
CXCR3 Provides a Competitive Advantage for Retention of Mycobacterium tuberculosis-Specific Tissue-Resident Memory T Cells Following a Mucosal Tuberculosis Vaccine


Abstract
1. Introduction
2. Materials and Methods
2.1. Mouse Strains
2.2. rIAV Vaccine and Immunization
2.3. Preparation of Single-Cell Suspensions
2.4. Antibodies
2.5. Flow Cytometry and Intracytoplasmic Cytokine Staining
2.6. Adoptive Transfer
2.7. IFNγ ELISpot
2.8. Statistical Analysis
3. Results
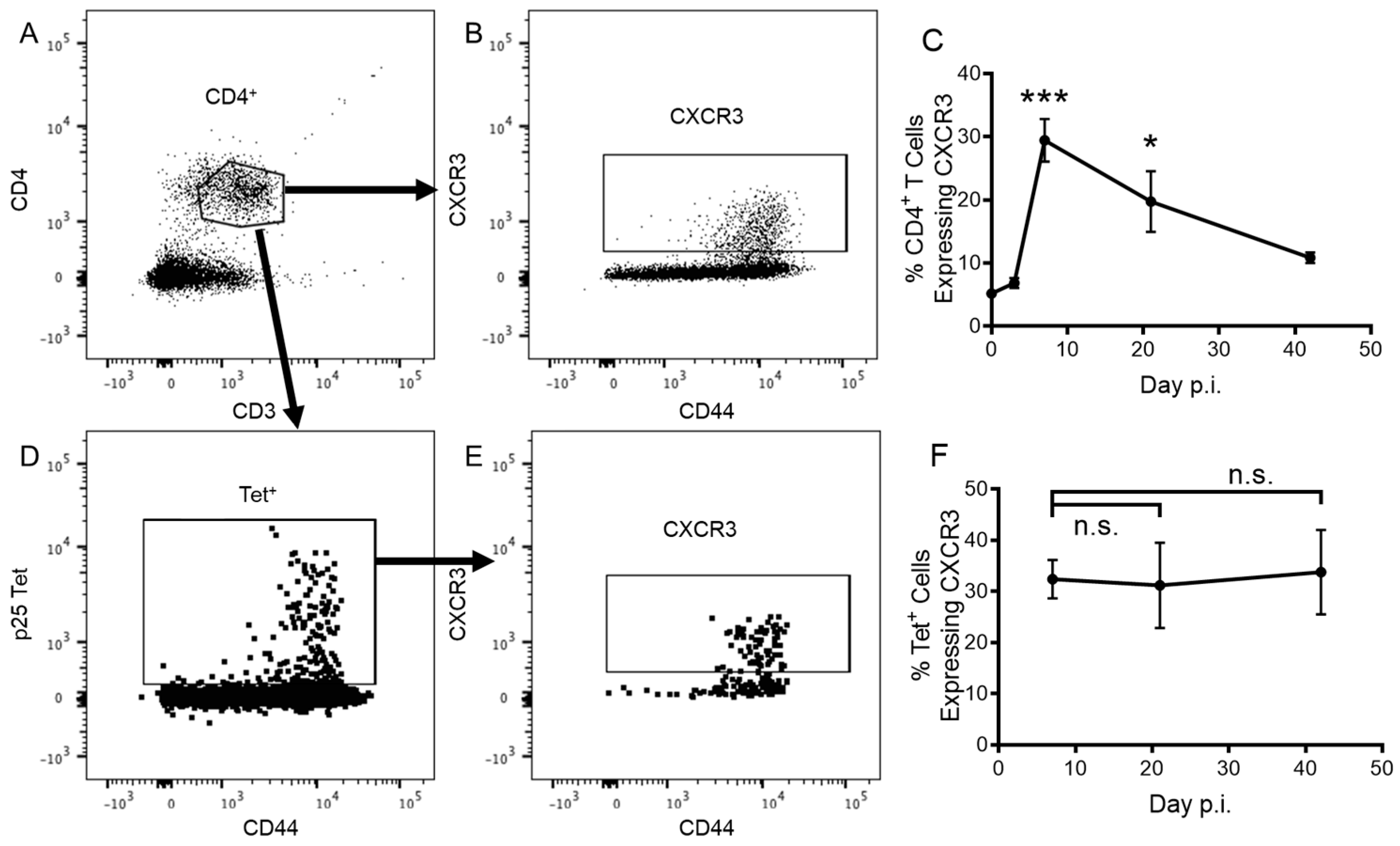
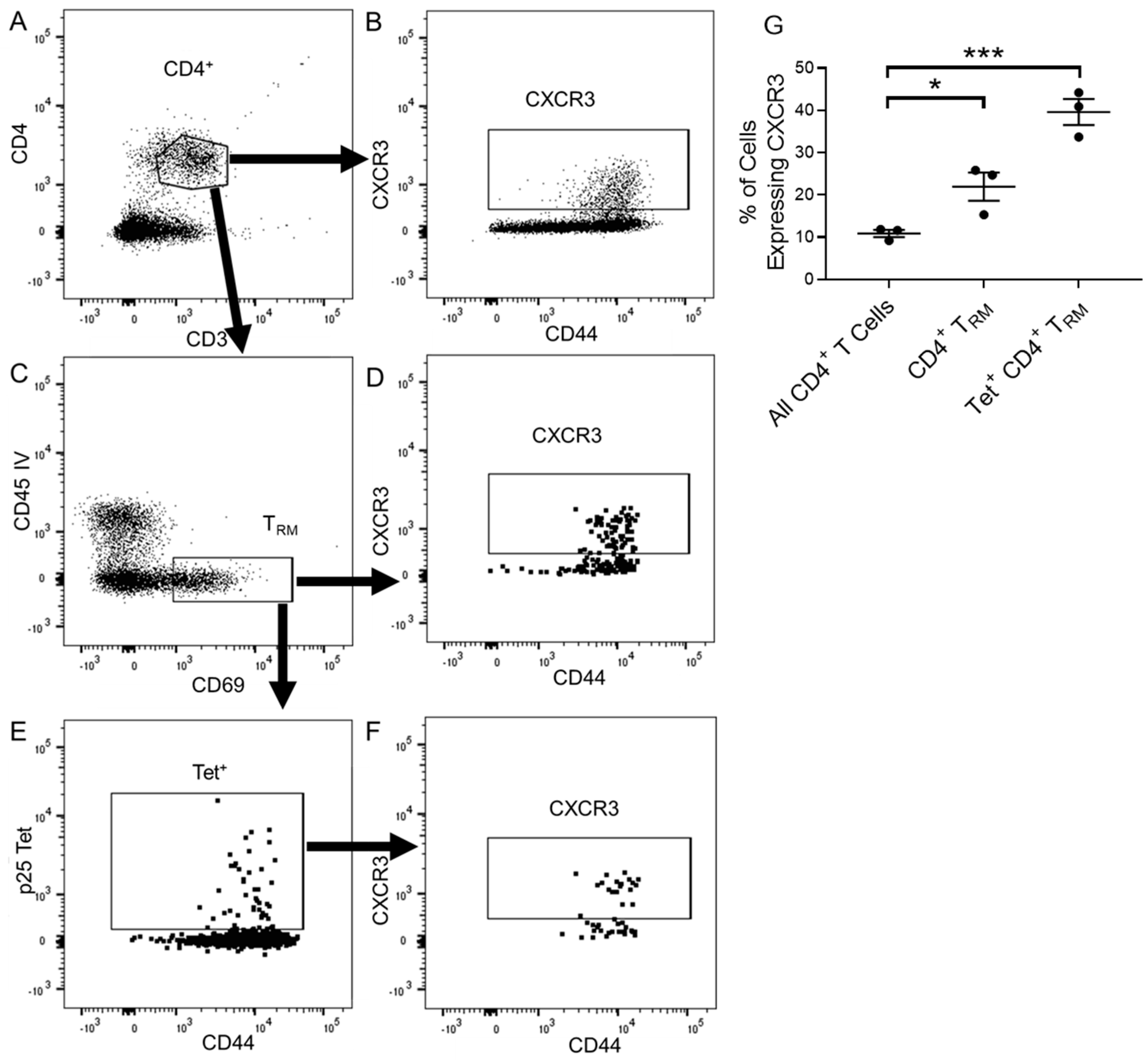
3.1. CD4+ T Cells Express CXCR3 in Response to Pulmonary Vaccination with rIAV
3.2. CXCR3 Is Not Required for the Recruitment of CD4+ T Cells to the Lungs Following rIAV Vaccination
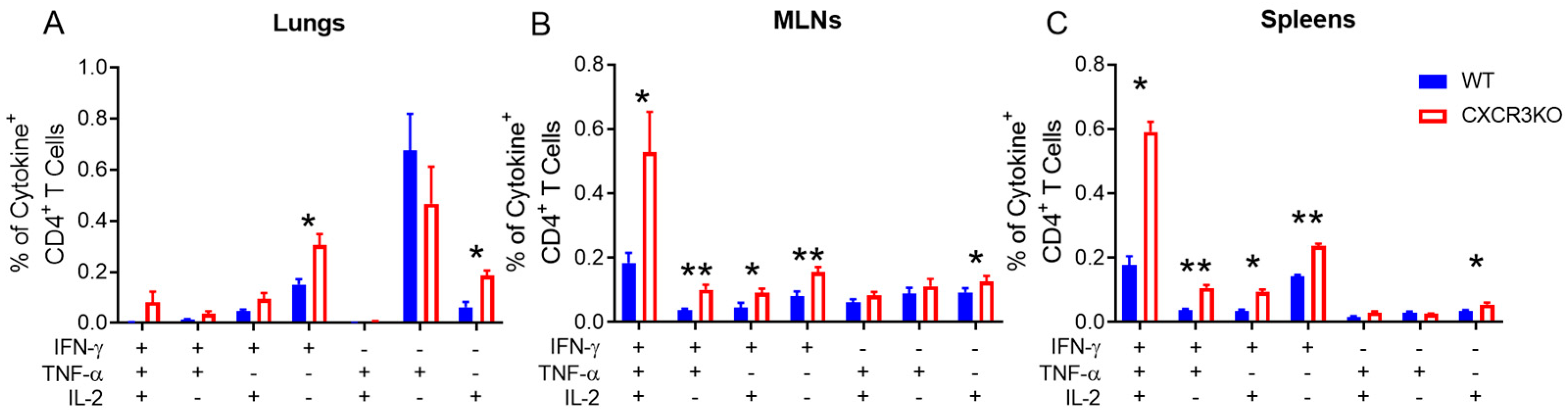
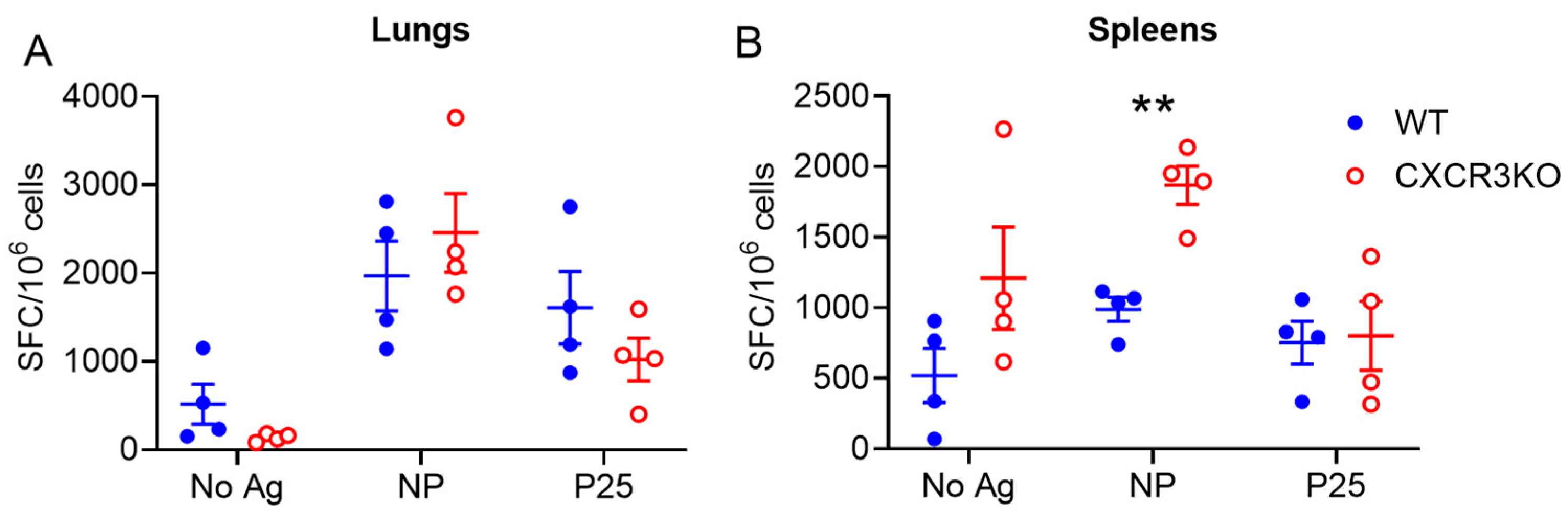
3.3. Pulmonary T Cell Cytokine Responses Are Independent of CXCR3 Following rIAV Vaccination
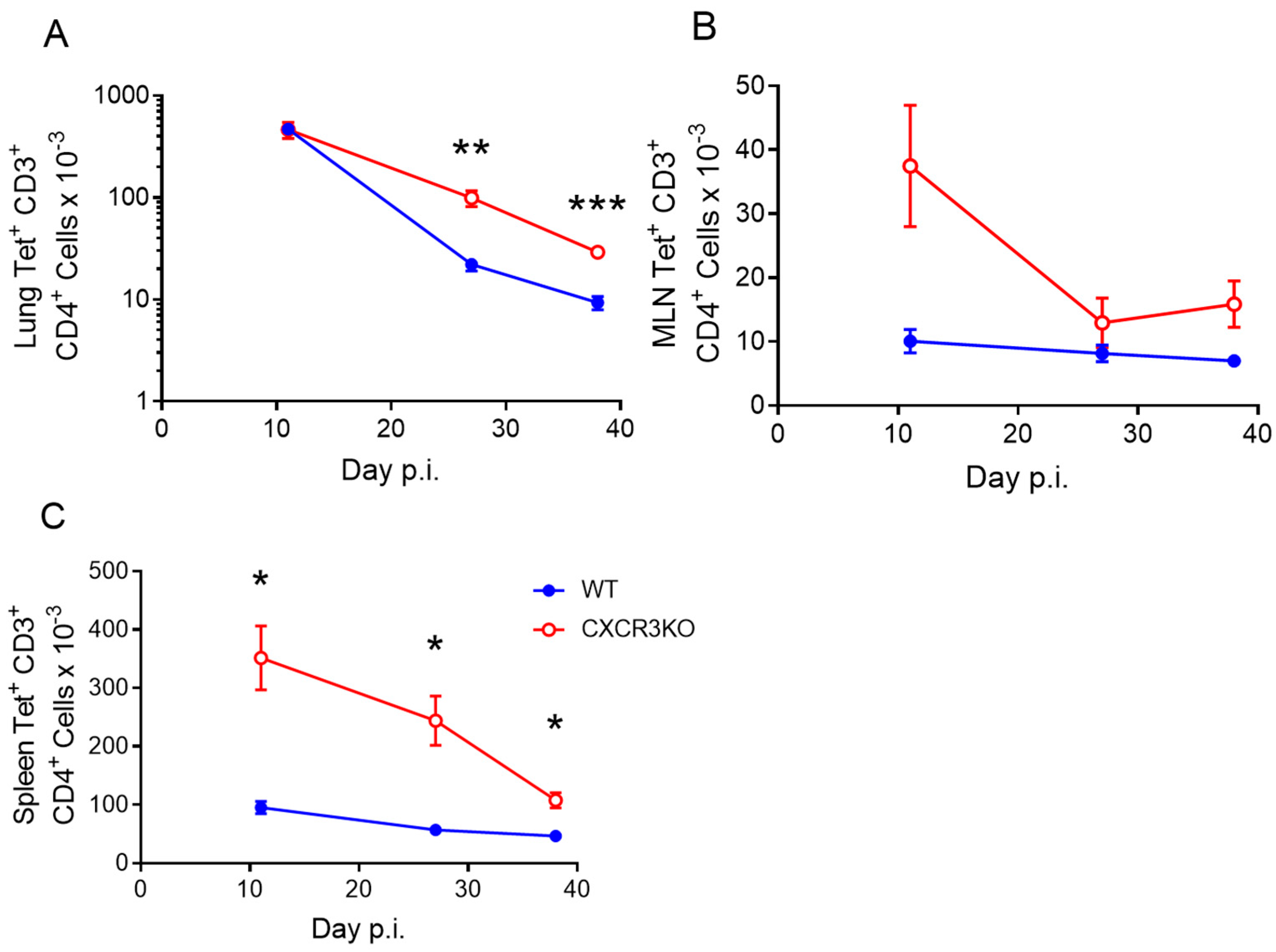
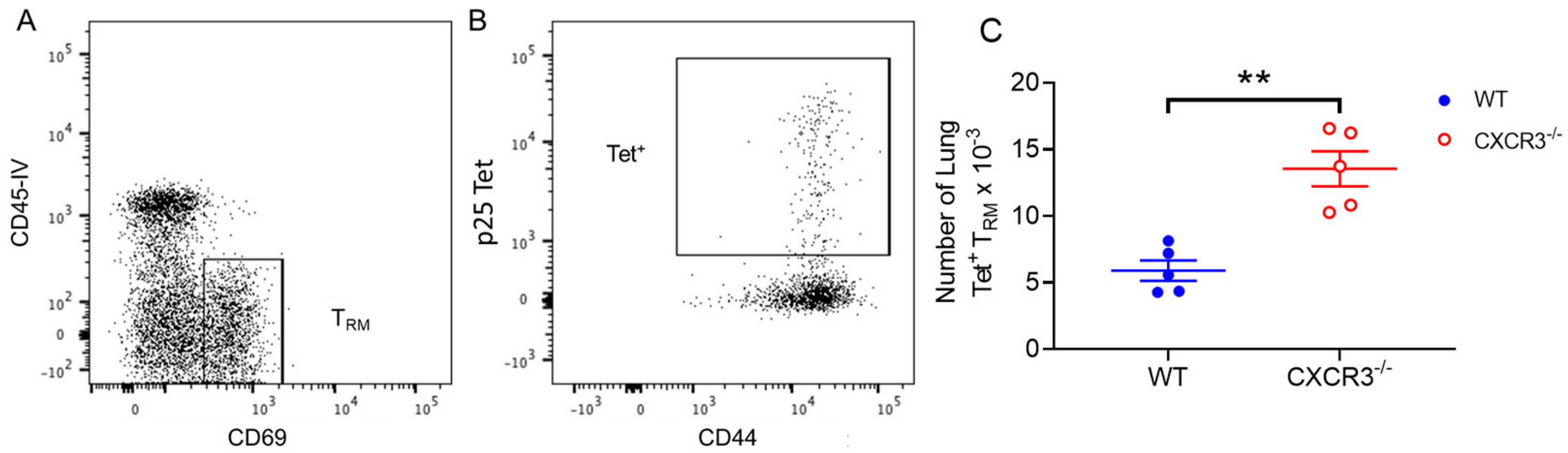
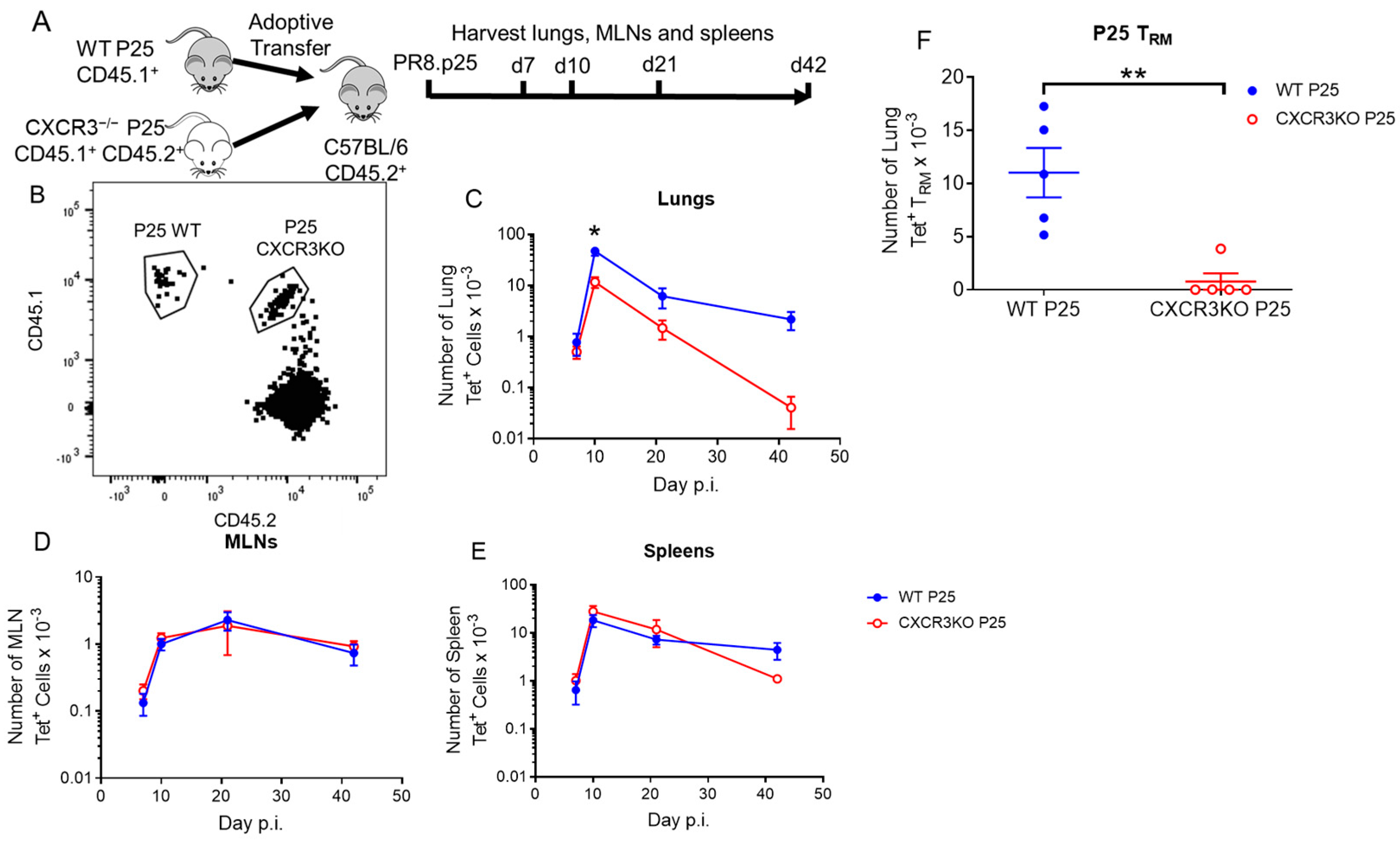
3.4. CXCR3 Provides a Competitive Advantage to CD4+ T Cell Responses to rIAV Vaccination
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report; World Health Organization: Genève, Switzerland, 2022. [Google Scholar]
- Trunz, B.B.; Fine, P.; Dye, C. Effect of BCG vaccination on childhood tuberculous meningitis and miliary tuberculosis worldwide: A meta-analysis and assessment of cost-effectiveness. Lancet 2006, 367, 1173–1180. [Google Scholar] [CrossRef]
- Whittaker, E.; Nicol, M.P.; Zar, H.J.; Tena-Coki, N.G.; Kampmann, B. Age-related waning of immune responses to BCG in healthy children supports the need for a booster dose of BCG in TB endemic countries. Sci. Rep. 2018, 8, 15309. [Google Scholar] [CrossRef]
- Flynn, J.L.; Chan, J.; Triebold, K.J.; Dalton, D.K.; Stewart, T.A.; Bloom, B.R. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 1993, 178, 2249–2254. [Google Scholar] [CrossRef] [PubMed]
- Dorman, S.E.; Holland, S.M. Interferon-gamma and interleukin-12 pathway defects and human disease. Cytokine Growth Factor Rev. 2000, 11, 321–333. [Google Scholar] [CrossRef]
- Kwan, C.K.; Ernst, J.D. HIV and tuberculosis: A deadly human syndemic. Clin. Microbiol. Rev. 2011, 24, 351–376. [Google Scholar] [CrossRef] [PubMed]
