
Oncolytic Viruses: An Inventory of Shedding Data from Clinical Trials and Elements for the Environmental Risk Assessment


Abstract
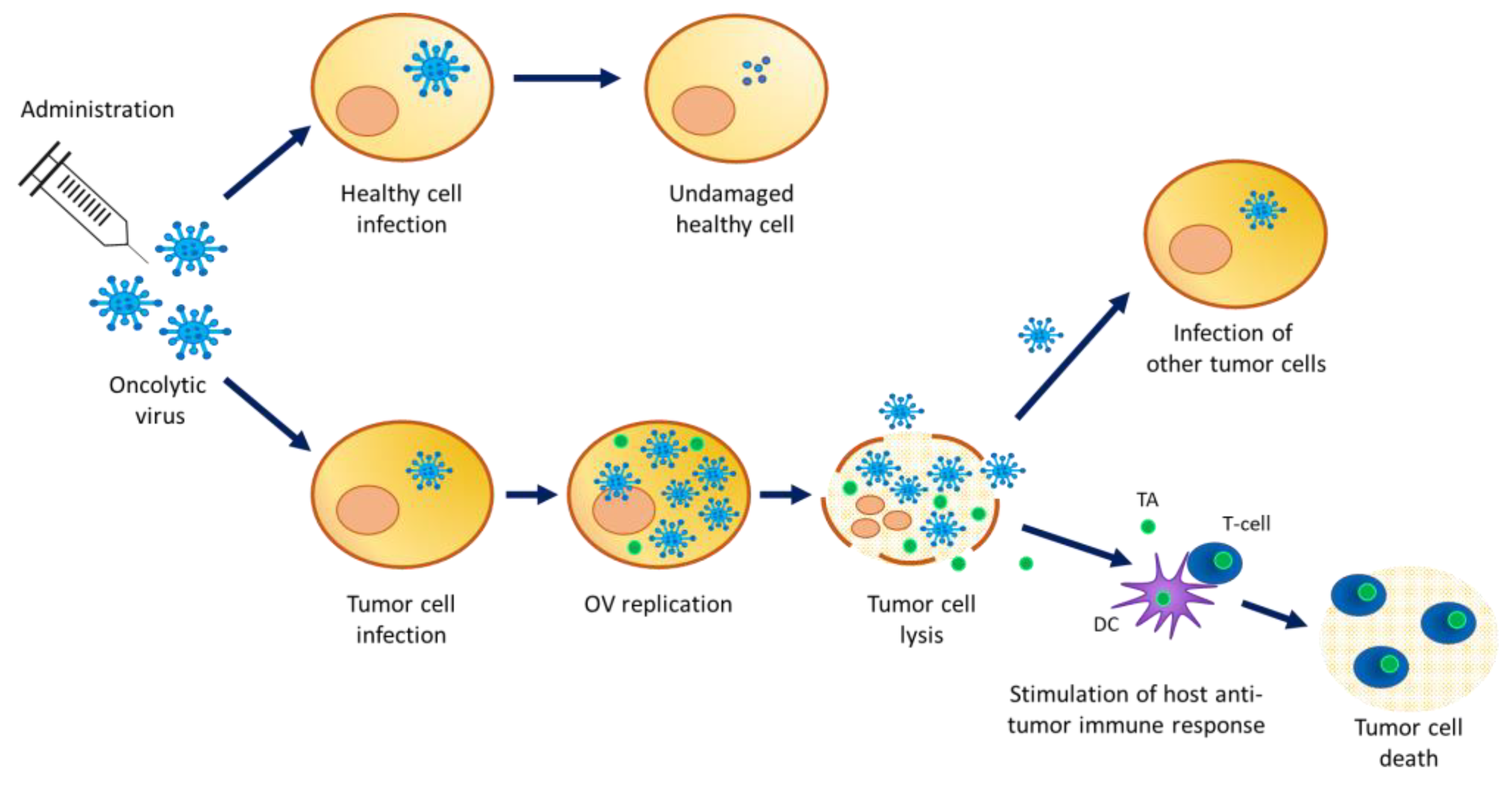
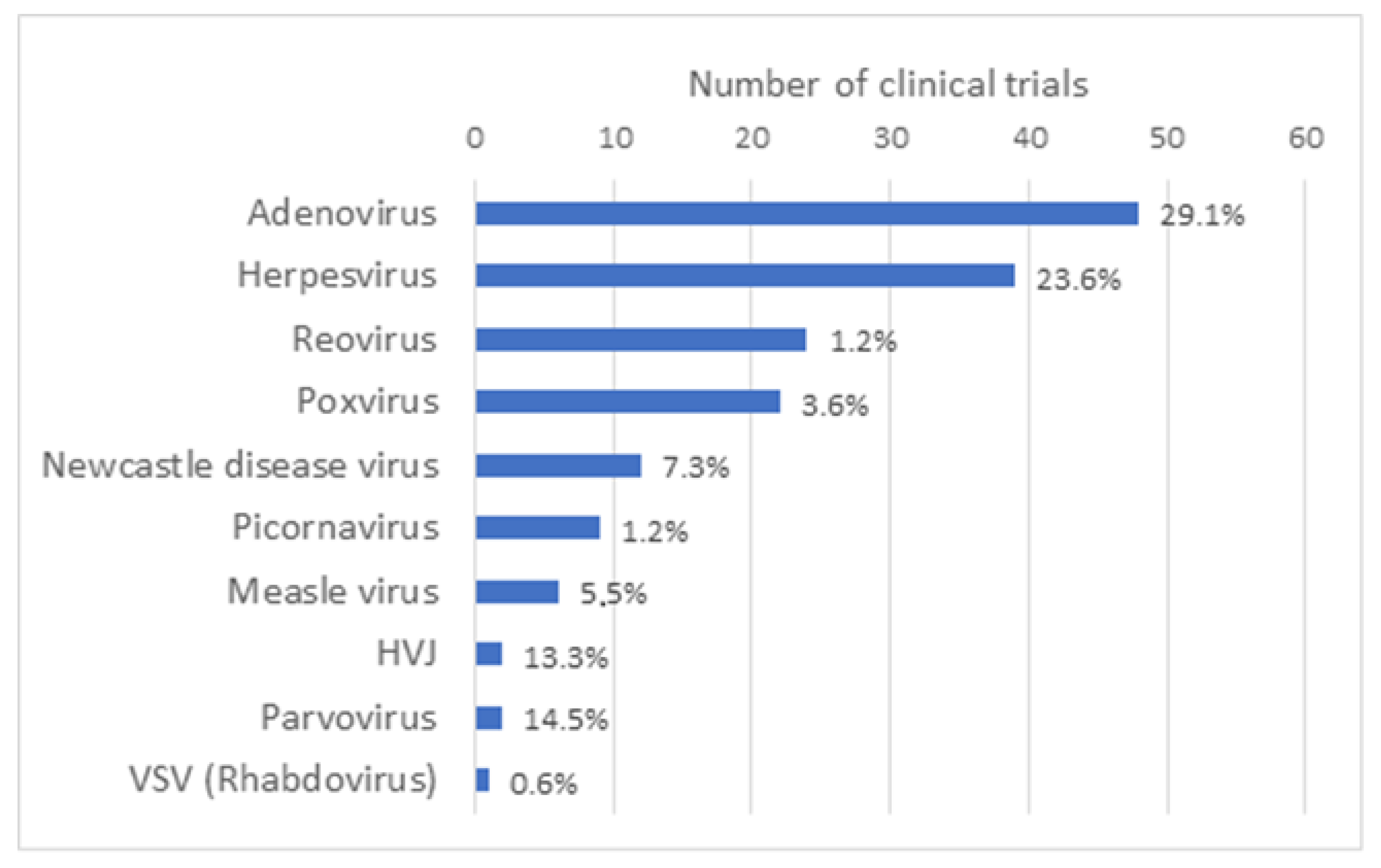
:1. Introduction
2. Materials and Methods
Literature Review
3. Environmental Risk Assessment
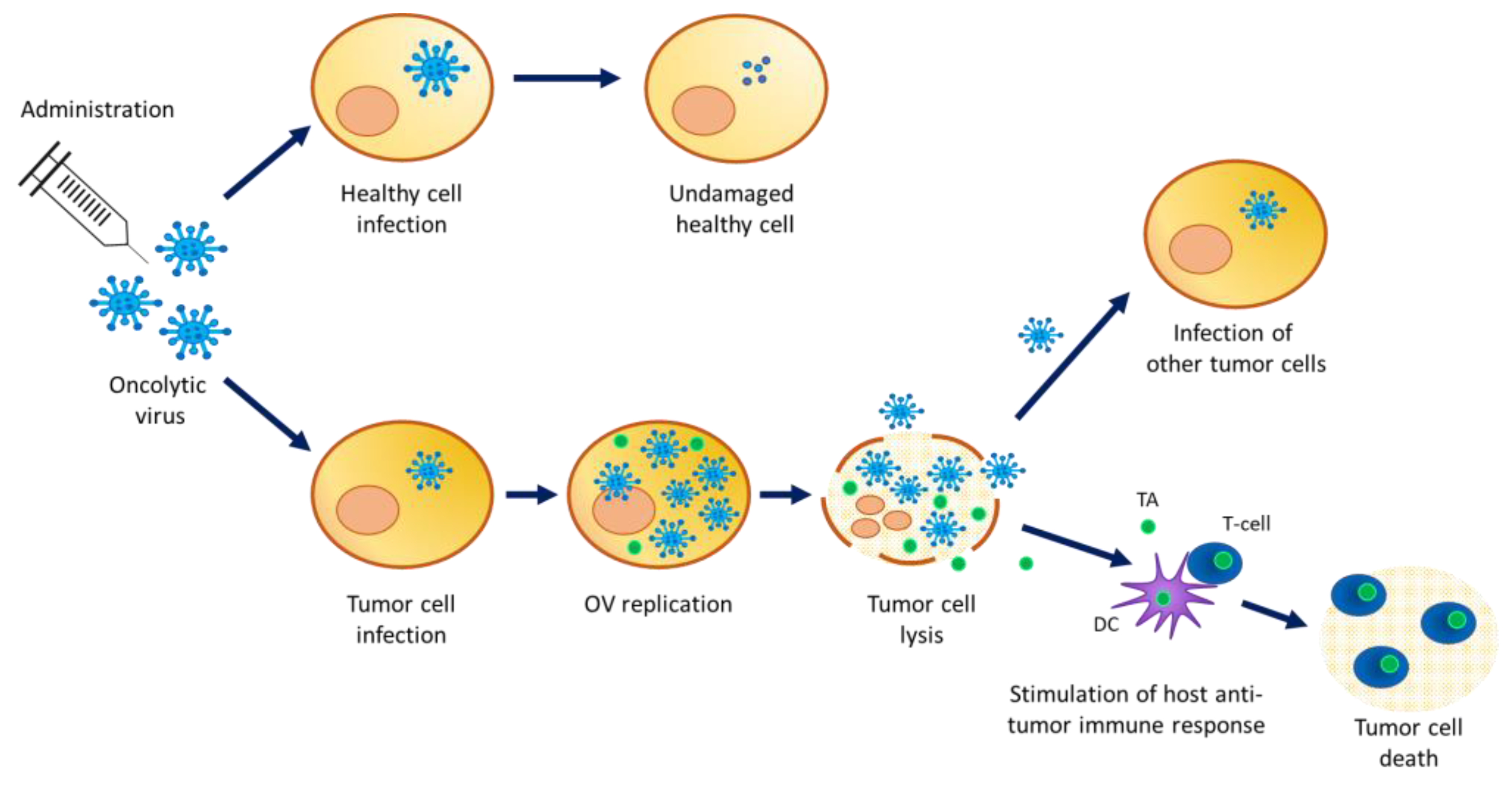
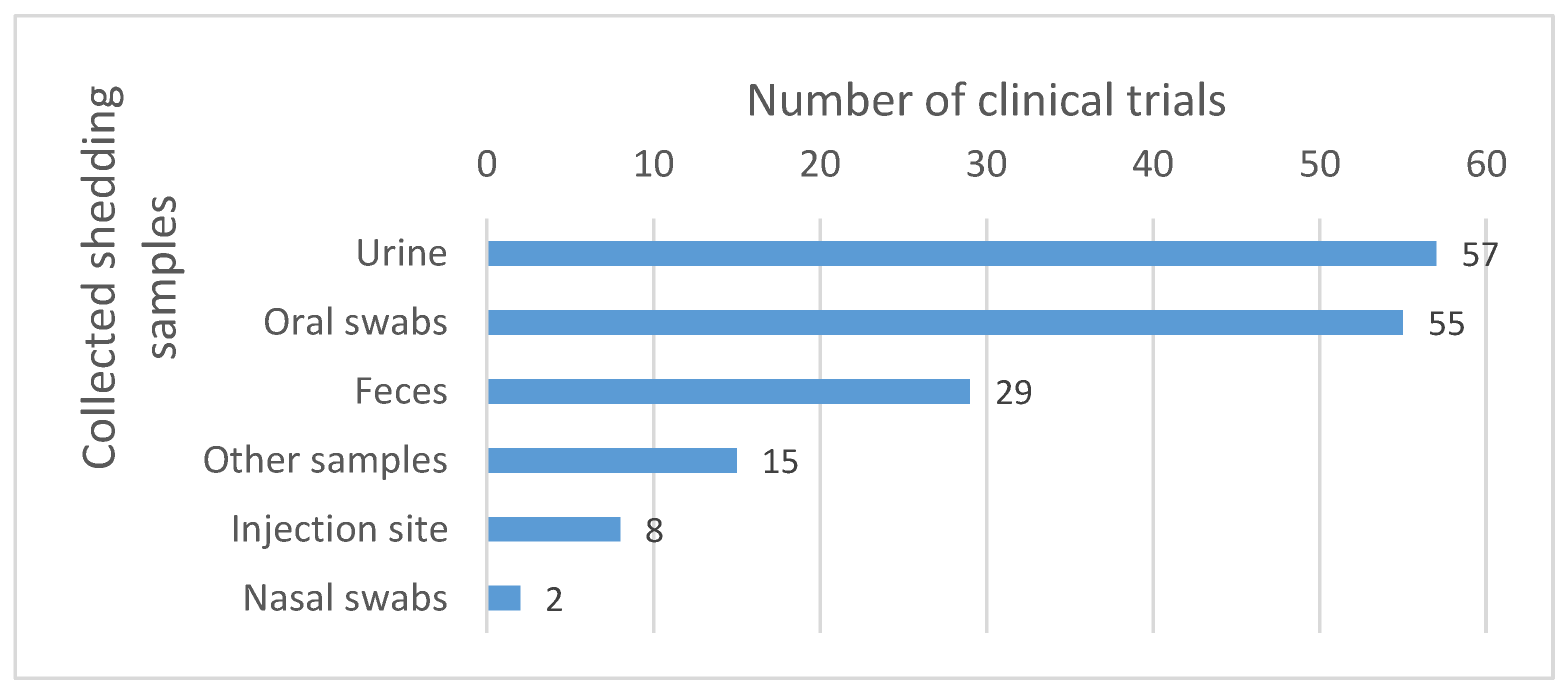
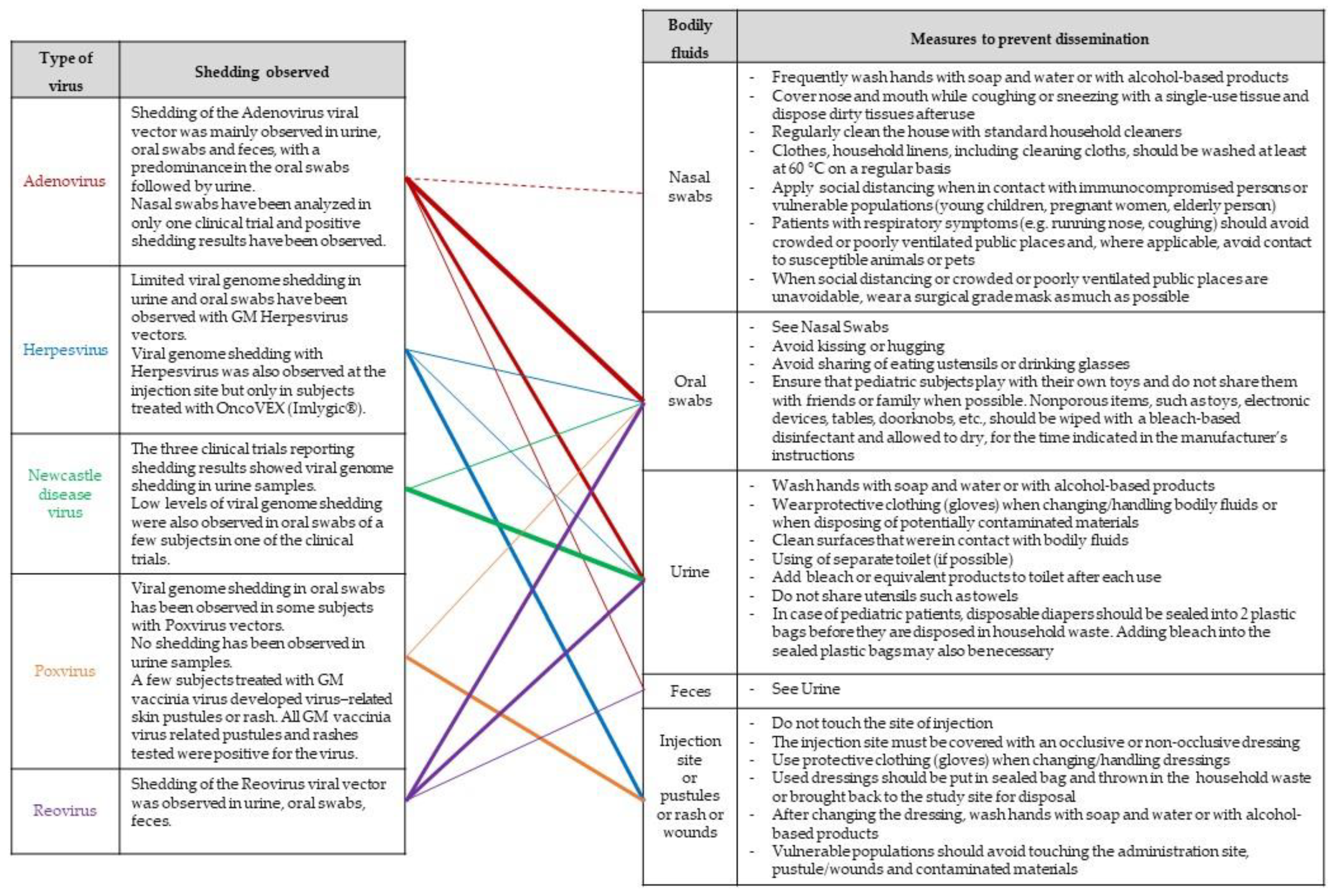
4. Shedding Analysis
4.1. Definition
