidobacterium longum | Glioblastoma | WT1 | Oral Bifidobacterium | Mouse | Shows Promise for Glioblastoma Treatment | [124] | |
| Bifidobacterium longum | Prostate cancer | WT1 | Oral Bifidobacterium | Mouse | Promising Oral Cancer Vaccine (B440) for Prostate Cancer Treatment | [125] | |
| Bifidobacterium longum | Wilms’ Tumour 1 | WT1 | Oral Bifidobacterium | Mouse | Induces Effective WT1-Specific Immunity | [125] | |
| Bifidobacterium longum | Leukaemia | WT1 | Oral Bifidobacterium | Mouse | Superior Antitumour Effect of WT1 Oral Cancer Vaccine Over Peptide Vaccine | [126] | |
| Bifidobacterium longum | Acute myeloid leukemia | WT1 | Oral Bifidobacterium | Mouse | Shows Strong Antitumour Activity | [127] | |
| Listeria monocytogenes | Cervical cancer | HPV-16 E7 | Oral Listeria | Mouse | Cervical Cancer Treatment | [110] | |
| Lactic acid bacteria | Cancers overexpressing carcinoembryonic antigen | CEA | Oral LAB | Mouse | LABs as Oral Vaccine Vector for CEA Antigen | [111] | |
| E. coli | Colorectal Cancer | Tumour antigen- and adjuvant-containing emulsions or liposomes | Mouse | Oral Bacteria Biohybrid-Based Vaccines for Colorectal Cancer Treatment | [112] | ||
| Salmonella typhimurium | Pancreatic cancer | VEGFR2 | Oral Salmonella | Human | Monthly Boost Vaccinations with VXM01 in Advanced Pancreatic Cancer Patients | [128] | |
| Salmonella typhimurium | targets cancers expressing the NY-ESO-1 tumour antigen | NY-ESO-1 | Oral Salmonella | Mouse | Salmonella Typhimurium-Based Vaccine for Cancer Immunotherapy | [113] | |
| Salmonella typhimurium | Cancer expressing ovalbumin (OVA) | OVA | Oral Salmonella | Mouse | H2O2-Inactivated Salmonella Typhimurium RE88 as a Novel Cancer Vaccine Carrier | [95] | |
| Salmonella typhimurium | Hepatocellular carcinoma (HCC) | AFP | Oral Salmonella | Mouse | Therapeutic Efficacy of an Oral DNA Vaccine Against Hepatocellular Carcinoma | [129] | |
| Nucleic acid-based vaccines | DNA vaccine | cancer-related angiogenesis | VEGFR2 | nanoparticle-coated bacterial vectors | Mouse | Nanoparticle-Coated Bacterial Vectors for DNA Vaccines | [87] |
| DNA vaccine | Lung | VEGFR-3 | Oral administration using attenuated Salmonella enterica serovar typhimurium strain SL3261 | Mouse | Oral VEGFR-3-Based Vaccine for Lung Cancer Inhibition | [114] | |
| DNA vaccine | Melanoma | Heat shock protein 70 (Hsp70) and tumour-associated antigens (TAA) | Oral administration using attenuated Salmonella typhimurium strain SL3261 as the carrier | Mouse | Oral Hsp70-TAA DNA Vaccine for Cancer Immunotherapy | [130] | |
| DNA vaccine | Breast | Murine endoglin (CD105) | Oral delivery using double attenuated Salmonella typhimurium (dam −, AroA −) | Mouse | Oral Endoglin DNA Vaccine for Breast Cancer | [131] | |
| DNA vaccine | Melanoma | Plasmid DNA gp100 | Oral delivery using a nanogel (Alg-Tat-gp100) | Mouse | Multi-Faceted Nanogel for Oral DNA Vaccine | [132] | |
| Virus-based vaccines | Adeno-Associated Viral Vector (AVV) | Breast cancer | neu | AAV | Mouse | Effective Antitumour Vaccination | [115,123] |
| Cell-based vaccines | Melanoma Colon cancer | Oral particulate | Mouse | Potential for Effective Antitumour Responses | [133] | ||
| Ovarian | Whole-cell lysate | enteric polymers | Mouse | Effective Against Ovarian Cancer | [116] | ||
| Prostate cancer | Whole-cell lysate | Aleuria aurantia lectin | Mouse | Effective Oral Vaccine Against Prostate Cancer | [134] | ||
| Prostate | TRAMP-C2 | Oral microparticulate vaccine | Mouse | Oral Microparticulate Vaccine Enhances Prostate Cancer Immunotherapy | [117] | ||
| Breast | microparticles combined with intraperitoneal administration of cyclophosphamide | Mouse | Effective Breast Cancer Immunotherapy | [135] |
6. Challenges in Oral Delivery: Gastrointestinal Barriers
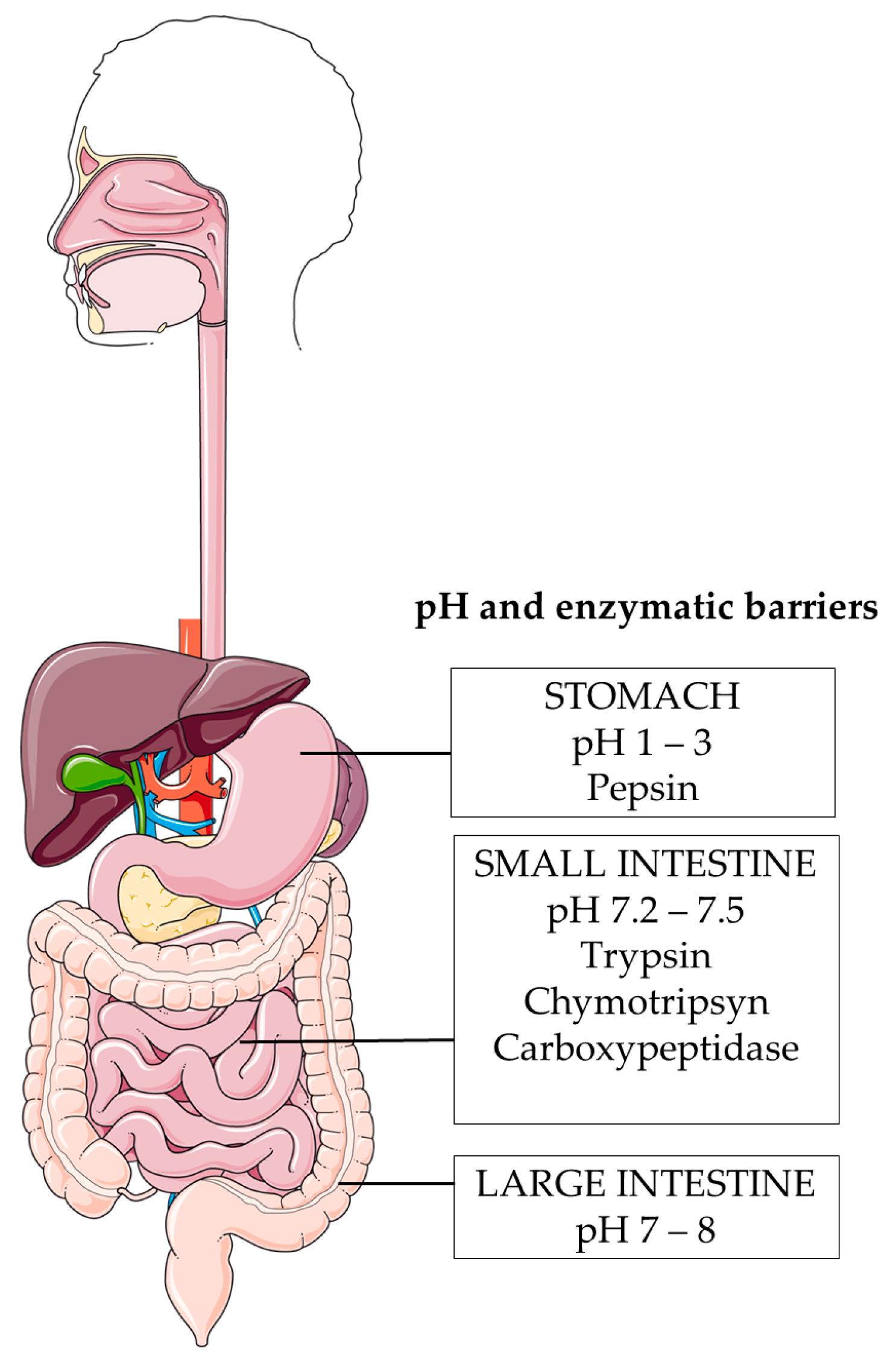
6.1. Harsh pH Environment
6.2. Enzymatic Degradation
6.3. Mucus Barrier
6.4. Epithelial Barrier
6.5. Gastrointestinal Microbiota
7. Strategies to Overcome the Challenges of Oral Administration of Cancer Vaccines
7.1. Delivery Systems
7.2. Targeting Strategies
Directing Attention towards DC Surface Receptors:
7.3. Adjuvants
| Type of Adjuvants | Subtypes | References |
|---|---|---|
| Toxin derivatives | Cholera toxin (CT) | [204] |
| labile enterotoxin (LT) | [205] | |
| Pattern-Recognition Receptor (PRR) ligands | Toll-like receptor (TLR) ligands | [206] |
| NOD-like receptor (NLR) ligands | [207] | |
| RIG-I-like receptor (RLR) ligands | [208] | |
| C-type lectin receptor (CLR) ligands | [209] | |
| Gut Peptide Cell Adjuvants | M-Cell Peptides/Ligands | [210,211,212] |
| Cytokine Adjuvants | IL-1β, IL-2, IL-12 | [213,214,215] |
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rappuoli, R.; Mandl, C.W.; Black, S.; De Gregorio, E. Vaccines for the twenty-first century society. Nat. Rev. Immunol. 2011, 11, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Firdous, J.; Badruddoza, A.Z.M.; Reesor, E.; Azad, M.; Hasan, A.; Lim, M.; Cao, W.; Guillemette, S.; Cho, C.S. M cell targeting engineered biomaterials for effective vaccination. Biomaterials 2019, 192, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, Y.; Sun, Y.; Cui, H.; Zhu, S.J.; Qiu, H.-J. Mucosal vaccines: Strategies and challenges. Immunol. Lett. 2020, 217, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Busato, D.; Mossenta, M.; Bo, M.D.; Macor, P.; Toffoli, G. The Proteoglycan Glypican-1 as a Possible Candidate for Innovative Targeted Therapeutic Strategies for Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2022, 23, 10279. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Al-Khadairi, G.; Roelands, J.; Hendrickx, W.; Dermime, S.; Bedognetti, D.; Decock, J. NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives. Front. Immunol. 2018, 9, 947. [Google Scholar] [CrossRef]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer vaccines as promising immuno-therapeutics: Platforms and current progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef]
- Darragh, L.B.; Karam, S.D. Amateur antigen-presenting cells in the tumor microenvironment. Mol. Carcinog. 2022, 61, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.H.; Nowicki, C.; Giurini, E.F.; Marzo, A.L.; Zloza, A. Bacterial-Based Cancer Therapy (BBCT): Recent Advances, Current Challenges, and Future Prospects for Cancer Immunotherapy. Vaccines 2021, 9, 1497. [Google Scholar] [CrossRef] [PubMed]
- Crunkhorn, S. A bacteria-derived oral tumour vaccine. Nat. Rev. Drug Discov. 2022, 21, 494. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tang, H.; Li, L.; Wang, X.; Yu, Z.; Li, J. Peptide-based therapeutic cancer vaccine: Current trends in clinical application. Cell Prolif. 2021, 54, e13025. [Google Scholar] [CrossRef] [PubMed]
- Vela Ramirez, J.E.; Sharpe, L.A.; Peppas, N.A. Current state and challenges in developing oral vaccines. Adv. Drug Deliv. Rev. 2017, 114, 116–131. [Google Scholar] [CrossRef]
