Abstract
Cutaneous Leishmaniasis (CL), a neglected vector-borne disease caused by protozoan parasite Leishmania major (L. major), is a major public health concern, and the development of new strategies to reduce the disease incidence has become a top priority. Advances in immunoinformatics and in-silico epitope prediction could be a promising approach to designing a finest vaccine candidate. In this study, we aimed to design a peptide-based vaccine against CL using computational tools and identified ten B-cell-derived T-cell epitopes from the glycoprotein gp63 of L. major. All of the potential immunodominant epitopes were used to design a vaccine construct along with a linker and an adjuvant at the N-terminal for enhancing its immunogenicity. Additionally, many characteristics of the proposed vaccine were examined, and it was confirmed to be non-allergenic, non-toxic, and thermally stable. To assess the vaccine interaction with the innate immune toll-like receptor-4 (TLR-4), a 3D structure of the vaccine construct was developed. Molecular docking and molecular dynamic simulation were used to confirm the binding and to assess the stability of the vaccine-TLR4 complex and interactions, respectively. In conclusion, our multi-epitope vaccine will provide a gateway to analyze the protein function of a potential vaccine candidate against CL.
1. Introduction
Leishmaniasis is caused by an obligatory intracellular parasite belonging to the genus Leishmania, which is transmitted via the bite of infected female phlebotomine sand-flies [1]. Approximately 20 different species of the sandfly can transmit the parasite to the mammalian host, either zoonotically or anthropologically [2,3], leading to a variety of disease patterns, particularly cutaneous leishmaniasis (CL), visceral leishmaniasis (VL), and muco-cutaneous leishmaniasis (MCL) [4,5,6]. Leishmaniasis is an important global health problem [7] and the seventh most neglected tropical infection, which is prevalent in 98 countries and affects 350 million people globally [8,9,10].
The most commonly used drugs against CL are pentavalent antimonials, paromomycin, liposomal amphotericin B (AmBisome, AmB) and oral miltefosine [11,12,13], which have multiple adverse effects. AmB has replaced antimony as a first-line therapy for treatment, but its use is limited due to the difficulty of administration, as well as its high cost [14,15,16]. Leishmania (L.) major, the causative agent of zoonotic CL, expresses three main types of molecules: glycosylphosphatidylinositol, lipophosphoglycan (LPG), and glycoproteins (GP). A 63 kDa surface proteinase (GP63), a glycoprotein, was identified as the major surface antigen [17,18,19], with more than 500,000 copies expressed and distributed throughout whole promastigote cell [20]. Its role in the survival of the parasite within macrophages promotes phagocytosis and takes control over the complement activation, which increases the parasite’s resistance to complement-mediated lysis [21]. Due to its abundance and ability to develop resistance, it has been suggested that GP63 could be a candidate for the vaccine against leishmaniasis [22].
The emergence of its resistance and the increasing rate of therapeutic failures has led to the critical need for novel anti-leishmanial treatment and the development of an effective vaccine against CL [23,24]. In the last few years, immunoinformatic tools have offered epitopes predictor programs to scan the whole genomes for the immunogenic epitopes and for the selection of potential proteins for vaccine development [25,26,27]. Recently, studies of the anti-leishmanial candidates for vaccine development have advanced due to the understanding of the cell-mediated immunological mechanisms for controlling the infection [28,29]. Minimal epitopes analogous to peptides are capable of inducing the T-cell-specific responses that are essential to eradicating the intracellular parasite [30]. Based on the understating of the mechanisms of immunology, several vaccines have been designed, but none of them have been found to have any remarkable efficacy. However, the major surface glycoprotein GP63 of L. major considered, a ligand involved in the interaction of the parasite with the immune system, is a potential vaccine candidate that might interact directly with the macrophages [31].
A DNA vaccine containing the GP63 protein of L. donovani T-cell epitopes was projected to reduce the parasite load in the liver and spleen of the tested mice [32]. The GP63 protein of L. infantum was also reported as a potent immuno-dominant epitope that is competent enough to induce an immune response and elicit the infection against L. infantum [33,34,35].
The recent advances and extensive research in vaccine designing and development have provided new insights for the Leishmania infection [36,37]. Moreover, it helps to design new therapeutics and epitope vaccines for the molecular targets with low cost [38] and providing a useful therapeutic tool to combat the infection [39]. The present study utilized a combination of immuno-informatics strategies to develop a subunit-epitope vaccine against CL, by obtaining antigenicity, allergenicity, as well as physiochemical properties, for the vaccine protein. To check the complex stability and binding energy, molecular docking and dynamic simulations of the vaccine constructs were also carried out. The GP63 protein of Leishmania major was used in this study to develop a novel vaccine construct that may help in preventing CL infection in human hosts.
2. Materials and Methods
2.1. Study Design
The design of a multi-epitope vaccine involved numerous technique steps. Figure 1 provides a summary of the general process utilized to design a multi-subunit vaccine and pipeline for the current study.
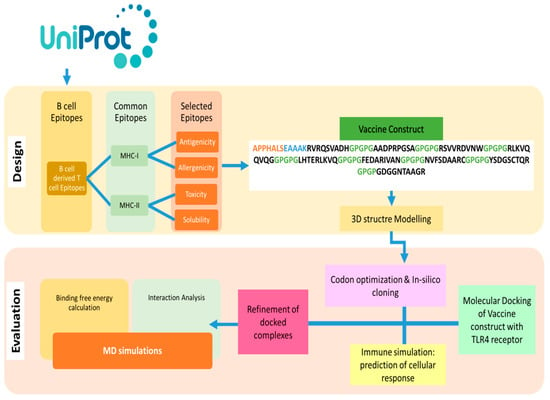
Figure 1.
Outline of methodology followed in this study to find B and T cell epitopes and designed MEVs using immunoinformatics methods is shown in the flow chart above. Additionally, biophysical investigation was carried out using integrated docking, modeling, and binding free energies methodologies to determine the vaccine’s affinity for immunological receptors.
2.2. Sequence Retrieval and Antigenicity Prediction
The FASTA formatted full amino acid sequence of the GP63 protein (ID: P08148) from L. major was retrieved from Uniprot at www.uniprot.org. The antigenic nature of a protein, or its capacity to generate an immunological response within the host body, was screened using the Vaxijen 2.0 antigen prediction service (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) (accessed on 2 March 2022). This server focuses on the auto cross-covariance (ACC) transformation and alignment-independent prediction, which both retain a predictive accuracy between 70–89% [40].
2.3. Immunoinformatics Analysis
2.3.1. B-Cell Epitope Prediction
The B-cell epitopes were predicted from the full-length protein sequences using the BCPreds method with a cutoff score of >0.8 (http://tools.iedb.org/main/bcell/) (accessed on 2 March 2022). The antigenicity of the predicted B-Cell epitopes was assessed using VaxiJen 2.0 with a threshold of 0.4 [41].
2.3.2. MHC-I and MHC-II Epitopes Prediction
The CTL epitopes (9-mer) were predicted through consensus approaches, using the EDB major histocompatibility complex MHC-I binding tool (http://tools.iedb.org/mhci/) (accessed on 4 March 2022) [42]. In this investigation, the MHC allele frequency was modified using the HLA allele reference set and the suggested algorithm from IEDB 2.1 [43]. The IEDB recommended technique was utilized to predict the HTL epitopes (15-mer) using the IEDB MHC-II binding tool (https://tools.iedb.org/mhcii/) (accessed on 4 March 2022) [44].
