
Persistent Enterovirus Infection: Little Deletions, Long Infections


Abstract
1. Introduction
2. Characteristics of Persistent Enterovirus Infections
2.1. Persistent Infections Can Occur Due to Host Immunodeficiencies
2.2. Carrier Cultures Produce Viruses in the Long Term
2.3. Defective Interfering Viruses Have Genomic Deletions
3. Discovery and Characterization of a Novel Late-Stage “Noninfectious” Persistent Enterovirus Mechanism
3.1. Structure of the Enterovirus Genome
3.2. Persistent Noncytopathic Enteroviruses in Hearts Are Infectious
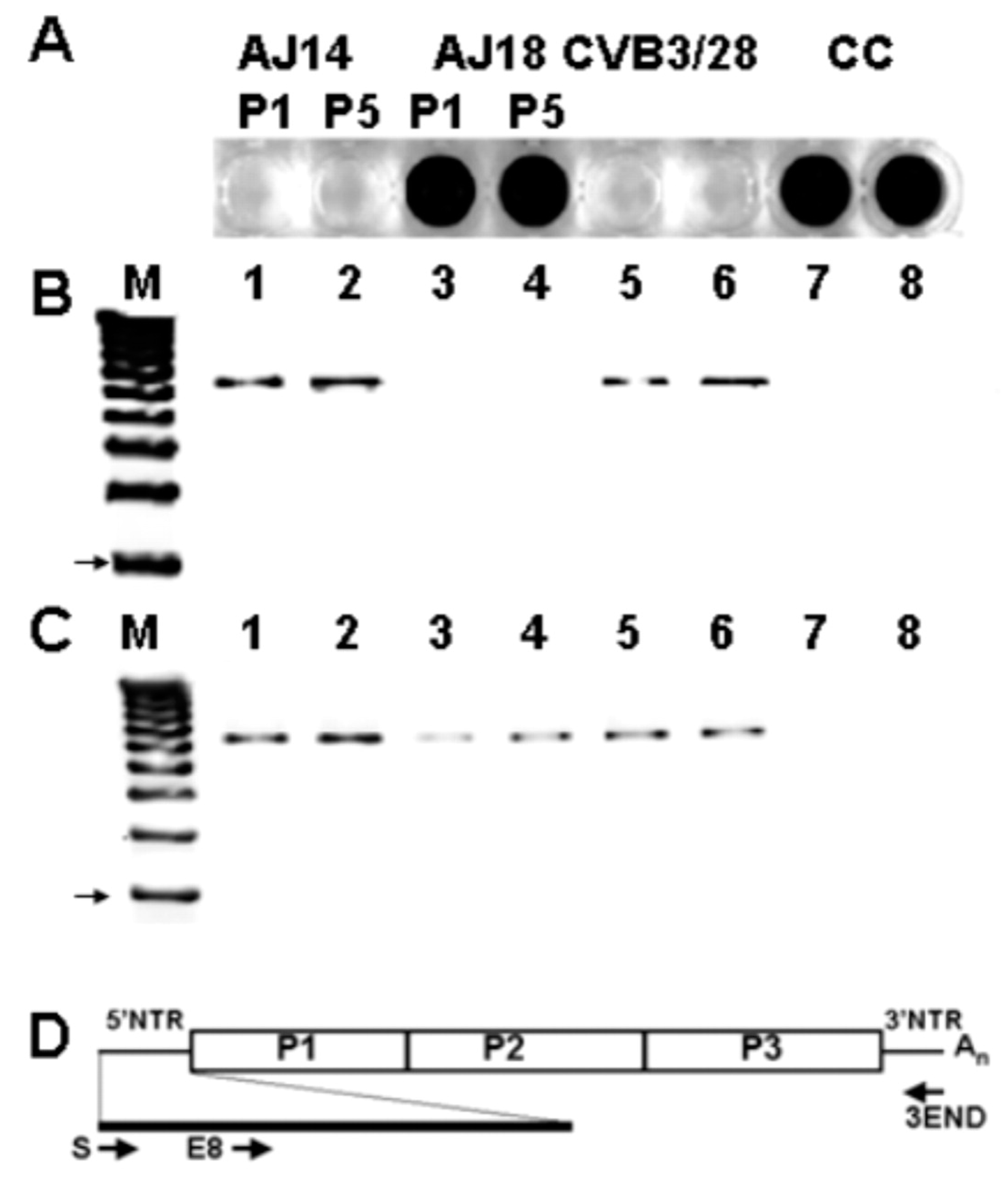
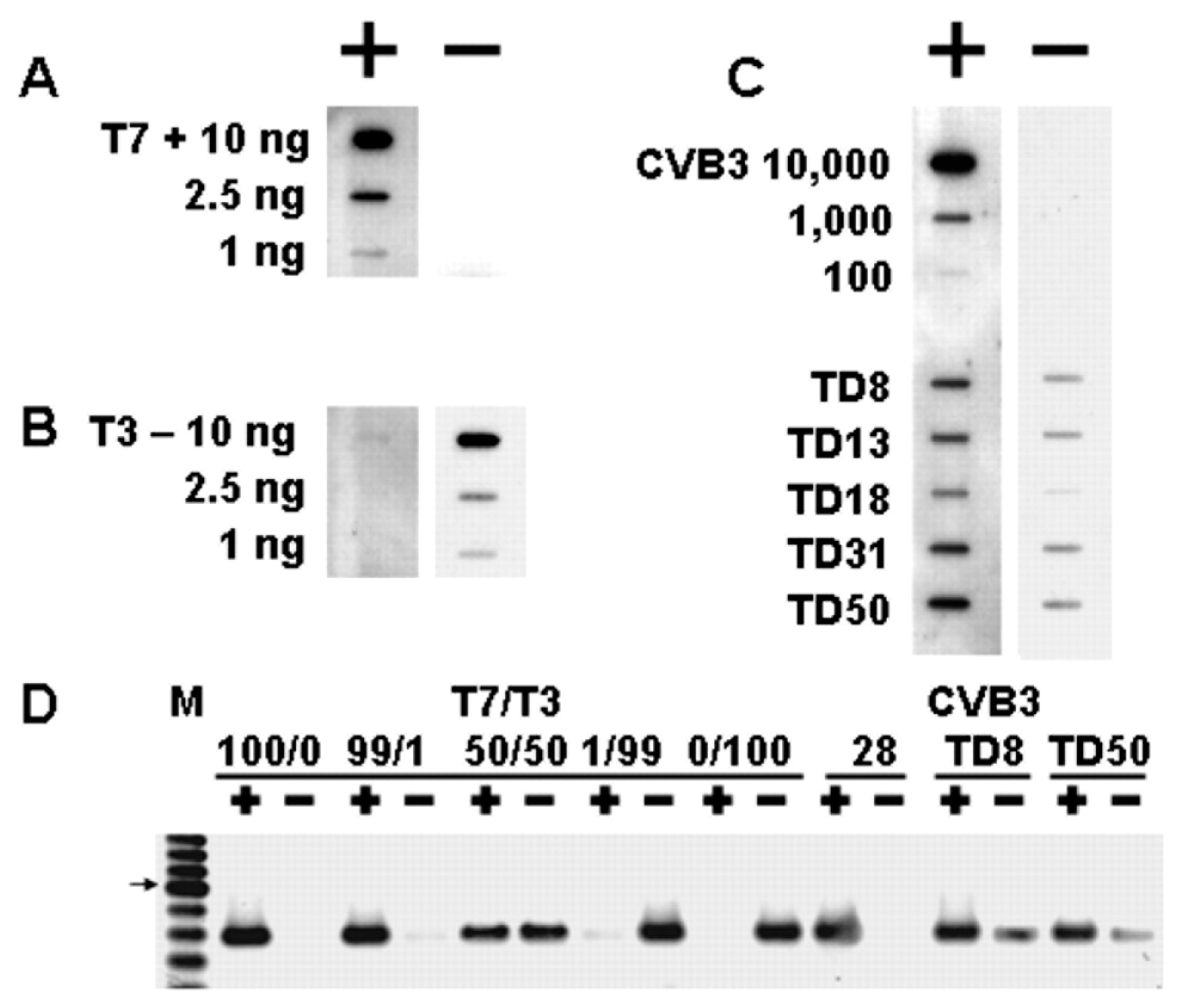
3.3. Persistent CVB Populations in Heart Tissue Demonstrated Deletions at the 5′ Genomic RNA Terminus
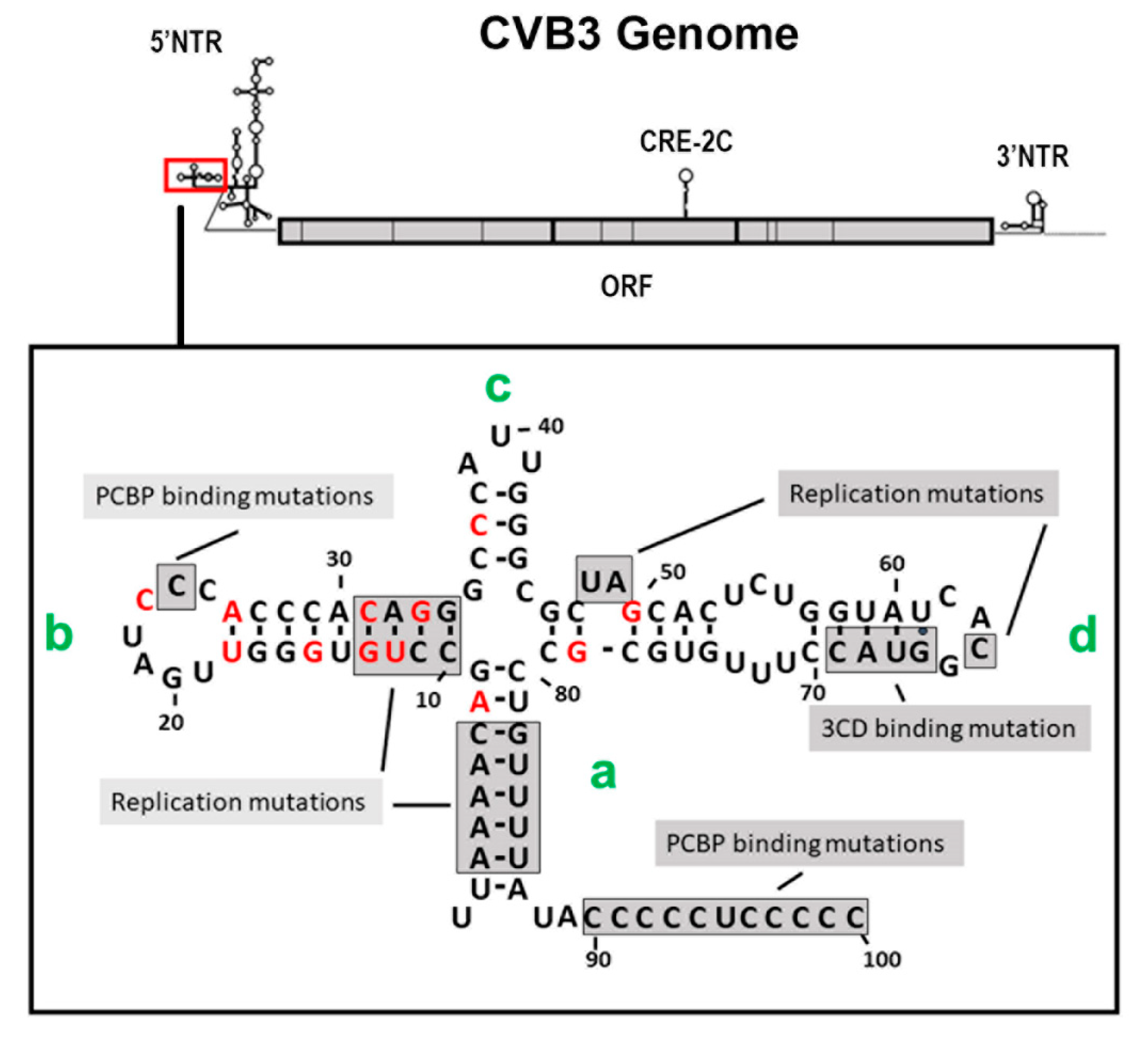