- Paston, S.J.; Brentville, V.A.; Symonds, P.; Durrant, L.G. Cancer Vaccines, Adjuvants, and Delivery Systems. Front. Immunol. 2021, 12, 627932. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Xu, J.; Li, Y.; Cheng, K.; Feng, Q.; Ma, X.; Ma, N.; Zhang, T.; Wang, X.; Zhao, X.; et al. Antigen-bearing outer membrane vesicles as tumour vaccines produced in situ by ingested genetically engineered bacteria. Nat. Biomed. Eng. 2022, 6, 898–909. [Google Scholar] [CrossRef]
- Tojjari, A.; Saeed, A.; Singh, M.; Cavalcante, L.; Sahin, I.H.; Saeed, A. A Comprehensive Review on Cancer Vaccines and Vaccine Strategies in Hepatocellular Carcinoma. Vaccines 2023, 11, 1357. [Google Scholar] [CrossRef]
- Igarashi, Y.; Sasada, T. Cancer Vaccines: Toward the Next Breakthrough in Cancer Immunotherapy. J. Immunol. Res. 2020, 2020, 5825401. [Google Scholar] [CrossRef]
- Sadeghi Najafabadi, S.A.; Bolhassani, A.; Aghasadeghi, M.R. Tumor cell-based vaccine: An effective strategy for eradication of cancer cells. Immunotherapy 2022, 14, 639–654. [Google Scholar] [CrossRef]
- Amin, M.K.; Boateng, J. Surface functionalization of PLGA nanoparticles for potential oral vaccine delivery targeting intestinal immune cells. Colloids Surf. B Biointerfaces 2023, 222, 113121. [Google Scholar] [CrossRef] [PubMed]
- Roda, G. Intestinal epithelial cells in inflammatory bowel diseases. World J. Gastroenterol. 2010, 16, 4264. [Google Scholar] [CrossRef] [PubMed]
- Artis, D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat. Rev. Immunol. 2008, 8, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Abraham, C.; Medzhitov, R. Interactions Between the Host Innate Immune System and Microbes in Inflammatory Bowel Disease. Gastroenterology 2011, 140, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Meresse, B.; Ripoche, J.; Heyman, M.; Cerf-Bensussan, N. Celiac disease: From oral tolerance to intestinal inflammation, autoimmunity and lymphomagenesis. Mucosal Immunol. 2009, 2, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.J.; Geuking, M.B.; McCoy, K.D. Immune responses that adapt the intestinal mucosa to commensal intestinal bacteria. Immunology 2005, 115, 153–162. [Google Scholar] [CrossRef]
- Spahn, T.W.; Weiner, H.L.; Rennert, P.D.; Lügering, N.; Fontana, A.; Domschke, W.; Kucharzik, T. Mesenteric lymph nodes are critical for the induction of high-dose oral tolerance in the absence of Peyer’s patches. Eur. J. Immunol. 2002, 32, 1109–1113. [Google Scholar] [CrossRef]
- Worbs, T.; Bode, U.; Yan, S.; Hoffmann, M.W.; Hintzen, G.; Bernhardt, G.; Förster, R.; Pabst, O. Oral tolerance originates in the intestinal immune system and relies on antigen carriage by dendritic cells. J. Exp. Med. 2006, 203, 519–527. [Google Scholar] [CrossRef]
- Suzuki, K.; Meek, B.; Doi, Y.; Muramatsu, M.; Chiba, T.; Honjo, T.; Fagarasan, S. Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc. Natl. Acad. Sci. USA 2004, 101, 1981–1986. [Google Scholar] [CrossRef]
- Hooper, L.V.; Macpherson, A.J. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol. 2010, 10, 159–169. [Google Scholar] [CrossRef]
- Pabst, O.; Herbrand, H.; Worbs, T.; Friedrichsen, M.; Yan, S.; Hoffmann, M.W.; Körner, H.; Bernhardt, G.; Pabst, R.; Förster, R. Cryptopatches and isolated lymphoid follicles: Dynamic lymphoid tissues dispensable for the generation of intraepithelial lymphocytes. Eur. J. Immunol. 2005, 35, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Fagarasan, S.; Kawamoto, S.; Kanagawa, O.; Suzuki, K. Adaptive Immune Regulation in the Gut: T Cell–Dependent and T Cell–Independent IgA Synthesis. Annu. Rev. Immunol. 2010, 28, 243–273. [Google Scholar] [CrossRef] [PubMed]
- Pabst, O.; Herbrand, H.; Friedrichsen, M.; Velaga, S.; Dorsch, M.; Berhardt, G.; Worbs, T.; Macpherson, A.J.; Förster, R. Adaptation of Solitary Intestinal Lymphoid Tissue in Response to Microbiota and Chemokine Receptor CCR7 Signaling. J. Immunol. 2006, 177, 6824–6832. [Google Scholar] [CrossRef] [PubMed]
- Cheroutre, H.; Lambolez, F.; Mucida, D. The light and dark sides of intestinal intraepithelial lymphocytes. Nat. Rev. Immunol. 2011, 11, 445–456. [Google Scholar] [CrossRef]
- Moens, E.; Veldhoen, M. Epithelial barrier biology: Good fences make good neighbours. Immunology 2012, 135, 1–8. [Google Scholar] [CrossRef]
- Shires, J.; Theodoridis, E.; Hayday, A.C. Biological Insights into TCRγδ+ and TCRαβ+ Intraepithelial Lymphocytes Provided by Serial Analysis of Gene Expression (SAGE). Immunity 2001, 15, 419–434. [Google Scholar] [CrossRef]
- Regnault, A.; Cumano, A.; Vassalli, P.; Guy-Grand, D.; Kourilsky, P. Oligoclonal repertoire of the CD8 alpha alpha and the CD8 alpha beta TCR-alpha/beta murine intestinal intraepithelial T lymphocytes: Evidence for the random emergence of T cells. J. Exp. Med. 1994, 180, 1345–1358. [Google Scholar] [CrossRef]
- Steege, J.C.A.T.; Buurman, W.A.; Forget, P.-P. The Neonatal Development of Intraepithelial and Lamina Propria Lymphocytes in the Murine Small Intestine. Dev. Immunol. 1997, 5, 121–128. [Google Scholar] [CrossRef]
- Leishman, A.J.; Gapin, L.; Capone, M.; Palmer, E.; MacDonald, H.; Kronenberg, M.; Cheroutre, H. Precursors of Functional MHC Class I- or Class II-Restricted CD8αα+ T Cells Are Positively Selected in the Thymus by Agonist Self-Peptides. Immunity 2002, 16, 355–364. [Google Scholar] [CrossRef]
- Staton, T.L.; Habtezion, A.; Winslow, M.M.; Sato, T.; Love, P.E.; Butcher, E.C. CD8+ recent thymic emigrants home to and efficiently repopulate the small intestine epithelium. Nat. Immunol. 2006, 7, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Shale, M.; Schiering, C.; Powrie, F. CD4+ T-cell subsets in intestinal inflammation. Immunol. Rev. 2013, 252, 164–182. [Google Scholar] [CrossRef] [PubMed]
- Persson, E.K.; Scott, C.L.; Mowat, A.M.; Agace, W.W. Dendritic cell subsets in the intestinal lamina propria: Ontogeny and function. Eur. J. Immunol. 2013, 43, 3098–3107. [Google Scholar] [CrossRef] [PubMed]
- Varol, C.; Vallon-Eberhard, A.; Elinav, E.; Aychek, T.; Shapira, Y.; Luche, H.; Fehling, H.J.; Hardt, W.-D.; Shakhar, G.; Jung, S. Intestinal Lamina Propria Dendritic Cell Subsets Have Different Origin and Functions. Immunity 2009, 31, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Bain, C.C.; Scott, C.L.; Uronen-Hansson, H.; Gudjonsson, S.; Jansson, O.; Grip, O.; Guilliams, M.; Malissen, B.; Agace, W.W.; Mowat, A.M. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 2013, 6, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Schlitzer, A.; McGovern, N.; Teo, P.; Zelante, T.; Atarashi, K.; Low, D.; Ho, A.W.S.; See, P.; Shin, A.; Wasan, P.S.; et al. IRF4 Transcription Factor-Dependent CD11b+ Dendritic Cells in Human and Mouse Control Mucosal IL-17 Cytokine Responses. Immunity 2013, 38, 970–983. [Google Scholar] [CrossRef] [PubMed]