2.3.3. Epitopes Mapping
In order to determine the binding affinity potential for the dominant HLA II DRB*0101, the chosen epitopes were then employed in MHCPred 2.0. Only those with IC50 values of 100 nM were determined to be excellent DRB*0101 binders. After setting the cut-off to >0.6, VirulentPred and Vaxijen 2.0 was used to highlight the antigenic epitopes. Two more online servers, AllerTOP v2.0 (https://www.ddg-pharmfac.net/AllerTOP/) (accessed on 4 March 2022) [45] and ToxinPred (https://webs.iiitd.edu.in/raghava/toxinpred/protein.php) (accessed on 4 March 2022) [46], were used to check the toxicity and allergenicity, respectively, and all of the parameters were left at their default settings to ensure an 88.9% prediction accuracy. We used the established peptide affinity measurements and, as the IC50 values 100nM are regarded as indicating the significant affinity, we used that value to select the epitopes for further consideration [47]. Furthermore, the non-toxic epitopes were tested using the IFN-epitope server (https://webs.iiitd.edu.in/raghava/ifnepitope/index.php) (accessed on 4 March 2022).
2.3.4. MEVC Designing and Post Analysis
The subunit vaccine was designed by using only the filtered epitopes, with GPGPG linkers placed at the intra-epitopic positions and APPHALS, a TLR4 peptide adjuvant, coming before it in the N-terminal and being joined by EAAK linkers [48]. The ProtParam tool of the EXPASSY server was used to examine the physiochemical characteristics of the designed MEVC and the SCRATCH protein server’s 3Dpro was used to model the three-dimensional (3D) structure of the vaccine construct from scratch [49]. Following that, loop modelling was carried out in the construct’s 3D structure using GlaxyLoop [32] from GlaxyWeb and improved using GlaxyRefine [50]. Disulphide engineering was used to improve the Design 2.0 model of the design because disulphide bonds increase the stability of the construction.
2.3.5. Codon Optimization and In-Silico Cloning
Additionally, the vaccine construct’s sequence was translated in a reversible manner to optimize the codon usage for the Escherichia coli (E. coli) K12 expression system and achieve a high rate of expression. The Java Codon Adaptation Tool (JCat) (CAI) was used to calculate the expression rate of the cloned vaccine construct and was subsequently cloned using SnapGene 4.2 (https://www.snapgene.com/snapgene-viewer/) (accessed on 6 March 2022) into the E. coli pET28a (+) vector.
2.4. Molecular Docking of Vaccine with TLR4 Receptor
The minimal TLR4 was chosen as a receptor from the RCSB PDB library (PDB ID: 4G8A), and the vaccination construct was utilized as a ligand (https://www.rcsb.org/) (accessed on 6 March 2022). For molecular docking, the PatchDock server was used to evaluate the binding affinity between the designed vaccine construct and the minimized TLR4 receptor. PatchDock’s effective rigid docking technique maximizes the complementarity between geometric shapes [51]. The clustering Root Mean Square Deviation (RMSD) was left at its default value of 4.0, and the Fast Interaction Refinement in Molecular Docking (FireDock) server was used to modify the output docked solutions for the interactions [52]. The refined complex with the lowest global energy was ranked first after the refined complexes were examined.
2.5. Molecular Dynamics Simulation with Vaccine-TLR4 Complex
The complex (vaccine-TLR4) comprising the best vaccine construct selected in the previous phase was the only one for which a molecular dynamics simulation investigation was conducted, using the previously described methods [53]. AMBER 20 was used for the molecular dynamics simulation to evaluate and measure the protein flexibility and analysis of the intermolecular interactions was conducted using the FF14SB force filed. Additionally, several parameters, such as the RMSD (root mean square deviation), RMSF (root mean square fluctuations), salt bridges analysis, simulated trajectories, and others, were investigated to assess the complex stability.
2.6. Free Energy of Binding and Decomposition
Using the MMPBSA.py module of AMBER20, the free energies of the binding and per-residue free-energy decomposition were calculated [54]. The following equations were used to estimate the free binding energy of the designed complex, Gbind:
ΔGbind, solv = ΔGbind, vaccum + ΔGsolv, complex − ΔGsolv, ligand − ΔGsolv, complex
ΔGsolv = ΔGelectrostatic(ϵ80−1) + ΔGhydrophobic
ΔGvaccum = ΔEmolecular mechanics − T.ΔGnormal mode analysis
The net free binding energy was decomposed into the individual residues to see which ones interacted and remained stable.
3. Results
3.1. Protein Antigenicity
The antigenic score acquired upon the running sequence of the protein through the Vaxijen 2.0 server was 0.5768, which signified that the protein is up to par immunogenicity. Hence, after checking its antigenic potential, the protein was considered to design the vaccine on the basis of its antigenic score, >0.5. The TMHMM server 2.0 transmembrane topology prediction tools predicted only one transmembrane helix for the selected protein.
3.2. B-Cell Epitope Prediction
The IEDB B-cell epitope prediction tool was used to predict the linear B-lymphocytes (LBL) epitopes from the chosen protein candidate GP63, and the benchmarks for the selection from the projected findings included the linear epitope, illustrated in Figure 2. Nine peptides with eight or fewer amino acid residues were disqualified from the results, yielding a total of nineteen peptide fragments. Table 1 displays the remaining nine epitope candidates. According to an antigenicity score of 1.3077, a 22-mer peptide with the sequence EVEDQGGAGSAGSHIKMRNAQD at positions 321–342 had the most antigenic potential.
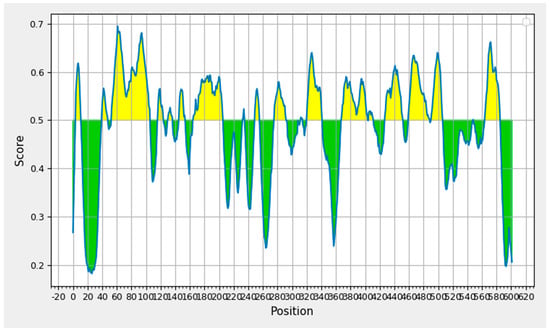
Figure 2.
B-cell epitope prediction based on prediction results obtained through Bepipred 2.0.

Table 1.
Selected B-cell epitopes based on linear epitope prediction method.
3.3. Prediction of MHC-I and MCH-II Binding Epitopes
These B-cell peptide sequences were evaluated for the T-cell epitope prediction and the binding sites for MHC-I and MHC-II were identified. The rapid immunological response caused by the CD+ T-cells’ recognition of the MHC-I molecules on the nucleated cell surface resulted in the death of the presenting cells. On the other hand, the MHC-II molecules were found on the antigen-presenting cells (APCs) and were recognized by the CD4+ T cells. Only those epitopes that are common to both classes were taken into consideration after filtering out the MHC-III predicted epitopes based on the percentile scores and comparing them with the MHC-I allele selected epitopes. The shortened 48 common MHC-I and MHC-II epitopes were tested for antigenicity. Here, the ability of the filtered T-cell epitopes produced from the B-cells to induce and bind with the products of adaptive immunity was examined. The 29 epitopes that were produced can bind with the most common DRB*0101, with an average IC50 score of 34.5, a maximum of 97.5, and a minimum of 3.24. In order to remove the allergic peptides that can result in allergic reactions, the antigenic epitopes underwent allergenicity validation. Nine epitopes were non-toxic and generated IFN-gamma, while eight allergic epitopes and nineteen non-allergenic epitopes were investigated. The final set of nine epitopes obtained through various rounds of the epitope mapping phase is given in Table 2, along with the additional information that six epitopes were likely non-antigens and six demonstrated poor solubility.
Table 2.
The filtered antigenic T-cell epitopes predicted for multi subunit peptide vaccine construct.
3.4. Construction of Multi-Epitope Peptide Vaccine (MEPVC)
The EAAK linker related to the adjuvant (50S ribosomal protein L7/L12, a TLR4 agonist) at the N-terminal of the vaccine construct in order to create a stable and coherent multi-epitope peptide vaccine construct. Then, a GPGPG linker was inserted between the epitope sequences to connect the prioritized B-cell-derived T-cell epitopes. The earlier studies have emphasized that TLR4 is a member of a larger class of toll-like receptor proteins that play a critical role in initiating the cascades of immune responses against an antigen, involving both the innate immune system and the adaptive immune system, and that the EAAAK linker amplifies the bioactivity of the vaccine protein. The schematic diagram of the chimera sequence is shown in Figure 3A and the final MEV construct composed of 119 amino acids residues are represented in Figure 3B,C.