- Tameris, M.D.; Hatherill, M.; Landry, B.S.; Scriba, T.J.; Snowden, M.A.; Lockhart, S.; Shea, J.E.; McClain, J.B.; Hussey, G.D.; Hanekom, W.A.; et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: A randomised, placebo-controlled phase 2b trial. Lancet 2013, 381, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Aguilo, N.; Alvarez-Arguedas, S.; Uranga, S.; Marinova, D.; Monzon, M.; Badiola, J.; Martin, C. Pulmonary but not subcutaneous delivery of BCG vaccine confers protection to tuberculosis-susceptible mice by an interleukin 17-dependent mechanism. J. Infect. Dis. 2016, 213, 831–839. [Google Scholar] [CrossRef]
- Florido, M.; Muflihah, H.; Lin, L.C.W.; Xia, Y.; Sierro, F.; Palendira, M.; Feng, C.G.; Bertolino, P.; Stambas, J.; Triccas, J.A.; et al. Pulmonary immunization with a recombinant influenza A virus vaccine induces lung-resident CD4(+) memory T cells that are associated with protection against tuberculosis. Mucosal Immunol. 2018, 11, 1743–1752. [Google Scholar] [CrossRef]
- Coler, R.N.; Day, T.A.; Ellis, R.; Piazza, F.M.; Beckmann, A.M.; Vergara, J.; Rolf, T.; Lu, L.; Alter, G.; Hokey, D.; et al. The TLR-4 agonist adjuvant, GLA-SE, improves magnitude and quality of immune responses elicited by the ID93 tuberculosis vaccine: First-in-human trial. NPJ Vaccines 2018, 3, 34. [Google Scholar] [CrossRef]
- Masopust, D.; Choo, D.; Vezys, V.; Wherry, E.J.; Duraiswamy, J.; Akondy, R.; Wang, J.; Casey, K.A.; Barber, D.L.; Kawamura, K.S.; et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J. Exp. Med. 2010, 207, 553–564. [Google Scholar] [CrossRef]
- Masopust, D.; Soerens, A.G. Tissue-resident T cells and other resident leukocytes. Annu. Rev. Immunol. 2019, 37, 521–546. [Google Scholar] [CrossRef] [PubMed]
- Sia, J.K.; Rengarajan, J. Immunology of Mycobacterium tuberculosis infections. Microbiol. Spectr. 2019, 7, GPP3-0022-2018. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Turner, D.; Pham, Q.; Wherry, E.J.; Lefrancois, L.; Farber, D.L. Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J. Immunol. 2011, 187, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Clark, R.A.; Liu, L.; Wagers, A.J.; Fuhlbrigge, R.C.; Kupper, T.S. Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature 2012, 483, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Peterson, A.C.; Brane, L.; Huppler, A.R.; Hernandez-Santos, N.; Whibley, N.; Garg, A.V.; Simpson-Abelson, M.R.; Gibson, G.A.; Mamo, A.J.; et al. Oral-resident natural Th17 cells and gammadelta T cells control opportunistic Candida albicans infections. J. Exp. Med. 2014, 211, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- Glennie, N.D.; Yeramilli, V.A.; Beiting, D.P.; Volk, S.W.; Weaver, C.T.; Scott, P. Skin-resident memory CD4+ T cells enhance protection against Leishmania major infection. J. Exp. Med. 2015, 212, 1405–1414. [Google Scholar] [CrossRef]
