

S Protein, ACE2 and Host Cell Proteases in SARS-CoV-2 Cell Entry and Infectivity; Is Soluble ACE2 a Two Blade Sword? A Narrative Review



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
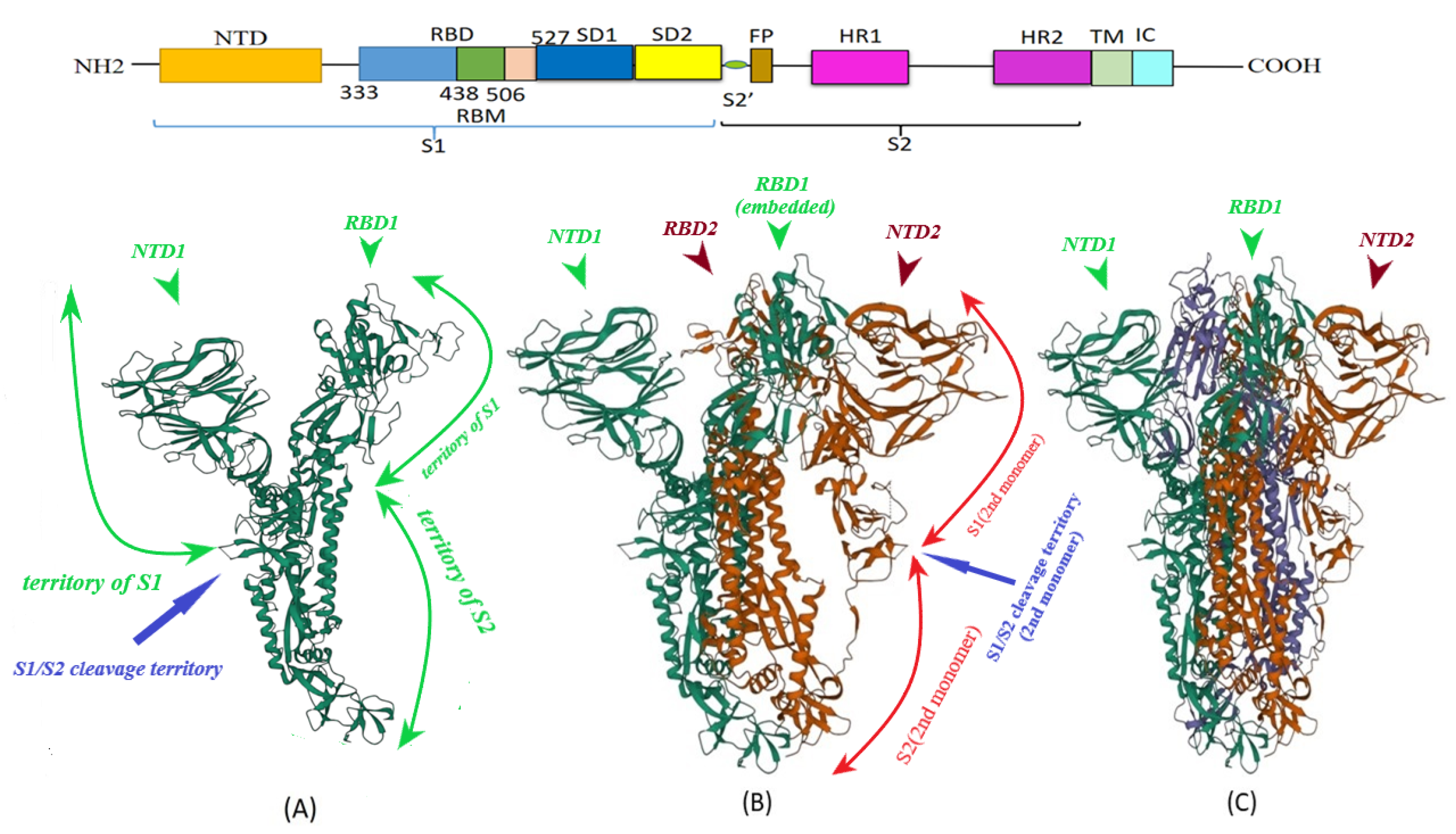
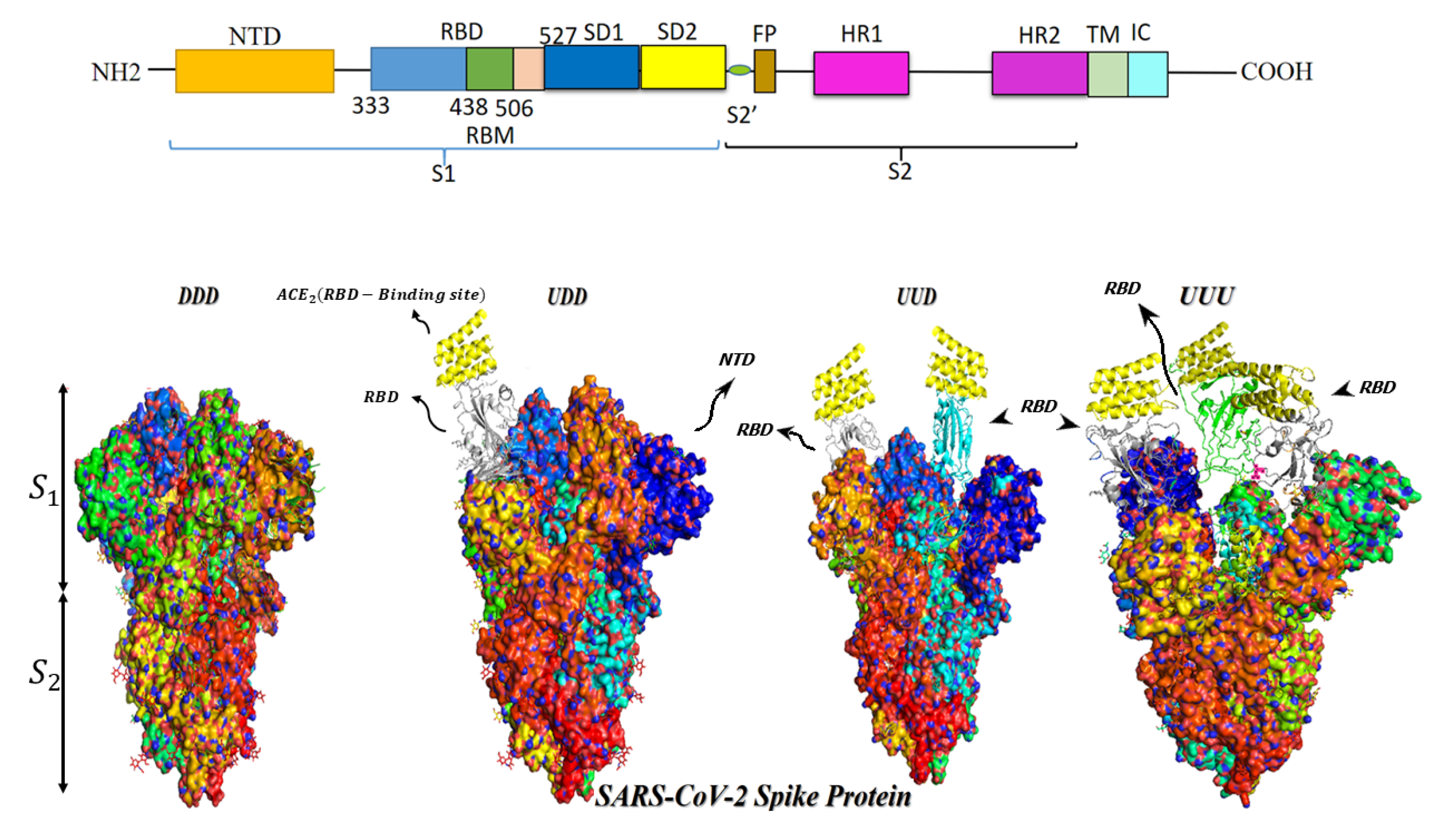
2. S Protein Structure
3. Binding of ACE2 with RBD Increases the Chance of Further Complexification of ACE2 and SARS-CoV-2
4. Proteases Play a Pivotal Role in Various Models of SARS-CoV-2 Cell Entry
5. Furin, a Ubiquitously Expressed Multifunctional Protease