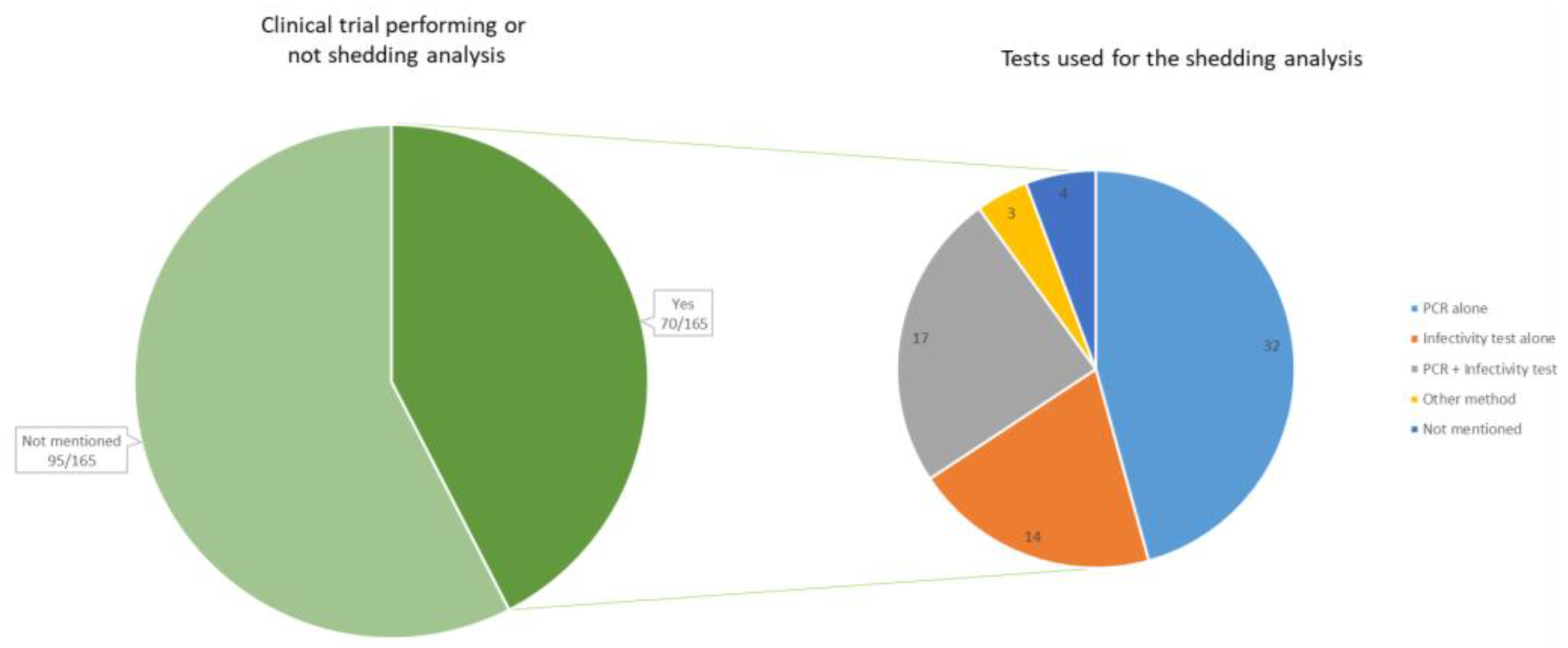
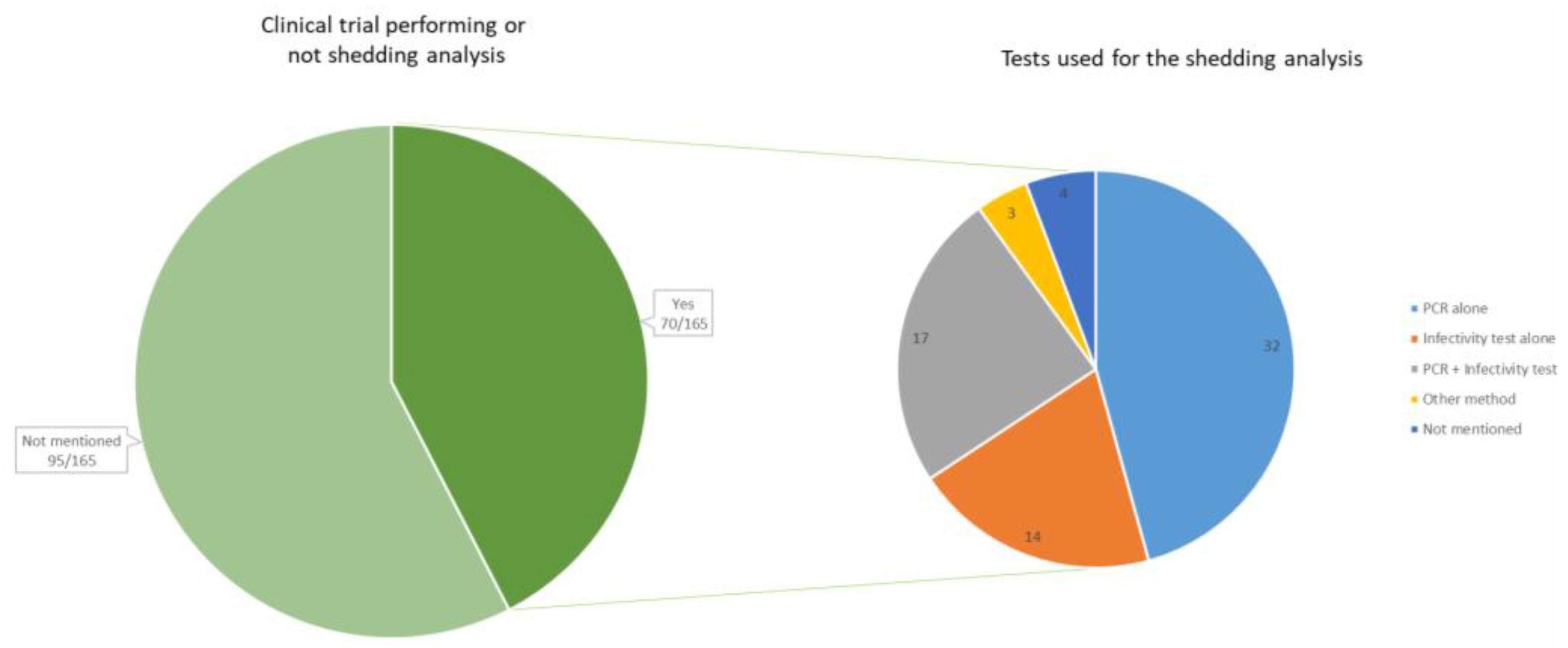
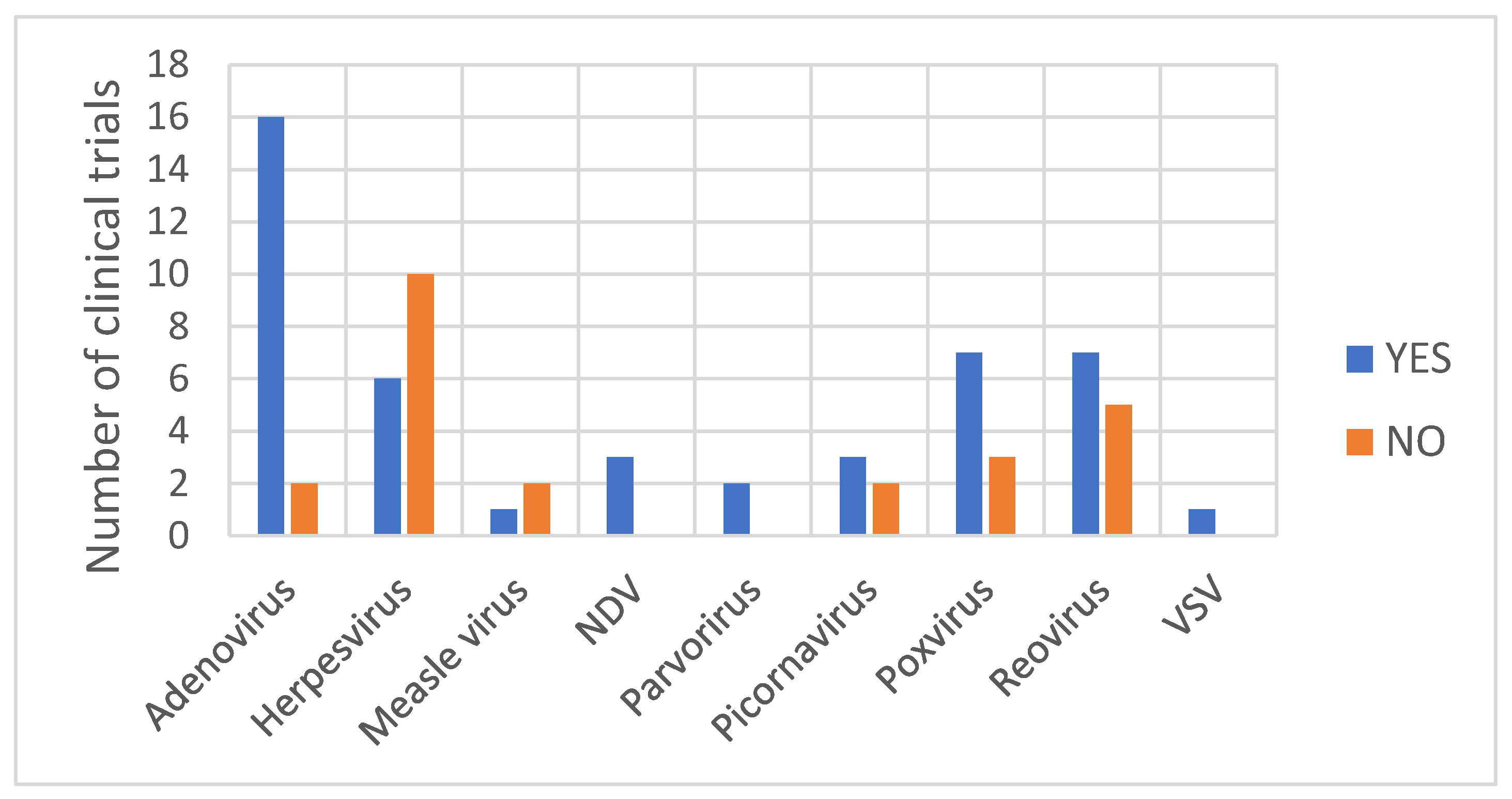
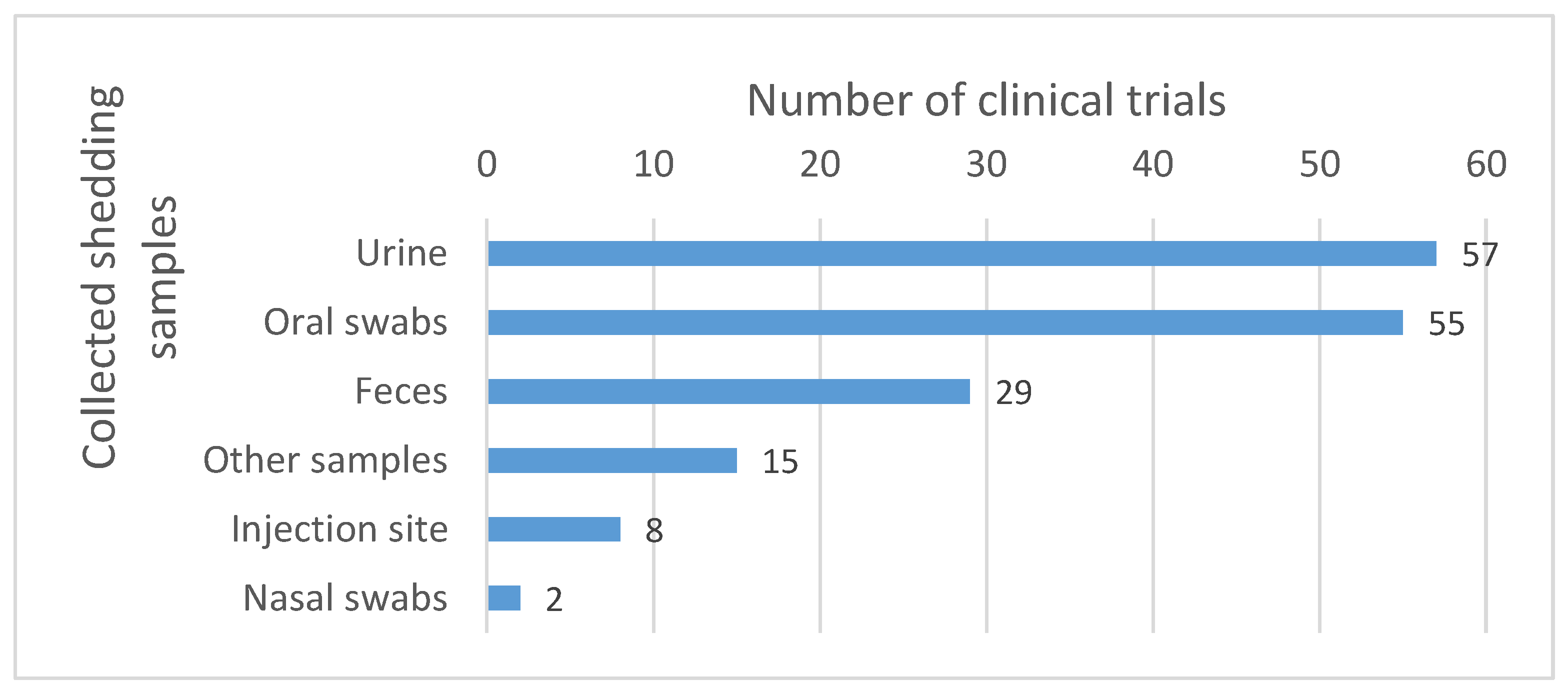
4.2. Detection
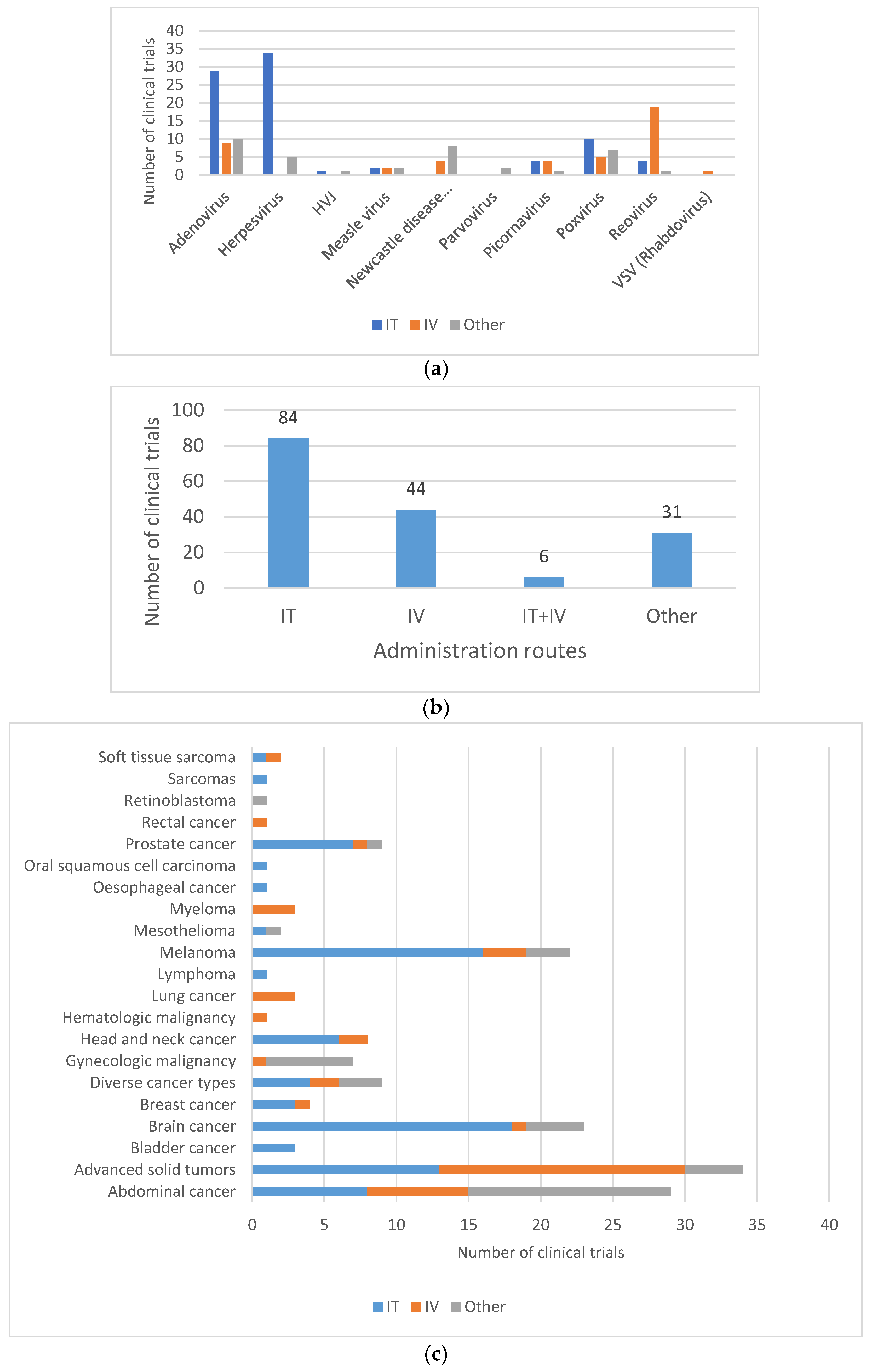
4.3. Aspects of the ERA Impacting Shedding
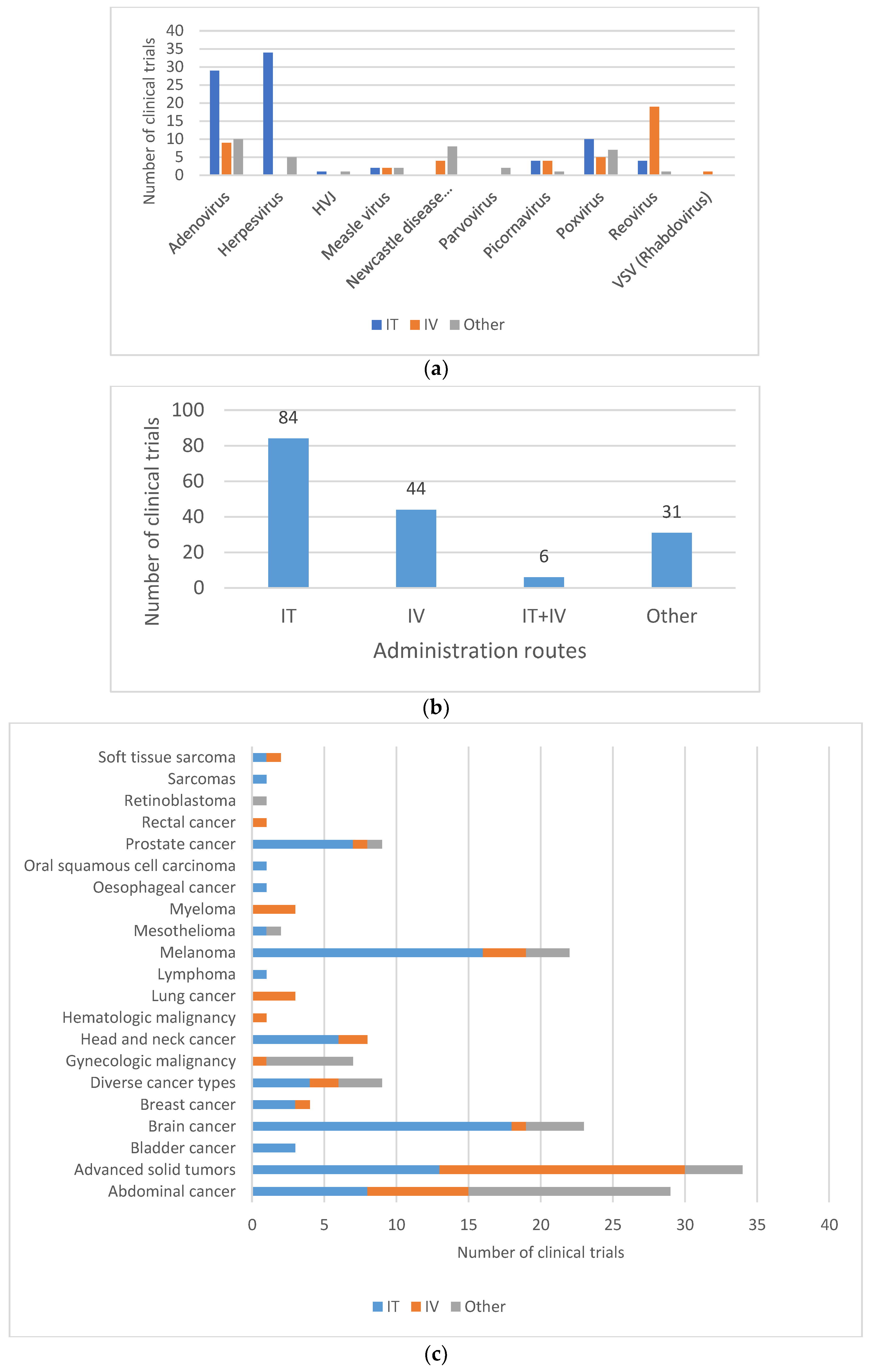
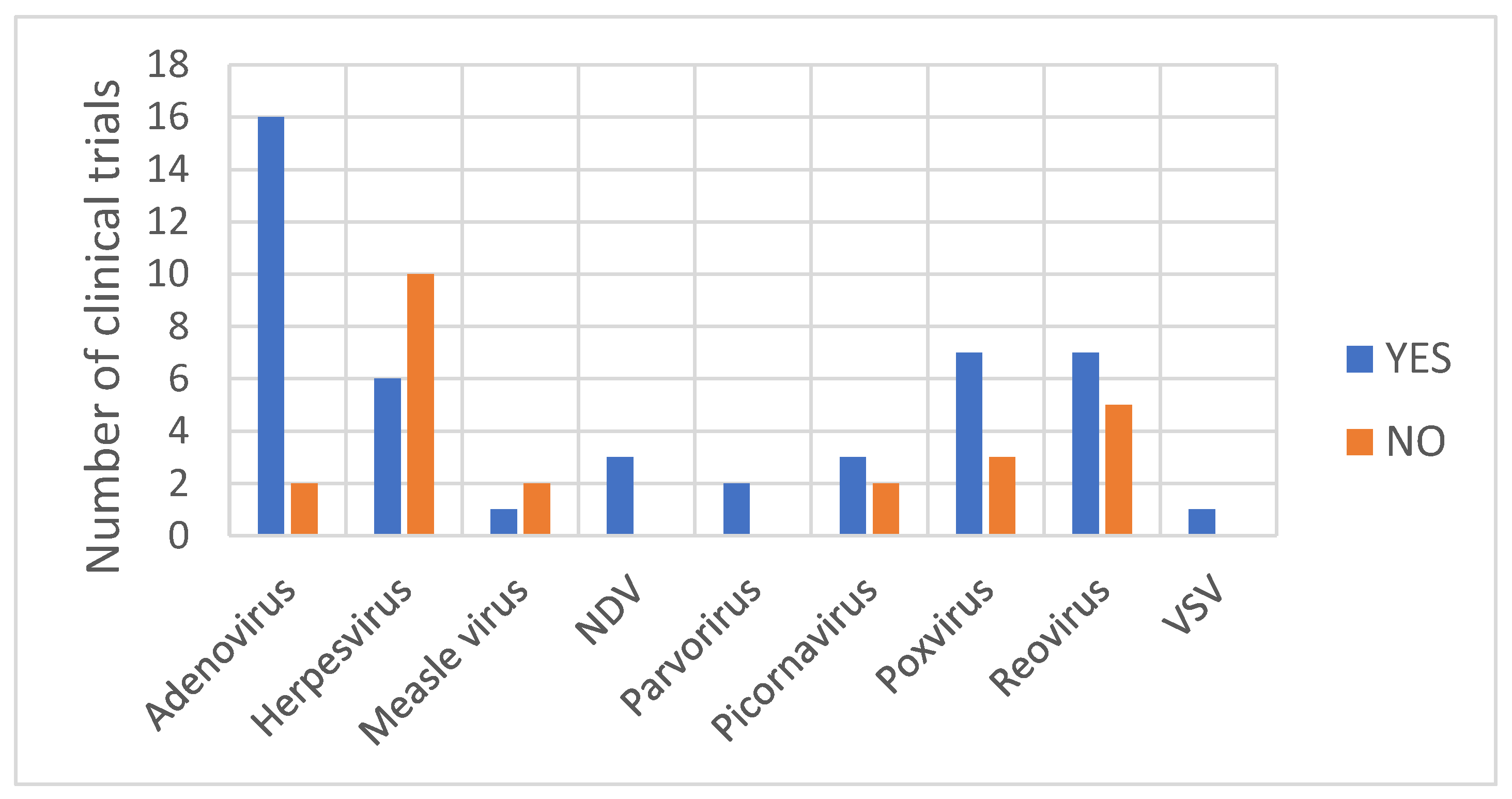
4.4. Shedding Study Design
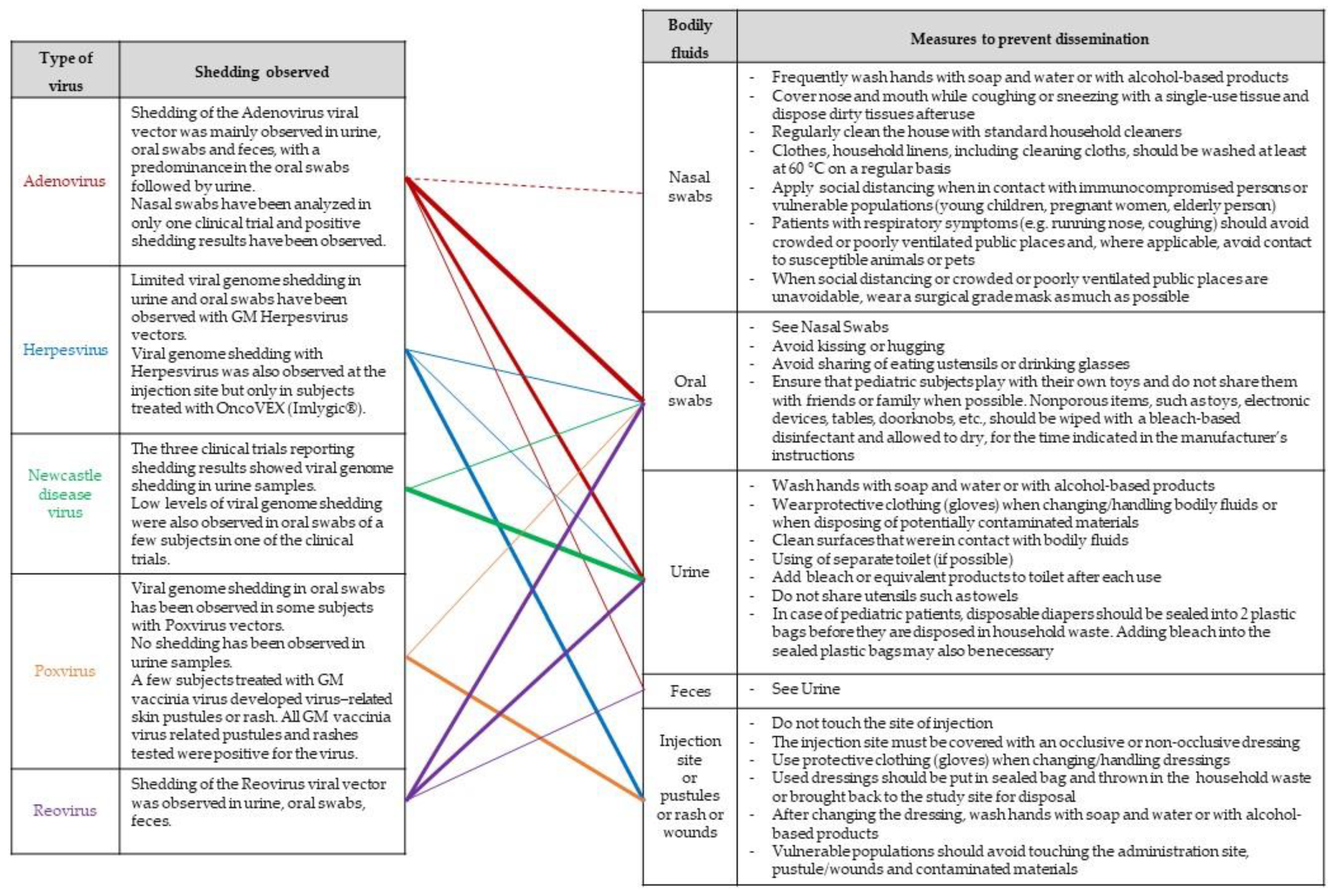
4.5. Risk Management Measures
5. Discussion and Recommendations