- Satpathy, A.T.; Briseño, C.G.; Lee, J.S.; Ng, D.; Manieri, N.A.; Kc, W.; Wu, X.; Thomas, S.R.; Lee, W.-L.; Turkoz, M.; et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat. Immunol. 2013, 14, 937–948. [Google Scholar] [CrossRef]
- Beitnes, A.C.; Ráki, M.; Lundin, K.E.A.; Jahnsen, J.; Sollid, L.M.; Jahnsen, F.L. Density of CD163+ CD11c+ Dendritic Cells Increases and CD103+ Dendritic Cells Decreases in the Coeliac Lesion. Scand. J. Immunol. 2011, 74, 186–194. [Google Scholar] [CrossRef]
- Jaensson, E.; Uronen-Hansson, H.; Pabst, O.; Eksteen, B.; Tian, J.; Coombes, J.L.; Berg, P.L.; Davidsson, T.; Powrie, F.; Johansson-Lindbom, B.; et al. Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J. Exp. Med. 2008, 205, 2139–2149. [Google Scholar] [CrossRef]
- Platt, A.M.; Bain, C.C.; Bordon, Y.; Sester, D.P.; Mowat, A.M. An Independent Subset of TLR Expressing CCR2-Dependent Macrophages Promotes Colonic Inflammation. J. Immunol. 2010, 184, 6843–6854. [Google Scholar] [CrossRef]
- Niess, J.H.; Brand, S.; Gu, X.; Landsman, L.; Jung, S.; McCormick, B.A.; Vyas, J.M.; Boes, M.; Ploegh, H.L.; Fox, J.G.; et al. CX3 CR1-Mediated Dendritic Cell Access to the Intestinal Lumen and Bacterial Clearance. Science 2005, 307, 254–258. [Google Scholar] [CrossRef]
- Zigmond, E.; Jung, S. Intestinal macrophages: Well educated exceptions from the rule. Trends Immunol. 2013, 34, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Bogunovic, M.; Ginhoux, F.; Helft, J.; Shang, L.; Hashimoto, D.; Greter, M.; Liu, K.; Jakubzick, C.; Ingersoll, M.A.; Leboeuf, M.; et al. Origin of the Lamina Propria Dendritic Cell Network. Immunity 2009, 31, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Tamoutounour, S.; Henri, S.; Lelouard, H.; de Bovis, B.; de Haar, C.; van der Woude, C.J.; Woltman, A.M.; Reyal, Y.; Bonnet, D.; Sichien, D.; et al. CD64 distinguishes macrophages from dendritic cells in the gut and reveals the T h1-inducing role of mesenteric lymph node macrophages during colitis. Eur. J. Immunol. 2012, 42, 3150–3166. [Google Scholar] [CrossRef] [PubMed]
- Zigmond, E.; Varol, C.; Farache, J.; Elmaliah, E.; Satpathy, A.T.; Friedlander, G.; Mack, M.; Shpigel, N.; Boneca, I.G.; Murphy, K.M.; et al. Ly6Chi Monocytes in the Inflamed Colon Give Rise to Proinflammatory Effector Cells and Migratory Antigen-Presenting Cells. Immunity 2012, 37, 1076–1090. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Kimura, S.; Hase, K. M cell-dependent antigen uptake on follicle-associated epithelium for mucosal immune surveillance. Inflamm. Regen. 2018, 38, 15. [Google Scholar] [CrossRef]
- Mabbott, N.A.; Donaldson, D.S.; Ohno, H.; Williams, I.R.; Mahajan, A. Microfold (M) cells: Important immunosurveillance posts in the intestinal epithelium. Mucosal Immunol. 2013, 6, 666–677. [Google Scholar] [CrossRef]
- Johansson-Lindbom, B.; Agace, W.W. Generation of gut-homing T cells and their localization to the small intestinal mucosa. Immunol. Rev. 2007, 215, 226–242. [Google Scholar] [CrossRef]
- Mowat, A.M. Anatomical basis of tolerance and immunity to intestinal antigens. Nat. Rev. Immunol. 2003, 3, 331–341. [Google Scholar] [CrossRef]
- Yamamoto, M.; Pascual, D.W.; Kiyono, H. M Cell-Targeted Mucosal Vaccine Strategies. In Mucosal Vaccines; Kozlowski, P.A., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 39–52. [Google Scholar]
- Nakamura, Y.; Mimuro, H.; Kunisawa, J.; Furusawa, Y.; Takahashi, D.; Fujimura, Y.; Kaisho, T.; Kiyono, H.; Hase, K. Microfold cell-dependent antigen transport alleviates infectious colitis by inducing antigen-specific cellular immunity. Mucosal Immunol. 2020, 13, 679–690. [Google Scholar] [CrossRef]
- Zhang, Z.; Lu, Y.; Qi, J.; Wu, W. An update on oral drug delivery via intestinal lymphatic transport. Acta Pharm. Sin. B 2021, 11, 2449–2468. [Google Scholar] [CrossRef] [PubMed]
- Coombes, J.L.; Siddiqui, K.R.; Arancibia-Cárcamo, C.V.; Hall, J.; Sun, C.-M.; Belkaid, Y.; Powrie, F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β– and retinoic acid–dependent mechanism. J. Exp. Med. 2007, 204, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-M.; Hall, J.A.; Blank, R.B.; Bouladoux, N.; Oukka, M.; Mora, J.R.; Belkaid, Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 2007, 204, 1775–1785. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, M.; Komatsu, N.; Kawamoto, S.; Suzuki, K.; Kanagawa, O.; Honjo, T.; Hori, S.; Fagarasan, S. Preferential Generation of Follicular B Helper T Cells from Foxp3+ T Cells in Gut Peyer’s Patches. Science 2009, 323, 1488–1492. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, A.; Zan, H.; Schaffer, A.; Bergsagel, L.; Harindranath, N.; Max, E.E.; Casali, P. CD40 ligand and appropriate cytokines induce switching to IgG, IgA, and IgE and coordinated germinal center and plasmacytoid phenotypic differentiation in a human monoclonal IgM+IgD+ B cell line. J. Immunol. 1998, 160, 2145–2157. [Google Scholar] [CrossRef]
- Tezuka, H.; Abe, Y.; Iwata, M.; Takeuchi, H.; Ishikawa, H.; Matsushita, M.; Shiohara, T.; Akira, S.; Ohteki, T. Regulation of IgA production by naturally occurring TNF/iNOS-producing dendritic cells. Nature 2007, 448, 929–933. [Google Scholar] [CrossRef]
- Suzuki, K.; Maruya, M.; Kawamoto, S.; Sitnik, K.; Kitamura, H.; Agace, W.W.; Fagarasan, S. The sensing of environmental stimuli by follicular dendritic cells promotes immunoglobulin A generation in the gut. Immunity 2010, 33, 71–83. [Google Scholar] [CrossRef]
- He, B.; Santamaria, R.; Xu, W.; Cols, M.; Chen, K.; Puga, I.; Shan, M.; Xiong, H.; Bussel, J.B.; Chiu, A.; et al. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat. Immunol. 2010, 11, 836–845. [Google Scholar] [CrossRef]
- Tsuji, M.; Suzuki, K.; Kitamura, H.; Maruya, M.; Kinoshita, K.; Ivanov, I.I.; Itoh, K.; Littman, D.R.; Fagarasan, S. Requirement for Lymphoid Tissue-Inducer Cells in Isolated Follicle Formation and T Cell-Independent Immunoglobulin A Generation in the Gut. Immunity 2008, 29, 261–271. [Google Scholar] [CrossRef]
- Uematsu, S.; Akira, S. Immune responses of TLR5+ lamina propria dendritic cells in enterobacterial infection. J. Gastroenterol. 2009, 44, 803–811. [Google Scholar] [CrossRef]
- Berkowska, M.A.; Driessen, G.J.A.; Bikos, V.; Grosserichter-Wagener, C.; Stamatopoulos, K.; Cerutti, A.; He, B.; Biermann, K.; Lange, J.F.; van der Burg, M.; et al. Human memory B cells originate from three distinct germinal center-dependent and -independent maturation pathways. Blood 2011, 118, 2150–2158. [Google Scholar] [CrossRef]
- Brandtzaeg, P. Secretory IgA: Designed for Anti-Microbial Defense. Front. Immunol. 2013, 4, 222. [Google Scholar] [CrossRef]
- Brandtzaeg, P. Induction of secretory immunity and memory at mucosal surfaces. Vaccine 2007, 25, 5467–5484. [Google Scholar] [CrossRef]
- Corthesy, B. Multi-Faceted Functions of Secretory IgA at Mucosal Surfaces. Front. Immunol. 2013, 4, 185. [Google Scholar] [CrossRef]