3.4. The 5′ Terminal Deletions Occur within the 5′ RNA Structural Domain Termed the Cloverleaf
3.5. Replication of CVB3-TD Genomes: Roles of Regions of the Cloverleaf
3.6. Evidence That 5′ Terminal Deletions (TDs) Produce Viruses That Replicate at Very Low Levels
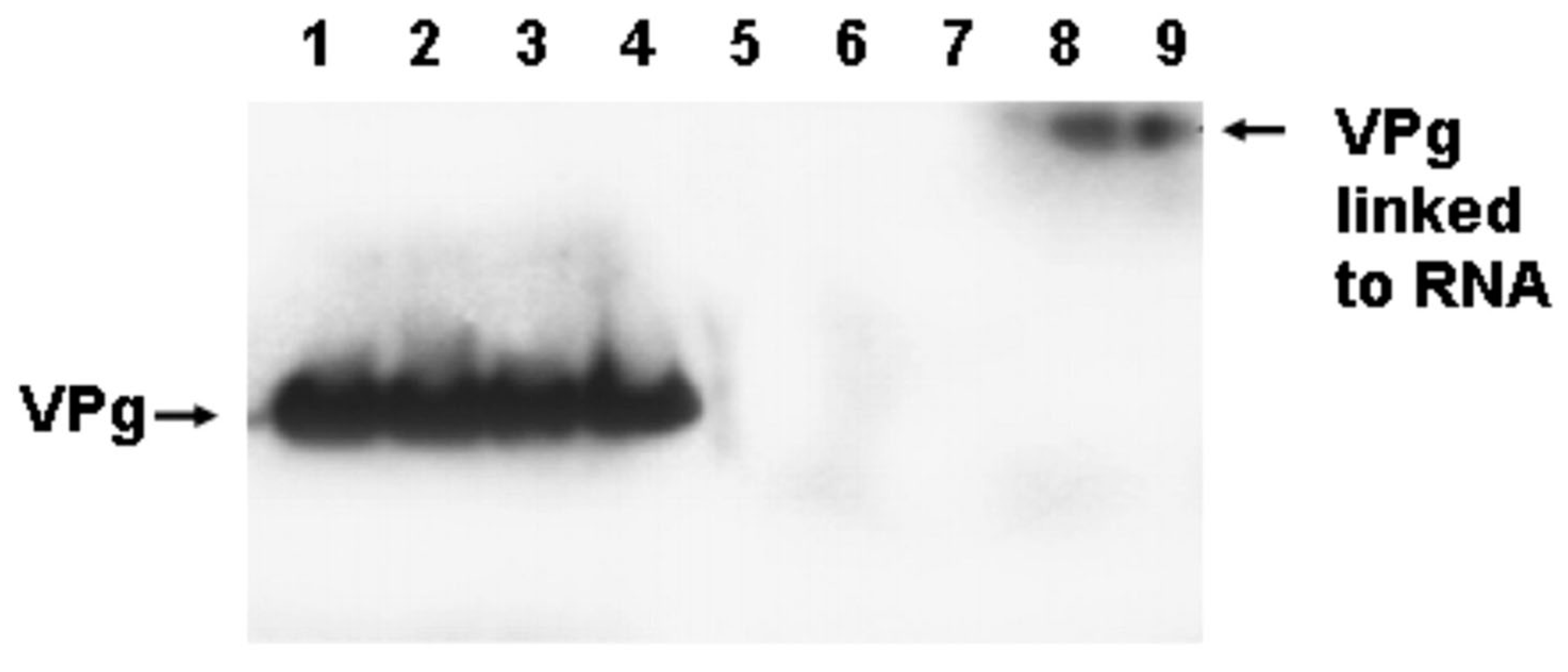
3.7. The Replication of CVB3-TD Strains Is Similar to Wildtype CVB3 in That the Initiation Process Produces RNA Covalently Linked to VPg