- Stary, G.; Olive, A.; Radovic-Moreno, A.F.; Gondek, D.; Alvarez, D.; Basto, P.A.; Perro, M.; Vrbanac, V.D.; Tager, A.M.; Shi, J.; et al. VACCINES. A mucosal vaccine against Chlamydia trachomatis generates two waves of protective memory T cells. Science 2015, 348, aaa8205. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, D.; Ng, W.Y.; Holz, L.E.; Ma, J.Z.; Zaid, A.; Wong, Y.C.; Lau, L.S.; Mollard, V.; Cozijnsen, A.; Collins, N.; et al. Liver-resident memory CD8(+) T cells form a front-line defense against malaria liver-stage infection. Immunity 2016, 45, 889–902. [Google Scholar] [CrossRef]
- Cha, S.B.; Kim, W.S.; Kim, J.S.; Kim, H.; Kwon, K.W.; Han, S.J.; Cho, S.N.; Coler, R.N.; Reed, S.G.; Shin, S.J. Pulmonary immunity and durable protection induced by the ID93/GLA-SE vaccine candidate against the hyper-virulent Korean Beijing Mycobacterium tuberculosis strain K. Vaccine 2016, 34, 2179–2187. [Google Scholar] [CrossRef]
- Perdomo, C.; Zedler, U.; Kuhl, A.A.; Lozza, L.; Saikali, P.; Sander, L.E.; Vogelzang, A.; Kaufmann, S.H.; Kupz, A. Mucosal BCG vaccination induces protective lung-resident memory T cell populations against tuberculosis. mBio 2016, 7, e01686-16. [Google Scholar] [CrossRef]
- Gupta, A.; Saqib, M.; Singh, B.; Pal, L.; Nishikanta, A.; Bhaskar, S. Mycobacterium indicus pranii Induced Memory T-Cells in Lung Airways Are Sentinels for Improved Protection against M.tb Infection. Front. Immunol. 2019, 10, 2359. [Google Scholar] [CrossRef] [PubMed]
- Groom, J.R. Regulators of T-cell fate: Integration of cell migration, differentiation and function. Immunol. Rev. 2019, 289, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Wein, A.N.; McMaster, S.R.; Takamura, S.; Dunbar, P.R.; Cartwright, E.K.; Hayward, S.L.; McManus, D.T.; Shimaoka, T.; Ueha, S.; Tsukui, T.; et al. CXCR6 regulates localization of tissue-resident memory CD8 T cells to the airways. J. Exp. Med. 2019, 216, 2748–2762. [Google Scholar] [CrossRef]
- Torraca, V.; Cui, C.; Boland, R.; Bebelman, J.P.; van der Sar, A.M.; Smit, M.J.; Siderius, M.; Spaink, H.P.; Meijer, A.H. The CXCR3-CXCL11 signaling axis mediates macrophage recruitment and dissemination of mycobacterial infection. Dis. Models Mech. 2015, 8, 253–269. [Google Scholar] [CrossRef]
- Carlin, L.E.; Hemann, E.A.; Zacharias, Z.R.; Heusel, J.W.; Legge, K.L. Natural killer cell recruitment to the lung during influenza A virus infection is dependent on CXCR3, CCR5, and virus exposure dose. Front. Immunol. 2018, 9, 781. [Google Scholar] [CrossRef]