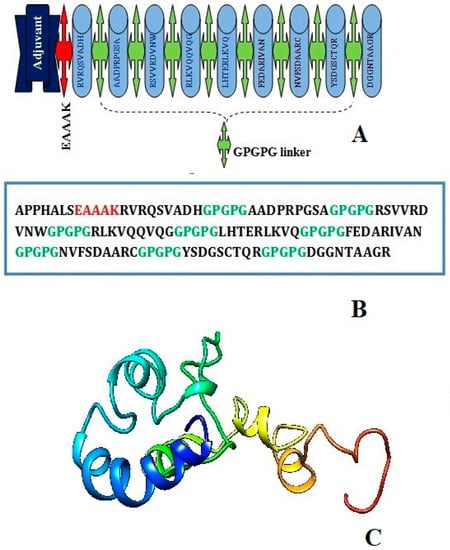
Figure 3.
Schematic diagram of construct comprised of 119 amino acid residues; out of which first seven amino acids are TLR4 adjuvant linked with five residues of linker followed by nine immunodominant epitopes joined together by GPGPG linker (A) chimera sequence (B) and 3D structure of original predicted vaccine construct (C).
3.5. Antigenic and Non-Allergic Evaluation of MEPVC
The conserved predicted epitopes from the preceding steps were further analyzed for allergenicity, antigenicity, and immunogenicity properties before conceding as the potential vaccine candidates. Thus, by following the analysis, we cut off allergenic, non-antigenic and toxic epitopes and the final eight epitopes from the above list were obtained by eliminating the allergen epitope. A 9-mer epitope, DGGNTAAGR, predicted allergen was discarded from further analysis. The AllerTOP 2.0, AlgPred, and AllergenFP 1.0 servers investigated the allergenicity of the multi-epitope vaccine that was ultimately developed. According to the results of AllerTOP 2.0, the designed build does not cause inflammatory reactions. According to ANTIGENpro’s and VaxiJen’s estimates of the probability of vaccination antigenicity, the MEPVC can effectively elicit cellular and humoral immune responses against the pathogens (0.6685 and 0.5872, respectively).
3.6. Physiochemical Assessment and Protein Stability
The Expassy server’s ProtParam tools revealed several important features, as shown in Table 3. The molecular weight of the vaccine construct was calculated to be around 11.8 kDa, and the theoretical pI of the protein was expected to be 9.68. Size exclusion chromatography can be used to separate such small size proteins and the projected pI value showed that the vaccine construct was substantially acidic in nature. There are 13 positively charged amino acid residues and 9 negatively charged amino acid residues in total. In addition, a half-life of 4.4 h in the mammalian reticulocytes (in vitro), >20 h in the yeast (in vivo), and >10 h in the Escherichia coli (in vivo) were calculated. The predicted instability index (II) was 25.96, as a value less than 40 is considered to be a stable protein, and this classifies the vaccine construct as stable. The construct’s aliphatic index was found to be 51.68, indicating it is thermo-stable. A high aliphatic index indicates that the protein is stable across a wide temperature range. Its GRAVY value was calculated to be −0.682; the negative score indicated that it is hydrophilic and has better contact with the water molecules around it. The Protein-sol and Solpro servers predicted the solubility of the vaccine with a high degree of accuracy [55]. The Protein-sol calculated 0.714 and the Sol-pro calculated 0.903, indicating that the proposed MEV is more soluble upon its overexpression in E. coli. To summarize, the developed vaccine is expected to be extremely acidic, thermo-stable, and hydrophilic.
Table 3.
Physiochemical properties of final vaccine construct.
3.7. Prediction of Secondary and Tertiary Structure and Validation
According to the RaptorX Property, the MEPVC consists of 9% α-helix, 12% β-sheets, and 78% coils. The predictions demonstrated that 75% of the constituent amino acid residues were exposed, 14% were medium, and 10% were buried in terms of solvent accessibility. As no suitable template for homology modeling and threading methods was available, the 3D model of the MEPVC was created using an ab initio SCRATCH Protein Predictor.
Furthermore, utilizing the GalaxyRefine server to refine a selected 3D structure of a multi-peptide vaccine, five 3D refined models were proposed. Model 5 had a higher Rama favored region (89.7) and overall acceptable GDT-HA (0.9769), RMSD (0.327), and MolProbity (2.341), as well as a lower clash score (16.5) and poor rotamers (1.2). As a result, this improved model was chosen as the best model for additional pool validation and was subjected to ProSA-web, Ramachandran Plot, and verified 3D model servers for the potential error evaluation. The refined model had a −2.37 z-score calculated through the ProSa-web, which is within a range of scores seen in native proteins of similar size (Figure 4A). According to the Ramachandran plot data, there were 66 (88%) residues in favorable, 8 (10.77%) residues in favored, 08 (10.7%) residues in allowed regions, and 1 (1.3%) residues in disallowed regions (Figure 4B). To assess the modelled structure, the ERRAT and verify 3D servers were used. The quality factor of the 3D refined model was 84.90 percent, according to the ERRAT findings (Figure 4C). The findings of the 3D score verification showed that 92.44% of the amino acid residues had a 3D-1D score >= 0.2. (Figure 4D) and in an improved 3D model, all of the residues were found to be in an acceptable side chain environment.
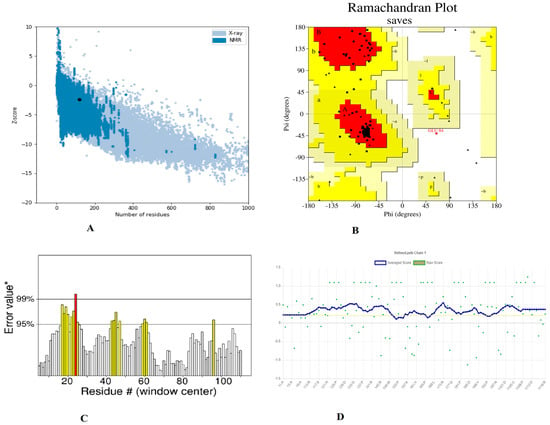
Figure 4.
Validation of the 3D structure model of refined vaccine construct (A). Z-core of construct model calculated −2.37 which in range of conformation scores of native protein (B). Ramachandran plot validation indicates; 88%, residues are in favored, 10.7% residues in allowed and 1.3% residues in disallowed region (C). ERRAT factor of final construct structure was 84.90%. In ERRAT plot, gray lines are showing regions of 3D model that can be rejected at 95% confidence level and yellow lines depicting regions that can be rejected at 99% level (D). The 3D score of the final model was 92.44% and amino acid residues with an average 3-1D score greater than zero are regarded as reliable.