3.8. Persistent, Nonlytic CVB3-TD Populations Demonstrated a Significantly Altered Positive to Negative RNA Strand Ratio
3.9. CVB3 TD Infections Encapsidate Both Negative and Positive Strands
4. Generation of TD Genomes Is Likely Due to a Defect in Positive-Strand Initiation
4.1. Wildtype Positive-Strand Replication
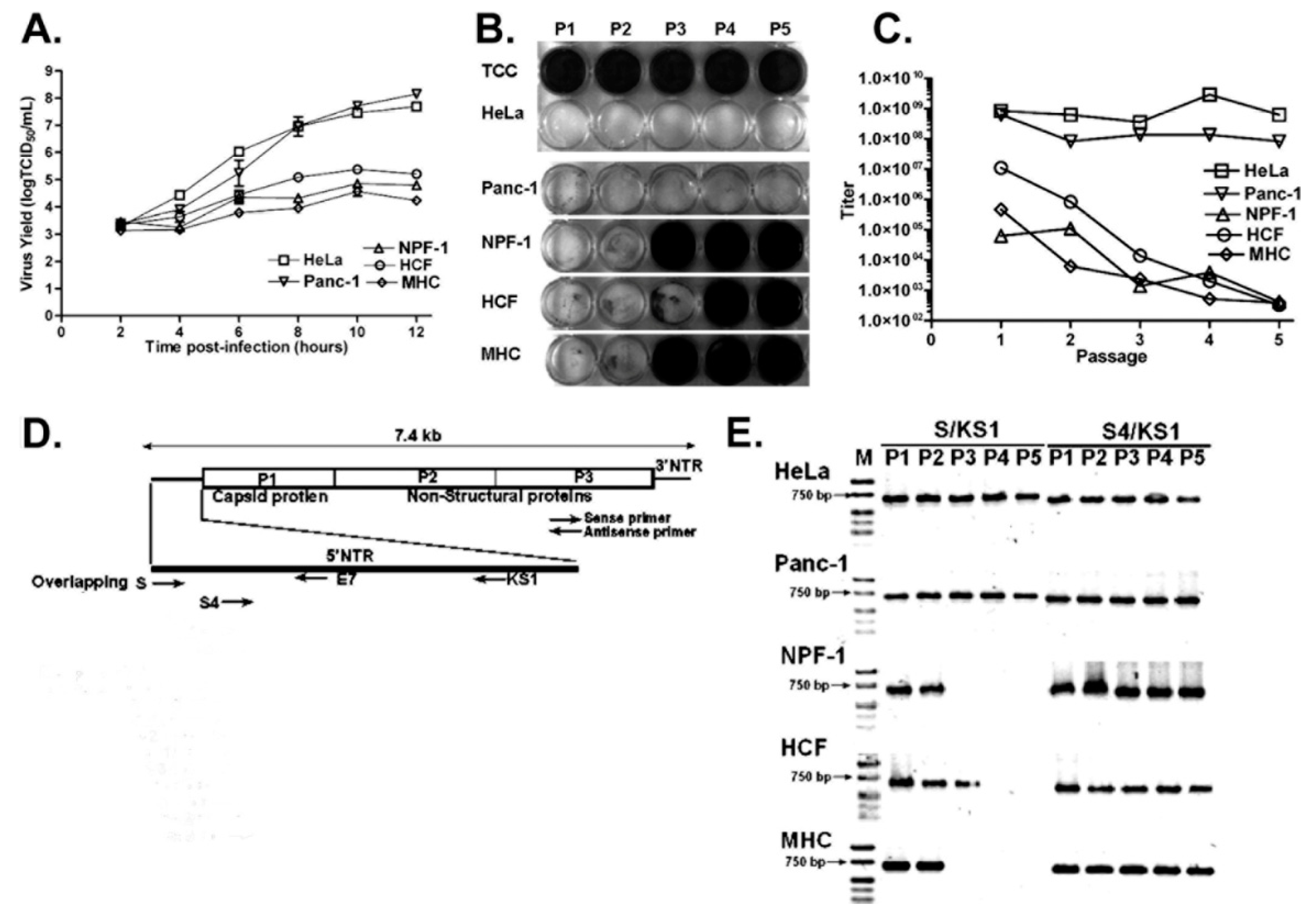