- Kohlmeier, J.E.; Cookenham, T.; Miller, S.C.; Roberts, A.D.; Christensen, J.P.; Thomsen, A.R.; Woodland, D.L. CXCR3 directs antigen-specific effector CD4+ T cell migration to the lung during parainfluenza virus infection. J. Immunol. 2009, 183, 4378–4384. [Google Scholar] [CrossRef]
- Fadel, S.A.; Bromley, S.K.; Medoff, B.D.; Luster, A.D. CXCR3-deficiency protects influenza-infected CCR5-deficient mice from mortality. Eur. J. Immunol. 2008, 38, 3376–3387. [Google Scholar] [CrossRef]
- Zhang, W. IP-10 for the diagnosis of tuberculosis in children: Protocol for a systematic review and meta-analysis. Medicine 2019, 98, e15977. [Google Scholar] [CrossRef]
- Jeyanathan, M.; Afkhami, S.; Khera, A.; Mandur, T.; Damjanovic, D.; Yao, Y.; Lai, R.; Haddadi, S.; Dvorkin-Gheva, A.; Jordana, M.; et al. CXCR3 signaling is required for restricted homing of parenteral tuberculosis vaccine-induced T cells to both the lung parenchyma and airway. J. Immunol. 2017, 199, 2555–2569. [Google Scholar] [CrossRef]
- Sakai, S.; Kauffman, K.D.; Schenkel, J.M.; McBerry, C.C.; Mayer-Barber, K.D.; Masopust, D.; Barber, D.L. Cutting edge: Control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J. Immunol. 2014, 192, 2965–2969. [Google Scholar] [CrossRef]
- Groom, J.R.; Richmond, J.; Murooka, T.T.; Sorensen, E.W.; Sung, J.H.; Bankert, K.; von Andrian, U.H.; Moon, J.J.; Mempel, T.R.; Luster, A.D. CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity 2012, 37, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.D.; Sallin, M.A.; Sakai, S.; Kamenyeva, O.; Kabat, J.; Weiner, D.; Sutphin, M.; Schimel, D.; Via, L.; Barry, C.E., 3rd; et al. Defective positioning in granulomas but not lung-homing limits CD4 T-cell interactions with Mycobacterium tuberculosis-infected macrophages in rhesus macaques. Mucosal Immunol. 2017, 11, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Florido, M.; Pillay, R.; Gillis, C.M.; Xia, Y.; Turner, S.J.; Triccas, J.A.; Stambas, J.; Britton, W.J. Epitope-specific CD4+, but not CD8+, T-cell responses induced by recombinant influenza A viruses protect against Mycobacterium tuberculosis infection. Eur. J. Immunol. 2015, 45, 780–793. [Google Scholar] [CrossRef]
- Hoffmann, E.; Krauss, S.; Perez, D.; Webby, R.; Webster, R.G. Eight-plasmid system for rapid generation of influenza virus vaccines. Vaccine 2002, 20, 3165–3170. [Google Scholar] [CrossRef]
- Kariyone, A.; Tamura, T.; Kano, H.; Iwakura, Y.; Takeda, K.; Akira, S.; Takatsu, K. Immunogenicity of peptide-25 of Ag85B in Th1 development: Role of IFN-gamma. Int. Immunol. 2003, 15, 1183–1194. [Google Scholar] [CrossRef]