6. Concluding Recommendations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Müller, L.; Berkeley, R.; Barr, T.; Ilett, E.; Errington-Mais, F. Past, Present and Future of Oncolytic Reovirus. Cancers 2020, 12, 3219. [Google Scholar] [CrossRef]
- Schirrmacher, V.; van Gool, S.; Stuecker, W. Breaking Therapy Resistance: An Update on Oncolytic Newcastle Disease Virus for Improvements of Cancer Therapy. Biomedicines 2019, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Nagalo, B.M. Immunovirotherapy Based on Recombinant Vesicular Stomatitis Virus: Where Are We? Front. Immunol. 2022, 13, 898631. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Opyrchal, M.; Domingo Musibay, E.; Galanis, E. Oncolytic Measles Virus Strains as Novel Anticancer Agents. Expert Opin. Biol. Ther. 2013, 13, 483–502. [Google Scholar] [CrossRef] [PubMed]
- Macedo, N.; Miller, D.M.; Haq, R.; Kaufman, H.L. Clinical Landscape of Oncolytic Virus Research in 2020. J. Immunother. Cancer 2020, 8, e001486. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; McKenna, M.K.; Rosewell Shaw, A.; Suzuki, M. Clinical CAR-T Cell and Oncolytic Virotherapy for Cancer Treatment. Mol. Ther. 2021, 29, 505–520. [Google Scholar] [CrossRef] [PubMed]
- FDA: U.S. Food & Drug Administration: Imlygic. STN: 125518. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/imlygic (accessed on 11 July 2023).