- Hutchings, A.B.; Helander, A.; Silvey, K.J.; Chandran, K.; Lucas, W.T.; Nibert, M.L.; Neutra, M.R. Secretory immunoglobulin A antibodies against the sigma1 outer capsid protein of reovirus type 1 Lang prevent infection of mouse Peyer’s patches. J. Virol. 2004, 78, 947–957. [Google Scholar] [CrossRef]
- Dickinson, B.L.; Badizadegan, K.; Wu, Z.; Ahouse, J.C.; Zhu, X.; Simister, N.E.; Blumberg, R.S.; Lencer, W.I. Bidirectional FcRn-dependent IgG transport in a polarized human intestinal epithelial cell line. J. Clin. Investig. 1999, 104, 903–911. [Google Scholar] [CrossRef]
- Yoshida, M.; Claypool, S.M.; Wagner, J.S.; Mizoguchi, E.; Mizoguchi, A.; Roopenian, D.C.; Lencer, W.I.; Blumberg, R.S. Human Neonatal Fc Receptor Mediates Transport of IgG into Luminal Secretions for Delivery of Antigens to Mucosal Dendritic Cells. Immunity 2004, 20, 769–783. [Google Scholar] [CrossRef]
- van Wijk, F.; Cheroutre, H. Mucosal T cells in gut homeostasis and inflammation. Expert Rev. Clin. Immunol. 2010, 6, 559–566. [Google Scholar] [CrossRef]
- Cheroutre, H.; Madakamutil, L. Acquired and natural memory T cells join forces at the mucosal front line. Nat. Rev. Immunol. 2004, 4, 290–300. [Google Scholar] [CrossRef]
- Müller, S.; Bühler-Jungo, M.; Mueller, C. Intestinal Intraepithelial Lymphocytes Exert Potent Protective Cytotoxic Activity During an Acute Virus Infection. J. Immunol. 2000, 164, 1986–1994. [Google Scholar] [CrossRef]
- Mora, J.R.; Iwata, M.; Eksteen, B.; Song, S.Y.; Junt, T.; Senman, B.; Otipoby, K.L.; Yokota, A.; Takeuchi, H.; Ricciardi-Castagnoli, P.; et al. Generation of Gut-Homing IgA-Secreting B Cells by Intestinal Dendritic Cells. Science 2006, 314, 1157–1160. [Google Scholar] [CrossRef]
- Campbell, D.J.; Butcher, E.C. Rapid Acquisition of Tissue-specific Homing Phenotypes by CD4+ T Cells Activated in Cutaneous or Mucosal Lymphoid Tissues. J. Exp. Med. 2002, 195, 135–141. [Google Scholar] [CrossRef]
- Kang, S.H.; Hong, S.J.; Lee, Y.-K.; Cho, S. Oral Vaccine Delivery for Intestinal Immunity—Biological Basis, Barriers, Delivery System, and M Cell Targeting. Polymers 2018, 10, 948. [Google Scholar] [CrossRef]
- Fox, B.E.; Vilander, A.C.; Gilfillan, D.; Dean, G.A.; Abdo, Z. Oral Vaccination Using a Probiotic Vaccine Platform Combined with Prebiotics Impacts Immune Response and the Microbiome. Vaccines 2022, 10, 1465. [Google Scholar] [CrossRef]
- Buonaguro, L.; Tagliamonte, M. Peptide-based vaccine for cancer therapies. Front. Immunol. 2023, 14, 1210044. [Google Scholar] [CrossRef]
- Hu, Q.; Wu, M.; Fang, C.; Cheng, C.; Zhao, M.; Fang, W.; Chu, P.K.; Ping, Y.; Tang, G. Engineering Nanoparticle-Coated Bacteria as Oral DNA Vaccines for Cancer Immunotherapy. Nano Lett. 2015, 15, 2732–2739. [Google Scholar] [CrossRef]
- Pietrzak, B.; Tomela, K.; Olejnik-Schmidt, A.; Mackiewicz, A.; Schmidt, M. Secretory IgA in Intestinal Mucosal Secretions as an Adaptive Barrier against Microbial Cells. Int. J. Mol. Sci. 2020, 21, 9254. [Google Scholar] [CrossRef]
- Stolfi, C.; Maresca, C.; Monteleone, G.; Laudisi, F. Implication of Intestinal Barrier Dysfunction in Gut Dysbiosis and Diseases. Biomedicines 2022, 10, 289. [Google Scholar] [CrossRef]
- Friedmann, K.S.; Kaschek, L.; Knörck, A.; Cappello, S.; Lünsmann, N.; Küchler, N.; Hoxha, C.; Schäfer, G.; Iden, S.; Bogeski, I.; et al. Interdependence of sequential cytotoxic T lymphocyte and natural killer cell cytotoxicity against melanoma cells. J. Physiol. 2022, 600, 5027–5054. [Google Scholar] [CrossRef]
- Schroeder, H.W.; Cavacini, L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52. [Google Scholar] [CrossRef]
- Cao, Y.; Rewatkar, P.; Wang, R.; Hasnain, S.Z.; Popat, A.; Kumeria, T. Nanocarriers for oral delivery of biologics: Small carriers for big payloads. Trends Pharmacol. Sci. 2021, 42, 957–972. [Google Scholar] [CrossRef]
- Mishra, N.; Tiwari, S.; Vaidya, B.; Agrawal, G.P.; Vyas, S.P. Lectin anchored PLGA nanoparticles for oral mucosal immunization against hepatitis B. J. Drug Target. 2011, 19, 67–78. [Google Scholar] [CrossRef]
- Sun, W.; Shi, J.; Wu, J.; Zhang, J.; Chen, H.; Li, Y.; Liu, S.; Wu, Y.; Tian, Z.; Cao, X.; et al. A modified HLA-A*0201-restricted CTL epitope from human oncoprotein (hPEBP4) induces more efficient antitumor responses. Cell. Mol. Immunol. 2018, 15, 768–781. [Google Scholar] [CrossRef]
- Fan, Y.; Bai, T.; Tian, Y.; Zhou, B.; Wang, Y.; Yang, L. H2O2-Inactivated Salmonella typhimurium RE88 Strain as a New Cancer Vaccine Carrier: Evaluation in a Mouse Model of Cancer. Drug Des. Dev. Ther. 2021, 15, 209–222. [Google Scholar] [CrossRef]
- Aldossary, A.M.; Ekweremadu, C.S.; Offe, I.M.; Alfassam, H.A.; Han, S.; Onyali, V.C.; Ozoude, C.H.; Ayeni, E.A.; Nwagwu, C.S.; Halwani, A.A.; et al. A guide to oral vaccination: Highlighting electrospraying as a promising manufacturing technique toward a successful oral vaccine development. Saudi Pharm. J. 2022, 30, 655–668. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.S.; Garon, J.; Seib, K.; Orenstein, W.A. Polio vaccination: Past, present and future. Future Microbiol. 2015, 10, 791–808. [Google Scholar] [CrossRef]
- New, R.R.C. Formulation technologies for oral vaccines. Clin. Exp. Immunol. 2019, 198, 153–169. [Google Scholar] [CrossRef]
- Freedman, D.O. Re-born in the USA: Another cholera vaccine for travellers. Travel Med. Infect. Dis. 2016, 14, 295–296. [Google Scholar] [CrossRef]
- Ward, R.L.; David, I. Bernstein, Rotarix: A Rotavirus Vaccine for the World. Clin. Infect. Dis. 2009, 48, 222–228. [Google Scholar] [CrossRef]
- Sadiq, A.; Bostan, N.; Khan, J.; Aziz, A. Effect of rotavirus genetic diversity on vaccine impact. Rev. Med. Virol. 2022, 32, e2259. [Google Scholar] [CrossRef]
- Shaikh, H.; Lynch, J.; Kim, J.; Excler, J.-L. Current and future cholera vaccines. Vaccine 2020, 38, A118–A126. [Google Scholar] [CrossRef] [PubMed]
- Naciute, M.; Niemi, V.; Kemp, R.A.; Hook, S. Lipid-encapsulated oral therapeutic peptide vaccines reduce tumour growth in an orthotopic mouse model of colorectal cancer. Eur. J. Pharm. Biopharm. 2020, 152, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Ueki, H.; Kitagawa, K.; Kato, M.; Yanase, S.; Okamura, Y.; Bando, Y.; Hara, T.; Terakawa, T.; Furukawa, J.; Nakano, Y.; et al. An oral cancer vaccine using Bifidobacterium vector augments combination of anti-PD-1 and anti-CTLA-4 antibodies in mouse renal cell carcinoma model. Sci. Rep. 2023, 13, 9994. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, M.; Nishimura, Y. The present status and future prospects of peptide-based cancer vaccines. Int. Immunol. 2016, 28, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Harao, M.; Mittendorf, E.A.; Radvanyi, L.G. Peptide-Based Vaccination and Induction of CD8+ T-Cell Responses Against Tumor Antigens in Breast Cancer. BioDrugs 2015, 29, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Rosalia, R.A.; Quakkelaar, E.D.; Redeker, A.; Khan, S.; Camps, M.; Drijfhout, J.W.; Silva, A.L.; Jiskoot, W.; van Hall, T.; van Veelen, P.A.; et al. Dendritic cells process synthetic long peptides better than whole protein, improving antigen presentation and T-cell activation. Eur. J. Immunol. 2013, 43, 2554–2565. [Google Scholar] [CrossRef]