3.8. Disulphide Engineering, Codon Optimization and In Silico Cloning Analysis
The MEVP was disulphide engineered to improve the molecular interactions and provide significant stability by obtaining the accurate geometric conformation. Thirteen pairs of residues were selected to be replaced with cysteine amino acids. These pairs are ALA1-GLY66 (χ3 angle, −65.07, energy value, 5.16 kcal/mol), PRO2-ARG45 (χ3 angle, +71.08, energy value, 2.6 kcal/mol), VAL14-VAL18 (χ3 angle, +112.54, energy value, 5.62 kcal/mol), GLY26-VAL43 (χ3 angle, −79.7, energy value, 2.69 kcal/mol), ALA27-GLY33 (χ3 angle, −107, energy value, 3.79 kcal/mol), PRO39-LYS57 (χ3 angle, −65.07, energy value, 5.16 kcal/mol), SER42-ARG73 (χ3 angle, +117.5, energy value, 4.85 kcal/mol), VAL47-GLN59 (χ3 angle, +98.63, energy value, 2.54 kcal/mol), PRO51-ARG55 (χ3 angle, +78.8, energy value, 2.63 kcal/mol), VAL61-GLY64 (χ3 angle, −114, energy value, 5.16 kcal/mol), GLY80-ASP85 (χ3 angle, +124.83, energy value, 4 kcal/mol), ASN91-ARG104 (χ3 angle, +101.81, energy value, 6.56 kcal/mol), PRO95-VAL98 (χ3 angle, +121.4, energy value, 2.42 kcal/mol). These residues have either a higher energy level i.e., >2 kcal/mol, or a χ3 angle out of range (<−79 and +71), and were selected on purpose for their stability. Disulphide bonds are a form of post-translational modification that often determines the protein structure and function. They also protect proteins against oxidants and proteolytic enzymes in extracellular environments, which can render proteins inactive. Disulfide linkages can increase the half-life of proteins and protect them from deterioration by stabilizing the proteins’ structure. Figure 5A depicts the MEPVC’s native and disulphide mutant structures. The native and mutant structures of the MEPVC were superimposed (Figure 5B), while the RMSD value for 76 pruned pairs is 0.650 Å, and across all 119 pairs, 7.128 Å. Codon optimization was then applied to the translated sequence using the JCat web server to produce a high-level protein expression in E. coli. Our optimized nucleotide sequence has a codon adaptation index (CAI) of 0.933 and a nucleotide sequence length of 64.426. These findings suggested that this optimized DNA sequence would have the highest level of expression in E. coli (Figure 6).
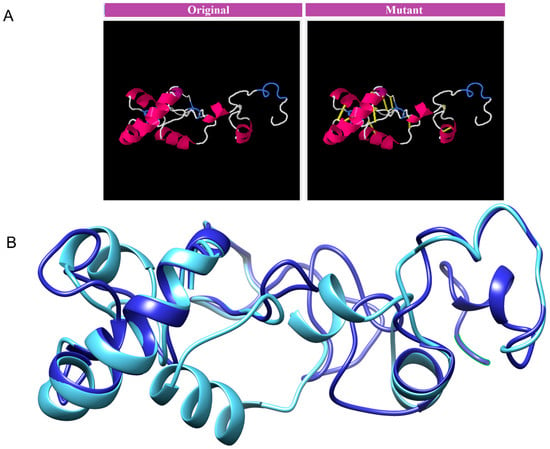
Figure 5.
The original and mutant disulphide structures of vaccine construct (A). Superimposed model for the vaccine model and its mutant (B).
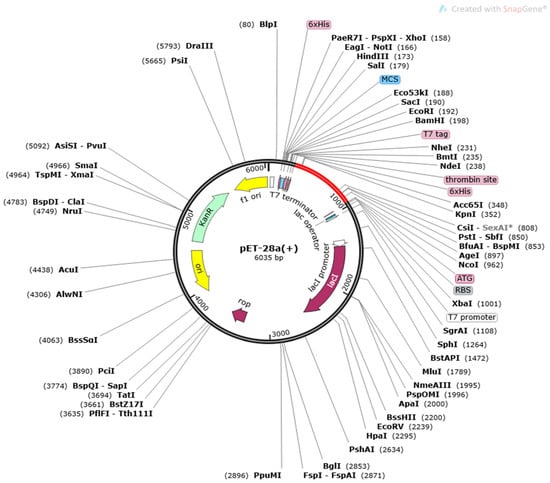
Figure 6.
In silico cloning of a vaccine construct into the pET28a (+) vector, with the region of interest highlighted in red and surrounded by XhoI (158) and NdeI (1788), and the vector highlighted in black lines.
3.9. Docking Interaction of MEPVC and TLR4 Receptor
In order to decipher the MEPVC’s potential for binding to induce the innate immune response, bioinformatics modeling-driven molecular docking of the proposed MEPVC to one representative innate immune response receptor (TLR4) was performed. The docking evaluation predicted the top 20 complexes, which were predominantly sorted based on the scoring function and the area size of the interacting molecules. The real rigid transformations of the complexes were then submitted to the FireDock online server for the refinement experiments. This permits a deep refinement of the predictions and makes it possible to minimize the docking procedure flexibility flaws, which lowers the possibility of false positive docking computations. With a net global energy of 8.12 kJ/mol, solution 5 was rated as having the highest level of energy. This energy is a combination of −0.57 kJ/mol hydrogen bond energy, 0.16 kJ/mol repulsive van der Waals, and −1.18 kJ/mol attractive van der Waals (vdW) (Table 4). The docked conformation of the MPEV with TLR4 and chemical interaction residues are illustrated in Figure 7. The visual assessment of the complex reveals deep MEPVC binding at the TLR4’s center, which favors weak van der Waals and rigorous hydrogen contacts with the other TLR4 residues.
Table 4.
Refined PatchDock complexes as an outcome of FireDock assay.
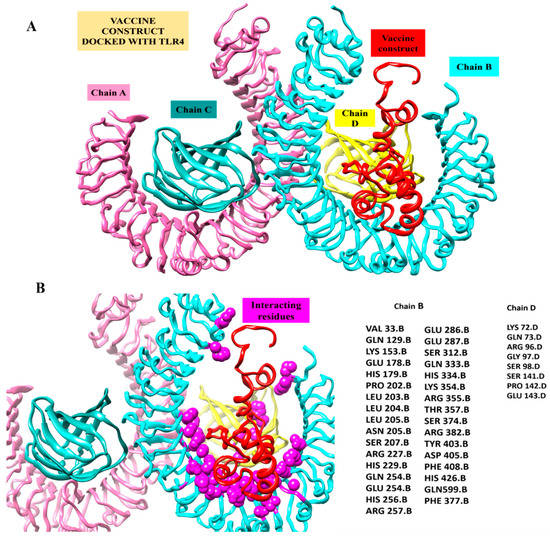
Figure 7.
Inspection of a proposed chimeric peptide vaccine construct and the TLR4 complex using molecular docking. (A) Vaccine construct’s predicted docked mode in relation to TLR4. The vaccine construct is shown in red using the New-Cartoon drawing approach, while the TLR4 chains are depicted using various colored beads: Chain A (plum) Chain B (cyan) chain C (dark cyan) and Chain D (yellow). (B) Vaccine construct’s interactions within five Angstrom region of TLR4 Receptor. Chains B (cyan) and D (Yellow) shows interaction with Vaccine construct. The vaccine design is shown in a red cartoon, while TLR4 chain interaction residues depicted as spheres in magenta color.
3.10. MD Simulation Assays to Study Conformational Stability and Residual Flexibility
Molecular Dynamics Simulation (MDS) is a widely used technique for examining micro-interactions between vaccine/ligand and receptor/protein complexes. To obtain a better understanding of the dynamics and stability, we ran 100 ns MD simulations of the vaccine ensemble docked-complex with TLR4 followed by RMSD and RMSF. Through a 100 ns MD simulation production run, the stability of the vaccine construct-TLR4 interaction and the complex’s dynamic behavior were clarified. Figure 8 illustrates the many statistical metrics used to assess the system stability and structural changes necessary to ensure that the vaccine construct adheres properly to the TLR4 binding site. By graphing the root mean square deviation (RMSD) over time, we were able to determine and define the complex’s conformational stability. The RMSD is the distance between the backbone carbon alpha atoms of the stacked proteins. The system exhibited a steadily growing RMSD, initially gradually rising until it reaches 40 ns, but then reached equilibrium for a short time, and the RMSD stayed uniform until 70 ns. The RMSD rose when the convergence between 80 and 100 ns was seen. However, no significant convergence indicated that the TLR4-vaccine complex is stable. Overall, the findings revealed that the complex exhibited stable behavior over the 100 ns simulation, as shown in Figure 8A. The RMSF was used to determine each complex’s residual flexibility. The residual fluctuation in the TLR4-vaccine complex was larger within residues 430–600 and 1000–1230. Increased residual fluctuation was seen in the complex. Overall, the results reveal that the docked complex has substantial behavior. Figure 8B shows the RMSFs of the complexes.