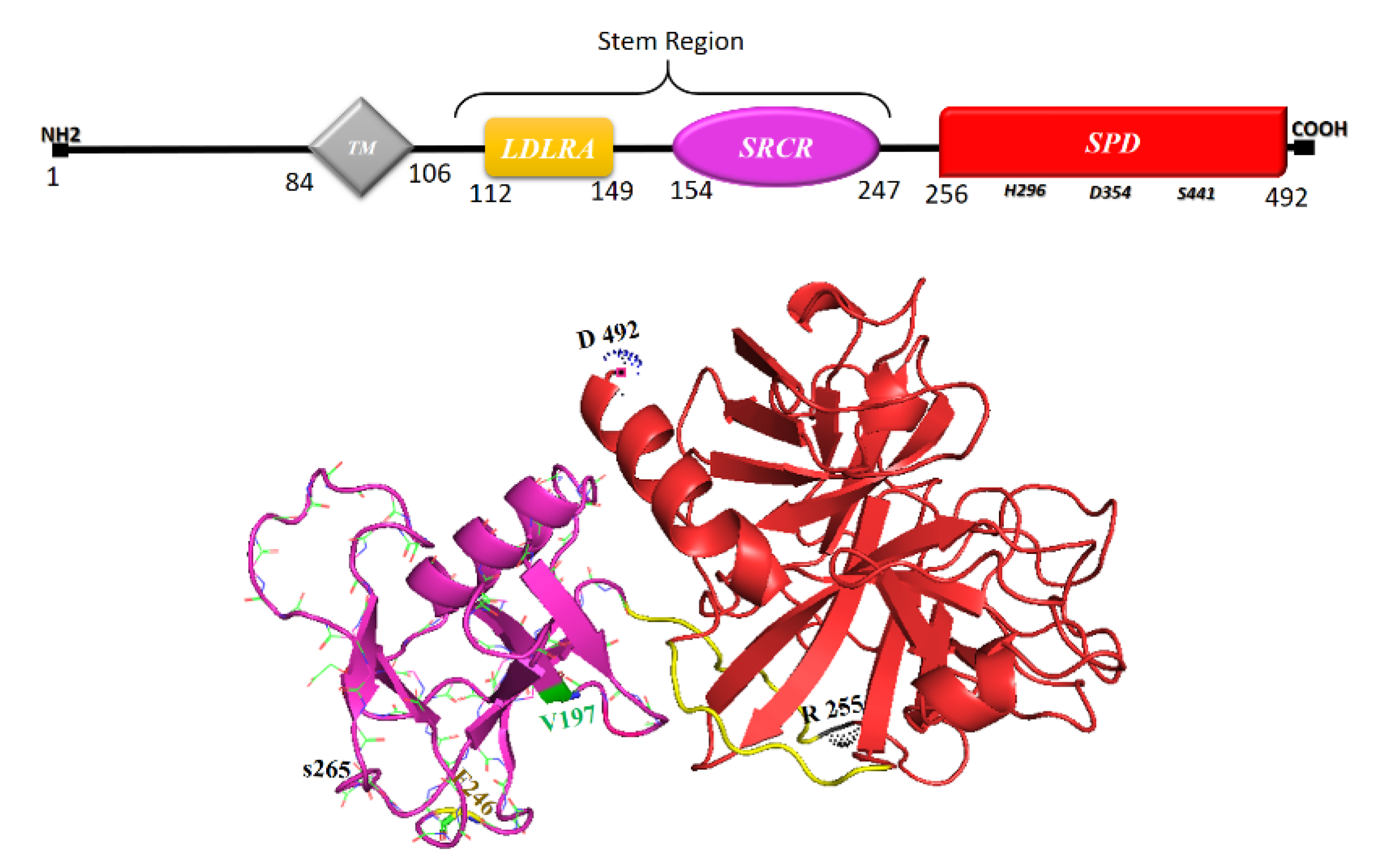
6. TMPRSS2, Its Soluble Protease Domain and Calcium
7. Major Activity of Cathepsins Occurs in the Membrane-Bound Intracellular Organelles
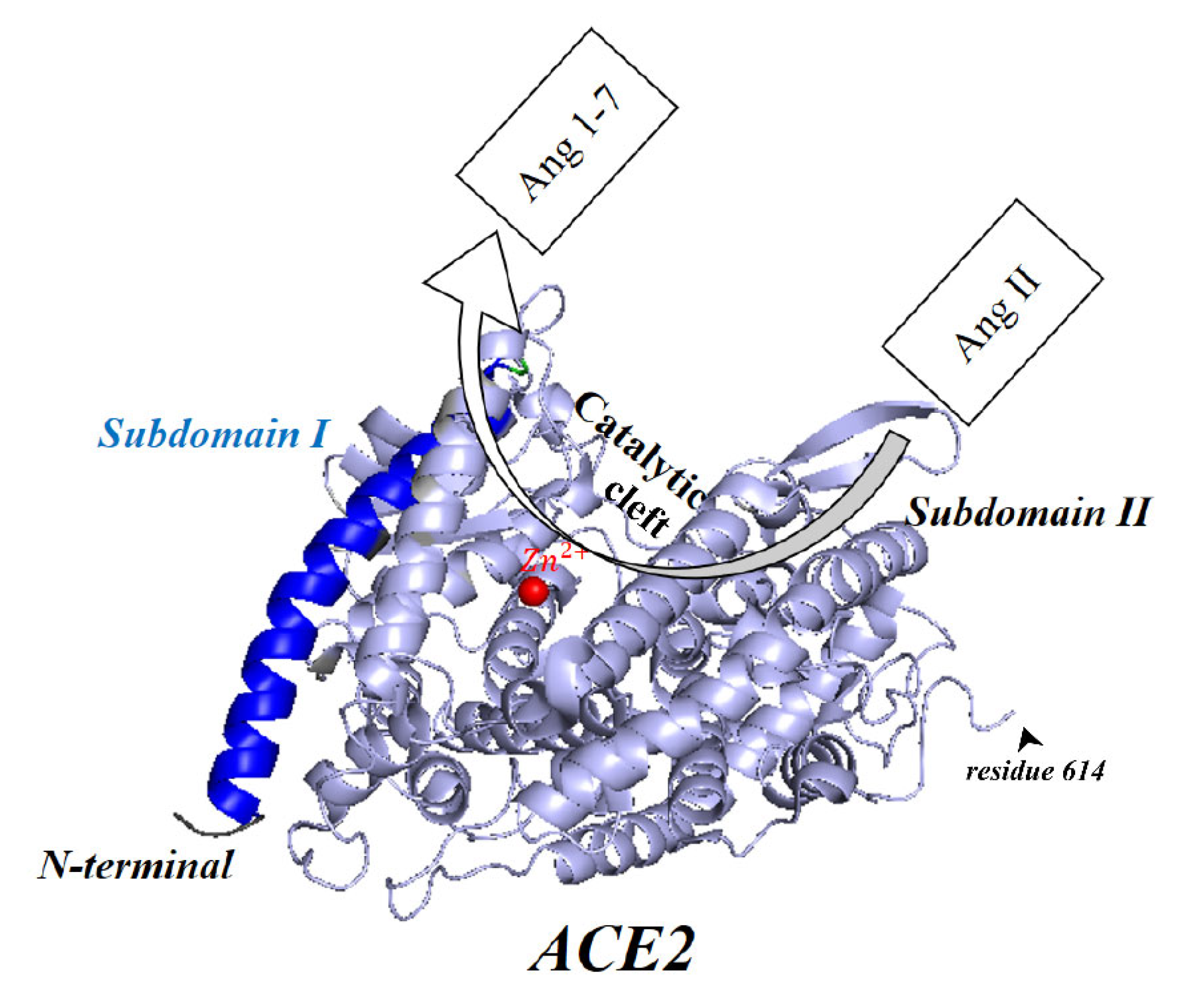
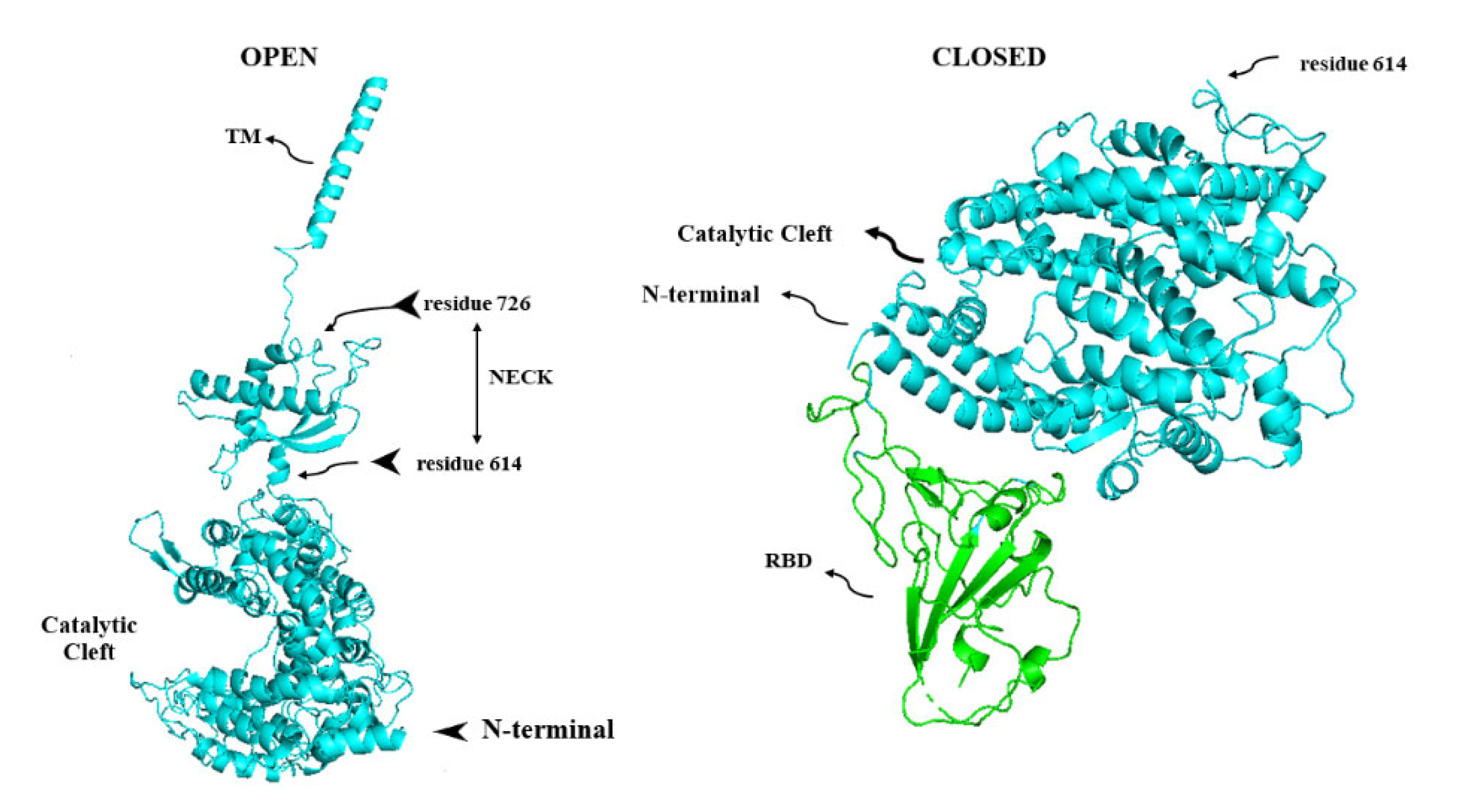
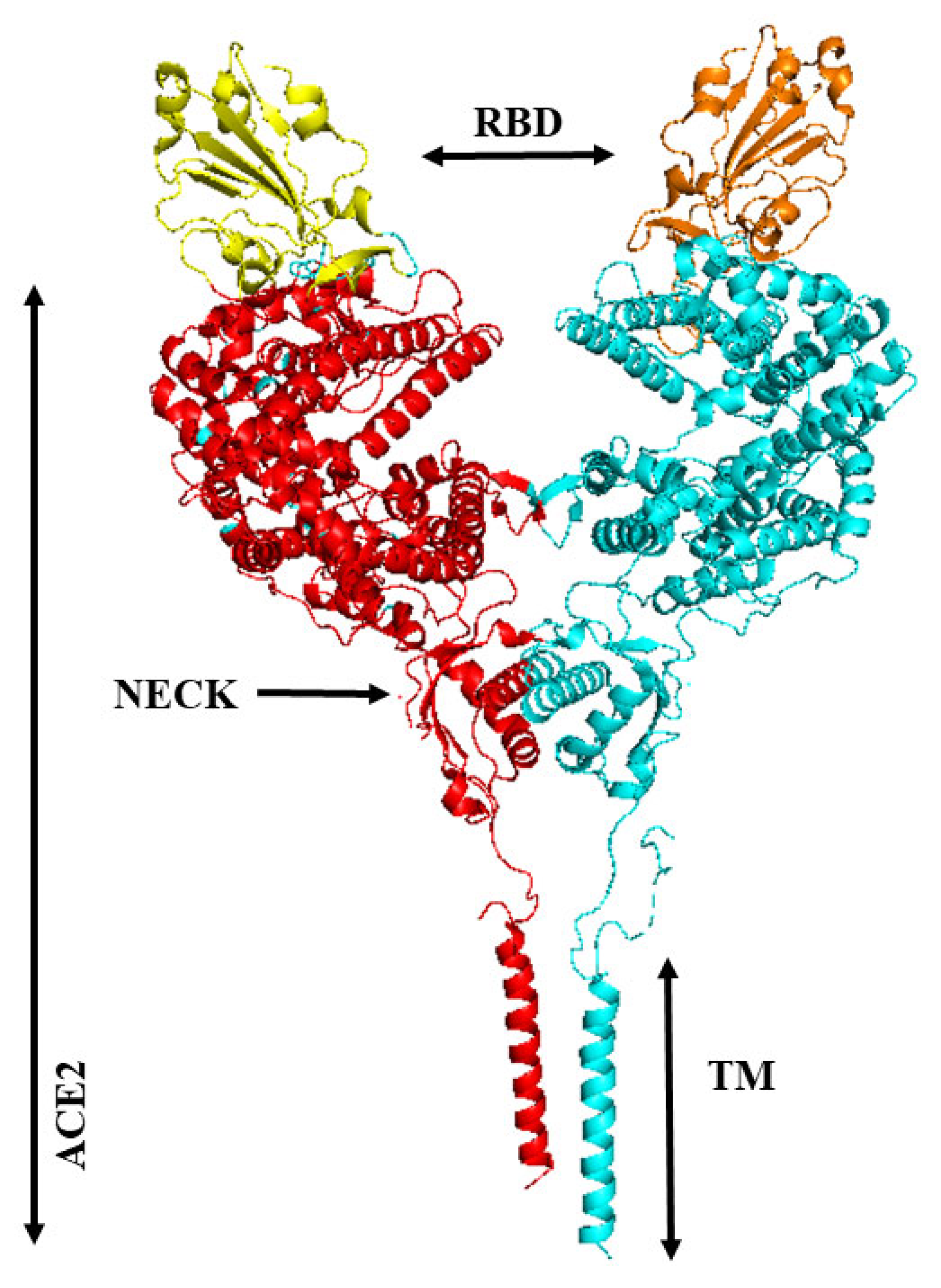
8. Membrane-Bound and Soluble ACE2
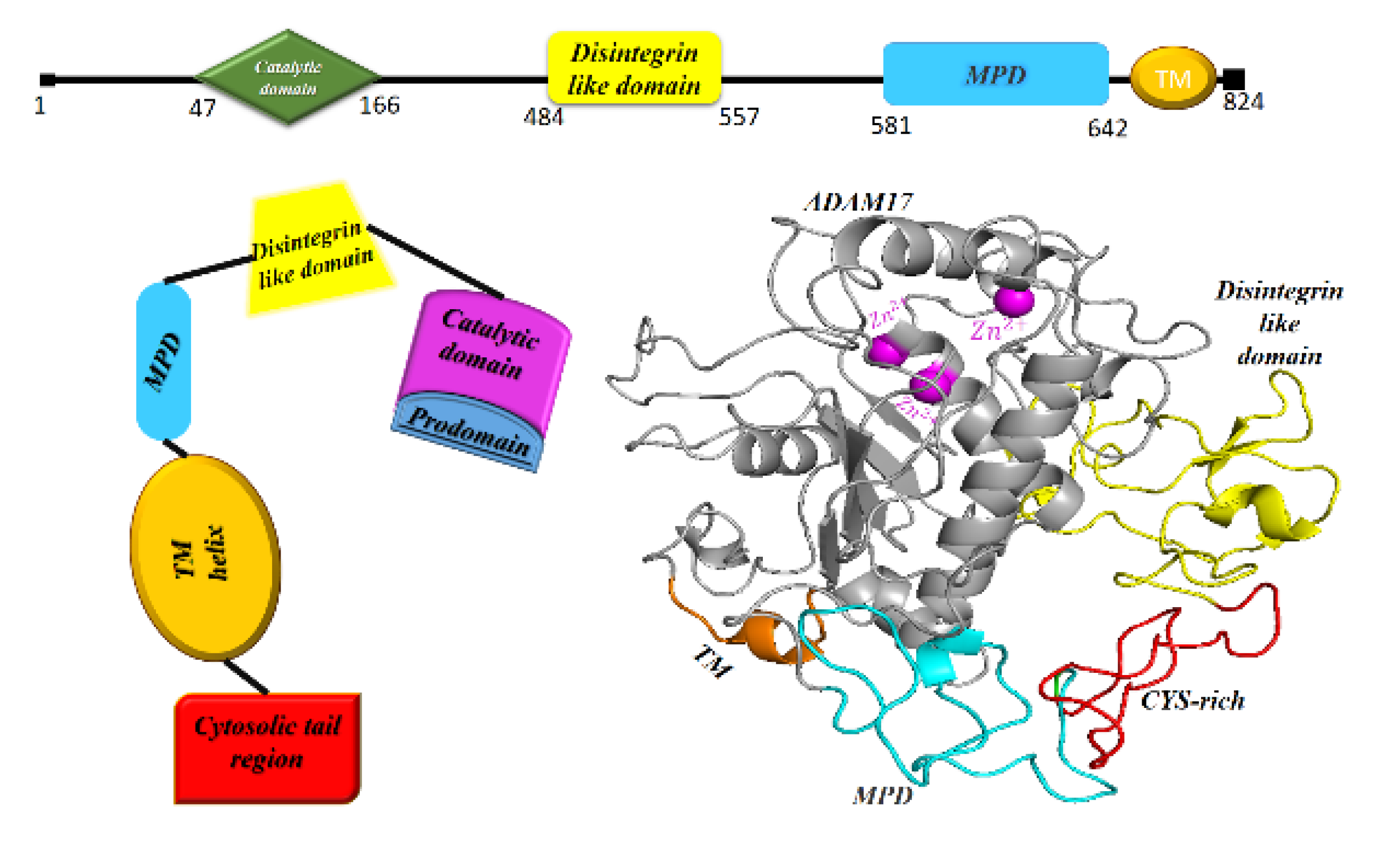
9. ADAM17, the Sheddase of ACE2
10. Shedding of ACE2 Ectodomain and sACE2 in SARS-CoV-2
11. sACE2: Attenuating Inflammatory Responses?