- Nelde, A.; Rammensee, H.-G.; Walz, J.S. The Peptide Vaccine of the Future. Mol. Cell. Proteom. 2021, 20, 100022. [Google Scholar] [CrossRef]
- Shahabi, V.; Maciag, P.C.; Rivera, S.; Wallecha, A. Live, attenuated strains of Listeria and Salmonella as vaccine vectors in cancer treatment. Bioeng. Bugs 2010, 1, 237–245. [Google Scholar] [CrossRef]
- Lin, C.W.; Lee, J.Y.; Tsao, Y.P.; Shen, C.P.; Lai, H.C.; Chen, S.L. Oral vaccination with recombinant Listeria monocytogenes expressing human papillomavirus type 16 E7 can cause tumor growth in mice to regress. Int. J. Cancer 2002, 102, 629–637. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, S.; Du, X.; Li, T.; Han, L.; Kong, J. Heterologous expression of carcinoembryonic antigen in Lactococcus lactis via LcsB-mediated surface displaying system for oral vaccine development. J. Microbiol. Immunol. Infect. 2016, 49, 851–858. [Google Scholar] [CrossRef]
- Naciute, M.; Kiwitt, T.; Kemp, R.A.; Hook, S. Bacteria biohybrid oral vaccines for colorectal cancer treatment reduce tumor growth and increase immune infiltration. Vaccine 2021, 39, 5589–5599. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H. In vivo antigen delivery by aSalmonella typhimurium type III secretion system for therapeutic cancer vaccines. J. Clin. Investig. 2006, 116, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, X.; Jin, C.G.; Zhou, Y.C.; Navab, R.; Jakobsen, K.R.; Chen, X.Q.; Li, J.; Li, T.T.; Luo, L.; et al. An orally administered DNA vaccine targeting vascular endothelial growth factor receptor-3 inhibits lung carcinoma growth. Tumor Biol. 2016, 37, 2395–2404. [Google Scholar] [CrossRef] [PubMed]
- Steel, J.C.; Di Pasquale, G.; Ramlogan, C.A.; Patel, V.; Chiorini, J.A.; Morris, J.C. Oral Vaccination with Adeno-associated Virus Vectors Expressing the Neu Oncogene Inhibits the Growth of Murine Breast Cancer. Mol. Ther. 2013, 21, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Tawde, S.A.; Chablani, L.; Akalkotkar, A.; D’Souza, C.; Chiriva-Internati, M.; Selvaraj, P.; D’Souza, M.J. Formulation and evaluation of oral microparticulate ovarian cancer vaccines. Vaccine 2012, 30, 5675–5681. [Google Scholar] [CrossRef] [PubMed]
- Parenky, A.C.; Akalkotkar, A.; Mulla, N.S.; D’Souza, M.J. Harnessing T-cell activity against prostate cancer: A therapeutic microparticulate oral cancer vaccine. Vaccine 2019, 37, 6085–6092. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, C.M.; Bernkop-Schnürch, A.; Friedl, J.D.; Préat, V.; Jannin, V. Oral delivery of non-viral nucleic acid-based therapeutics—Do we have the guts for this? Eur. J. Pharm. Sci. 2019, 133, 190–204. [Google Scholar] [CrossRef]
- Schwendener, R.A. Liposomes as vaccine delivery systems: A review of the recent advances. Ther. Adv. Vaccines 2014, 2, 159–182. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, N.; Liu, C.; Zhuo, Y.; Liang, L.; Gan, Y.; Yu, M. Oral delivery of nucleic acid therapeutics: Challenges, strategies, and opportunities. Drug Discov. Today 2023, 28, 103507. [Google Scholar] [CrossRef]
- He, L.; Bai, Y.; Xia, L.; Pan, J.; Sun, X.; Zhu, Z.; Ding, J.; Qi, C.; Tang, C. Oral administration of a whole glucan particle (WGP)-based therapeutic cancer vaccine targeting macrophages inhibits tumor growth. Cancer Immunol. Immunother. 2022, 71, 2007–2028. [Google Scholar] [CrossRef]
- Harnack, U.; Eckert, K.; Fichtner, I.; Pecher, G. Oral administration of a soluble 1–3, 1–6 β-glucan during prophylactic survivin peptide vaccination diminishes growth of a B cell lymphoma in mice. Int. Immunopharmacol. 2009, 9, 1298–1303. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, K.; Tatsumi, M.; Kato, M.; Komai, S.; Doi, H.; Hashii, Y.; Katayama, T.; Fujisawa, M.; Shirakawa, T. An oral cancer vaccine using a Bifidobacterium vector suppresses tumor growth in a syngeneic mouse bladder cancer model. Mol. Ther.-Oncolytics 2021, 22, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, H.; Hashii, Y.; Nakagawa, N.; Nakajima, H.; Oka, Y.; Katayama, T.; Shirakawa, T.; Ozono, K. The anti-tumor effect of oral cancer vaccine using Bifidobacterium as a platform for displaying Wilms’ tumor 1 protein. J. Clin. Oncol. 2019, 37 (Suppl. S8), 72. [Google Scholar] [CrossRef]
- Kitagawa, K.; Gonoi, R.; Tatsumi, M.; Kadowaki, M.; Katayama, T.; Hashii, Y.; Fujisawa, M.; Shirakawa, T. Preclinical Development of a WT1 Oral Cancer Vaccine Using a Bacterial Vector to Treat Castration-Resistant Prostate Cancer. Mol. Cancer Ther. 2019, 18, 980–990. [Google Scholar] [CrossRef]
- Shirakawa, T.; Kitagawa, K. Antitumor effect of oral cancer vaccine with Bifidobacterium delivering WT1 protein to gut immune system is superior to WT1 peptide vaccine. Hum. Vaccines Immunother. 2018, 14, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, N.; Hashii, Y.; Kayama, H.; Okumura, R.; Nakajima, H.; Minagawa, H.; Morimoto, S.; Fujiki, F.; Nakata, J.; Shirakawa, T.; et al. An oral WT1 protein vaccine composed of WT1-anchored, genetically engineered Bifidobacterium longum allows for intestinal immunity in mice with acute myeloid leukemia. Cancer Immunol. Immunother. 2023, 72, 39–53. [Google Scholar] [CrossRef]
- Schmitz-Winnenthal, F.H.; Hohmann, N.; Schmidt, T.; Podola, L.; Friedrich, T.; Lubenau, H.; Springer, M.; Wieckowski, S.; Breiner, K.M.; Mikus, G.; et al. A phase 1 trial extension to assess immunologic efficacy and safety of prime-boost vaccination with VXM01, an oral T cell vaccine against VEGFR2, in patients with advanced pancreatic cancer. OncoImmunology 2018, 7, e1303584. [Google Scholar] [CrossRef]
- Chou, C.-K.; Hung, J.-Y.; Liu, J.-C.; Chen, C.-T.; Hung, M.-C. An attenuated Salmonella oral DNA vaccine prevents the growth of hepatocellular carcinoma and colon cancer that express α-fetoprotein. Cancer Gene Ther. 2006, 13, 746–752. [Google Scholar] [CrossRef]
- Zhu, X.; Cai, J.; Huang, J.; Jiang, X.; Ren, D. The Treatment and Prevention of Mouse Melanoma with an Oral DNA Vaccine Carried by Attenuated Salmonella Typhimurium. J. Immunother. 2010, 33, 453–460. [Google Scholar] [CrossRef]
- Lee, S.-H.; Mizutani, N.; Mizutani, M.; Luo, Y.; Zhou, H.; Kaplan, C.; Kim, S.-W.; Xiang, R.; Reisfeld, R.A. Endoglin (CD105) is a target for an oral DNA vaccine against breast cancer. Cancer Immunol. Immunother. 2006, 55, 1565–1574. [Google Scholar] [CrossRef]
- Shen, Y.; Qiu, L. Effective oral delivery of gp100 plasmid vaccine against metastatic melanoma through multi-faceted blending-by-blending nanogels. Nanomed. Nanotechnol. Biol. Med. 2019, 22, 102114. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Zhang, H.; Yin, X.; Liu, Y.; Chen, D.; Liang, X.; Jin, X.; Lv, J.; Ma, J.; Tang, K.; et al. Oral delivery of tumor microparticle vaccines activates NOD2 signaling pathway in ileac epithelium rendering potent antitumor T cell immunity. OncoImmunology 2017, 6, e1282589. [Google Scholar] [CrossRef] [PubMed]