- Olsen, A.W.; Hansen, P.R.; Holm, A.; Andersen, P. Efficient protection against Mycobacterium tuberculosis by vaccination with a single subdominant epitope from the ESAT-6 antigen. Eur. J. Immunol. 2000, 30, 1724–1732. [Google Scholar] [CrossRef]
- Anderson, K.G.; Mayer-Barber, K.; Sung, H.; Beura, L.; James, B.R.; Taylor, J.J.; Qunaj, L.; Griffith, T.S.; Vezys, V.; Barber, D.L.; et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat. Protoc. 2014, 9, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.; Copland, A.; Diogo, G.R.; Harris, S.; Spallek, R.; Oehlmann, W.; Singh, M.; Basile, J.; Rottenberg, M.; Paul, M.J.; et al. Nanoparticle-fusion protein complexes protect against Mycobacterium tuberculosis infection. Mol. Ther. 2018, 26, 822–833. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, M.; Chen, Q.; Chen, W.; Wei, C.; Qiao, K.; Ye, T.; Deng, G.; Li, J.; Zhu, J.; et al. Cutting Edge: Characterization of Human Tissue-Resident Memory T Cells at Different Infection Sites in Patients with Tuberculosis. J. Immunol. 2020, 204, 2331–2336. [Google Scholar] [CrossRef]
- Pan, Y.; Tian, T.; Park, C.O.; Lofftus, S.Y.; Mei, S.; Liu, X.; Luo, C.; O’Malley, J.T.; Gehad, A.; Teague, J.E.; et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 2017, 543, 252–256. [Google Scholar] [CrossRef]
- Steinert, E.M.; Schenkel, J.M.; Fraser, K.A.; Beura, L.K.; Manlove, L.S.; Igyarto, B.Z.; Southern, P.J.; Masopust, D. Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell 2015, 161, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Ogongo, P.; Porterfield, J.Z.; Leslie, A. Lung Tissue Resident Memory T-Cells in the Immune Response to Mycobacterium tuberculosis. Front. Immunol. 2019, 10, 992. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, E.J.; Boisvert, J.; Murphy, K.; Vierra, M.A.; Genovese, M.C.; Wardlaw, A.J.; Greenberg, H.B.; Hodge, M.R.; Wu, L.; Butcher, E.C.; et al. Expression of the chemokine receptors CCR4, CCR5, and CXCR3 by human tissue-infiltrating lymphocytes. Am. J. Pathol. 2002, 160, 347–355. [Google Scholar] [CrossRef] [PubMed]
- McMaster, S.R.; Wilson, J.J.; Wang, H.; Kohlmeier, J.E. Airway-resident memory CD8 T cells provide antigen-specific protection against respiratory virus challenge through rapid IFN-gamma production. J. Immunol. 2015, 195, 203–209. [Google Scholar] [CrossRef]
- Loetscher, M.; Loetscher, P.; Brass, N.; Meese, E.; Moser, B. Lymphocyte-specific chemokine receptor CXCR3: Regulation, chemokine binding and gene localization. Eur. J. Immunol. 1998, 28, 3696–3705. [Google Scholar] [CrossRef]