12. sACE2: Increasing SARS-CoV-2 Infectivity?
13. Clinical Evidence Showing Benefit of Recombinant Human Soluble ACE2
14. Future Directions
15. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, J.; Petitjean, S.J.; Koehler, M.; Zhang, Q.; Dumitru, A.C.; Chen, W.; Derclaye, S.; Vincent, S.P.; Soumillion, P.; Alsteens, D. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Glebov, O.O. Understanding SARS-CoV-2 endocytosis for COVID-19 drug repurposing. FEBS J. 2020, 287, 3664–3671. [Google Scholar] [CrossRef]
- Bayati, A.; Kumar, R.; Francis, V.; McPherson, P.S. SARS-CoV-2 infects cells after viral entry via clathrin-mediated endocytosis. J. Biol. Chem. 2021, 296, 100306. [Google Scholar] [CrossRef]
- Knyazev, E.; Nersisyan, S.; Tonevitsky, A. Endocytosis and transcytosis of SARS-CoV-2 across the intestinal epithelium and other tissue barriers. Front. Immunol. 2021, 12, 636966. [Google Scholar] [CrossRef]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. SARS coronavirus entry into host cells through a novel clathrin-and caveolae-independent endocytic pathway. Cell Res. 2008, 18, 290–301. [Google Scholar] [CrossRef]
- Shen, X.-R.; Geng, R.; Li, Q.; Chen, Y.; Li, S.-F.; Wang, Q.; Min, J.; Yang, Y.; Li, B.; Jiang, R.-D. ACE2-independent infection of T lymphocytes by SARS-CoV-2. Signal Transduct. Target. Ther. 2022, 7, 1–11. [Google Scholar] [CrossRef]
- Junqueira, C.; Crespo, Â.; Ranjbar, S.; de Lacerda, L.B.; Lewandrowski, M.; Ingber, J.; Parry, B.; Ravid, S.; Clark, S.; Schrimpf, M.R. FcγR-mediated SARS-CoV-2 infection of monocytes activates inflammation. Nature 2022, 606, 576–584. [Google Scholar] [CrossRef]
- Zipeto, D.; Palmeira, J.d.F.; Argañaraz, G.A.; Argañaraz, E.R. ACE2/ADAM17/TMPRSS2 interplay may be the main risk factor for COVID-19. Front. Immunol. 2020, 11, 2642. [Google Scholar] [CrossRef]
- Fagyas, M.; Fejes, Z.; Sütő, R.; Nagy, Z.; Székely, B.; Pócsi, M.; Ivády, G.; Bíró, E.; Bekő, G.; Nagy, A. Circulating ACE2 activity predicts mortality and disease severity in hospitalized COVID-19 patients. Int. J. Infect. Dis. 2022, 115, 8–16. [Google Scholar] [CrossRef]
- Fuentes-Prior, P. Priming of SARS-CoV-2 S protein by several membrane-bound serine proteinases could explain enhanced viral infectivity and systemic COVID-19 infection. J. Biol. Chem. 2021, 296, 100135. [Google Scholar] [CrossRef]
- Oubahmane, M.; Hdoufane, I.; Bjij, I.; Lahcen, N.A.; Villemin, D.; Daoud, R.; Allali, A.E.; Cherqaoui, D. Host cell proteases mediating SARS-CoV-2 entry: An overview. Curr. Top. Med. Chem. 2022, 22, 1776–1792. [Google Scholar]
- Stevens, C.S.; Oguntuyo, K.Y.; Lee, B. Proteases and variants: Context matters for SARS-CoV-2 entry assays. Curr. Opin. Virol. 2021, 50, 49–58. [Google Scholar] [CrossRef]
- Guizani, I.; Fourti, N.; Zidi, W.; Feki, M.; Allal-Elasmi, M. SARS-CoV-2 and pathological matrix remodeling mediators. Inflamm. Res. 2021, 70, 847–858. [Google Scholar] [CrossRef]
- Benlarbi, M.; Laroche, G.; Fink, C.; Fu, K.; Mulloy, R.P.; Phan, A.; Ariana, A.; Stewart, C.M.; Prévost, J.; Beaudoin-Bussières, G. Identification of a SARS-CoV-2 host metalloproteinase-dependent entry pathway differentially used by SARS-CoV-2 and variants of concern Alpha, Delta, and Omicron. bioRxiv 2022. [Google Scholar]
- Boon, L.; Ugarte-Berzal, E.; Vandooren, J.; Opdenakker, G. Protease propeptide structures, mechanisms of activation, and functions. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 111–165. [Google Scholar] [CrossRef]
- Fraser, B.J.; Beldar, S.; Seitova, A.; Hutchinson, A.; Mannar, D.; Li, Y.; Kwon, D.; Tan, R.; Wilson, R.P.; Leopold, K. Structure and activity of human TMPRSS2 protease implicated in SARS-CoV-2 activation. Nat. Chem. Biol. 2022, 18, 963–971. [Google Scholar] [CrossRef]
- Verma, S.; Dixit, R.; Pandey, K.C. Cysteine proteases: Modes of activation and future prospects as pharmacological targets. Front. Pharmacol. 2016, 7, 107. [Google Scholar] [CrossRef]
- Anderson, E.D.; VanSlyke, J.K.; Thulin, C.D.; Jean, F.; Thomas, G. Activation of the furin endoprotease is a multiple-step process: Requirements for acidification and internal propeptide cleavage. EMBO J. 1997, 16, 1508–1518. [Google Scholar] [CrossRef]
- Lorenzen, I.; Lokau, J.; Korpys, Y.; Oldefest, M.; Flynn, C.M.; Künzel, U.; Garbers, C.; Freeman, M.; Grötzinger, J.; Düsterhöft, S. Control of ADAM17 activity by regulation of its cellular localisation. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-Y.; Zhao, R.; Gao, L.-J.; Gao, X.-F.; Wang, D.-P.; Cao, J.-M. SARS-CoV-2: Structure, biology, and structure-based therapeutics development. Front. Cell. Infect. Microbiol. 2020, 10, 587269. [Google Scholar] [CrossRef] [PubMed]
- Berger, I.; Schaffitzel, C. The SARS-CoV-2 spike protein: Balancing stability and infectivity. Cell Res. 2020, 30, 1059–1060. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Shah, P.; Canziani, G.A.; Carter, E.P.; Chaiken, I. The case for S2: The potential benefits of the S2 subunit of the SARS-CoV-2 spike protein as an immunogen in fighting the COVID-19 pandemic. Front. Immunol. 2021, 12, 637651. [Google Scholar] [CrossRef]
- Wu, Y.; Zhao, S. Furin cleavage sites naturally occur in coronaviruses. Stem Cell Res. 2021, 50, 102115. [Google Scholar] [CrossRef]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef]
- Buchanan, C.J.; Gaunt, B.; Harrison, P.J.; Yang, Y.; Liu, J.; Khan, A.; Giltrap, A.M.; Le Bas, A.; Ward, P.N.; Gupta, K. Pathogen-sugar interactions revealed by universal saturation transfer analysis. Science 2022, 377, eabm3125. [Google Scholar] [CrossRef]
- Mehdipour, A.R.; Hummer, G. Dual nature of human ACE2 glycosylation in binding to SARS-CoV-2 spike. Proc. Natl. Acad. Sci. USA 2021, 118, e2100425118. [Google Scholar] [CrossRef]
- Zhu, C.; He, G.; Yin, Q.; Zeng, L.; Ye, X.; Shi, Y.; Xu, W. Molecular biology of the SARs-CoV-2 spike protein: A review of current knowledge. J. Med. Virol. 2021, 93, 5729–5741. [Google Scholar] [CrossRef]
- Khare, S.; Azevedo, M.; Parajuli, P.; Gokulan, K. Conformational changes of the receptor binding domain of sars-cov-2 spike protein and prediction of a b-cell antigenic epitope using structural data. Front. Artif. Intell. 2021, 4, 31. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C. Structural and functional analysis of the D614G SARS-CoV-2 spike protein variant. Cell 2020, 183, 739–751.e738. [Google Scholar] [CrossRef]
- Giron, C.C.; Laaksonen, A.; Barroso da Silva, F.L. Up state of the SARS-COV-2 spike homotrimer favors an increased virulence for new variants. Front. Med. Technol. 2021, 3, 694347. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B. Tracking changes in SARS-CoV-2 spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827.e819. [Google Scholar] [CrossRef]
- Jordan, M. The meaning of affinity and the importance of identity in the designed world. Interactions 2010, 17, 6–11. [Google Scholar] [CrossRef]
- Delgado, J.M.; Duro, N.; Rogers, D.M.; Tkatchenko, A.; Pandit, S.A.; Varma, S. Molecular basis for higher affinity of SARS-CoV-2 spike RBD for human ACE2 receptor. Proteins Struct. Funct. Bioinform. 2021, 89, 1134–1144. [Google Scholar] [CrossRef]
- Wu, L.; Zhou, L.; Mo, M.; Liu, T.; Wu, C.; Gong, C.; Lu, K.; Gong, L.; Zhu, W.; Xu, Z. SARS-CoV-2 Omicron RBD shows weaker binding affinity than the currently dominant Delta variant to human ACE2. Signal Transduct. Target. Ther. 2022, 7, 1–3. [Google Scholar] [CrossRef]
- Stalls, V.; Lindenberger, J.; Gobeil, S.M.-C.; Henderson, R.; Parks, R.; Barr, M.; Deyton, M.; Martin, M.; Janowska, K.; Huang, X. Cryo-EM structures of SARS-CoV-2 Omicron BA. 2 spike. Cell Rep. 2022, 39, 111009. [Google Scholar] [CrossRef]
- Gobeil, S.M.-C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Manne, K.; Stalls, V.; Kopp, M.F.; Henderson, R.; Edwards, R.J. D614G mutation alters SARS-CoV-2 spike conformation and enhances protease cleavage at the S1/S2 junction. Cell Rep. 2021, 34, 108630. [Google Scholar] [CrossRef]
- Henderson, R.; Edwards, R.J.; Mansouri, K.; Janowska, K.; Stalls, V.; Gobeil, S.; Kopp, M.; Li, D.; Parks, R.; Hsu, A.L. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020, 27, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Gobeil, S.M.-C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Stalls, V.; Kopp, M.F.; Manne, K.; Li, D.; Wiehe, K. Effect of natural mutations of SARS-CoV-2 on spike structure, conformation, and antigenicity. Science 2021, 373, eabi6226. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.-f.; Xu, W.; Liu, S.-w. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Saputri, D.S.; Li, S.; Van Eerden, F.J.; Rozewicki, J.; Xu, Z.; Ismanto, H.S.; Davila, A.; Teraguchi, S.; Katoh, K.; Standley, D.M. Flexible, functional, and familiar: Characteristics of SARS-CoV-2 spike protein evolution. Front. Microbiol. 2020, 11, 2112. [Google Scholar] [CrossRef]
- Xia, S.; Lan, Q.; Su, S.; Wang, X.; Xu, W.; Liu, Z.; Zhu, Y.; Wang, Q.; Lu, L.; Jiang, S. The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin. Signal Transduct. Target. Ther. 2020, 5, 1–3. [Google Scholar] [CrossRef]
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, I.I.I.K.H.; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell 2020, 182, 429–446.e414. [Google Scholar] [CrossRef]
- Örd, M.; Faustova, I.; Loog, M. The sequence at Spike S1/S2 site enables cleavage by furin and phospho-regulation in SARS-CoV2 but not in SARS-CoV1 or MERS-CoV. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Xia, X. Domains and functions of spike protein in Sars-Cov-2 in the context of vaccine design. Viruses 2021, 13, 109. [Google Scholar] [CrossRef]
- Lemmin, T.; Kalbermatter, D.; Harder, D.; Plattet, P.; Fotiadis, D. Structures and dynamics of the novel S1/S2 protease cleavage site loop of the SARS-CoV-2 spike glycoprotein. J. Struct. Biol. X 2020, 4, 100038. [Google Scholar] [CrossRef]
- Yu, S.; Zheng, X.; Zhou, B.; Li, J.; Chen, M.; Deng, R.; Wong, G.; Lavillette, D.; Meng, G. SARS-CoV-2 spike engagement of ACE2 primes S2′ site cleavage and fusion initiation. Proc. Natl. Acad. Sci. USA 2022, 119, e2111199119. [Google Scholar] [CrossRef]
- Sternberg, A.; Naujokat, C. Structural features of coronavirus SARS-CoV-2 spike protein: Targets for vaccination. Life Sci. 2020, 257, 118056. [Google Scholar] [CrossRef]
- Tang, T.; Jaimes, J.A.; Bidon, M.K.; Straus, M.R.; Daniel, S.; Whittaker, G.R. Proteolytic activation of SARS-CoV-2 spike at the S1/S2 boundary: Potential role of proteases beyond furin. ACS Infect. Dis. 2021, 7, 264–272. [Google Scholar] [CrossRef]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; Rohde, C. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef]