- Akalkotkar, A.; Tawde, S.A.; Chablani, L.; D’Souza, M.J. Oral delivery of particulate prostate cancer vaccine: In vitro and in vivo evaluation. J. Drug Target. 2012, 20, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Mulla, N.; Chablani, L.; Parenky, A.C.; D’souza, M.J. Boosting In-Vivo Anti-Tumor Immunity with an Oral Microparticulate Breast Cancer Vaccine and Low-Dose Cyclophosphamide. Vaccines 2023, 11, 543. [Google Scholar] [CrossRef] [PubMed]
- Jou, J.; Harrington, K.J.; Zocca, M.-B.; Ehrnrooth, E.; Cohen, E.E. The Changing Landscape of Therapeutic Cancer Vaccines—Novel Platforms and Neoantigen Identification. Clin. Cancer Res. 2021, 27, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Maurer, J.M.; Schellekens, R.C.A.; van Rieke, H.M.; Wanke, C.; Iordanov, V.; Stellaard, F.; Wutzke, K.D.; Dijkstra, G.; van der Zee, M.; Woerdenbag, H.J.; et al. Gastrointestinal pH and Transit Time Profiling in Healthy Volunteers Using the IntelliCap System Confirms Ileo-Colonic Release of ColoPulse Tablets. PLoS ONE 2015, 10, e0129076. [Google Scholar] [CrossRef] [PubMed]
- Homayun, B.; Lin, X.; Choi, H.-J. Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals. Pharmaceutics 2019, 11, 129. [Google Scholar] [CrossRef]
- Mooij, M.G.; de Koning, B.A.; Huijsman, M.L.; de Wildt, S.N. Ontogeny of oral drug absorption processes in children. Expert Opin. Drug Metab. Toxicol. 2012, 8, 1293–1303. [Google Scholar] [CrossRef]
- Zhu, Q.; Chen, Z.; Paul, P.K.; Lu, Y.; Wu, W.; Qi, J. Oral delivery of proteins and peptides: Challenges, status quo and future perspectives. Acta Pharm. Sin. B 2021, 11, 2416–2448. [Google Scholar] [CrossRef]
- Fallingborg, J.; Christensen, L.A.; Jacobsen, B.A.; Rasmussen, S.N. Very low intraluminal colonic pH in patients with active ulcerative colitis. Dig. Dis. Sci. 1993, 38, 1989–1993. [Google Scholar] [CrossRef]
- Sasaki, Y.; Hada, R.; Nakajima, H.; Fukuda, S.; Munakata, A. Improved localizing method of radiopill in measurement of entire gastrointestinal pH profiles: Colonic luminal pH in normal subjects and patients with Crohn’s disease. Am. J. Gastroenterol. 1997, 92, 114–118. [Google Scholar] [PubMed]
- Pridgen, E.M.; Alexis, F.; Farokhzad, O.C. Polymeric Nanoparticle Technologies for Oral Drug Delivery. Clin. Gastroenterol. Hepatol. 2014, 12, 1605–1610. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, I.; Ruiz, L.; Gueimonde, M.; Margolles, A.; Sánchez, B. Factors involved in the colonization and survival of bifidobacteria in the gastrointestinal tract. FEMS Microbiol. Lett. 2013, 340, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Janiak, M.C. Digestive enzymes of human and nonhuman primates: Digestive enzymes of human and nonhuman primates. Evol. Anthropol. Issues News Rev. 2016, 25, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Haddadzadegan, S.; Dorkoosh, F.; Bernkop-Schnürch, A. Oral delivery of therapeutic peptides and proteins: Technology landscape of lipid-based nanocarriers. Adv. Drug Deliv. Rev. 2022, 182, 114097. [Google Scholar] [CrossRef] [PubMed]
- Miner-Williams, W.M.; Stevens, B.R.; Moughan, P.J. Are intact peptides absorbed from the healthy gut in the adult human? Nutr. Res. Rev. 2014, 27, 308–329. [Google Scholar] [CrossRef]
- Paone, P.; Cani, P.D. Mucus barrier, mucins and gut microbiota: The expected slimy partners? Gut 2020, 69, 2232–2243. [Google Scholar] [CrossRef]
- Javitt, G.; Khmelnitsky, L.; Albert, L.; Bigman, L.S.; Elad, N.; Morgenstern, D.; Ilani, T.; Levy, Y.; Diskin, R.; Fass, D. Assembly Mechanism of Mucin and von Willebrand Factor Polymers. Cell 2020, 183, 717–729.e16. [Google Scholar] [CrossRef]
- Parrish, A.; Boudaud, M.; Kuehn, A.; Ollert, M.; Desai, M.S. Intestinal mucus barrier: A missing piece of the puzzle in food allergy. Trends Mol. Med. 2022, 28, 36–50. [Google Scholar] [CrossRef]
- Johansson, M.E.V.; Hansson, G.C. Immunological aspects of intestinal mucus and mucins. Nat. Rev. Immunol. 2016, 16, 639–649. [Google Scholar] [CrossRef]
- Birchenough, G.M.H.; Johansson, M.E.; Gustafsson, J.K.; Bergström, J.H.; Hansson, G.C. New developments in goblet cell mucus secretion and function. Mucosal Immunol. 2015, 8, 712–719. [Google Scholar] [CrossRef] [PubMed]
- McShane, A.; Bath, J.; Jaramillo, A.M.; Ridley, C.; Walsh, A.A.; Evans, C.M.; Thornton, D.J.; Ribbeck, K. Mucus. Curr. Biol. 2021, 31, R938–R945. [Google Scholar] [CrossRef] [PubMed]
- Shan, M.; Gentile, M.; Yeiser, J.R.; Walland, A.C.; Bornstein, V.U.; Chen, K.; He, B.; Cassis, L.; Bigas, A.; Cols, M.; et al. Mucus Enhances Gut Homeostasis and Oral Tolerance by Delivering Immunoregulatory Signals. Science 2013, 342, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, J.; Bohr, S.-R.; Harloff-Helleberg, S.; Hatzakis, N.; Saaby, L.; Nielsen, H. Physical and barrier changes in gastrointestinal mucus induced by the permeation enhancer sodium 8- [(2-hydroxybenzoyl)amino]octanoate (SNAC). J. Control. Release 2022, 352, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Sogias, I.A.; Williams, A.C.; Khutoryanskiy, V.V. Why is Chitosan Mucoadhesive? Biomacromolecules 2008, 9, 1837–1842. [Google Scholar] [CrossRef] [PubMed]
- Chelakkot, C.; Ghim, J.; Ryu, S.H. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T. Regulation of the intestinal barrier by nutrients: The role of tight junctions. Anim. Sci. J. 2020, 91, e13357. [Google Scholar] [CrossRef]
- Dhurjad, P.; Dhavaliker, C.; Gupta, K.; Sonti, R. Exploring Drug Metabolism by the Gut Microbiota: Modes of Metabolism and Experimental Approaches. Drug Metab. Dispos. 2022, 50, 224–234. [Google Scholar] [CrossRef]
- Li, H.; He, J.; Jia, W. The influence of gut microbiota on drug metabolism and toxicity. Expert Opin. Drug Metab. Toxicol. 2016, 12, 31–40. [Google Scholar] [CrossRef]
- Westman, E.L.; Canova, M.J.; Radhi, I.J.; Koteva, K.; Kireeva, I.; Waglechner, N.; Wright, G.D. Bacterial Inactivation of the Anticancer Drug Doxorubicin. Chem. Biol. 2012, 19, 1255–1264. [Google Scholar] [CrossRef]
- Van der Weken, H.; Cox, E.; Devriendt, B. Advances in Oral Subunit Vaccine Design. Vaccines 2020, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Trovato, M. Novel antigen delivery systems. World J. Virol. 2015, 4, 156. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Xu, R. Trends in orally viral vector gene delivery and therapy. In Nanostructures for Oral Medicine; Elsevier: Amsterdam, The Netherlands, 2017; pp. 123–146. [Google Scholar]
- Mohsen, M.O.; Bachmann, M.F. Virus-like particle vaccinology, from bench to bedside. Cell. Mol. Immunol. 2022, 19, 993–1011. [Google Scholar] [CrossRef] [PubMed]
- Serradell, M.C.; Rupil, L.L.; Martino, R.A.; Prucca, C.G.; Carranza, P.G.; Saura, A.; Fernández, E.A.; Gargantini, P.R.; Tenaglia, A.H.; Petiti, J.P.; et al. Efficient oral vaccination by bioengineering virus-like particles with protozoan surface proteins. Nat. Commun. 2019, 10, 361. [Google Scholar] [CrossRef] [PubMed]