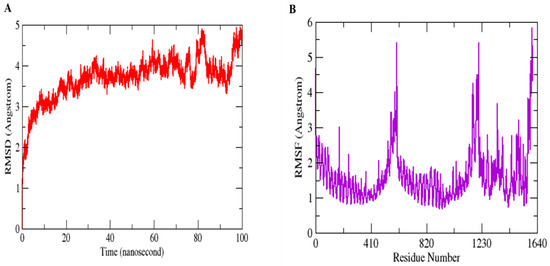
Figure 8.
MD simulation paths are statistically analyzed. RMSD (A) and RMSF (B) are the two output values shown here.
3.11. Determination of the Binding Free Energy of TLR4-Vaccie Ensemble Complexes
The MM-PBSA was utilized as a post-simulation processing to verify the vaccine construct’s affinity for TLR4, and the MD simulation trajectories were used to determine the molecules’ free energies in solution. As an end state free energy computation method, MM-PBSA.py was used because it is user-friendly, more accurate than docking scoring, and cheaper than free energy perturbation. The various binding free energies discovered using the GB and PB techniques are summarized in Table 5 and Table 6. The MM-PBSA analysis found that the net delta energy in GB was −272.2354 kcal/mol and in PB was −410.5471 kcal/mol. The delta energies of the complex, TLR4, and vaccine construct are −144,847.5215 kcal/mol, −109,303.9694 kcal/mol, and −35,271.3167 kcal/mol, respectively, in GB. The vaccine design contributed the most to PB (−35,207.3949 kcal/mol), followed by the complex (−144,514.7558 kcal/mol) and the TLR4 receptor (−108,896.8137 kcal/mol). In both GB and PB, the net electrostatic energy is substantially dominant and contributes favorably to the net binding energy. The system is projected to provide a net electrostatic energy contribution of −4443.1483 kal/mol to both GB and PB. In both GB and PB, the electrostatic contribution of the vaccine construct (−113,422.7858 kcal/mol) to the net PB is much greater than that of the receptor TLR4 (−81,782.776 kcal/mol) and the complex (−27,196.8614 kcal/mol). Additionally, the van der Waals energy is advantageous to the total free energy. This energy is −467.0527 kcal/mol for both GB and PB (complex = −13,331.9017 kcal/mol, TLR4 receptor = −10,091.7946 kcal/mol, vaccine construct = −2773.0544 kcal/mol). The net solvation free energy is found to be less than the total energy in both GB (4637.9657 kcal/mol) and PB (4499.654 kcal/mol), owing mostly to the polar energy (GB = 4705.7822 kcal/mol and PB = 4552.4332 kcal/mol). In comparison, non-polar salvation seems to contribute just a little amount, as GB has a −67.8165 kcal/mol and PB has a −52.7792 kcal/mol.
Table 5.
Calculation of the generalized Born ESURF utilizing ‘LCPO’ surface areas. Each value is given in kcal/mol.
Table 6.
Calculations of the Poisson Boltzmann equations are carried out utilizing sander’s internal PBSA solver. Each value is given in kcal/mole.
In order to specify the TLR4 residues that serve as a hotspot for binding or stabilizing the vaccine construct at the docked location, the net free energy of the binding in both PB and GB was further deconstructed into each TLR4 residue. For the purpose of learning more about the local interactions in a system, free energy must be decomposed. It enables the user to figure out how much each residue contributes to the net total free energy. The TLR4 and vaccine design residues in GB and PB that significantly contribute to the stability of the complex are listed in Table 7.
Table 7.
Hotspot residues from TLR4 and Vaccine ensemble highly contributes to complex stabilization.
3.12. TLR4-MEPVC Stability and Salt Bridges
When two ionized states come into contact, salt bridges, which are non-covalent structures, arise. These interactions involve both a hydrogen bond and an electrostatic contact. In salt bridges, glutamine or aspartate serves as the acid and lysine or arginine serves as the base. The bridge is created when a proton can move from the carboxylic acid group to the amine groups of guanidine and arginine. The strongest non-covalent contacts are salt bridges, which are important for bimolecular stability. As shown in Figure 9, there were six salt bridges detected between the TLR4 (Lys732, Glu757, Arg806, Glu955, Glu1004, Lys1056) and the vaccine ensemble (Asp1506, Arg1517, Arg1527, Glu1558 and 1570 within the cut-off distance of 3.2 Å).
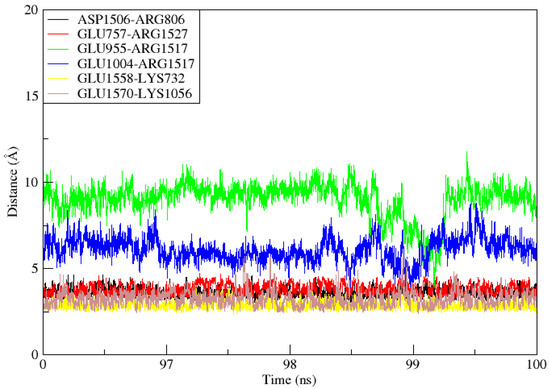
Figure 9.
During the 100ns simulation period, salt bridges developed between TLR4 and Vaccine ensemble.
4. Discussion
The process of finding a new vaccine candidate and validating it in vitro and in vivo is costly and very time consuming [56]. However, immunoinformatics and bioinformatics technologies are now more effective than one antigen or classic deactivated pathogen vaccines and it is time saving to design multi-epitope peptide-based vaccinations [57]. Some significant studies have also demonstrated the benefits and validity of vaccines developed using these methods. These approaches are quite useful for swiftly screening antigenic vaccine compounds and many peptide-based vaccines for infectious diseases developed with immunoinformatics technologies have been experimentally confirmed and are now in use as effective vaccines [58].
In the present study, we designed a peptide-based vaccine using immunoinformatics tools against the L. major parasite that causes CL. Based on earlier research, we predicted several epitopes derived from the L. major antigenic heat shock protein, GP63. Heat shock proteins (HSPs) are intracellular proteins that are extremely conserved molecules that perform key roles in protein complex formation, protein folding, and protein translocation in parasite cells, as well as being involved in a variety of immunological processes [59]. GP63 was previously identified as a CL vaccine target in an immunoproteomics study that showed above 90% sequence similarity with various other Leishmania species, including L. infantum and L. donovani. On the surface of Leishmania, there is a zinc-dependent metalloprotease known as GP63, also known as leishmanolysin, which causes human humoral reactions [60,61]. It has been discovered that metalloproteases GP63, the main Leishmania surface antigen, serve a variety of vital roles in parasite’s survival. Multiple genes, whose copy counts change significantly between various species, encode GP63. One of the several vaccine possibilities being tested, primarily against CL, is Gp63 protein and numerous studies point to this surface-expressed virulence factor’s crucial function. The L. donovani GP63 surface protein’s possibly immunogenic T cell epitopes were designed utilizing the EpiMatrix tool kit in a study [1,12]. Additionally, in preliminary research, Gp63 antigens and a novel recombinant vaccine against L. infantum created using computational methods were chosen as promising immunodominant epitopes to elicit immunological responses. Hence, the current in-silico study was designed to identify the immunogenic epitopes of Gp63of L. major as a basis for future vaccinology studies.