- Juraszek, J.; Rutten, L.; Blokland, S.; Bouchier, P.; Voorzaat, R.; Ritschel, T.; Bakkers, M.J.; Renault, L.L.; Langedijk, J.P. Stabilizing the closed SARS-CoV-2 spike trimer. Nat. Commun. 2021, 12, 1–8. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Belouzard, S.; Madu, I.; Whittaker, G.R. Elastase-mediated activation of the severe acute respiratory syndrome coronavirus spike protein at discrete sites within the S2 domain. J. Biol. Chem. 2010, 285, 22758–22763. [Google Scholar] [CrossRef]
- Fan, X.; Cao, D.; Kong, L.; Zhang, X. Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Koppisetti, R.K.; Fulcher, Y.G.; Van Doren, S.R. Fusion peptide of SARS-CoV-2 spike rearranges into a wedge inserted in bilayered micelles. J. Am. Chem. Soc. 2021, 143, 13205–13211. [Google Scholar] [CrossRef]
- Arbeitman, C.R.; Rojas, P.; Ojeda-May, P.; Garcia, M.E. The SARS-CoV-2 spike protein is vulnerable to moderate electric fields. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Walls, A.C.; Tortorici, M.A.; Bosch, B.-J.; Frenz, B.; Rottier, P.J.; DiMaio, F.; Rey, F.A.; Veesler, D. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 2016, 531, 114–117. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell 2020, 78, 779–. [Google Scholar] [CrossRef]
- Hörnich, B.F.; Großkopf, A.K.; Schlagowski, S.; Tenbusch, M.; Kleine-Weber, H.; Neipel, F.; Stahl-Hennig, C.; Hahn, A.S. SARS-CoV-2 and SARS-CoV spike-mediated cell-cell fusion differ in their requirements for receptor expression and proteolytic activation. J. Virol. 2021, 95, e00002–e00021. [Google Scholar] [CrossRef] [PubMed]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin-Ortolá, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Zheng, Q.; Zhang, H.; Niu, Y.; Lou, Y.; Wang, H. The SARS-CoV-2 spike glycoprotein biosynthesis, structure, function, and antigenicity: Implications for the design of spike-based vaccine immunogens. Front. Immunol. 2020, 11, 2593. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Pöhlmann, S. How SARS-CoV-2 makes the cut. Nat. Microbiol. 2021, 6, 828–829. [Google Scholar] [CrossRef]
- Ohradanova-Repic, A.; Skrabana, R.; Gebetsberger, L.; Tajti, G.; Baráth, P.; Ondrovičová, G.; Praženicová, R.; Jantova, N.; Hrasnova, P.; Stockinger, H. Blockade of TMPRSS2-mediated priming of SARS-CoV-2 by lactoferricin. Front. Immunol. 2022, 13, 958581. [Google Scholar] [CrossRef]
- Wettstein, L.; Knaff, P.M.; Kersten, C.; Müller, P.; Weil, T.; Conzelmann, C.; Müller, J.A.; Brückner, M.; Hoffmann, M.; Pöhlmann, S. Peptidomimetic inhibitors of TMPRSS2 block SARS-CoV-2 infection in cell culture. Commun. Biol. 2022, 5, 1–12. [Google Scholar] [CrossRef]
- Harte, J.V.; Wakerlin, S.L.; Lindsay, A.J.; McCarthy, J.V.; Cloeman-Vaughan, C. Metalloprotease-Dependent S2’-Activation Promotes Cell–Cell Fusion and Syncytiation of SARS-CoV-2. Viruses 2022, 14, 2094. [Google Scholar] [CrossRef]
- Yamamoto, M.; Gohda, J.; Kobayashi, A.; Tomita, K.; Hirayama, Y.; Koshikawa, N.; Seiki, M.; Semba, K.; Akiyama, T.; Kawaguchi, Y. Metalloproteinase-dependent and TMPRSS2-independent cell surface entry pathway of SARS-CoV-2 requires the furin cleavage site and the S2 domain of spike protein. Mbio 2022, 13, e00519–e00522. [Google Scholar] [CrossRef]
- Jocher, G.; Grass, V.; Tschirner, S.K.; Riepler, L.; Breimann, S.; Kaya, T.; Oelsner, M.; Hamad, M.S.; Hofmann, L.I.; Blobel, C.P. ADAM10 and ADAM17 promote SARS-CoV-2 cell entry and spike protein-mediated lung cell fusion. EMBO Rep. 2022, 23, e54305. [Google Scholar] [CrossRef]
- Xiu, S.; Dick, A.; Ju, H.; Mirzaie, S.; Abdi, F.; Cocklin, S.; Zhan, P.; Liu, X. Inhibitors of SARS-CoV-2 entry: Current and future opportunities. J. Med. Chem. 2020, 63, 12256–12274. [Google Scholar] [CrossRef]
- Petersen, O.H.; Gerasimenko, O.V.; Gerasimenko, J.V. Endocytic uptake of SARS-CoV-2: The critical roles of pH, Ca2+, and NAADP. Function 2020, 1, zqaa003. [Google Scholar] [CrossRef]
- Khan, N.; Chen, X.; Geiger, J.D. Role of endolysosomes in severe acute respiratory syndrome coronavirus-2 infection and coronavirus disease 2019 pathogenesis: Implications for potential treatments. Front. Pharmacol. 2020, 11, 595888. [Google Scholar] [CrossRef]
- Padmanabhan, P.; Desikan, R.; Dixit, N.M. Targeting TMPRSS2 and Cathepsin B/L together may be synergistic against SARS-CoV-2 infection. PLoS Comput. Biol. 2020, 16, e1008461. [Google Scholar] [CrossRef]
- Hashimoto, R.; Sakamoto, A.; Deguchi, S.; Yi, R.; Sano, E.; Hotta, A.; Takahashi, K.; Yamanaka, S.; Takayama, K. Dual inhibition of TMPRSS2 and Cathepsin B prevents SARS-CoV-2 infection in iPS cells. Mol. Ther. Nucleic Acids 2021, 26, 1107–1114. [Google Scholar] [CrossRef]
- Zhao, M.-M.; Yang, W.-L.; Yang, F.-Y.; Zhang, L.; Huang, W.-J.; Hou, W.; Fan, C.-F.; Jin, R.-H.; Feng, Y.-M.; Wang, Y.-C. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct. Target. Ther. 2021, 6, 1–12. [Google Scholar] [CrossRef]
- Schapiro, F.B.; Soe, T.T.; Mallet, W.G.; Maxfield, F.R. Role of cytoplasmic domain serines in intracellular trafficking of furin. Mol. Biol. Cell 2004, 15, 2884–2894. [Google Scholar] [CrossRef]
- Thomas, G. Furin at the cutting edge: From protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 2002, 3, 753–766. [Google Scholar] [CrossRef]
- Fitzgerald, K. Furin protease: From SARS Cov-2 to anthrax, diabetes, and hypertension. Perm. J. 2020, 24, 187. [Google Scholar] [CrossRef]
- Gu, M.; Rappapor, J.; Leppla, S.H. Furin is important but not essential for the proteolytic maturation of gp160 of HIV-1. FEBS Lett. 1995, 365, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Gawlik, K.; Shiryaev, S.A.; Zhu, W.; Motamedchaboki, K.; Desjardins, R.; Day, R.; Remacle, A.G.; Stec, B.; Strongin, A.Y. Autocatalytic activation of the furin zymogen requires removal of the emerging enzyme’s N-terminus from the active site. PLoS ONE 2009, 4, e5031. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, C.; Rysä, J.; Almgren, P.; Nilsson, J.; Engström, G.; Orho-Melander, M.; Ruskoaho, H.; Melander, O. Plasma levels of the proprotein convertase furin and incidence of diabetes and mortality. J. Intern. Med. 2018, 284, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Duhamel, M.; Rodet, F.; Salzet, M. The role of proprotein convertases in the regulation of the function of immune cells in the oncoimmune response. Front. Immunol. 2021, 12, 667850. [Google Scholar] [CrossRef]
- Srour, N.; Lebel, A.; McMahon, S.; Fournier, I.; Fugère, M.; Day, R.; Dubois, C.M. TACE/ADAM-17 maturation and activation of sheddase activity require proprotein convertase activity. FEBS Lett. 2003, 554, 275–283. [Google Scholar] [CrossRef]
- Sato, H.; Kinoshita, T.; Takino, T.; Nakayama, K.; Seiki, M. Activation of a recombinant membrane type 1-matrix metalloproteinase (MT1-MMP) by furin and its interaction with tissue inhibitor of metalloproteinases (TIMP)-2. FEBS Lett. 1996, 393, 101–104. [Google Scholar] [CrossRef]
- Pesu, M.; Watford, W.T.; Wei, L.; Xu, L.; Fuss, I.; Strober, W.; Andersson, J.; Shevach, E.M.; Quezado, M.; Bouladoux, N. T-cell-expressed proprotein convertase furin is essential for maintenance of peripheral immune tolerance. Nature 2008, 455, 246–250. [Google Scholar] [CrossRef]
- Rawley, O.; Lillicrap, D. Functional roles of the von Willebrand factor propeptide. Hämostaseologie 2021, 41, 063–068. [Google Scholar] [CrossRef]
- Vankadari, N. Structure of furin protease binding to SARS-CoV-2 spike glycoprotein and implications for potential targets and virulence. J. Phys. Chem. Lett. 2020, 11, 6655–6663. [Google Scholar] [CrossRef]
- Zhang, L.; Mann, M.; Syed, Z.A.; Reynolds, H.M.; Tian, E.; Samara, N.L.; Zeldin, D.C.; Tabak, L.A.; Ten Hagen, K.G. Furin cleavage of the SARS-CoV-2 spike is modulated by O-glycosylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2109905118. [Google Scholar] [CrossRef]
- Johnson, B.A.; Xie, X.; Bailey, A.L.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 2021, 591, 293–299. [Google Scholar] [CrossRef]
- Kocyigit, A.; Sogut, O.; Durmus, E.; Kanimdan, E.; Guler, E.M.; Kaplan, O.; Yenigun, V.B.; Eren, C.; Ozman, Z.; Yasar, O. Circulating furin, IL-6, and presepsin levels and disease severity in SARS-CoV-2–infected patients. Sci. Prog. 2021, 104 (Suppl. S2), 00368504211026119. [Google Scholar] [CrossRef]
- Declercq, J.; Creemers, J.W.M. Furin. In Handbook of Proteolytic Enzymes, 3rd ed.; Rawlings, N.D., Saavedra, G.S., Eds.; Academic Press: London, UK, 2012; pp. 3281–3285. [Google Scholar]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ sensing: Its role in calcium homeostasis and signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef]
- Atchison, D.K.; Beierwaltes, W.H. The influence of extracellular and intracellular calcium on the secretion of renin. Pflügers Arch. Eur. J. Physiol. 2013, 465, 59–69. [Google Scholar] [CrossRef]
- Thier, S.O. Potassium physiology. Am. J. Med. 1986, 80, 3–7. [Google Scholar] [CrossRef]
- Vidricaire, G.; Denault, J.-B.; Leduc, R. Characterization of a secreted form of human furin endoprotease. Biochem. Biophys. Res. Commun. 1993, 195, 1011–1018. [Google Scholar] [CrossRef]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W. SARS-CoV-2 receptor ACE 2 and TMPRSS 2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020, 39, e105114. [Google Scholar] [CrossRef]
- Qiao, Y.; Wang, X.-M.; Mannan, R.; Pitchiaya, S.; Zhang, Y.; Wotring, J.W.; Xiao, L.; Robinson, D.R.; Wu, Y.-M.; Tien, J.C.-Y. Targeting transcriptional regulation of SARS-CoV-2 entry factors ACE2 and TMPRSS2. Proc. Natl. Acad. Sci. USA 2021, 118, e2021450118. [Google Scholar] [CrossRef]
- Rokni, M.; Heidari Nia, M.; Sarhadi, M.; Mirinejad, S.; Sargazi, S.; Moudi, M.; Saravani, R.; Rahdar, S.; Kargar, M. Association of TMPRSS2 gene polymorphisms with COVID-19 severity and mortality: A case-control study with computational analyses. Appl. Biochem. Biotechnol. 2022, 194, 3507–3526. [Google Scholar] [CrossRef]
- Dong, M.; Zhang, J.; Ma, X.; Tan, J.; Chen, L.; Liu, S.; Xin, Y.; Zhuang, L. ACE2, TMPRSS2 distribution and extrapulmonary organ injury in patients with COVID-19. Biomed. Pharmacother. 2020, 131, 110678. [Google Scholar] [CrossRef]
- Thunders, M.; Delahunt, B. Gene of the month: TMPRSS2 (transmembrane serine protease 2). J. Clin. Pathol. 2020, 73, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Jabeen, N.; Amanullah, A.; Baig, A.A.; Aziz, B.; Shabbir, S.; Raza, F.; Uddin, N. Molecular docking between human TMPRSS2 and SARS-CoV-2 spike protein: Conformation and intermolecular interactions. AIMS Microbiol. 2020, 6, 350. [Google Scholar] [CrossRef] [PubMed]
- Reichhardt, M.P.; Loimaranta, V.; Lea, S.M.; Johnson, S. Structures of SALSA/DMBT1 SRCR domains reveal the conserved ligand-binding mechanism of the ancient SRCR fold. Life Sci. Alliance 2020, 3, e201900502. [Google Scholar] [CrossRef] [PubMed]