- Choukaife, H.; Seyam, S.; Alallam, B.; Doolaanea, A.A.; Alfatama, M. Current Advances in Chitosan Nanoparticles Based Oral Drug Delivery for Colorectal Cancer Treatment. Int. J. Nanomed. 2022, 17, 3933–3966. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; McDowell, A.; Ishii, Y.; Hook, S. Liposomal α-galactosylceramide is taken up by gut-associated lymphoid tissue and stimulates local and systemic immune responses. J. Pharm. Pharmacol. 2017, 69, 1724–1735. [Google Scholar] [CrossRef] [PubMed]
- Crennell, S.; Garman, E.; Laver, G.; Vimr, E.; Taylor, G. Crystal structure of Vibrio cholerae neuraminidase reveals dual lectin-like domains in addition to the catalytic domain. Structure 1994, 2, 535–544. [Google Scholar] [CrossRef]
- Roth-Walter, F.; Schöll, I.; Untersmayr, E.; Fuchs, R.; Boltz-Nitulescu, G.; Weissenböck, A.; Scheiner, O.; Gabor, F.; Jensen-Jarolim, E. M cell targeting with Aleuria aurantia lectin as a novel approach for oral allergen immunotherapy. J. Allergy Clin. Immunol. 2004, 114, 1362–1368. [Google Scholar] [CrossRef]
- Marasini, N.; Ghaffar, K.A.; Skwarczynski, M.; Toth, I. Liposomes as a Vaccine Delivery System. In Micro and Nanotechnology in Vaccine Development; Elsevier: Amsterdam, The Netherlands, 2017; pp. 221–239. [Google Scholar]
- Mallakpour, S.; Azadi, E.; Hussain, C.M. Chitosan, alginate, hyaluronic acid, gums, and β-glucan as potent adjuvants and vaccine delivery systems for viral threats including SARS-CoV-2: A review. Int. J. Biol. Macromol. 2021, 182, 1931–1940. [Google Scholar] [CrossRef]
- Vetvicka, V.; Vannucci, L.; Sima, P. β-glucan as a new tool in vaccine development. Scand. J. Immunol. 2020, 91, e12833. [Google Scholar] [CrossRef]
- Wang, M.; Gao, Z.; Zhang, Z.; Pan, L.; Zhang, Y. Roles of M cells in infection and mucosal vaccines. Hum. Vaccines Immunother. 2014, 10, 3544–3551. [Google Scholar] [CrossRef] [PubMed]
- Ohno, H.; Hase, K. Glycoprotein 2 (GP2): Grabbing the FimH+ bacteria into M cells for mucosal immunity. Gut Microbes 2010, 1, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Giannasca, P.J.; Giannasca, K.T.; Leichtner, A.M.; Neutra, M.R. Human Intestinal M Cells Display the Sialyl Lewis A Antigen. Infect. Immun. 1999, 67, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jung, D.; Yang, I.; Kim, J.; Lee, K.; Nochi, T.; Kiyono, H.; Jang, Y. M cells expressing the complement C5a receptor are efficient targets for mucosal vaccine delivery. Eur. J. Immunol. 2011, 41, 3219–3229. [Google Scholar] [CrossRef] [PubMed]
- Pridgen, E.M.; Alexis, F.; Kuo, T.T.; Levy-Nissenbaum, E.; Karnik, R.; Blumberg, R.S.; Langer, R.; Farokhzad, O.C. Transepithelial Transport of Fc-Targeted Nanoparticles by the Neonatal Fc Receptor for Oral Delivery. Sci. Transl. Med. 2013, 5, 213ra167. [Google Scholar] [CrossRef] [PubMed]
- Gou, S.; Wang, S.; Liu, W.; Chen, G.; Zhang, D.; Du, J.; Yan, Z.; Wang, H.; Zhai, W.; Sui, X.; et al. Adjuvant-free peptide vaccine targeting Clec9a on dendritic cells can induce robust antitumor immune response through Syk/IL-21 axis. Theranostics 2021, 11, 7308–7321. [Google Scholar] [CrossRef]
- Silva, M.O.; Almeida, B.S.; Sales, N.S.; Diniz, M.O.; Aps, L.R.; Rodrigues, K.B.; Silva, J.R.; Moreno, A.C.R.; Porchia, B.F.; Sulczewski, F.B.; et al. Antigen Delivery to DEC205(+) Dendritic Cells Induces Immunological Memory and Protective Therapeutic Effects against HPV-Associated Tumors at Different Anatomical Sites. Int. J. Biol. Sci. 2021, 17, 2944–2956. [Google Scholar] [CrossRef]
- Lahoud, M.H.; Ahmet, F.; Zhang, J.-G.; Meuter, S.; Policheni, A.N.; Kitsoulis, S.; Lee, C.-N.; O’keeffe, M.; Sullivan, L.C.; Brooks, A.G.; et al. DEC-205 is a cell surface receptor for CpG oligonucleotides. Proc. Natl. Acad. Sci. USA 2012, 109, 16270–16275. [Google Scholar] [CrossRef]
- Cohn, L.; Chatterjee, B.; Esselborn, F.; Smed-Sörensen, A.; Nakamura, N.; Chalouni, C.; Lee, B.-C.; Vandlen, R.; Keler, T.; Lauer, P.; et al. Antigen delivery to early endosomes eliminates the superiority of human blood BDCA3+ dendritic cells at cross presentation. J. Exp. Med. 2013, 210, 1049–1063. [Google Scholar] [CrossRef]
- Jiang, W.; Swiggard, W.J.; Heufler, C.; Peng, M.; Mirza, A.; Steinman, R.M.; Nussenzweig, M.C. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature 1995, 375, 151–155. [Google Scholar] [CrossRef]
- Mahnke, K.; Guo, M.; Lee, S.; Sepulveda, H.; Swain, S.L.; Nussenzweig, M.; Steinman, R.M. The dendritic cell receptor for endocytosis, DEC-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J. Cell Biol. 2000, 151, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Birkholz, K.; Schwenkert, M.; Kellner, C.; Gross, S.; Fey, G.; Schuler-Thurner, B.; Schuler, G.; Schaft, N.; Dörrie, J. Targeting of DEC-205 on human dendritic cells results in efficient MHC class II-restricted antigen presentation. Blood 2010, 116, 2277–2285. [Google Scholar] [CrossRef] [PubMed]
- Bonifaz, L.C.; Bonnyay, D.P.; Charalambous, A.; Darguste, D.I.; Fujii, S.-I.; Soares, H.; Brimnes, M.K.; Moltedo, B.; Moran, T.M.; Steinman, R.M. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J. Exp. Med. 2004, 199, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Lahoud, M.H.; Ahmet, F.; Kitsoulis, S.; Wan, S.S.; Vremec, D.; Lee, C.-N.; Phipson, B.; Shi, W.; Smyth, G.K.; Lew, A.M.; et al. Targeting antigen to mouse dendritic cells via Clec9A induces potent CD4 T cell responses biased toward a follicular helper phenotype. J. Immunol. 2011, 187, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, K.; Qian, Y.; Fondel, S.; Brueck, J.; Becker, C.; Enk, A.H. Targeting of antigens to activated dendritic cells in vivo cures metastatic melanoma in mice. Cancer Res. 2005, 65, 7007–7012. [Google Scholar] [CrossRef]
- Johnson, T.S.; Mahnke, K.; Storn, V.; Schönfeld, K.; Ring, S.; Nettelbeck, D.M.; Haisma, H.J.; Le Gall, F.; Kontermann, R.E.; Enk, A.H. Inhibition of melanoma growth by targeting of antigen to dendritic cells via an anti-DEC-205 single-chain fragment variable molecule. Clin. Cancer Res. 2008, 14, 8169–8177. [Google Scholar] [CrossRef]
- Caminschi, I.; Proietto, A.I.; Ahmet, F.; Kitsoulis, S.; Shin Teh, J.; Lo, J.C.Y.; Rizzitelli, A.; Wu, L.; Vremec, D.; Van Dommelen, S.L.H.; et al. The dendritic cell subtype-restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood 2008, 112, 3264–3273. [Google Scholar] [CrossRef]
- Sancho, D.; Mourão-Sá, D.; Joffre, O.P.; Schulz, O.; Rogers, N.C.; Pennington, D.J.; Carlyle, J.R.; Reis E Sousa, C. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J. Clin. Investig. 2008, 118, 2098–2110. [Google Scholar] [CrossRef]
- Huysamen, C.; Willment, J.A.; Dennehy, K.M.; Brown, G.D. CLEC9A is a novel activation C-type lectin-like receptor expressed on BDCA3+ dendritic cells and a subset of monocytes. J. Biol. Chem. 2008, 283, 16693–16701. [Google Scholar] [CrossRef]