- Koch, M.A.; Tucker-Heard, G.; Perdue, N.R.; Killebrew, J.R.; Urdahl, K.B.; Campbell, D.J. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 2009, 10, 595–602. [Google Scholar] [CrossRef]
- Lindgren, G.; Ols, S.; Liang, F.; Thompson, E.A.; Lin, A.; Hellgren, F.; Bahl, K.; John, S.; Yuzhakov, O.; Hassett, K.J.; et al. Induction of Robust B Cell Responses after Influenza mRNA vaccination is accompanied by circulating hemagglutinin-specific ICOS+ PD-1+ CXCR3+ T follicular helper cells. Front. Immunol. 2017, 8, 1539. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, X.; Zhang, X.; Yu, Q.; Zhao, P.; Wang, J.; Duan, C.; Li, J.; Johnson, H.; Feng, X.; et al. IP-10 and RANTES as biomarkers for pulmonary tuberculosis diagnosis and monitoring. Tuberculosis 2018, 111, 45–53. [Google Scholar] [CrossRef]
- Chung, W.Y.; Lee, K.S.; Jung, Y.J.; Lee, H.L.; Kim, Y.S.; Park, J.H.; Sheen, S.S.; Park, K.J. A TB antigen-stimulated CXCR3 ligand assay for the diagnosis of active pulmonary TB. Chest 2014, 146, 283–291. [Google Scholar] [CrossRef]
- Chung, W.Y.; Yoon, D.; Lee, K.S.; Jung, Y.J.; Kim, Y.S.; Sheen, S.S.; Park, K.J. The usefulness of serum CXCR3 ligands for evaluating the early treatment response in tuberculosis: A longitudinal cohort study. Medicine 2016, 95, e3575. [Google Scholar] [CrossRef]
- Rivino, L.; Messi, M.; Jarrossay, D.; Lanzavecchia, A.; Sallusto, F.; Geginat, J. Chemokine receptor expression identifies Pre-T helper (Th)1, Pre-Th2, and nonpolarized cells among human CD4+ central memory T cells. J. Exp. Med. 2004, 200, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Hikono, H.; Kohlmeier, J.E.; Takamura, S.; Wittmer, S.T.; Roberts, A.D.; Woodland, D.L. Activation phenotype, rather than central- or effector-memory phenotype, predicts the recall efficacy of memory CD8+ T cells. J. Exp. Med. 2007, 204, 1625–1636. [Google Scholar] [CrossRef]
- Shanmugasundaram, U.; Bucsan, A.N.; Ganatra, S.R.; Ibegbu, C.; Quezada, M.; Blair, R.V.; Alvarez, X.; Velu, V.; Kaushal, D.; Rengarajan, J. Pulmonary Mycobacterium tuberculosis control associates with CXCR3- and CCR6-expressing antigen-specific Th1 and Th17 cell recruitment. JCI Insight 2020, 5, e137858. [Google Scholar] [CrossRef]
- Chakravarty, S.D.; Xu, J.; Lu, B.; Gerard, C.; Flynn, J.; Chan, J. The chemokine receptor CXCR3 attenuates the control of chronic Mycobacterium tuberculosis infection in BALB/c mice. J. Immunol. 2007, 178, 1723–1735. [Google Scholar] [CrossRef] [PubMed]