- Paoloni-Giacobino, A.; Chen, H.; Peitsch, M.C.; Rossier, C.; Antonarakis, S.E. Cloning of the TMPRSS2 gene, which encodes a novel serine protease with transmembrane, LDLRA, and SRCR domains and maps to 21q22. 3. Genomics 1997, 44, 309–320. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef]
- Afar, D.E.; Vivanco, I.; Hubert, R.S.; Kuo, J.; Chen, E.; Saffran, D.C.; Raitano, A.B.; Jakobovits, A. Catalytic cleavage of the androgen-regulated TMPRSS2 protease results in its secretion by prostate and prostate cancer epithelia. Cancer Res. 2001, 61, 1686–1692. [Google Scholar]
- David, A.; Parkinson, N.; Peacock, T.P.; Pairo-Castineira, E.; Khanna, T.; Cobat, A.; Tenesa, A.; Sancho-Shimizu, V.; Casanova, J.-L.; Abel, L. A common TMPRSS2 variant has a protective effect against severe COVID-19. Curr. Res. Transl. Med. 2022, 70, 103333. [Google Scholar] [CrossRef]
- Wettstein, L.; Kirchhoff, F.; Münch, J. The transmembrane protease TMPRSS2 as a therapeutic target for COVID-19 treatment. Int. J. Mol. Sci. 2022, 23, 1351. [Google Scholar] [CrossRef]
- Epstein, R.J. The secret identities of TMPRSS2: Fertility factor, virus trafficker, inflammation moderator, prostate protector and tumor suppressor. Tumor Biol. 2021, 43, 159–176. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- Papaleo, E.; Fantucci, P.; De Gioia, L. Effects of calcium binding on structure and autolysis regulation in trypsins. A molecular dynamics investigation. J. Chem. Theory Comput. 2005, 1, 1286–1297. [Google Scholar] [CrossRef]
- Liu, S.-Q.; Tao, Y.; Meng, Z.-H.; Fu, Y.-X.; Zhang, K.-Q. The effect of calciums on molecular motions of proteinase K. J. Mol. Model. 2011, 17, 289–300. [Google Scholar] [CrossRef]
- Patel, S.; Homaei, A.; El-Seedi, H.R.; Akhtar, N. Cathepsins: Proteases that are vital for survival but can also be fatal. Biomed. Pharmacother. 2018, 105, 526–532. [Google Scholar] [CrossRef]
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The ins and outs of cathepsins: Physiological function and role in disease management. Cells 2020, 9, 1679. [Google Scholar] [CrossRef]
- Gomes, C.P.; Fernandes, D.E.; Casimiro, F.; da Mata, G.F.; Passos, M.T.; Varela, P.; Mastroianni-Kirsztajn, G.; Pesquero, J.B. Cathepsin L in COVID-19, From Pharmacological Evidences to Genetics. Front. Cell. Infect. Microbiol. 2020, 10, 589505. [Google Scholar] [CrossRef]
- Ou, T.; Mou, H.; Zhang, L.; Ojha, A.; Choe, H.; Farzan, M. Hydroxychloroquine-mediated inhibition of SARS-CoV-2 entry is attenuated by TMPRSS2. PLoS Pathog. 2021, 17, e1009212. [Google Scholar] [CrossRef]
- Turner, A. ACE2 cell biology, regulation, and physiological functions. In The Protective Arm of the Renin Angiotensin System (RAS): Functional Aspects and Therapeutic Implications; Unger, T., Steckelings, U.M., Eds.; Academic Press: London, UK, 2015; pp. 185–189. [Google Scholar]
- Scialo, F.; Daniele, A.; Amato, F.; Pastore, L.; Matera, M.G.; Cazzola, M.; Castaldo, G.; Bianco, A. ACE2: The major cell entry receptor for SARS-CoV-2. Lung 2020, 198, 867–877. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Ohto-Nakanishi, T.; Penninger, J.M. Trilogy of ACE2: A peptidase in the renin–angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol. Ther. 2010, 128, 119–128. [Google Scholar] [CrossRef]
- Hu, H.; Zhang, R.; Dong, L.; Chen, E.; Ying, K. Overexpression of ACE2 prevents hypoxia-induced pulmonary hypertension in rats by inhibiting proliferation and immigration of PASMCs. Eur. Rev. Med. Pharm. Sci. 2020, 24, 3968–3980. [Google Scholar]
- Li, M.-Y.; Li, L.; Zhang, Y.; Wang, X.-S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Neimi, M.E.K.; Karjalainen, J.; Liao, R.G.; Neale, B.M.; Daly, M.; Ganna, A.; Pathak, G.A.; Andrews, S.J.; Kanai, M.; Veerapen, K.; et al. Mapping the human genetic architecture of COVID-19. Nature 2021, 600, 472–477. [Google Scholar]
- Luo, Y.; Liu, C.; Guan, T.; Li, Y.; Lai, Y.; Li, F.; Zhao, H.; Maimaiti, T.; Zeyaweiding, A. Association of ACE2 genetic polymorphisms with hypertension-related target organ damages in south Xinjiang. Hypertens. Res. 2019, 42, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Li, L.; Feng, Z.; Wan, S.; Huang, P.; Sun, X.; Wen, F.; Huang, X.; Ning, G.; Wang, W. Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discov. 2020, 6, 1–4. [Google Scholar] [CrossRef]
- Singh, H.; Choudhari, R.; Nema, V.; Khan, A.A. ACE2 and TMPRSS2 polymorphisms in various diseases with special reference to its impact on COVID-19 disease. Microb. Pathog. 2021, 150, 104621. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Liu, Y.; Zhang, Z.; Zhai, Y.; Dai, Y.; Wu, Z.; Nie, X.; Du, L. Polymorphisms and mutations of ACE2 and TMPRSS2 genes are associated with COVID-19, a systematic review. Eur. J. Med. Res. 2022, 27, 1–10. [Google Scholar] [CrossRef]
- Lanjanian, H.; Moazzam-Jazi, M.; Hedayati, M.; Akbarzadeh, M.; Guity, K.; Sedaghati-Khayat, B.; Azizi, F.; Daneshpour, M.S. SARS-CoV-2 infection susceptibility influenced by ACE2 genetic polymorphisms: Insights from Tehran Cardio-Metabolic Genetic Study. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Senapati, S.; Banerjee, P.; Bhagavatula, S.; Kushwaha, P.P.; Kumar, S. Contributions of human ACE2 and TMPRSS2 in determining host–pathogen interaction of COVID-19. J. Genet. 2021, 100, 1–16. [Google Scholar] [CrossRef]
- Devaux, C.A.; Rolain, J.-M.; Raoult, D. ACE2 receptor polymorphism: Susceptibility to SARS-CoV-2, hypertension, multi-organ failure, and COVID-19 disease outcome. J. Microbiol. Immunol. Infect. 2020, 53, 425–435. [Google Scholar] [CrossRef]
- Badawi, S.; Ali, B.R. ACE2 Nascence, trafficking, and SARS-CoV-2 pathogenesis: The saga continues. Hum. Genom. 2021, 15, 1–14. [Google Scholar]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.-C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: Celebrating the 20th anniversary of the discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R. A novel angiotensin-converting enzyme–related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, e1–e9. [Google Scholar] [CrossRef]
- Haga, S.; Yamamoto, N.; Nakai-Murakami, C.; Osawa, Y.; Tokunaga, K.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. Modulation of TNF-α-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-α production and facilitates viral entry. Proc. Natl. Acad. Sci. USA 2008, 105, 7809–7814. [Google Scholar] [CrossRef]
- Kliche, J.; Kuss, H.; Ali, M.; Ivarsson, Y. Cytoplasmic short linear motifs in ACE2 and integrin β3 link SARS-CoV-2 host cell receptors to mediators of endocytosis and autophagy. Sci. Signal. 2021, 14, eabf1117. [Google Scholar] [CrossRef]
- Karthika, T.; Joseph, J.; Das, V.; Nair, N.; Charulekha, P.; Roji, M.D.; Raj, V.S. SARS-CoV-2 cellular entry is independent of the ACE2 cytoplasmic domain signaling. Cells 2021, 10, 1814. [Google Scholar] [CrossRef]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef]
- Villalobos, L.A.; Hipólito-Luengo, S.; Ramos-González, M.; Cercas, E.; Vallejo, S.; Romero, A.; Romacho, T.; Carraro, R.; Sánchez-Ferrer, C.F.; Peiró, C. The angiotensin-(1-7)/mas axis counteracts angiotensin II-dependent and-independent pro-inflammatory signaling in human vascular smooth muscle cells. Front. Pharmacol. 2016, 7, 482. [Google Scholar] [CrossRef]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef]
- Koka, V.; Huang, X.R.; Chung, A.C.; Wang, W.; Truong, L.D.; Lan, H.Y. Angiotensin II up-regulates angiotensin I-converting enzyme (ACE), but down-regulates ACE2 via the AT1-ERK/p38 MAP kinase pathway. Am. J. Pathol. 2008, 172, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, P.E.; Ferrario, C.M.; Tallant, E.A. MAP kinase/phosphatase pathway mediates the regulation of ACE2 by angiotensin peptides. Am. J. Physiol. Cell Physiol. 2008, 295, C1169–C1174. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.A.; Wei, H. Comparative docking studies to understand the binding affinity of nicotine with soluble ACE2 (sACE2)-SARS-CoV-2 complex over sACE2. Toxicol. Rep. 2020, 7, 1366–1372. [Google Scholar]
- Eguchi, S.; Kawai, T.; Scalia, R.; Rizzo, V. Understanding angiotensin II type 1 receptor signaling in vascular pathophysiology. Hypertension 2018, 71, 804–810. [Google Scholar] [CrossRef]
- Patel, V.B.; Clarke, N.; Wang, Z.; Fan, D.; Parajuli, N.; Basu, R.; Putko, B.; Kassiri, Z.; Turner, A.J.; Oudit, G.Y. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: A positive feedback mechanism in the RAS. J. Mol. Cell. Cardiol. 2014, 66, 167–176. [Google Scholar] [CrossRef]
- Chu, K.Y.; Leung, P.S. Angiotensin II in type 2 diabetes mellitus. Curr. Protein Pept. Sci. 2009, 10, 75–84. [Google Scholar] [CrossRef]
- Ferder, L.; Inserra, F.; Martínez-Maldonado, M. Inflammation and the metabolic syndrome: Role of angiotensin II and oxidative stress. Curr. Hypertens. Rep. 2006, 8, 191–198. [Google Scholar] [CrossRef]
- Ortiz-Pérez, J.T.; Riera, M.; Bosch, X.; De Caralt, T.M.; Perea, R.J.; Pascual, J.; Soler, M.J. Role of circulating angiotensin converting enzyme 2 in left ventricular remodeling following myocardial infarction: A prospective controlled study. PLoS ONE 2013, 8, e61695. [Google Scholar] [CrossRef]
- Hayashida, K.; Bartlett, A.H.; Chen, Y.; Park, P.W. Molecular and cellular mechanisms of ectodomain shedding. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2010, 293, 925–937. [Google Scholar] [CrossRef]
- Marczynska, J.; Ozga, A.; Wlodarczyk, A.; Majchrzak-Gorecka, M.; Kulig, P.; Banas, M.; Michalczyk-Wetula, D.; Majewski, P.; Hutloff, A.; Schwarz, J. The role of metalloproteinase ADAM17 in regulating ICOS Ligand–mediated humoral immune responses. J. Immunol. 2014, 193, 2753–2763. [Google Scholar] [CrossRef]
- Grötzinger, J.; Lorenzen, I.; Düsterhöft, S. Molecular insights into the multilayered regulation of ADAM17: The role of the extracellular region. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2088–2095. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Garbers, C.; Rose-John, S. ADAM17: A molecular switch to control inflammation and tissue regeneration. Trends Immunol. 2011, 32, 380–387. [Google Scholar] [CrossRef]
- Dreymueller, D.; Pruessmeyer, J.; Groth, E.; Ludwig, A. The role of ADAM-mediated shedding in vascular biology. Eur. J. Cell Biol. 2012, 91, 472–485. [Google Scholar] [CrossRef]
- De Queiroz, T.M.; Lakkappa, N.; Lazartigues, E. ADAM17-mediated shedding of inflammatory cytokines in hypertension. Front. Pharmacol. 2020, 11, 1154. [Google Scholar] [CrossRef]
- Schlöndorff, J.; Becherer, J.D.; Blobel, C.P. Intracellular maturation and localization of the tumour necrosis factor α convertase (TACE). Biochem. J. 2000, 347, 131–138. [Google Scholar] [CrossRef]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. BBA Mol. Cell Res. 2017, 1864, 2059–2070. [Google Scholar] [CrossRef]