4.2. CVB3-TD Populations Arise and Become Dominant in Quiescent Primary Cell Cultures and in Nondividing Tissue In Vivo
4.3. Host Cell Factors Binding to the 3′ Nucleotides of the Negative Strand May Play a Critical Role in Localization of the Positive-Strand RNA Initiation
4.4. Heterogeneous Nuclear Ribonucleoprotein-C (hnRNP-C) Is a Likely Candidate for a Host Factor in Positive-Strand Initiation
5. A Cis-Acting Replication Element (CRE-2C) Plays a Role in Localization of the Positive-Strand Initiation to the 3′ End of the Negative Strand
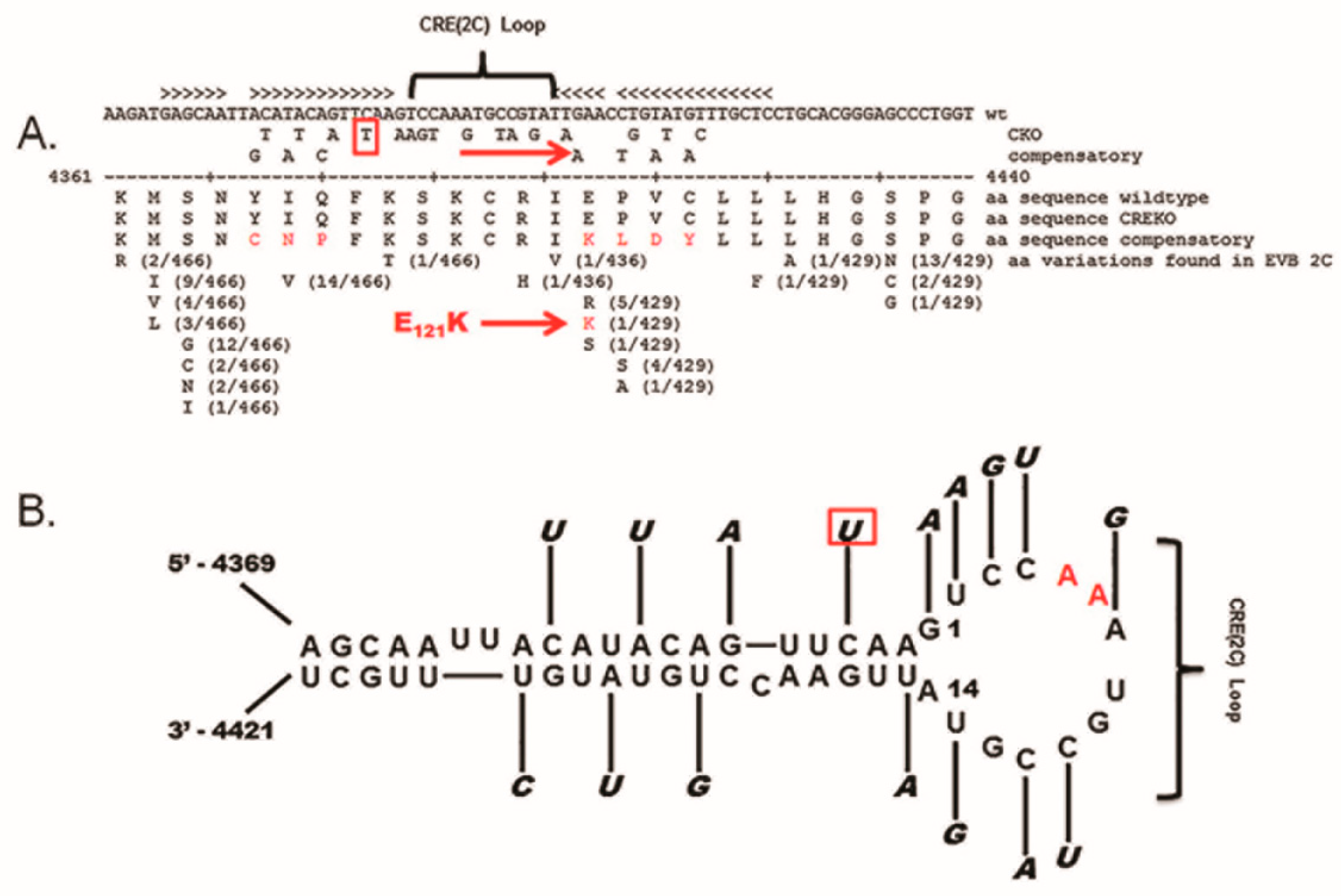
5.1. CVB3-TD Viruses Are Generated by Replication of CVB3 with Multiple Mutations of the CRE-2C
5.2. Reversion of the Mutated CRE-2C to Wildtype in CVB3-TD Genomes Indicates That the Functions of This Structure Are Required for TD Genome Replication