In the immune system, T-cells detect peptide epitopes, which are provided by MHC molecules. These molecules are antigen-presenting cell surface proteins that are recognized by T-cell receptors (TCRs) and are divided into two classes. Almost all nucleated cells include Class I MHC molecules, which present processed proteins to CTLs via the cytosolic pathway. Class II MHC molecules, on the other hand, are present on antigen-presenting cells and represent pathogen surface proteins that are delivered to CD4+ T cells and helper T lymphocytes via the endocytic pathways [62,63]. We predicted B-cells-derived T-cells that promote the humoral and cellular immune system. Based on the structural characteristics and physiochemical properties, the immunoinformatics study revealed that our proposed multi-epitope vaccine has a large number of high affinities epitopes. Although multi-epitope vaccines offer many advantages, their most considerable disadvantage is their low immunogenicity [64]. To solve this problem, adjuvants are added to the N-terminal of the construct involved in immune system stimulation. However, several clinical patterns suggest that the T-cell responses, particularly the Th1 effector mechanisms, appear to be involved in the acquired resistance to the leishmaniasis.
Consequently, designing a successful vaccination can be possible if the suitable antigens are chosen and combined with adjuvants that stimulate a Th1 immune response [65] TLR4 is unique among the other TLRs as it engages both the MyD88/MAL and TRIF/TRAM signaling pathways and triggers TFN generation and NF-κB induction at the same time [66]. This adjuvant, on the other hand, secretes IFN- and IL2-components that fight against protozoan parasites by activating CD+ and CD8+ T-cells [67]. We also examined whether the multi-epitope peptide vaccine had a substantial affinity for the TLR4 receptor, and MD modelling confirmed the stability of the vaccine-TLR4 complex. As a result, the vaccine-TLR4 complex may trigger the TLR4-dependent signaling pathways that protect against the infection of Leishmania. In order to promote the bioactivity, stability and in peptide structure of the vaccine, EAAAK also linked the adjuvant TLR4 to the start point. The EAAAK linker is quite rigid, with a helical structure that is used to create and maintain space between the functional domains [68]. Linkers, which play both functional and structural roles in vaccine construction, are an important component of the multi-epitope peptide vaccine [69,70]. Additionally, GPGPG linker was used to join the assembled protein in this study. This flexible linker, which is made up of tiny non-polar amino acids similar to glycine and polar amino acids i.e., threonine and serine, was used to connect the functional domains that required inter-domain interactions. These linkers also provide flexibility and mobility to the multi-epitope vaccine construct [71].
Further, we assessed the physiochemical features, such as the toxic potential and allergic nature, of the final vaccine construct. It was found that the designed vaccine was highly antigenic, non-allergic, thermostable, and non-toxic. Thus, the secondary and the tertiary structures were investigated. The secondary structure is made up of −helix (9%), sheets (12%), and coils (78%) and the final construct’s 3D model was evaluated and confirmed to have a stable 3D structure. The interaction of the vaccination with an innate immune receptor (TLR4) was examined using this improved docking mode. TLRs are cell-surface receptors that are present on dendritic cells, macrophages, and some other immune cells and are able to recognize specific epitopic regions on parasites [72]. To combat the infection, it forms a complex and starts a downstream cascade. It has been observed that the TLR4 receptor had a considerable affinity for MEVS, and MD simulations also validated the stability of the vaccine-TLR4 complex. As a result, the vaccine-TLR4 complex may trigger the TLR4-dependent signaling pathways that protect against the infection of Leishmania. This in silico-developed vaccine has significant immunogenic potential and should be evaluated for an in vitro experimental study in the next phase of research. The current in silico study obtained thoroughly screened potential immunogenic epitopes for the L. major gp63 protein that could be used alone or in combination with other candidate antigens/epitopes to engineer a finely tuned, multi-epitope vaccine construct to be tested against CL in the ongoing vaccinology studies.
5. Conclusions
On the basis of this in silico study, future in-vitro and in-vivo studies should confirm the studied vaccine candidate’s performance in terms of the effective dose, cross-reaction, lymphocyte proliferation, cytokine production assays, as well as any potential toxicity in the relevant animal model challenges.
Author Contributions
S.W.A. and S.N. participated in study design, conception, interpretation of result and overall supervised the research work. A.A. performed the analysis. Y.O., A.C., E.A.Ş. and S.T. made critical revisions and provide technical help. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data for this study are contained within the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
CL (cutaneous leishmaniasis), TLR-4 (toll-like receptor-4), VL (visceral leishmaniasis), MCL (muco-cutaneous leishmaniasis), AmB (amphotericin B), LPG (lipophosphoglycan), GP (glycoproteins), ACC (auto cross-covariance), CTL (cytotoxic T-cells), JCat (Java Codon Adaptation), RMSD (root mean square deviation), RMSF (root mean square fluctuations), LBL (linear B-lymphocytes), CAI (codon adaptation index), HSPs (Heat shock proteins).
References
- Elmahallawy, E.K.; Martínez, A.S.; Rodriguez-Granger, J.; Hoyos-Mallecot, Y.; Agil, A.; Mari, J.M.N.; Fernández, J.G. Diagnosis of leishmaniasis. JIDC 2014, 8, 961–972. [Google Scholar] [CrossRef] [PubMed]
- De Freitas, E.O.; Leoratti, F.M.D.S.; Freire-de-Lima, C.G.; Morrot, A.; Feijó, D.F. The contribution of immune evasive mechanisms to parasite persistence in visceral leishmaniasis. Front. Immunol. 2016, 7, 153. [Google Scholar] [CrossRef] [PubMed]
- Sunyoto, T.; Potet, J.; Boelaert, M. Why miltefosine—A life-saving drug for leishmaniasis—Is unavailable to people who need it the most. BMJ Glob. Health 2018, 3, e000709. [Google Scholar] [CrossRef] [PubMed]
- Desjeux, P. Leishmaniasis: Current situation and new perspectives. Comp. Immunol. Microbiol. Infect. Dis. 2004, 27, 305–318. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Health Statistics; World Health Organization: Geneva, Switzerland, 2010. [Google Scholar]
- Pashaei, A.; Ghatee, M.; Sajedi, H. Convolution neural network joint with mixture of extreme learning machines for feature extraction and classification of accident images. J. Real Time Image Process. 2020, 17, 1051–1066. [Google Scholar] [CrossRef]
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M.; Team, W.L.C. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef]