- Zelenay, S.; Keller, A.M.; Whitney, P.G.; Schraml, B.U.; Deddouche, S.; Rogers, N.C.; Schulz, O.; Sancho, D.; e Sousa, C.R. The dendritic cell receptor DNGR-1 controls endocytic handling of necrotic cell antigens to favor cross-priming of CTLs in virus-infected mice. J. Clin. Investig. 2012, 122, 1615–1627. [Google Scholar] [CrossRef]
- Sancho, D.; Joffre, O.P.; Keller, A.M.; Rogers, N.C.; Martínez, D.; Hernanz-Falcón, P.; Rosewell, I.; Reis E Sousa, C. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 2009, 458, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, S.; Zelenay, S.; Sancho, D.; Hanč, P.; Kjær, S.; Feest, C.; Fletcher, G.; Durkin, C.; Postigo, A.; Skehel, M.; et al. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 2012, 36, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Picco, G.; Beatson, R.; Taylor-Papadimitriou, J.; Burchell, J.M. Targeting DNGR-1 (CLEC9A) with antibody/MUC1 peptide conjugates as a vaccine for carcinomas. Eur. J. Immunol. 2014, 44, 1947–1955. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.P.; Sancho, D.; Zelenay, S.; Keller, A.M.; Reis e Sousa, C. Efficient and versatile manipulation of the peripheral CD4+ T-cell compartment by antigen targeting to DNGR-1/CLEC9A. Eur. J. Immunol. 2010, 40, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen Recognition by the Innate Immune System. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gusti, V.; Saraswati, A.; Lo, D.D. Convergent and Divergent Development among M Cell Lineages in Mouse Mucosal Epithelium. J. Immunol. 2011, 187, 5277–5285. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Staudt, L.M. Toll-Like Receptor Signaling. Cold Spring Harb. Perspect. Biol. 2013, 5, a011247. [Google Scholar] [CrossRef]
- Snäkä, T.; Fasel, N. Behind the Scenes: Nod-Like Receptor X1 Controls Inflammation and Metabolism. Front. Cell. Infect. Microbiol. 2020, 10, 609812. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Brown, G.D.; Willment, J.A.; Whitehead, L. C-type lectins in immunity and homeostasis. Nat. Rev. Immunol. 2018, 18, 374–389. [Google Scholar] [CrossRef]
- Basset, C.; Thiam, F.; Di Martino, C.; Holton, J.; Clements, J.D.; Kohli, E. Cholera-Like Enterotoxins and Regulatory T cells. Toxins 2010, 2, 1774–1795. [Google Scholar] [CrossRef] [PubMed]
- Tregoning, J.S.; Russell, R.F.; Kinnear, E. Adjuvanted influenza vaccines. Hum. Vaccines Immunother. 2018, 14, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Luchner, M.; Reinke, S.; Milicic, A. TLR Agonists as Vaccine Adjuvants Targeting Cancer and Infectious Diseases. Pharmaceutics 2021, 13, 142. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, C.; Bertholet, S.; Philpott, D.J.; De Gregorio, E. Unleashing the potential of NOD- and Toll-like agonists as vaccine adjuvants. Proc. Natl. Acad. Sci. USA 2014, 111, 12294–12299. [Google Scholar] [CrossRef] [PubMed]
- Månsson Kvarnhammar, A.; Tengroth, L.; Adner, M.; Cardell, L.O. Innate Immune Receptors in Human Airway Smooth Muscle Cells: Activation by TLR1/2, TLR3, TLR4, TLR7 and NOD1 Agonists. PLoS ONE 2013, 8, e68701. [Google Scholar] [CrossRef] [PubMed]
- Johannssen, T.; Lepenies, B. Identification and Characterization of Carbohydrate-Based Adjuvants. In Carbohydrate-Based Vaccines; Lepenies, B., Ed.; Springer: New York, NY, USA, 2015; pp. 173–187. [Google Scholar]
- Kim, S.-H.; Seo, K.-W.; Kim, J.; Lee, K.-Y.; Jang, Y.-S. The M Cell-Targeting Ligand Promotes Antigen Delivery and Induces Antigen-Specific Immune Responses in Mucosal Vaccination. J. Immunol. 2010, 185, 5787–5795. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.N.; Wang, L.; Zheng, D.Z.; Chen, S.; Shi, W.; Qiao, X.Y.; Jiang, Y.P.; Tang, L.J.; Xu, Y.G.; Li, Y.J. Oral immunization with a Lactobacillus casei-Based anti-porcine epidemic diarrhoea virus (PEDV) vaccine expressing microfold cell-targeting peptide Co1 fused with the COE antigen of PEDV. J. Appl. Microbiol. 2018, 124, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.-H.; Kim, S.-H.; Jeon, J.-H.; Kim, E.B.; Lee, N.-K.; Beck, S.; Choi, Y.-J.; Kang, S.-K. Cytoplasmic expression of a model antigen with M Cell-Targeting moiety in lactic acid bacteria and implication of the mechanism as a mucosal vaccine via oral route. Vaccine 2021, 39, 4072–4081. [Google Scholar] [CrossRef]
- Potocki, W.; Negri, A.; Peszyńska-Sularz, G.; Hinc, K.; Obuchowski, M.; Iwanicki, A. IL-1 Fragment Modulates Immune Response Elicited by Recombinant Bacillus subtilis Spores Presenting an Antigen/Adjuvant Chimeric Protein. Mol. Biotechnol. 2018, 60, 810–819. [Google Scholar] [CrossRef]
- Deng, Z.; Geng, Y.; Wang, K.; Yu, Z.; Yang, P.O.; Yang, Z.; He, C.; Huang, C.; Yin, L.; He, M.; et al. Adjuvant effects of interleukin-2 co-expression with VP60 in an oral vaccine delivered by attenuated Salmonella typhimurium against rabbit hemorrhagic disease. Vet. Microbiol. 2019, 230, 49–55. [Google Scholar] [CrossRef]
- Hugentobler, F.; Di Roberto, R.B.; Gillard, J.; Cousineau, B. Oral immunization using live Lactococcus lactis co-expressing LACK and IL-12 protects BALB/c mice against Leishmania major infection. Vaccine 2012, 30, 5726–5732. [Google Scholar] [CrossRef] [PubMed]

| Disease | Vaccine Type | Manufacturer | Trade Name | References |
|---|---|---|---|---|
| Poliomyelitis | Poliovirus vaccine inactivated | Sanofi Pasteur, Paris, France | IPOL® | [26,29] |
| Rotavirus | Live attenuated monovalent human rotavirus strain | GlaxoSmithKline | Rotarix® | [100] |
| Pentavalent live vaccine (lyophilized) | Serum Institute (India) | Rotasiil® | [101] | |
| Pentavalent live vaccine | Merck and Co., Inc. | RotaTeq® | [102] | |
| Cholera | Cholera toxin B subunit and inactivated V. cholerae 01 whole cells | Valneva | Dukoral® | [102] |
| Live attenuated V. cholerae 01 strain (CVD 103.HgR) | PaxVax | Vaxchora® | [102] | |
| Typhoid | Ty21a live attenuated vaccine | Previously PaxVax Berna GmbH, now Emergent Biosolutions | Vivotif® | [98] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambirasi, M.; Safa, A.; Vruzhaj, I.; Giacomin, A.; Sartor, F.; Toffoli, G. Oral Administration of Cancer Vaccines: Challenges and Future Perspectives. Vaccines 2024, 12, 26. https://doi.org/10.3390/vaccines12010026
Gambirasi M, Safa A, Vruzhaj I, Giacomin A, Sartor F, Toffoli G. Oral Administration of Cancer Vaccines: Challenges and Future Perspectives. Vaccines. 2024; 12(1):26. https://doi.org/10.3390/vaccines12010026
Chicago/Turabian StyleGambirasi, Marta, Amin Safa, Idris Vruzhaj, Aurora Giacomin, Franca Sartor, and Giuseppe Toffoli. 2024. "Oral Administration of Cancer Vaccines: Challenges and Future Perspectives" Vaccines 12, no. 1: 26. https://doi.org/10.3390/vaccines12010026
APA StyleGambirasi, M., Safa, A., Vruzhaj, I., Giacomin, A., Sartor, F., & Toffoli, G. (2024). Oral Administration of Cancer Vaccines: Challenges and Future Perspectives. Vaccines, 12(1), 26. https://doi.org/10.3390/vaccines12010026