- European Medicines Agency: Imlygic (Talimogene Laherparepvec). EMEA/H/C/002771. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/imlygic (accessed on 11 July 2023).
- European Commission: Public Health—Union Register of Medicinal Products—Imlygic: EU/1/15/1064. Active Substance: Talimogene Laherparepvec. Available online: https://ec.europa.eu/health/documents/community-register/html/h1064.htm (accessed on 11 July 2023).
- Alberts, P.; Tilgase, A.; Rasa, A.; Bandere, K.; Venskus, D. The Advent of Oncolytic Virotherapy in Oncology: The Rigvir®® Story. Eur. J. Pharmacol. 2018, 837, 117–126. [Google Scholar] [CrossRef] [PubMed]
- State Agency of Medicines Republic of Latvia: Rigvir Marketing Authorisation Suspended; Information for Current Patients. 3 July 2019. Available online: https://www.zva.gov.lv/en/news-and-publications/news/rigvir-marketing-authorisation-suspended-information-current-patients (accessed on 11 July 2023).
- Wei, D.; Xu, J.; Liu, X.-Y.; Chen, Z.-N.; Bian, H. Fighting Cancer with Viruses: Oncolytic Virus Therapy in China. Hum. Gene Ther. 2018, 29, 151–159. [Google Scholar] [CrossRef]
- Report on the Deliberation Results from the Committee on Regenerative Medicine Products and Biotechnology. Brand Name: Delytact Injection. 24 May 2021. Available online: https://www.pmda.go.jp/files/000242808.pdf (accessed on 11 July 2023).
- Schulze, T.; Kemmner, W.; Weitz, J.; Wernecke, K.-D.; Schirrmacher, V.; Schlag, P.M. Efficiency of Adjuvant Active Specific Immunization with Newcastle Disease Virus Modified Tumor Cells in Colorectal Cancer Patients Following Resection of Liver Metastases: Results of a Prospective Randomized Trial. Cancer Immunol. Immunother. 2009, 58, 61–69. [Google Scholar] [CrossRef]
- Liang, W.; Wang, H.; Sun, T.-M.; Yao, W.-Q.; Chen, L.-L.; Jin, Y.; Li, C.-L.; Meng, F.-J. Application of Autologous Tumor Cell Vaccine and NDV Vaccine in Treatment of Tumors of Digestive Tract. World J. Gastroenterol. 2003, 9, 495–498. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Galle, P.R.; Chao, Y.; Brown, K.T.; Heo, J.; Borad, M.J.; Luca, A.; Pelusio, A.; Agathon, D.; Lusky, M.; et al. PHOCUS: A Phase 3 Randomized, Open-Label Study Comparing the Oncolytic Immunotherapy Pexa-Vec Followed by Sorafenib (SOR) vs. SOR in Patients with Advanced Hepatocellular Carcinoma (HCC) without Prior Systemic Therapy. JCO 2016, 34, TPS4146. [Google Scholar] [CrossRef]
- U.S. National Institutes of Health, ClinicalTrials.gov. Efficacy Study of REOLYSIN®® in Combination with Paclitaxel and Carboplatin in Platinum-Refractory Head and Neck Cancers. Official Title: Randomized, Double-Blind, Multicenter Two-Stage Adaptive Phase 3 Study of Intravenous Administration of REOLYSIN (Reovirus Type 3 Dearing) in Combination with Paclitaxel and Carboplatin Versus the Chemotherapy Alone in Patients with Metastatic or Recurrent Squamous Cell Carcinoma of the Head and Neck Who Have Progressed on or After Prior Platinum-Based Chemotherapy. Identifier: NCT01166542. Available online: https://clinicaltrials.gov/study/NCT01166542 (accessed on 11 July 2023).
- U.S. National Institutes of Health, ClinicalTrials.gov. An Integrated Phase II/III, Open Label, Randomized and Controlled Study of the Safety and Efficacy of CG0070 Adenovirus 679 Vector Expressing GM-CSF in Patients With NMIBC With Carcinoma In Situ Disease Who Have Failed BCG. Identifier: NCT01438112. Available online: https://clinicaltrials.gov/study/NCT01438112 (accessed on 11 July 2023).
- Schenk-Braat, E.A.M.; van Mierlo, M.M.K.B.; Wagemaker, G.; Bangma, C.H.; Kaptein, L.C.M. An Inventory of Shedding Data from Clinical Gene Therapy Trials. J. Gene Med. 2007, 9, 910–921. [Google Scholar] [CrossRef]
- Cook, M.; Chauhan, A. Clinical Application of Oncolytic Viruses: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 7505. [Google Scholar] [CrossRef] [PubMed]
- EMEA 2008. Committee for the Medicinal Product for Human Use (CHMP)—Guideline on Scientific Requirements for the Environmental Risk Assessment of Gene Therapy Medicinal Products. EMEA/CHMP/GTMP/125491/2006. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-scientific-requirements-environmental-risk-assessment-gene-therapy-medicinal-products_en.pdf (accessed on 11 July 2023).
- Baldo, A.; van den Akker, E.; Bergmans, H.E.; Lim, F.; Pauwels, K. General Considerations on the Biosafety of Virus-Derived Vectors Used in Gene Therapy and Vaccination. Curr. Gene Ther. 2013, 13, 385–394. [Google Scholar] [CrossRef]
- EC Commission Decision 2002/623/EC of 24 July 2002 Establishing Guidance Notes Supplementing Annex II to Directive 2001/18/EC of the European Parliament and of the Council on the Deliberate Release into the Environment of Genetically Modified Organisms and Repealing Council Directive 90/220/EEC. Official Journal L 200, 30/07/2002 P. 0022-0033. Available online: http://data.europa.eu/eli/dec/2002/623/oj (accessed on 11 July 2023).
- van den Akker, E.; van der Vlugt, C.J.B.; Bleijs, D.A.; Bergmans, H.E. Environmental Risk Assessment of Replication Competent Viral Vectors Applied in Clinical Trials: Potential Effects of Inserted Sequences. Curr. Gene Ther. 2013, 13, 395–412. [Google Scholar] [CrossRef] [PubMed]
- FDA 2015. Design and Analysis of Shedding Studies for Virus or Bacteria-Based Gene Therapy and Oncolytic Products—Guidance for Industry. Available online: https://www.fda.gov/media/89036/download (accessed on 11 July 2023).
- EMEA 2009. IHC Considerations: General Principles to Address Virus and Vector Shedding. EMEA/CHMP/ICH/449035/2009. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-10.pdf (accessed on 11 July 2023).
- Oncolytic Viruses: Considerations for Evaluation of Shedding. Version 2 January 2022 European Commission. Available online: https://health.ec.europa.eu/system/files/2022-01/oncolytic_evaluation_en.pdf (accessed on 11 July 2023).