- Ashour, A.; Atia, A.; Akash, N.; Jumaa, E.; Alkhishrabi, A. Cutaneous Leishmaniasis in Al-Jabal Al-Gharbi, Libya: Incidence and Epidemiology. Khalij-Libya J. Dent. Med. Res. 2022, 6, 81–85. [Google Scholar] [CrossRef]
- de Souza Machado, A.A.; Kloas, W.; Zarfl, C.; Hempel, S.; Rillig, M.C. Microplastics as an emerging threat to terrestrial ecosystems. Glob. Chang. Biol. 2018, 24, 1405–1416. [Google Scholar] [CrossRef]
- Zijlstra, E.E. Biomarkers in post-kala-azar dermal leishmaniasis. Front. Cell. Infect. Microbiol. 2019, 9, 228. [Google Scholar] [CrossRef]
- Akhtari, J.; Soosaraei, M.; Ziaei, H.; Fakhar, M. Last decade developments on Leishmania vaccines with emphasis on nanovaccines. J. Maz. Univ. Med. Sci. 2017, 26, 232–253. [Google Scholar]
- Abu Ammar, A.; Nasereddin, A.; Ereqat, S.; Dan-Goor, M.; Jaffe, C.L.; Zussman, E.; Abdeen, Z. Amphotericin B-loaded nanoparticles for local treatment of cutaneous leishmaniasis. Drug Deliv. Transl. Res. 2019, 9, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Kip, A.E.; Schellens, J.H.; Beijnen, J.H.; Dorlo, T.P. Clinical pharmacokinetics of systemically administered antileishmanial drugs. Clin. Pharmacokinet. 2018, 57, 151–176. [Google Scholar] [CrossRef] [PubMed]
- Alexandrino-Junior, F.; Silva, K.G.D.H.E.; Freire, M.C.L.C.; Lione, V.D.O.F.; Cardoso, E.A.; Marcelino, H.R.; Genre, J.; Oliveira, A.G.D.; Egito, E.S.T.D. A functional wound dressing as a potential treatment for cutaneous leishmaniasis. Pharmaceutics 2019, 11, 200. [Google Scholar] [CrossRef] [PubMed]
- Minodier, P.; Parola, P. Cutaneous leishmaniasis treatment. Travel Med. Infect. Dis. 2007, 5, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Chakravarty, J. An update on pharmacotherapy for leishmaniasis. Expert Opin. Pharmacother. 2015, 16, 237–252. [Google Scholar] [CrossRef]
- dos Santos Nogueira, F.; Avino, V.C.; Galvis-Ovallos, F.; Pereira-Chioccola, V.L.; Moreira, M.A.B.; Romariz, A.P.P.L.; Molla, L.M.; Menz, I. Use of miltefosine to treat canine visceral leishmaniasis caused by Leishmania infantum in Brazil. Parasites Vectors 2019, 12, 79. [Google Scholar] [CrossRef]
- Osorio, Y.; Travi, B.L.; Renslo, A.R.; Peniche, A.G.; Melby, P.C. Identification of small molecule lead compounds for visceral leishmaniasis using a novel ex vivo splenic explant model system. PLoS Negl. Trop. Dis. 2011, 5, e962. [Google Scholar] [CrossRef]
- Lieke, T.; Nylen, S.; Eidsmo, L.; McMaster, W.; Mohammadi, A.; Khamesipour, A.; Berg, L.; Akuffo, H. Leishmania surface protein gp63 binds directly to human natural killer cells and inhibits proliferation. Clin. Exp. Immunol. 2008, 153, 221–230. [Google Scholar] [CrossRef]
- Mangoni, M.L.; Saugar, J.M.; Dellisanti, M.; Barra, D.; Simmaco, M.; Rivas, L. Temporins, small antimicrobial peptides with leishmanicidal activity. J. Biol. Chem. 2005, 280, 984–990. [Google Scholar] [CrossRef]
- Isnard, A.; Shio, M.; Olivier, M. Impact of Leishmania metalloprotease GP63 on macrophage signaling. Front. Cell. Infect. Microbiol. 2012, 2, 72. [Google Scholar] [CrossRef]
- Berberich, C.; Ramírez-Pineda, J.R.; Hambrecht, C.; Alber, G.; Skeiky, Y.A.; Moll, H. Dendritic cell (DC)-based protection against an intracellular pathogen is dependent upon DC-derived IL-12 and can be induced by molecularly defined antigens. J. Immunol. Res. 2003, 170, 3171–3179. [Google Scholar] [CrossRef] [PubMed]
- Sacks, D.L. Vaccines against tropical parasitic diseases: A persisting answer to a persisting problem. Nat. Immunol. 2014, 15, 403–405. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Chakravarty, J.; Meena, L.P. Leishmaniasis: Treatment, drug resistance and emerging therapies. Expert Opin. Orphan Drugs. 2019, 7, 1–10. [Google Scholar] [CrossRef]
- Bahrami, A.A.; Payandeh, Z.; Khalili, S.; Zakeri, A.; Bandehpour, M. Immunoinformatics: In silico approaches and computational design of a multi-epitope, immunogenic protein. Int. Rev. Immunol. 2019, 38, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Raoufi, E.; Hemmati, M.; Eftekhari, S.; Khaksaran, K.; Mahmodi, Z.; Farajollahi, M.M.; Mohsenzadegan, M. Epitope prediction by novel immunoinformatics approach: A state-of-the-art review. Int. J. Pept. Res. Ther. 2020, 26, 1155–1163. [Google Scholar] [CrossRef]
- Xu, C.; Ye, B.; Han, Z.; Huang, M.; Zhu, Y. Comparison of transcriptional profiles between CD4+ and CD8+ T cells in HIV type 1-infected patients. AIDS Res. Hum. Retrovir. 2014, 30, 134–141. [Google Scholar] [CrossRef]
- Mansueto, P.; Vitale, G.; Di Lorenzo, G.; Rini, G.; Mansueto, S.; Cillari, E. Immunopathology of leishmaniasis: An update. Int. J. Immunopathol. Pharmacol. 2007, 20, 435–445. [Google Scholar] [CrossRef]
- Rhaiem, R.B.; Houimel, M. Targeting Leishmania major parasite with peptides derived from a combinatorial phage display library. Acta Trop. 2016, 159, 11–19. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Toth, I. Peptide-based synthetic vaccines. Chem. Sci. 2016, 7, 842–854. [Google Scholar] [CrossRef]
- Conceicao, J.; Davis, R.; Carneiro, P.P.; Giudice, A.; Muniz, A.C.; Wilson, M.E.; Carvalho, E.M.; Bacellar, O. Characterization of neutrophil function in human cutaneous leishmaniasis caused by Leishmania braziliensis. PLoS Negl. Trop. Dis. 2016, 10, e0004715. [Google Scholar] [CrossRef]
- Sachdeva, R.; Banerjea, A.C.; Malla, N.; Dubey, M.L. Immunogenicity and efficacy of single antigen Gp63, polytope and polytopeHSP70 DNA vaccines against visceral Leishmaniasis in experimental mouse model. PLoS ONE 2009, 4, e7880. [Google Scholar] [CrossRef] [PubMed]
- Hashemzadeh, P.; Ghorbanzadeh, V.; Lashgarian, H.E.; Kheirandish, F.; Dariushnejad, H. Harnessing Bioinformatic approaches to design novel multi-epitope subunit vaccine against Leishmania infantum. Int. J. Pept. Res. Ther. 2020, 26, 1417–1428. [Google Scholar] [CrossRef]
- Firouzmand, H.; Sahranavard, M.; Badiee, A.; Khamesipour, A.; Alavizadeh, S.H.; Samiei, A.; Soroush, D.; Tavassoti Kheiri, M.; Mahboudi, F.; Jaafari, M.R. The role of LPD-nanoparticles containing recombinant major surface glycoprotein of Leishmania (rgp63) in protection against leishmaniasis in murine model. Immunopharmacol. Immunotoxicol. 2018, 40, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Castro Neto, A.L.; Brito, A.N.; Rezende, A.M.; Magalhães, F.B.; de Melo Neto, O.P. In silico characterization of multiple genes encoding the GP63 virulence protein from Leishmania braziliensis: Identification of sources of variation and putative roles in immune evasion. BMC Genom. 2019, 20, 118. [Google Scholar] [CrossRef]
- Foroutan, M.; Ghaffarifar, F.; Sharifi, Z.; Dalimi, A.; Pirestani, M. Bioinformatics analysis of ROP8 protein to improve vaccine design against Toxoplasma gondii. Infect. Genet. Evol. 2018, 62, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Rasti, S.; Ghorbanzadeh, B.; Kheirandish, F.; Mousavi, S.G.; Pirozmand, A.; Hooshyar, H.; Abani, B. Comparison of molecular, microscopic, and culture methods for diagnosis of cutaneous leishmaniasis. J. Clin. Lab. Anal. 2016, 30, 610–615. [Google Scholar] [CrossRef]
- Moafi, M.; Rezvan, H.; Sherkat, R.; Taleban, R. Leishmania vaccines entered in clinical trials: A review of literature. Int. J. Prev. Med. 2019, 10, 95. [Google Scholar]