- Bull, N.C.; Stylianou, E.; Kaveh, D.A.; Pinpathomrat, N.; Pasricha, J.; Harrington-Kandt, R.; Garcia-Pelayo, M.C.; Hogarth, P.J.; McShane, H. Enhanced protection conferred by mucosal BCG vaccination associates with presence of antigen-specific lung tissue-resident PD-1(+) KLRG1(-) CD4(+) T cells. Mucosal Immunol. 2019, 12, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Lyon, M.F. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 1961, 190, 372–373. [Google Scholar] [CrossRef]
- Sallusto, F.; Lenig, D.; Mackay, C.R.; Lanzavecchia, A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J. Exp. Med. 1998, 187, 875–883. [Google Scholar] [CrossRef]
- Kim, C.H.; Rott, L.; Kunkel, E.J.; Genovese, M.C.; Andrew, D.P.; Wu, L.; Butcher, E.C. Rules of chemokine receptor association with T cell polarization in vivo. J. Clin. Investig. 2001, 108, 1331–1339. [Google Scholar] [CrossRef]
- Xie, J.H.; Nomura, N.; Lu, M.; Chen, S.L.; Koch, G.E.; Weng, Y.; Rosa, R.; Di Salvo, J.; Mudgett, J.; Peterson, L.B.; et al. Antibody-mediated blockade of the CXCR3 chemokine receptor results in diminished recruitment of T helper 1 cells into sites of inflammation. J. Leukoc. Biol. 2003, 73, 771–780. [Google Scholar] [CrossRef]
- Li, R.; Zhang, N.; Tian, M.; Ran, Z.; Zhu, M.; Zhu, H.; Han, F.; Yin, J.; Zhong, J. Temporary CXCR3 and CCR5 antagonism following vaccination enhances memory CD8 T cell immune responses. Mol. Med. 2016, 22, 497–507. [Google Scholar] [CrossRef]
- Dhume, K.; Finn, C.M.; Strutt, T.M.; Sell, S.; McKinstry, K.K. T-bet optimizes CD4 T-cell responses against influenza through CXCR3-dependent lung trafficking but not functional programming. Mucosal Immunol. 2019, 15, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.L. Vaccination for Mycobacterium tuberculosis infection: Reprogramming CD4 T-cell homing into the lung. Mucosal Immunol. 2017, 10, 318–321. [Google Scholar] [CrossRef]
- Woodworth, J.S.; Cohen, S.B.; Moguche, A.O.; Plumlee, C.R.; Agger, E.M.; Urdahl, K.B.; Andersen, P. Subunit vaccine H56/CAF01 induces a population of circulating CD4 T cells that traffic into the Mycobacterium tuberculosis-infected lung. Mucosal Immunol. 2017, 10, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Lindenstrom, T.; Knudsen, N.P.; Agger, E.M.; Andersen, P. Control of chronic mycobacterium tuberculosis infection by CD4 KLRG1- IL-2-secreting central memory cells. J. Immunol. 2013, 190, 6311–6319. [Google Scholar] [CrossRef] [PubMed]
- Kagina, B.M.; Abel, B.; Scriba, T.J.; Hughes, E.J.; Keyser, A.; Soares, A.; Gamieldien, H.; Sidibana, M.; Hatherill, M.; Gelderbloem, S.; et al. Specific T cell frequency and cytokine expression profile do not correlate with protection against tuberculosis after bacillus Calmette-Guerin vaccination of newborns. Am. J. Respir. Crit. Care Med. 2010, 182, 1073–1079. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | Marker | Fluorophore | Clone | Manufacturer |