- Small, E.J.; Carducci, M.A.; Burke, J.M.; Rodriguez, R.; Fong, L.; van Ummersen, L.; Yu, D.C.; Aimi, J.; Ando, D.; Working, P.; et al. A Phase I Trial of Intravenous CG7870, a Replication-Selective, Prostate-Specific Antigen-Targeted Oncolytic Adenovirus, for the Treatment of Hormone-Refractory, Metastatic Prostate Cancer. Mol. Ther. 2006, 14, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Ranki, T.; Pesonen, S.; Hemminki, A.; Partanen, K.; Kairemo, K.; Alanko, T.; Lundin, J.; Linder, N.; Turkki, R.; Ristimäki, A.; et al. Phase I Study with ONCOS-102 for the Treatment of Solid Tumors—An Evaluation of Clinical Response and Exploratory Analyses of Immune Markers. J. Immunother. Cancer 2016, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Amatruda, T.; Nemunaitis, J.; Zager, J.S.; Walker, J.; Chesney, J.A.; Liu, K.; Hsu, C.-P.; Pickett, C.A.; Mehnert, J.M. Biodistribution, Shedding, and Transmissibility of the Oncolytic Virus Talimogene Laherparepvec in Patients with Melanoma. EBioMedicine 2019, 47, 89–97. [Google Scholar] [CrossRef]
- Hajda, J.; Leuchs, B.; Angelova, A.L.; Frehtman, V.; Rommelaere, J.; Mertens, M.; Pilz, M.; Kieser, M.; Krebs, O.; Dahm, M.; et al. Phase 2 Trial of Oncolytic H-1 Parvovirus Therapy Shows Safety and Signs of Immune System Activation in Patients With Metastatic Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2021, 27, 5546–5556. [Google Scholar] [CrossRef]
- Rudin, C.M.; Poirier, J.T.; Senzer, N.N.; Stephenson, J.; Loesch, D.; Burroughs, K.D.; Reddy, P.S.; Hann, C.L.; Hallenbeck, P.L. Phase I Clinical Study of Seneca Valley Virus (SVV-001), a Replication-Competent Picornavirus, in Advanced Solid Tumors with Neuroendocrine Features. Clin. Cancer Res. 2011, 17, 888–895. [Google Scholar] [CrossRef]
- Mell, L.K.; Brumund, K.T.; Daniels, G.A.; Advani, S.J.; Zakeri, K.; Wright, M.E.; Onyeama, S.-J.; Weisman, R.A.; Sanghvi, P.R.; Martin, P.J.; et al. Phase I Trial of Intravenous Oncolytic Vaccinia Virus (GL-ONC1) with Cisplatin and Radiotherapy in Patients with Locoregionally Advanced Head and Neck Carcinoma. Clin. Cancer Res. 2017, 23, 5696–5702. [Google Scholar] [CrossRef] [PubMed]
- Bergmans, H.; Logie, C.; Van Maanen, K.; Hermsen, H.; Meredyth, M.; Van Der Vlugt, C. Identification of Potentially Hazardous Human Gene Products in GMO Risk Assessment. Environ. Biosaf. Res. 2008, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Wang, X.; Lian, B.; Ji, Q.; Zhou, L.; Chi, Z.; Si, L.; Sheng, X.; Kong, Y.; Yu, J.; et al. OrienX010, an Oncolytic Virus, in Patients with Unresectable Stage IIIC-IV Melanoma: A Phase Ib Study. J. Immunother. Cancer 2022, 10, e004307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Huang, J.; Tang, J.; Hu, S.; Luo, S.; Luo, Z.; Zhou, F.; Tan, S.; Ying, J.; Chang, Q.; et al. Intratumoral OH2, an Oncolytic Herpes Simplex Virus 2, in Patients with Advanced Solid Tumors: A Multicenter, Phase I/II Clinical Trial. J. Immunother. Cancer 2021, 9, e002224. [Google Scholar] [CrossRef] [PubMed]
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.-D.; Totsch, S.; et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 2021, 384, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Markert, J.M.; Medlock, M.D.; Rabkin, S.D.; Gillespie, G.Y.; Todo, T.; Hunter, W.D.; Palmer, C.A.; Feigenbaum, F.; Tornatore, C.; Tufaro, F.; et al. Conditionally Replicating Herpes Simplex Virus Mutant, G207 for the Treatment of Malignant Glioma: Results of a Phase I Trial. Gene Ther. 2000, 7, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Markert, J.M.; Liechty, P.G.; Wang, W.; Gaston, S.; Braz, E.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Lakeman, A.D.; Palmer, C.A.; et al. Phase Ib Trial of Mutant Herpes Simplex Virus G207 Inoculated Pre-and Post-Tumor Resection for Recurrent GBM. Mol. Ther. 2009, 17, 199–207. [Google Scholar] [CrossRef]
- Todo, T.; Ino, Y.; Ohtsu, H.; Shibahara, J.; Tanaka, M. A Phase I/II Study of Triple-Mutated Oncolytic Herpes Virus G47∆ in Patients with Progressive Glioblastoma. Nat. Commun. 2022, 13, 4119. [Google Scholar] [CrossRef]
- Kasuya, H.; Kodera, Y.; Nakao, A.; Yamamura, K.; Gewen, T.; Zhiwen, W.; Hotta, Y.; Yamada, S.; Fujii, T.; Fukuda, S.; et al. Phase I Dose-Escalation Clinical Trial of HF10 Oncolytic Herpes Virus in 17 Japanese Patients with Advanced Cancer. Hepatogastroenterology 2014, 61, 599–605. [Google Scholar]
- Danson, S.J.; Conner, J.; Edwards, J.G.; Blyth, K.G.; Fisher, P.M.; Muthana, M.; Salawu, A.; Taylor, F.; Hodgkinson, E.; Joyce, P.; et al. Oncolytic Herpesvirus Therapy for Mesothelioma—A Phase I/IIa Trial of Intrapleural Administration of HSV1716. Lung Cancer 2020, 150, 145–151. [Google Scholar] [CrossRef]
- Streby, K.A.; Geller, J.I.; Currier, M.A.; Warren, P.S.; Racadio, J.M.; Towbin, A.J.; Vaughan, M.R.; Triplet, M.; Ott-Napier, K.; Dishman, D.J.; et al. Intratumoral Injection of HSV1716, an Oncolytic Herpes Virus, Is Safe and Shows Evidence of Immune Response and Viral Replication in Young Cancer Patients. Clin. Cancer Res. 2017, 23, 3566–3574. [Google Scholar] [CrossRef] [PubMed]
- Menotti, L.; Avitabile, E. Herpes Simplex Virus Oncolytic Immunovirotherapy: The Blossoming Branch of Multimodal Therapy. Int. J. Mol. Sci. 2020, 21, 8310. [Google Scholar] [CrossRef] [PubMed]
- Fong, Y.; Kim, T.; Bhargava, A.; Schwartz, L.; Brown, K.; Brody, L.; Covey, A.; Karrasch, M.; Getrajdman, G.; Mescheder, A.; et al. A Herpes Oncolytic Virus Can Be Delivered via the Vasculature to Produce Biologic Changes in Human Colorectal Cancer. Mol. Ther. 2009, 17, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Geevarghese, S.K.; Geller, D.A.; de Haan, H.A.; Hörer, M.; Knoll, A.E.; Mescheder, A.; Nemunaitis, J.; Reid, T.R.; Sze, D.Y.; Tanabe, K.K.; et al. Phase I/II Study of Oncolytic Herpes Simplex Virus NV1020 in Patients with Extensively Pretreated Refractory Colorectal Cancer Metastatic to the Liver. Hum. Gene Ther. 2010, 21, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Annels, N.E.; Mansfield, D.; Arif, M.; Ballesteros-Merino, C.; Simpson, G.R.; Denyer, M.; Sandhu, S.S.; Melcher, A.A.; Harrington, K.J.; Davies, B.; et al. Phase I Trial of an ICAM-1-Targeted Immunotherapeutic-Coxsackievirus A21 (CVA21) as an Oncolytic Agent Against Non Muscle-Invasive Bladder Cancer. Clin. Cancer Res. 2019, 25, 5818–5831. [Google Scholar] [CrossRef]
- Burke, M.J.; Ahern, C.; Weigel, B.J.; Poirier, J.T.; Rudin, C.M.; Chen, Y.; Cripe, T.P.; Bernhardt, M.B.; Blaney, S.M. Phase I Trial of Seneca Valley Virus (NTX-010) in Children with Relapsed/Refractory Solid Tumors: A Report of the Children’s Oncology Group. Pediatr. Blood Cancer 2015, 62, 743–750. [Google Scholar] [CrossRef]
- Desjardins, A.; Gromeier, M.; Herndon, J.E.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- Beasley, G.M.; Nair, S.K.; Farrow, N.E.; Landa, K.; Selim, M.A.; Wiggs, C.A.; Jung, S.-H.; Bigner, D.D.; True Kelly, A.; Gromeier, M.; et al. Phase I Trial of Intratumoral PVSRIPO in Patients with Unresectable, Treatment-Refractory Melanoma. J. Immunother. Cancer 2021, 9, e002203. [Google Scholar] [CrossRef]
- Velazquez-Salinas, L.; Naik, S.; Pauszek, S.J.; Peng, K.-W.; Russell, S.J.; Rodriguez, L.L. Oncolytic Recombinant Vesicular Stomatitis Virus (VSV) Is Nonpathogenic and Nontransmissible in Pigs, a Natural Host of VSV. Hum. Gene Ther. Clin Dev 2017, 28, 108–115. [Google Scholar] [CrossRef]
- Naik, S.; Galyon, G.D.; Jenks, N.J.; Steele, M.B.; Miller, A.C.; Allstadt, S.D.; Suksanpaisan, L.; Peng, K.W.; Federspiel, M.J.; Russell, S.J.; et al. Comparative Oncology Evaluation of Intravenous Recombinant Oncolytic Vesicular Stomatitis Virus Therapy in Spontaneous Canine Cancer. Mol. Cancer Ther. 2018, 17, 316–326. [Google Scholar] [CrossRef]
- Cook, J.; Peng, K.-W.; Witzig, T.E.; Broski, S.M.; Villasboas, J.C.; Paludo, J.; Patnaik, M.; Rajkumar, V.; Dispenzieri, A.; Leung, N.; et al. Clinical Activity of Single-Dose Systemic Oncolytic VSV Virotherapy in Patients with Relapsed Refractory T-Cell Lymphoma. Blood Adv. 2022, 6, 3268–3279. [Google Scholar] [CrossRef] [PubMed]
- Jenks, N.; Myers, R.; Greiner, S.M.; Thompson, J.; Mader, E.K.; Greenslade, A.; Griesmann, G.E.; Federspiel, M.J.; Rakela, J.; Borad, M.J.; et al. Safety Studies on Intrahepatic or Intratumoral Injection of Oncolytic Vesicular Stomatitis Virus Expressing Interferon-Beta in Rodents and Nonhuman Primates. Hum. Gene Ther. 2010, 21, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.-O.; Schöning, T.; Hüsing, J.; Beelte, B.; et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol. Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Atherton, P.J.; Maurer, M.J.; Knutson, K.L.; Dowdy, S.C.; Cliby, W.A.; Haluska, P.; Long, H.J.; Oberg, A.; Aderca, I.; et al. Oncolytic Measles Virus Expressing the Sodium Iodide Symporter to Treat Drug-Resistant Ovarian Cancer. Cancer Res. 2015, 75, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A.; Tong, C.; LaPlant, B.; Lacy, M.Q.; Laumann, K.; Dingli, D.; Zhou, Y.; Federspiel, M.J.; Gertz, M.A.; Hayman, S.; et al. Phase I Trial of Systemic Administration of Edmonston Strain of Measles Virus Genetically Engineered to Express the Sodium Iodide Symporter in Patients with Recurrent or Refractory Multiple Myeloma. Leukemia 2017, 31, 2791–2798. [Google Scholar] [CrossRef] [PubMed]