- Tahir ul Qamar, M.; Rehman, A.; Tusleem, K.; Ashfaq, U.A.; Qasim, M.; Zhu, X.; Fatima, I.; Shahid, F.; Chen, L.-L. Designing of a next generation multiepitope based vaccine (MEV) against SARS-COV-2: Immunoinformatics and in silico approaches. PLoS ONE 2020, 15, e0244176. [Google Scholar] [CrossRef]
- Esboei, B.R.; Mohebali, M.; Mousavi, P.; Fakhar, M.; Akhoundi, B. Potent antileishmanial activity of chitosan against Iranian strain of Leishmania major (MRHO/IR/75/ER): In vitro and in vivo assay. J. Vector Borne Dis. 2018, 55, 111. [Google Scholar]
- Mahendran, R.; Jeyabaskar, S.; Sitharaman, G.; Michael, R.D.; Paul, A.V. Computer-aided vaccine designing approach against fish pathogens Edwardsiella tarda and Flavobacterium columnare using bioinformatics softwares. Drug Des. Dev. Ther. 2016, 10, 1703. [Google Scholar] [CrossRef]
- Caro-Gomez, E.; Gazi, M.; Goez, Y.; Valbuena, G. Discovery of novel cross-protective Rickettsia prowazekii T-cell antigens using a combined reverse vaccinology and in vivo screening approach. Vaccine 2014, 32, 4968–4976. [Google Scholar] [CrossRef]
- Gaafar, B.; Ali, S.A.; Abd-Elrahman, K.A.; Almofti, Y.A. Immunoinformatics approach for multiepitope vaccine prediction from H, M, F, and N proteins of Peste des Petits ruminants virus. J. Immunol. Res. 2019, 2019, 6124030. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, E.; Crusat, M.; Nieto, J.; Chicharro, C.; del Carmen Thomas, M.; Martínez, E.; Valladares, B.; Cañavate, C.; Requena, J.M.; López, M.C. Immunogenicity of HSP-70, KMP-11 and PFR-2 leishmanial antigens in the experimental model of canine visceral leishmaniasis. Vaccine 2008, 26, 1902–1911. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rafati, S.; Gholami, E.; Hassani, N.; Ghaemimanesh, F.; Taslimi, Y.; Taheri, T.; Soong, L. Leishmania major heat shock protein 70 (HSP70) is not protective in murine models of cutaneous leishmaniasis and stimulates strong humoral responses in cutaneous and visceral leishmaniasis patients. Vaccine 2007, 25, 4159–4169. [Google Scholar] [CrossRef]
- Mazumder, S.; Maji, M.; Das, A.; Ali, N. Potency, efficacy and durability of DNA/DNA, DNA/protein and protein/protein based vaccination using gp63 against Leishmania donovani in BALB/c mice. PLoS ONE 2011, 6, e14644. [Google Scholar] [CrossRef] [PubMed]
- Elfaki, M.E.; Khalil, E.A.; De Groot, A.S.; Musa, A.M.; Gutierrez, A.; Younis, B.M.; Salih, K.A.; El-Hassan, A.M. Immunogenicity and immune modulatory effects of in silico predicted L. donovani candidate peptide vaccines. Hum. Vaccin. Immunother. 2012, 8, 1769–1774. [Google Scholar] [CrossRef]
- Lang, T.; de Chastellier, C.; Frehel, C.; Hellio, R.; Metezeau, P.; Leao, S.; Antoine, J.-C. Distribution of MHC class I and of MHC class II molecules in macrophages infected with Leishmania amazonensis. J. Cell Sci. 1994, 107, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 8285. [Google Scholar] [CrossRef] [PubMed]
- Groot, A.S.D.; Moise, L.; McMurry, J.A.; Martin, W. Epitope-based immunome-derived vaccines: A strategy for improved design and safety. Clin. Appl. Immunol. 2009, 2, 39–69. [Google Scholar]
- Reed, S.G.; Coler, R.N.; Campos-Neto, A. Development of a leishmaniasis vaccine: The importance of MPL. Expert Rev. Vaccines 2003, 2, 239–252. [Google Scholar] [CrossRef]
- Duthie, M.S.; Windish, H.P.; Fox, C.B.; Reed, S.G. Use of defined TLR ligands as adjuvants within human vaccines. Immunol. Rev. 2011, 239, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kim, W.S.; Choi, H.G.; Jang, B.; Lee, K.; Park, J.H.; Kim, H.J.; Cho, S.N.; Shin, S.J. Mycobacterium tuberculosis RpfB drives Th1-type T cell immunity via a TLR4-dependent activation of dendritic cells. J. Leukoc. Biol. 2013, 94, 733–749. [Google Scholar] [CrossRef] [PubMed]
- Le-Barillec, K.; Magalhaes, J.G.; Corcuff, E.; Thuizat, A.; Sansonetti, P.J.; Phalipon, A.; Di Santo, J.P. Roles for T and NK cells in the innate immune response to Shigella flexneri. J. Immunol. 2005, 175, 1735–1740. [Google Scholar] [CrossRef]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: Accurate sequence-based prediction of protein solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef] [PubMed]
- Setrerrahmane, S.; Yu, J.; Hao, J.; Zheng, H.; Xu, H. Novel production method of innovative antiangiogenic and antitumor small peptides in Escherichia coli. Drug Des. Dev. Ther. 2017, 11, 3207. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Ismail, S.; Ahmad, S.; Azam, S.S. Immunoinformatics characterization of SARS-CoV-2 spike glycoprotein for prioritization of epitope based multivalent peptide vaccine. J. Mol. Liq. 2020, 314, 113612. [Google Scholar] [CrossRef]
- Calis, J.J.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef]
- Yadav, S.; Prakash, J.; Shukla, H.; Das, K.C.; Tripathi, T.; Dubey, V.K. Design of a multi-epitope subunit vaccine for immune-protection against Leishmania parasite. Pathog. Glob. Health 2020, 114, 471–481. [Google Scholar] [CrossRef]
- ul Qamar, M.T.; Ahmad, S.; Fatima, I.; Ahmad, F.; Shahid, F.; Naz, A.; Abbasi, S.W.; Khan, A.; Mirza, M.U.; Ashfaq, U.A. Designing multi-epitope vaccine against Staphylococcus aureus by employing subtractive proteomics, reverse vaccinology and immuno-informatics approaches. Comput. Biol. Med. 2021, 132, 104389. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Consortium, O.S.D.D.; Raghava, G.P. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [PubMed]
- Hoque, H.; Islam, R.; Ghosh, S.; Rahaman, M.M.; Jewel, N.A.; Miah, M.A. Implementation of in silico methods to predict common epitopes for vaccine development against Chikungunya and Mayaro viruses. Heliyon 2021, 7, e06396. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.A.; Ami, J.Q.; Faisal, K.; Chowdhury, R.; Ghosh, P.; Hossain, F.; Abd El Wahed, A.; Mondal, D. An immunoinformatic approach driven by experimental proteomics: In silico design of a subunit candidate vaccine targeting secretory proteins of Leishmania donovani amastigotes. Parasites Vectors 2020, 13, 196. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, 72–76. [Google Scholar] [CrossRef]
- Giardine, B.; Riemer, C.; Hardison, R.C.; Burhans, R.; Elnitski, L.; Shah, P.; Zhang, Y.; Blankenberg, D.; Albert, I.; Taylor, J.; et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005, 15, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, 384–388. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. Geometry-based flexible and symmetric protein docking. Proteins Struct. Funct. Genet 2005, 60, 224–231. [Google Scholar] [CrossRef]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast interaction refinement in molecular docking. Proteins Struct. Funct. Genet. 2007, 69, 139–159. [Google Scholar] [CrossRef]
- Heinzelmann, G.; Gilson, M.K. Automated docking refinement and virtual compound screening with absolute binding free energy calculations. BioRxiv 2020. [Google Scholar] [CrossRef]
- Naz, S.; Ahmad, S.; Walton, S.; Abbasi, S.W. Multi-epito.pe based vaccine design against Sarcoptes scabiei paramyosin using immunoinformatics approach. J. Mol. Liq. 2020, 319, 114105. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).