- Lorenzen, I.; Lokau, J.; Düsterhöft, S.; Trad, A.; Garbers, C.; Scheller, J.; Rose-John, S.; Grötzinger, J. The membrane-proximal domain of A Disintegrin and Metalloprotease 17 (ADAM17) is responsible for recognition of the interleukin-6 receptor and interleukin-1 receptor II. FEBS Lett. 2012, 586, 1093–1100. [Google Scholar] [CrossRef]
- Weskamp, G.; Tüshaus, J.; Li, D.; Feederle, R.; Maretzky, T.; Swendemann, S.; Falck-Pedersen, E.; McIlwain, D.R.; Mak, T.W.; Salmon, J.E. ADAM17 stabilizes its interacting partner inactive Rhomboid 2 (iRhom2) but not inactive Rhomboid 1 (iRhom1). J. Biol. Chem. 2020, 295, 4350–4358. [Google Scholar] [CrossRef]
- Al-Salihi, M.A.; Lang, P.A. iRhom2: An emerging adaptor regulating immunity and disease. Int. J. Mol. Sci. 2020, 21, 6570. [Google Scholar] [CrossRef]
- Saad, M.I.; Alhayyani, S.; McLeod, L.; Yu, L.; Alanazi, M.; Deswaerte, V.; Tang, K.; Jarde, T.; Smith, J.A.; Prodanovic, Z. ADAM 17 selectively activates the IL-6 trans-signaling/ERK MAPK axis in KRAS-addicted lung cancer. EMBO Mol. Med. 2019, 11, e9976. [Google Scholar] [CrossRef]
- Fan, D.; Kassiri, Z. Biology of tissue inhibitor of metalloproteinase 3 (TIMP3), and its therapeutic implications in cardiovascular pathology. Front. Physiol. 2020, 11, 661. [Google Scholar] [CrossRef] [PubMed]
- Ingram, R.N.; Orth, P.; Strickland, C.L.; Le, H.V.; Madison, V.; Beyer, B.M. Stabilization of the autoproteolysis of TNF-alpha converting enzyme (TACE) results in a novel crystal form suitable for structure-based drug design studies. Protein Eng. Des. Sel. 2006, 19, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.-W.; Li, S.; Li, C.; Zheng, Z.-Q.; Cao, P.; Tong, Z.; Lian, H.; Wang, S.-Y.; Shu, H.-B.; Wang, Y.-Y. iRhom2 is essential for innate immunity to RNA virus by antagonizing ER-and mitochondria-associated degradation of VISA. PLoS Pathog. 2017, 13, e1006693. [Google Scholar] [CrossRef] [PubMed]
- Adrain, C.; Zettl, M.; Christova, Y.; Taylor, N.; Freeman, M. Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 2012, 335, 225–228. [Google Scholar] [CrossRef]
- Chao-Chu, J.; Murtough, S.; Zaman, N.; Pennington, D.J.; Blaydon, D.C.; Kelsell, D.P. iRHOM2: A regulator of palmoplantar biology, inflammation, and viral susceptibility. J. Investig. Dermatol. 2021, 141, 722–726. [Google Scholar] [CrossRef]
- Yoda, M.; Kimura, T.; Tohmonda, T.; Morioka, H.; Matsumoto, M.; Okada, Y.; Toyama, Y.; Horiuchi, K. Systemic overexpression of TNFα-converting enzyme does not lead to enhanced shedding activity in vivo. PLoS ONE 2013, 8, e54412. [Google Scholar] [CrossRef]
- Le Gall, S.M.; Maretzky, T.; Issuree, P.D.; Niu, X.-D.; Reiss, K.; Saftig, P.; Khokha, R.; Lundell, D.; Blobel, C.P. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J. Cell Sci. 2010, 123, 3913–3922. [Google Scholar] [CrossRef]
- Zeng, S.-y.; Yang, L.; Hong, C.-l.; Lu, H.-q.; Yan, Q.-j.; Chen, Y.; Qin, X.-p. Evidence That ADAM17 mediates the protective action of CGRP against angiotensin II-induced inflammation in vascular smooth muscle cells. Mediat. Inflamm. 2018, 2018, 2109352. [Google Scholar] [CrossRef]
- Guo, D.F.; Sun, Y.L.; Hamet, P.; Inagami, T. The angiotensin II type 1 receptor and receptor-associated proteins. Cell Res. 2001, 11, 165–180. [Google Scholar] [CrossRef]
- Nejat, R.; Sadr, A.S.; Torshizi, M.F.; Najafi, D.J. GPCRs of diverse physiologic and pathologic effects with fingerprints in COVID-19. Biol. Life Sci. Forum 2021, 7, 19. [Google Scholar]
- Fuller, A.J.; Hauschild, B.C.; Gonzalez-Villalobos, R.; Awayda, M.S.; Imig, J.D.; Inscho, E.W.; Navar, L.G. Calcium and chloride channel activation by angiotensin II-AT1 receptors in preglomerular vascular smooth muscle cells. Am. J. Physiol. Ren. Physiol. 2005, 289, F760–F767. [Google Scholar] [CrossRef]
- Vivar, R.; Soto, C.; Copaja, M.; Mateluna, F.; Aranguiz, P.; Munoz, J.P.; Chiong, M.; Garcia, L.; Letelier, A.; Thomas, W.G. Phospholipase C/protein kinase C pathway mediates angiotensin II-dependent apoptosis in neonatal rat cardiac fibroblasts expressing AT1 receptor. J. Cardiovasc. Pharmacol. 2008, 52, 184–190. [Google Scholar] [CrossRef]
- Fischer, O.M.; Hart, S.; Gschwind, A.; Prenzel, N.; Ullrich, A. Oxidative and osmotic stress signaling in tumor cells is mediated by ADAM proteases and heparin-binding epidermal growth factor. Mol. Cell. Biol. 2004, 24, 5172–5183. [Google Scholar] [CrossRef]
- Brill, A.; Chauhan, A.K.; Canault, M.; Walsh, M.T.; Bergmeier, W.; Wagner, D.D. Oxidative stress activates ADAM17/TACE and induces its target receptor shedding in platelets in a p38-dependent fashion. Cardiovasc. Res. 2009, 84, 137–144. [Google Scholar] [CrossRef]
- Ford, B.M.; Eid, A.A.; Göőz, M.; Barnes, J.L.; Gorin, Y.C.; Abboud, H.E. ADAM17 mediates Nox4 expression and NADPH oxidase activity in the kidney cortex of OVE26 mice. Am. J. Physiol. Ren. Physiol. 2013, 305, F323–F332. [Google Scholar] [CrossRef]
- Chatterjee, S. Oxidative stress, inflammation, and disease. In Oxidative Stress and Biomaterials; Elsevier: Amsterdam, The Netherlands, 2016; pp. 35–58. [Google Scholar]
- Forcados, G.E.; Muhammad, A.; Oladipo, O.O.; Makama, S.; Meseko, C.A. Metabolic implications of oxidative stress and inflammatory process in SARS-CoV-2 pathogenesis: Therapeutic potential of natural antioxidants. Front. Cell. Infect. Microbiol. 2021, 11, 654813. [Google Scholar] [CrossRef]
- De Costa, R.A.; Granato, D.C.; Trino, L.D.; Yokoo, S.; Carnielli, C.M.; Kawahara, R.; Domingues, R.R.; Pauletti, B.A.; Neves, L.X.; Santana, A.G. ADAM17 cytoplasmic domain modulates Thioredoxin-1 conformation and activity. Redox Biol. 2020, 37, 101735. [Google Scholar] [CrossRef]
- Granato, D.C.; e Costa, R.A.; Kawahara, R.; Yokoo, S.; Aragao, A.Z.; Domingues, R.R.; Pauletti, B.A.; Honorato, R.V.; Fattori, J.; Figueira, A.C.M. Thioredoxin-1 negatively modulates ADAM17 activity through direct binding and indirect reductive activity. Antioxid. Redox Signal. 2018, 29, 717–734. [Google Scholar] [CrossRef]
- Fang, Q.; Liu, X.; Abe, S.; Kobayashi, T.; Wang, X.; Kohyama, T.; Hashimoto, M.; Wyatt, T.; Rennard, S. Thrombin induces collagen gel contraction partially through PAR1 activation and PKC-ε. Eur. Respir. J. 2004, 24, 918–924. [Google Scholar] [CrossRef]
- Yang, C.-C.; Hsiao, L.-D.; Shih, Y.-F.; Hsu, C.-K.; Hu, C.-Y.; Yang, C.-M. Thrombin induces COX-2 and PGE2 expression via PAR1/PKCalpha/MAPK-dependent NF-kappaB activation in human tracheal smooth muscle cells. Mediat. Inflamm. 2022, 2022, 4600029. [Google Scholar] [CrossRef]
- He, R.-Q.; Tang, X.-F.; Zhang, B.-L.; Li, X.-D.; Hong, M.-N.; Chen, Q.-Z.; Han, W.-Q.; Gao, P.-J. Protease-activated receptor 1 and 2 contribute to angiotensin II-induced activation of adventitial fibroblasts from rat aorta. Biochem. Biophys. Res. Commun. 2016, 473, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Menghini, R.; Fiorentino, L.; Casagrande, V.; Lauro, R.; Federici, M. The role of ADAM17 in metabolic inflammation. Atherosclerosis 2013, 228, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Zamel, I.A.; Palakkott, A.; Ashraf, A.; Iratni, R.; Ayoub, M.A. Interplay between angiotensin II type 1 receptor and thrombin receptor revealed by bioluminescence resonance energy transfer assay. Front. Pharmacol. 2020, 11, 1283. [Google Scholar] [CrossRef]
- Hall, K.C.; Blobel, C.P. Interleukin-1 stimulates ADAM17 through a mechanism independent of its cytoplasmic domain or phosphorylation at threonine 735. PLoS ONE 2012, 7, e31600. [Google Scholar] [CrossRef] [PubMed]
- Pearl, M.H.; Grotts, J.; Rossetti, M.; Zhang, Q.; Gjertson, D.W.; Weng, P.; Elashoff, D.; Reed, E.F.; Chambers, E.T. Cytokine profiles associated with angiotensin II type 1 receptor antibodies. Kidney Int. Rep. 2019, 4, 541–550. [Google Scholar] [CrossRef]
- Cardoso, V.G.; Gonçalves, G.L.; Costa-Pessoa, J.M.; Thieme, K.; Lins, B.B.; Casare, F.A.M.; de Ponte, M.C.; Camara, N.O.S.; Oliveira-Souza, M. Angiotensin II-induced podocyte apoptosis is mediated by endoplasmic reticulum stress/PKC-δ/p38 MAPK pathway activation and trough increased Na+/H+ exchanger isoform 1 activity. BMC Nephrol. 2018, 19, 179–191. [Google Scholar] [CrossRef]
- Zhu, G.; Liu, J.; Wang, Y.; Jia, N.; Wang, W.; Zhang, J.; Liang, Y.; Tian, H.; Zhang, J. ADAM17 mediates hypoxia-induced keratinocyte migration via the p38/MAPK pathway. BioMed Res. Int. 2021, 2021, 8328216. [Google Scholar] [CrossRef]
- Xu, P.; Derynck, R. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol. Cell 2010, 37, 551–566. [Google Scholar] [CrossRef]
- Horiuchi, K.; Le Gall, S.; Schulte, M.; Yamaguchi, T.; Reiss, K.; Murphy, G.; Toyama, Y.; Hartmann, D.; Saftig, P.; Blobel, C.P. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol. Biol. Cell 2007, 18, 176–188. [Google Scholar] [CrossRef]
- Kadurin, I.; Dahimene, S.; Page, K.M.; Ellaway, J.I.; Chaggar, K.; Troeberg, L.; Nagase, H.; Dolphin, A.C. ADAM17 mediates proteolytic maturation of voltage-gated calcium channel auxiliary α2δ subunits, and enables calcium current enhancement. Function 2022, 3, zqac013. [Google Scholar] [CrossRef]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-α convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef]
- Cheng, J.; Xue, F.; Cheng, C.; Sui, W.; Zhang, M.; Qiao, L.; Ma, J.; Ji, X.; Chen, W.; Yu, X. ADAM17 knockdown mitigates while ADAM17 overexpression aggravates cardiac fibrosis and dysfunction via regulating ACE2 shedding and myofibroblast transformation. Front. Pharmacol. 2022, 13, 997916. [Google Scholar] [CrossRef]
- Kumar, R.; Khandelwal, N.; Thachamvally, R.; Tripathi, B.N.; Barua, S.; Kashyap, S.K.; Maherchandani, S.; Kumar, N. Role of MAPK/MNK1 signaling in virus replication. Virus Res. 2018, 253, 48–61. [Google Scholar] [CrossRef]
- Pleschka, S. RNA viruses and the mitogenic Raf/MEK/ERK signal transduction cascade. Biol. Chem. 2008, 389, 1273–1282. [Google Scholar] [CrossRef]
- Huang, C.; Feng, F.; Shi, Y.; Li, W.; Wang, Z.; Zhu, Y.; Yuan, S.; Hu, D.; Dai, J.; Jiang, Q. Protein Kinase C inhibitors reduce SARS-CoV-2 replication in cultured cells. Microbiol. Spectr. 2022, 10, e0105622. [Google Scholar] [CrossRef]
- Liu, M.; Yang, Y.; Gu, C.; Yue, Y.; Wu, K.K.; Wu, J.; Zhu, Y. Spike protein of SARS-CoV stimulates cyclooxygenase-2 expression via both calcium-dependent and calcium-independent protein kinase C pathways. FASEB J. 2007, 21, 1586–1596. [Google Scholar] [CrossRef]
- Wieczfinska, J.; Kleniewska, P.; Pawliczak, R. Oxidative stress-related mechanisms in SARS-CoV-2 infections. Oxidative Med. Cell. Longev. 2022, 2022, 5589089. [Google Scholar] [CrossRef]