- Lauer, U.M.; Schell, M.; Beil, J.; Berchtold, S.; Koppenhöfer, U.; Glatzle, J.; Königsrainer, A.; Möhle, R.; Nann, D.; Fend, F.; et al. Phase I Study of Oncolytic Vaccinia Virus GL-ONC1 in Patients with Peritoneal Carcinomatosis. Clin. Cancer Res. 2018, 24, 4388–4398. [Google Scholar] [CrossRef]
- Park, B.-H.; Hwang, T.; Liu, T.-C.; Sze, D.Y.; Kim, J.-S.; Kwon, H.-C.; Oh, S.Y.; Han, S.-Y.; Yoon, J.-H.; Hong, S.-H.; et al. Use of a Targeted Oncolytic Poxvirus, JX-594, in Patients with Refractory Primary or Metastatic Liver Cancer: A Phase I Trial. Lancet Oncol. 2008, 9, 533–542. [Google Scholar] [CrossRef]
- Moehler, M.; Heo, J.; Lee, H.C.; Tak, W.Y.; Chao, Y.; Paik, S.W.; Yim, H.J.; Byun, K.S.; Baron, A.; Ungerechts, G.; et al. Vaccinia-Based Oncolytic Immunotherapy Pexastimogene Devacirepvec in Patients with Advanced Hepatocellular Carcinoma after Sorafenib Failure: A Randomized Multicenter Phase IIb Trial (TRAVERSE). Oncoimmunology 2019, 8, 1615817. [Google Scholar] [CrossRef]
- Dunn, G.; Klapsa, D.; Wilton, T.; Stone, L.; Minor, P.D.; Martin, J. Twenty-Eight Years of Poliovirus Replication in an Immunodeficient Individual: Impact on the Global Polio Eradication Initiative. PLoS Pathog. 2015, 11, e1005114. [Google Scholar] [CrossRef]
- Weil, M.; Shulman, L.M.; Heiman, S.; Stauber, T.; Alfandari, J.; Weiss, L.; Silberstein, I.; Indenbaum, V.; Mendelson, E.; Sofer, D. Prolonged Excretion of Type-2 Poliovirus from a Primary Immune Deficient Patient during the Transition to a Type-2 Poliovirus-Free World, Israel, 2016. Eurosurveillance 2016, 21, 30408. [Google Scholar] [CrossRef]
- Roulstone, V.; Khan, K.; Pandha, H.S.; Rudman, S.; Coffey, M.; Gill, G.M.; Melcher, A.A.; Vile, R.; Harrington, K.J.; de Bono, J.; et al. Phase I Trial of Cyclophosphamide as an Immune Modulator for Optimizing Oncolytic Reovirus Delivery to Solid Tumors. Clin. Cancer Res. 2015, 21, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.J.; Hingorani, M.; Tanay, M.A.; Hickey, J.; Bhide, S.A.; Clarke, P.M.; Renouf, L.C.; Thway, K.; Sibtain, A.; McNeish, I.A.; et al. Phase I/II Study of Oncolytic HSV GM-CSF in Combination with Radiotherapy and Cisplatin in Untreated Stage III/IV Squamous Cell Cancer of the Head and Neck. Clin. Cancer Res. 2010, 16, 4005–4015. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.C.C.; Coffin, R.S.; Davis, C.J.; Graham, N.J.; Groves, N.; Guest, P.J.; Harrington, K.J.; James, N.D.; Love, C.A.; McNeish, I.; et al. A Phase I Study of OncoVEXGM-CSF, a Second-Generation Oncolytic Herpes Simplex Virus Expressing Granulocyte Macrophage Colony-Stimulating Factor. Clin. Cancer Res. 2006, 12, 6737–6747. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.N.; Kaufman, H.L.; Amatruda, T.; Nemunaitis, M.; Reid, T.; Daniels, G.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; et al. Phase II Clinical Trial of a Granulocyte-Macrophage Colony-Stimulating Factor-Encoding, Second-Generation Oncolytic Herpesvirus in Patients with Unresectable Metastatic Melanoma. J. Clin. Oncol. 2009, 27, 5763–5771. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Smith, K.M.; McKinney, B.; Palmer, C.A.; Rosenfeld, S.; Lillehei, K.; Hamilton, A.; DeMasters, B.K.; Judy, K.; Kirn, D. A Phase I Trial of Ad.HIFN-Beta Gene Therapy for Glioma. Mol. Ther. 2008, 16, 618–626. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Tong, A.W.; Nemunaitis, M.; Senzer, N.; Phadke, A.P.; Bedell, C.; Adams, N.; Zhang, Y.-A.; Maples, P.B.; Chen, S.; et al. A Phase I Study of Telomerase-Specific Replication Competent Oncolytic Adenovirus (Telomelysin) for Various Solid Tumors. Mol. Ther. 2010, 18, 429–434. [Google Scholar] [CrossRef]
- Imlygic®® Clinical Overview and Handling Guide (USA, 2019). Available online: https://cdn.imlygichcp.com/cdn/917dac5d-ff46-4382-bb1e-8a4041ae951b/en/1/20201222t152404z/imlygic-clinical-overview.pdf (accessed on 11 July 2023).
- U.S. National Institutes of Health, ClinicalTrials.gov. Postmarketing Prospective Study of Melanoma Patients Treated With IMLYGIC®® to Characterize Risk of Herpetic Infection. Identifier: NCT02910557. Available online: https://www.clinicaltrials.gov/study/NCT02910557 (accessed on 11 July 2023).
- Laurie, S.A.; Bell, J.C.; Atkins, H.L.; Roach, J.; Bamat, M.K.; O’Neil, J.D.; Roberts, M.S.; Groene, W.S.; Lorence, R.M. A Phase 1 Clinical Study of Intravenous Administration of PV701, an Oncolytic Virus, Using Two-Step Desensitization. Clin. Cancer Res. 2006, 12, 2555–2562. [Google Scholar] [CrossRef]
- Hotte, S.J.; Lorence, R.M.; Hirte, H.W.; Polawski, S.R.; Bamat, M.K.; O’Neil, J.D.; Roberts, M.S.; Groene, W.S.; Major, P.P. An Optimized Clinical Regimen for the Oncolytic Virus PV701. Clin. Cancer Res. 2007, 13, 977–985. [Google Scholar] [CrossRef]
- Pecora, A.L.; Rizvi, N.; Cohen, G.I.; Meropol, N.J.; Sterman, D.; Marshall, J.L.; Goldberg, S.; Gross, P.; O’Neil, J.D.; Groene, W.S.; et al. Phase I Trial of Intravenous Administration of PV701, an Oncolytic Virus, in Patients with Advanced Solid Cancers. J. Clin. Oncol. 2002, 20, 2251–2266. [Google Scholar] [CrossRef]
- Tell, J.G.; Coller, B.-A.G.; Dubey, S.A.; Jenal, U.; Lapps, W.; Wang, L.; Wolf, J. Environmental Risk Assessment for RVSVΔG-ZEBOV-GP, a Genetically Modified Live Vaccine for Ebola Virus Disease. Vaccines 2020, 8, 779. [Google Scholar] [CrossRef]
- European Commission GMO: Deliberate Release into the Environment of Other than Plants GMOs for Any Other Purposes than Placing on the Market (Experimental Releases). List of SNIFs Submitted to the Member State’s Competent Authorities under Directive 2001/18/EC (after 17 October 2002). Keyword VSV. Available online: https://webgate.ec.europa.eu/fip/GMO_Registers/GMO_Part_B_Others.php?Keyword=VSV (accessed on 11 July 2023).
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Palero, F.; González-Candelas, F. Recombination in Viruses: Mechanisms, Methods of Study, and Evolutionary Consequences. Infect. Genet. Evol. 2015, 30, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Buijs, P.R.A.; Verhagen, J.H.E.; van Eijck, C.H.J.; van den Hoogen, B.G. Oncolytic Viruses: From Bench to Bedside with a Focus on Safety. Hum. Vaccin. Immunother. 2015, 11, 1573–1584. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Herpesvirus Vectors | Genetic Modifications [44] | Administration Route | Viral Genome Shedding | References |
|---|---|---|---|---|
| Imlygic®® | Armed recombinant HSV 1 with GM-CSF 2 transgen | IT 3 | Yes | [30] |
| OrienX010 | Armed recombinant HSV with GM-CSF transgen | IT | Yes | [35] |
| OH2 | Armed recombinant HSV with GM-CSF transgen | IT | No | [36] |
| G207 | Conditionally replicating HSV with multiple mutations | IT | No | [37,38,39] |
| G47Δ | Conditionally replicating HSV with multiple mutations | IT | No | [40] |
| HF10 | Conditionally replicating HSV with multiple mutations | IT | No | [41] |
| HSV1716 | Conditionally replicating HSV with multiple mutations | IT | No | [42,43] |
| NV1020 | Conditionally replicating HSV with multiple mutations | Hepatic arterial injection | Yes and No | [45,46] |
| Preventing Measures | PPE |
|
| Needle preparation |
| |
| Spill kits |
| |
| Risk Management Measures | Skin contact |
|
| Mucus membrane or eye contact |
| |
| Accidental spill | In the case of accidental spills or breakage of a vial containing the GMO:
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onnockx, S.; Baldo, A.; Pauwels, K. Oncolytic Viruses: An Inventory of Shedding Data from Clinical Trials and Elements for the Environmental Risk Assessment. Vaccines 2023, 11, 1448. https://doi.org/10.3390/vaccines11091448
Onnockx S, Baldo A, Pauwels K. Oncolytic Viruses: An Inventory of Shedding Data from Clinical Trials and Elements for the Environmental Risk Assessment. Vaccines. 2023; 11(9):1448. https://doi.org/10.3390/vaccines11091448
Chicago/Turabian StyleOnnockx, Sheela, Aline Baldo, and Katia Pauwels. 2023. "Oncolytic Viruses: An Inventory of Shedding Data from Clinical Trials and Elements for the Environmental Risk Assessment" Vaccines 11, no. 9: 1448. https://doi.org/10.3390/vaccines11091448
APA StyleOnnockx, S., Baldo, A., & Pauwels, K. (2023). Oncolytic Viruses: An Inventory of Shedding Data from Clinical Trials and Elements for the Environmental Risk Assessment. Vaccines, 11(9), 1448. https://doi.org/10.3390/vaccines11091448