|---|---|---|---|---|
| Flow Cytometry | CD3 | PerCP-Cy5.5 | 145-2C11 | BioLegend (San Diego, CA, USA) |
| CD3 | PE-Cy7 | 145-2C11 | BD Biosciences (Sydney, Australia) | |
| CD3 | N/A | 145-2C11 | BD Biosciences | |
| CD28 | N/A | 37.51 | BD Biosciences | |
| CD4 | AF700 | RM4-5 | BD Biosciences | |
| CD44 | FITC | IM7 | BD Biosciences | |
| CD45.1 | Biotin | A20 | BD Biosciences | |
| CD45.2 | PerCP-Cy5.5 | 104 | BioLegend | |
| CD62L | eF450 | MEL-14 | eBioscience (San Diego, CA, USA) | |
| CD69 | PE | H1.2F3 | BD Biosciences | |
| IFN-γ | PE | XMG1.2 | BD Biosciences | |
| IFN-γ | FITC | XMG1.2 | BD Biosciences | |
| TNF | APC | MP6-XT22 | BD Biosciences | |
| TNF | PE | MP6-XT22 | BioLegend | |
| IL-2 | APC | JES6-5H4 | BioLegend | |
| UV LIVE/DEAD® | UV LIVE/DEAD® | N/A | BioLegend | |
| Biotin | Pacific orange | N/A | Invitrogen (Waltham, MA, USA) | |
| p25 tetramer | APC | N/A | NIH Tetramer Core Facility | |
| ELISpot | IFN-γ | N/A | AN18 | Produced in house |
| IFN-γ | N/A | XMG1.2 | Produced in house |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armitage, E.; Quan, D.; Flórido, M.; Palendira, U.; Triccas, J.A.; Britton, W.J. CXCR3 Provides a Competitive Advantage for Retention of Mycobacterium tuberculosis-Specific Tissue-Resident Memory T Cells Following a Mucosal Tuberculosis Vaccine. Vaccines 2023, 11, 1549. https://doi.org/10.3390/vaccines11101549
Armitage E, Quan D, Flórido M, Palendira U, Triccas JA, Britton WJ. CXCR3 Provides a Competitive Advantage for Retention of Mycobacterium tuberculosis-Specific Tissue-Resident Memory T Cells Following a Mucosal Tuberculosis Vaccine. Vaccines. 2023; 11(10):1549. https://doi.org/10.3390/vaccines11101549
Chicago/Turabian StyleArmitage, Ellis, Diana Quan, Manuela Flórido, Umaimainthan Palendira, James A. Triccas, and Warwick J. Britton. 2023. "CXCR3 Provides a Competitive Advantage for Retention of Mycobacterium tuberculosis-Specific Tissue-Resident Memory T Cells Following a Mucosal Tuberculosis Vaccine" Vaccines 11, no. 10: 1549. https://doi.org/10.3390/vaccines11101549
APA StyleArmitage, E., Quan, D., Flórido, M., Palendira, U., Triccas, J. A., & Britton, W. J. (2023). CXCR3 Provides a Competitive Advantage for Retention of Mycobacterium tuberculosis-Specific Tissue-Resident Memory T Cells Following a Mucosal Tuberculosis Vaccine. Vaccines, 11(10), 1549. https://doi.org/10.3390/vaccines11101549