- Suhail, S.; Zajac, J.; Fossum, C.; Lowater, H.; McCracken, C.; Severson, N.; Laatsch, B.; Narkiewicz-Jodko, A.; Johnson, B.; Liebau, J. Role of oxidative stress on SARS-CoV (SARS) and SARS-CoV-2 (COVID-19) infection: A review. Protein J. 2020, 39, 644–656. [Google Scholar] [CrossRef]
- Berlansky, S.; Sallinger, M.; Grabmayr, H.; Humer, C.; Bernhard, A.; Fahrner, M.; Frischauf, I. Calcium signals during SARS-CoV-2 infection: Assessing the potential of emerging therapies. Cells 2022, 11, 253. [Google Scholar] [CrossRef]
- Vollbracht, C.; Kraft, K. Oxidative stress and hyper-inflammation as major drivers of severe COVID-19 and long COVID: Implications for the benefit of high-dose intravenous vitamin C. Front. Pharmacol. 2022, 13, 899198. [Google Scholar] [CrossRef]
- Aiello, E.; Cingolani, H. Angiotensin II stimulates cardiac L-type Ca2+ current by a Ca2+-and protein kinase C-dependent mechanism. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1528–H1536. [Google Scholar] [CrossRef] [PubMed]
- Verma, K.; Pant, M.; Paliwal, S.; Dwivedi, J.; Sharma, S. An insight on multicentric signaling of angiotensin II in cardiovascular system: A Recent Update. Front. Pharmacol. 2021, 12, 734917. [Google Scholar] [CrossRef] [PubMed]
- Palacios, Y.; Ruiz, A.; Ramon-Luing, L.A.; Ocaña-Guzman, R.; Barreto-Rodriguez, O.; Sanchez-Moncivais, A.; Tecuatzi-Cadena, B.; Regalado-García, A.G.; Pineda-Gudiño, R.D.; García-Martínez, A. Severe COVID-19 patients show an increase in soluble TNFR1 and ADAM17, with a relationship to mortality. Int. J. Mol. Sci. 2021, 22, 8423. [Google Scholar] [CrossRef] [PubMed]
- Vieira, C.; Nery, L.; Martins, L.; Jabour, L.; Dias, R.; Simões e Silva, A.C. Downregulation of membrane-bound angiotensin converting enzyme 2 (ACE2) receptor has a pivotal role in COVID-19 immunopathology. Curr. Drug Targets 2021, 22, 254–281. [Google Scholar] [CrossRef] [PubMed]
- Ramos, S.G.; da Cruz Rattis, B.A.; Ottaviani, G.; Celes, M.R.N.; Dias, E.P. ACE2 down-regulation may act as a transient molecular disease causing RAAS dysregulation and tissue damage in the microcirculatory environment among COVID-19 patients. Am. J. Pathol. 2021, 191, 1154–1164. [Google Scholar] [CrossRef]
- Yalcin, H.C.; Sukumaran, V.; Al-Ruweidi, M.K.A.; Shurbaji, S. Do changes in ace-2 expression affect SARS-CoV-2 virulence and related complications: A closer look into membrane-bound and soluble forms. Int. J. Mol. Sci. 2021, 22, 6703. [Google Scholar] [CrossRef]
- Tseng, Y.H.; Yang, R.C.; Lu, T.S. Two hits to the renin-angiotensin system may play a key role in severe COVID-19. Kaohsiung J. Med. Sci. 2020, 36, 389–392. [Google Scholar] [CrossRef]
- Tang, Q.; Wang, Y.; Ou, L.; Li, J.; Zheng, K.; Zhan, H.; Gu, J.; Zhou, G.; Xie, S.; Zhang, J. Downregulation of ACE2 expression by SARS-CoV-2 worsens the prognosis of KIRC and KIRP patients via metabolism and immunoregulation. Int. J. Biol. Sci. 2021, 17, 1925. [Google Scholar] [CrossRef]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Haga, S.; Nagata, N.; Okamura, T.; Yamamoto, N.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. TACE antagonists blocking ACE2 shedding caused by the spike protein of SARS-CoV are candidate antiviral compounds. Antivir. Res. 2010, 85, 551–555. [Google Scholar] [CrossRef]
- Kornilov, S.A.; Lucas, I.; Jade, K.; Dai, C.L.; Lovejoy, J.C.; Magis, A.T. Plasma levels of soluble ACE2are associated with sex, Metabolic Syndrome, and its biomarkers in a large cohort, pointing to a possible mechanism for increased severity in COVID-19. Crit. Care 2020, 24, 1–3. [Google Scholar] [CrossRef]
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B., Jr. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L84–L96. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, H.; An, Y. ACE2 Shedding and the Role in COVID-19. Front. Cell. Infect. Microbiol. 2022, 11, 789180. [Google Scholar] [CrossRef]
- Paul, M.; Poyan Mehr, A.; Kreutz, R. Physiology of local renin-angiotensin systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef]
- Bitker, L.; Burrell, L.M. Classic and nonclassic renin-angiotensin systems in the critically ill. Crit. Care Clin. 2019, 35, 213–227. [Google Scholar] [CrossRef]
- Tepasse, P.-R.; Vollenberg, R.; Steinebrey, N.; König, S. The Dysregulation of the Renin–Angiotensin System in COVID-19 Studied by Serum Proteomics: Angiotensinogen increases with disease severity. Molecules 2022, 27, 2495. [Google Scholar] [CrossRef]
- Camargo, R.L.; Bombassaro, B.; Monfort-Pires, M.; Mansour, E.; Palma, A.C.; Ribeiro, L.C.; Ulaf, R.G.; Bernardes, A.F.; Nunes, T.A.; Agrela, M.V. Plasma Angiotensin II is increased in critical coronavirus disease 2019. Front. Cardiovasc. Med. 2022, 145, 112420. [Google Scholar] [CrossRef]
- Wu, Z.; Hu, R.; Zhang, C.; Ren, W.; Yu, A.; Zhou, X. Elevation of plasma angiotensin II level is a potential pathogenesis for the critically ill COVID-19 patients. Crit. Care 2020, 24, 1–3. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef]
- Osman, I.O.; Melenotte, C.; Brouqui, P.; Million, M.; Lagier, J.-C.; Parola, P.; Stein, A.; La Scola, B.; Meddeb, L.; Mege, J.-L. Expression of ACE2, soluble ACE2, angiotensin I, angiotensin II and angiotensin-(1-7) is modulated in COVID-19 patients. Front. Immunol. 2021, 12, 2350. [Google Scholar] [CrossRef]
- van Lier, D.; Kox, M.; Santos, K.; van der Hoeven, H.; Pillay, J.; Pickkers, P. Increased blood angiotensin converting enzyme 2 activity in critically ill COVID-19 patients. ERJ Open Res. 2021, 7, 00848–02020. [Google Scholar] [CrossRef] [PubMed]
- Elrayess, M.; Zedan, H.T.; Alattar, R.A.; Abusriwil, H.; Al-Ruweidi, M.A.A.; Almuraikhy, S.; Parengal, J.; Alhariri, B.; Yassine, H.S.; Hssain, A.A.; et al. Soluble ACE2 and angiotensin II levels are modulated in hypertensive COVID-19 patients treated with different antihypertension drugs. Blood Press. 2022, 31, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Kragstrup, T.W.; Singh, H.S.; Grundberg, I.; Nielsen, A.L.-L.; Rivellese, F.; Mehta, A.; Goldberg, M.B.; Filbin, M.R.; Qvist, P.; Bibby, B.M. Plasma ACE2 predicts outcome of COVID-19 in hospitalized patients. PLoS ONE 2021, 16, e0252799. [Google Scholar] [CrossRef] [PubMed]
- Mariappan, V.; Ranganadin, P.; Shanmugam, L.; Rao, S.; Pillai, A.B. Early shedding of membrane-bounded ACE2 could be an indicator for disease severity in SARS-CoV-2. Biochimie 2022, 201, 139–147. [Google Scholar] [CrossRef]
- Hedges, J.F.; Snyder, D.T.; Robison, A.; Grifka-Walk, H.M.; Blackwell, K.; Shepardson, K.; Kominsky, D.; Rynda-Apple, A.; Walcheck, B.; Jutila, M.A. An ADAM17-neutralizing antibody reduces inflammation and mortality while increasing viral burden in a COVID-19 mouse model. Front. Immunol. 2022, 13, 918881. [Google Scholar] [CrossRef]
- Patel, S.K.; Juno, J.A.; Lee, W.S.; Wragg, K.M.; Hogarth, P.M.; Kent, S.J.; Burrell, L.M. Plasma ACE2 activity is persistently elevated following SARS-CoV-2 infection: Implications for COVID-19 pathogenesis and consequences. Eur. Respir. J. 2021, 57, 2003730. [Google Scholar] [CrossRef]
- Lundström, A.; Ziegler, L.; Havervall, S.; Rudberg, A.S.; Von Meijenfeldt, F.; Lisman, T.; Mackman, N.; Sandén, P.; Thålin, C. Soluble angiotensin-converting enzyme 2 is transiently elevated in COVID-19 and correlates with specific inflammatory and endothelial markers. J. Med. Virol. 2021, 93, 5908–5916. [Google Scholar] [CrossRef]
- Said, E.A.; Al-Reesi, I.; Al-Shizawi, N.; Jaju, S.; Al-Balushi, M.S.; Koh, C.Y.; Al-Jabri, A.A.; Jeyaseelan, L. Defining IL-6 levels in healthy individuals: A meta-analysis. J. Med. Virol. 2021, 93, 3915–3924. [Google Scholar] [CrossRef]
- Yan, I.; Schwarz, J.; Lücke, K.; Schumacher, N.; Schumacher, V.; Schmidt, S.; Rabe, B.; Saftig, P.; Donners, M.; Rose-John, S. ADAM17 controls IL-6 signaling by cleavage of the murine IL-6Rα from the cell surface of leukocytes during inflammatory responses. J. Leukoc. Biol. 2016, 99, 749–760. [Google Scholar] [CrossRef]
- Pedersen, K.B.; Chodavarapu, H.; Porretta, C.; Robinson, L.K.; Lazartigues, E. Dynamics of ADAM17-mediated shedding of ACE2 applied to pancreatic islets of male db/db mice. Endocrinology 2015, 156, 4411–4425. [Google Scholar] [CrossRef]
- Contini, C.; Caselli, E.; Martini, F.; Maritati, M.; Torreggiani, E.; Seraceni, S.; Vesce, F.; Perri, P.; Rizzo, L.; Tognon, M. COVID-19 is a multifaceted challenging pandemic which needs urgent public health interventions. Microorganisms 2020, 8, 1228. [Google Scholar] [CrossRef]
- Quinaglia, T.; Shabani, M.; Breder, I.; Silber, H.A.; Lima, J.A.; Sposito, A.C. Coronavirus disease-19: The multi-level, multi-faceted vasculopathy. Atherosclerosis 2021, 322, 39–50. [Google Scholar] [CrossRef]
- Daniell, H.; Nair, S.K.; Shi, Y.; Wang, P.; Montone, K.T.; Shaw, P.A.; Choi, G.H.; Ghani, D.; Weaver, J.; Rader, D.J. Decrease in Angiotensin-Converting Enzyme activity but not concentration in plasma/lungs in COVID-19 patients offers clues for diagnosis/treatment. Mol. Ther. Methods Clin. Dev. 2022, 26, 266–278. [Google Scholar] [CrossRef]
- Carrera, E.; Linares, A.; Joachim, A.; Jean-Baptiste, M.; Speth, R. MCA-APK (Dnp) is not a selective substrate of angiotensin-converting enzyme-2 (1067.6). FASEB J. 2014, 28, 1067. [Google Scholar] [CrossRef]
- Elased, K.M.; Cunha, T.S.; Gurley, S.B.; Coffman, T.M.; Morris, M. New mass spectrometric assay for angiotensin-converting enzyme 2 activity. Hypertension 2006, 47, 1010–1017. [Google Scholar] [CrossRef]
- Chappell, M.C.; Pirro, N.T.; Sykes, A.; Ferrario, C.M. Metabolism of angiotensin-(1–7) by angiotensin-converting enzyme. Hypertension 1998, 31, 362–367. [Google Scholar] [CrossRef]
- Chappell, M.C. The angiotensin-(1-7) axis: Formation and metabolism pathways. In Angiotensin-(1-7): A Comprehensive Review; Dos Santos, R.A.S., Ed.; Springer: Cham, Switzerland, 2019; pp. 1–26. [Google Scholar]
- Chappell, M.C.; Pirro, N.T.; South, A.M.; Gwathmey, T.M. Concerns on the Specificity of Commercial ELISAs for the Measurement of Angiotensin (1–7) and Angiotensin II in Human Plasma. Hypertension 2021, 77, e29–e31. [Google Scholar] [CrossRef]
- Maiti, B.K. Bioengineered angiotensin-converting-enzyme-2: A potential therapeutic option against SARS-CoV-2 infection. J. Hum. Hypertens. 2022, 36, 488–492. [Google Scholar] [CrossRef]
- Kiseleva, A.A.; Troisi, E.M.; Hensley, S.E.; Kohli, R.M.; Epstein, J.A. SARS-CoV-2 spike protein binding selectively accelerates substrate-specific catalytic activity of ACE2. J. Biochem. 2021, 170, 299–306. [Google Scholar] [CrossRef]
- Pedersen, K.B.; Sriramula, S.; Chhabra, K.H.; Xia, H.; Lazartigues, E. Species-specific inhibitor sensitivity of angiotensin-converting enzyme 2 (ACE2) and its implication for ACE2 activity assays. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1293–R1299. [Google Scholar] [CrossRef]
- Oz, M.; Lorke, D.E. Multifunctional angiotensin converting enzyme 2, the SARS-CoV-2 entry receptor, and critical appraisal of its role in acute lung injury. Biomed. Pharmacother. 2021, 136, 111193. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B., Jr.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg9 bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sun, L.; Ullah, I.; Beaudoin-Bussières, G.; Anand, S.P.; Hederman, A.P.; Tolbert, W.D.; Sherburn, R.; Nguyen, D.N.; Marchitto, L. Engineered ACE2-Fc counters murine lethal SARS-CoV-2 infection through direct neutralization and Fc-effector activities. Sci. Adv. 2022, 8, eabn4188. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.P.; Chen, Y.; Prévost, J.; Gasser, R.; Beaudoin-Bussières, G.; Abrams, C.F.; Pazgier, M.; Finzi, A. Interaction of human ACE2 to membrane-bound SARS-CoV-1 and SARS-CoV-2 S glycoproteins. Viruses 2020, 12, 1104. [Google Scholar] [CrossRef]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef]
- Rahman, M.M.; Hasan, M.; Ahmed, A. Potential detrimental role of soluble ACE2 in severe COVID-19 comorbid patients. Rev. Med. Virol. 2021, 31, 1–12. [Google Scholar] [CrossRef]
- Kiani, A.; Dhuli, K.; Anpilogov, K.; Bressan, S.; Dautaj, A.; Dundar, M.; Beccari, T.; Ergoren, M.; Bertelli, M. Natural compounds as inhibitors of SARS-CoV-2 endocytosis: A promising approach against COVID-19. Acta Bio-medica: Atenei Parmensis 2020, 91, e2020008. [Google Scholar]
- Yeung, M.L.; Teng, J.L.L.; Jia, L.; Zhang, C.; Huang, C.; Cai, J.-P.; Zhou, R.; Chan, K.-H.; Zhao, H.; Zhu, L. Soluble ACE2-mediated cell entry of SARS-CoV-2 via interaction with proteins related to the renin-angiotensin system. Cell 2021, 184, 2212–2228.e2212. [Google Scholar] [CrossRef]
- Zoufaly, A.; Poglitsch, M.; Aberle, J.H.; Hoepler, W.; Seitz, T.; Traugott, M.; Grieb, A.; Pawelka, E.; Laferl, H.; Wenisch, C. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir. Med. 2020, 8, 1154–1158. [Google Scholar] [CrossRef]
- Cocozza, F.; Névo, N.; Piovesana, E.; Lahaye, X.; Buchrieser, J.; Schwartz, O.; Manel, N.; Tkach, M.; Théry, C.; Martin-Jaular, L. Extracellular vesicles containing ACE2 efficiently prevent infection by SARS-CoV-2 Spike protein-containing virus. J. Extracell. Vesicles 2020, 10, e12050. [Google Scholar] [CrossRef]
- Zhu, J.; Su, Y.; Tang, Y. Disrupting ACE2 dimerization mitigates the infection by SARS-CoV-2 Pseudovirus. Front. Virol. 2022, 2, 916700. [Google Scholar] [CrossRef]
- Xiao, T.; Lu, J.; Zhang, J.; Johnson, R.I.; McKay, L.G.; Storm, N.; Lavine, C.L.; Peng, H.; Cai, Y.; Rits-Volloch, S. A trimeric human angiotensin-converting enzyme 2 as an anti-SARS-CoV-2 agent. Nat. Struct. Mol. Biol. 2021, 28, 202–209. [Google Scholar] [CrossRef]
- Nesci, S. SARS-CoV-2 first contact: Spike–ACE2 interactions in COVID-19. Chem. Biol. Drug Des. 2021, 98, 207–211. [Google Scholar] [CrossRef]
- Chan, K.K.; Dorosky, D.; Sharma, P.; Abbasi, S.A.; Dye, J.M.; Kranz, D.M.; Herbert, A.S.; Procko, E. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science 2020, 369, 1261–1265. [Google Scholar] [CrossRef]
- Kayabolen, A.; Akcan, U.; Ozturan, D.; Ulbegi-Polat, H.; Sahin, G.; Degirmenci, N.; Bayraktar, C.; Soyler, G.; Sarayloo, E.; Nurtop, E. Protein scaffold-based multimerization of soluble ACE2 efficiently blocks SARS-CoV-2 infection in vitro and in vivo. Adv. Sci. 2021, 9, 2201294. [Google Scholar] [CrossRef]
- Raghuvamsi, P.V.; Tulsian, N.K.; Samsudin, F.; Qian, X.; Purushotorman, K.; Yue, G.; Kozma, M.M.; Hwa, W.Y.; Lescar, J.; Bond, P.J. SARS-CoV-2 S protein: ACE2 interaction reveals novel allosteric targets. Elife 2021, 10, e63646. [Google Scholar] [CrossRef]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef]
- Feng, F.; Chen, J.; Zhao, J.; Li, Y.; Li, M.; Sun, C. Killing two birds with one stone by administration of soluble ACE2: A promising strategy to treat both cardiovascular diseases and SARS-CoV-2 infection. Viruses 2021, 13, 2243. [Google Scholar] [CrossRef]
- Basu, R.; Poglitsch, M.; Yogasundaram, H.; Thomas, J.; Rowe, B.H.; Oudit, G.Y. Roles of angiotensin peptides and recombinant human ACE2 in heart failure. J. Am. Coll. Cardiol. 2017, 69, 805–819. [Google Scholar] [CrossRef]
- Iwasaki, M.; Saito, J.; Zhao, H.; Sakamoto, A.; Hirota, K.; Ma, D. Inflammation triggered by SARS-CoV-2 and ACE2 augment drives multiple organ failure of severe COVID-19: Molecular mechanisms and implications. Inflammation 2021, 44, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues Prestes, T.R.; Rocha, N.P.; Miranda, A.S.; Teixeira, A.L.; Simoes-e-Silva, A.C. The anti-inflammatory potential of ACE2/angiotensin-(1-7)/Mas receptor axis: Evidence from basic and clinical research. Curr. Drug Targets 2017, 18, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Haschke, M.; Schuster, M.; Poglitsch, M.; Loibner, H.; Salzberg, M.; Bruggisser, M.; Penninger, J.; Krähenbühl, S. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin-converting enzyme 2 in healthy human subjects. Clin. Pharmacokinet. 2013, 52, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Gu, H.; Xie, Z.; Li, T.; Zhang, S.; Lai, C.; Zhu, P.; Wang, K.; Han, L.; Duan, Y.; Zhao, Z. Angiotensin-converting enzyme 2 inhibits lung injury induced by respiratory syncytial virus. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Zhang, H.; Baker, A. Recombinant human ACE2: Acing out angiotensin II in ARDS therapy. Crit. Care 2017, 21, 1–3. [Google Scholar] [CrossRef]
- Roshanravan, N.; Ghaffari, S.; Hedayati, M. Angiotensin converting enzyme-2 as therapeutic target in COVID-19. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 637–639. [Google Scholar] [CrossRef]
- Basit, A.; Ali, T.; Rehman, S.U. Truncated human angiotensin converting enzyme 2; a potential inhibitor of SARS-CoV-2 spike glycoprotein and potent COVID-19 therapeutic agent. J. Biomol. Struct. Dyn. 2021, 39, 3605–3614. [Google Scholar] [CrossRef]
- Ferrari, M.; Mekkaoui, L.; Ilca, F.T.; Akbar, Z.; Bughda, R.; Lamb, K.; Ward, K.; Parekh, F.; Karattil, R.; Allen, C. Characterization of a novel ACE2-based therapeutic with enhanced rather than reduced activity against SARS-CoV-2 variants. J. Virol. 2021, 95, e0068521. [Google Scholar] [CrossRef]
- Vitiello, A.; Ferrara, F. Pharmacotherapy based on ACE2 targeting and COVID-19 infection. Int. J. Mol. Sci. 2022, 23, 6644. [Google Scholar] [CrossRef]
- Krishnamurthy, S.; Lockey, R.; Kolliputi, N. Soluble ACE2 as a potential therapy for COVID-19. Am. J. Physiol. Cell Physiol. 2021, 320, C279–C281. [Google Scholar] [CrossRef]
- Iwanaga, N.; Cooper, L.; Rong, L.; Maness, N.J.; Beddingfield, B.; Qin, Z.; Crabtree, J.; Tripp, R.A.; Yang, H.; Blair, R. ACE2-IgG1 fusions with improved in vitro and in vivo activity against SARS-CoV-2. Iscience 2022, 25, 103670. [Google Scholar] [CrossRef]
- Zhang, Z.; Zeng, E.; Zhang, L.; Wang, W.; Jin, Y.; Sun, J.; Huang, S.; Yin, W.; Dai, J.; Zhuang, Z. Potent prophylactic and therapeutic efficacy of recombinant human ACE2-Fc against SARS-CoV-2 infection in vivo. Cell Discov. 2021, 7, 1–15. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.; Qin, X.; Jiang, X.; Zhang, J.; Mao, L.; Jiang, Z.; Jiang, Y.; Liu, G.; Qiu, J. Recombinant ACE2 protein protects against acute lung injury induced by SARS-CoV-2 spike RBD protein. Crit. Care 2022, 26, 172–193. [Google Scholar] [CrossRef]
- Nejat, R.; Sadr, A. Are losartan and imatinib effective against SARS-CoV2 pathogenesis? A pathophysiologic-based in silico study. In Silico Pharmacol. 2021, 9, 1–22. [Google Scholar] [CrossRef]
- Daniell, H.; Nair, S.K.; Esmaeili, N.; Wakade, G.; Shahid, N.; Ganesan, P.K.; Islam, M.R.; Shepley-McTaggart, A.; Feng, S.; Gary, E.N. Debulking SARS-CoV-2 in saliva using angiotensin converting enzyme 2 in chewing gum to decrease oral virus transmission and infection. Mol. Ther. 2022, 30, 1966–1978. [Google Scholar] [CrossRef]
- Malik, J.A.; Ahmed, S.; Mir, A.; Shinde, M.; Bender, O.; Alshammari, F.; Ansari, M.; Anwar, S. The SARS-CoV-2 mutations versus vaccine effectiveness: New opportunities to new challenges. J. Infect. Public Health 2022, 15, 228–240. [Google Scholar] [CrossRef]
- Thakur, S.; Sasi, S.; Pillai, S.; Nag, A.; Shukla, D.; Singhal, R.; Phalke, S.; Velu, G. SARS-CoV-2 mutations and their impact on diagnostics, therapeutics and vaccines. Front. Med. 2022, 9, 815389. [Google Scholar] [CrossRef]
- Mohammadzadeh, N.; Asl, N.S.M.; Forouharnejad, K.; Ghadimi, K.; Parsa, S.; Mohammadi, S.; Omidi, A. Mechanism and adverse effects of COVID-19 drugs: A basic review. Int. J. Physiol. Pathophysiol. Pharmacol. 2021, 13, 102. [Google Scholar]
- Khalatbari, A.; Aghazadeh, Z.; Ji, C. Adverse effects of anti-Covid-19 drug candidates and alcohol on cellular stress responses of hepatocytes. Hepatol. Commun. 2022, 6, 1262. [Google Scholar] [CrossRef]
- Liu, D.; Zeng, X.; Ding, Z.; Lv, F.; Mehta, J.; Wang, X. Adverse cardiovascular effects of anti-COVID-19 drugs. Front. Pharmacol. 2021, 12, 699949. [Google Scholar] [CrossRef]
- Izcovich, A.; Siemieniuk, R.A.; Bartoszko, J.J.; Ge, L.; Zeraatkar, D.; Kum, E.; Qasim, A.; Khamis, A.M.; Rochwerg, B.; Agoritsas, T. Adverse effects of remdesivir, hydroxychloroquine and lopinavir/ritonavir when used for COVID-19, systematic review and meta-analysis of randomised trials. BMJ Open 2022, 12, e048502. [Google Scholar] [CrossRef]
- Consortium, W.S.T. Remdesivir and three other drugs for hospitalised patients with COVID-19: Final results of the WHO Solidarity randomised trial and updated meta-analyses. Lancet 2022, 399, 1941–1953. [Google Scholar]
- Delano, W.L. PyMol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nejat, R.; Torshizi, M.F.; Najafi, D.J. S Protein, ACE2 and Host Cell Proteases in SARS-CoV-2 Cell Entry and Infectivity; Is Soluble ACE2 a Two Blade Sword? A Narrative Review. Vaccines 2023, 11, 204. https://doi.org/10.3390/vaccines11020204
Nejat R, Torshizi MF, Najafi DJ. S Protein, ACE2 and Host Cell Proteases in SARS-CoV-2 Cell Entry and Infectivity; Is Soluble ACE2 a Two Blade Sword? A Narrative Review. Vaccines. 2023; 11(2):204. https://doi.org/10.3390/vaccines11020204
Chicago/Turabian StyleNejat, Reza, Maziar Fayaz Torshizi, and David J. Najafi. 2023. "S Protein, ACE2 and Host Cell Proteases in SARS-CoV-2 Cell Entry and Infectivity; Is Soluble ACE2 a Two Blade Sword? A Narrative Review" Vaccines 11, no. 2: 204. https://doi.org/10.3390/vaccines11020204
APA StyleNejat, R., Torshizi, M. F., & Najafi, D. J. (2023). S Protein, ACE2 and Host Cell Proteases in SARS-CoV-2 Cell Entry and Infectivity; Is Soluble ACE2 a Two Blade Sword? A Narrative Review. Vaccines, 11(2), 204. https://doi.org/10.3390/vaccines11020204