6. Enteroviral TDs as Defective Interfering RNA?
6.1. TD Viruses Are Produced in Populations That Include Wildtype Virus
6.2. The Presence of TD Virus in a Mixed Population Lowers the Level of Replication of the Wildtype Genomes
6.3. Nonlytic Production of Virus via Exosomes May Allow Mixed Populations to Be Transferred in New Infections in Tissues
7. Production of TDs as Part of Wildtype Infection in Some Cells: Is There a Cooperative Advantage?
8. TDs and the Immune Response
8.1. TDs Produce Viral Proteins When Levels of RNA Replication Are Low
8.2. The Adaptive Immune Response That Is Very Likely to Be Induced in Humans in the Course of the Acute Infection, Does Not Clear TD Persistence
8.3. Can the Presence of TD Enterovirus-Infected Cells Trigger Continuing Immune-Mediated Inflammation?
9. Treatment
10. Final Thoughts on TD and Enterovirus Persistence
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| cpe | cytopathic effect |
| CRE-2C | cis-acting replication element |
| CVB3 | coxsackievirus B3 |
| CVB3-TDn | CVB3 TD with 5′ nucleotide n |
| DCM | dilated cardiomyopathy |
| DI | defective interfering |
| hnRNP-C | heterogeneous nuclear ribonucleoprotein C |
| IHC | immunohistochemistry |
| IRES | internal ribosome entry site |
| ISH | in situ hybridization |
| MOI | multiplicity of infection |
| NTR | nontranslated region |
| ORF | open reading frame |
| P1 | capsid coding |
| PCBP2 | polyC-binding protein 2 |
| p.i. | post-inoculation |
| PV | poliovirus |
| RACE | rapid amplification of cDNA ends |
| RI | replication intermediate |
| RF | replicative form |
| RT-PCR | reverse transcription-polymerase chain reaction |
| TD | 5′ terminal deletions |
| T1D | type 1 diabetes |
References
- Racaniello, V.R. Picornaviridae: The Viruses and their Replication. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Cohen, J.I., Griffin, D.E., Lamb, R.A., Martin, M.A., Racaniello, V.R., Roizman, B., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 453–489. [Google Scholar]
- Pallansch, M.A.; Oberste, M.S.; Whitton, J.L. Enteroviruses: Polioviruses, Coxsackieviruses, Echoviruses, and Newer Enteroviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Cohen, J.I., Griffin, D.E., Lamb, R.A., Martin, M.A., Racaniello, V.R., Roizman, B., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; pp. 490–530. [Google Scholar]
- Kim, K.-S.; Chapman, N.; Tracy, S. The group B coxsackieviruses and myocarditis. Rev. Med. Virol. 2001, 11, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.E.; Tamhankar, M.; Lernmark, A. Enteroviruses and Type 1 Diabetes: Multiple Mechanisms and Factors? Annu. Rev. Med. 2022, 73, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Heim, A.; Canu, A.; Kirschner, P.; Simon, T.; Mall, G.; Hofschneider, P.H.; Kandolf, R. Synergistic Interaction of Interferon- and Interferon- in Coxsackievirus B3-Infected Carrier Cultures of Human Myocardial Fibroblasts. J. Infect. Dis. 1992, 166, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Matteucci, D.; Paglianti, M.; Giangregorio, A.M.; Capobianchi, M.R.; Dianzani, F.; Bendinelli, M. Group B coxsackieviruses readily establish persistent infections in human lymphoid cell lines. J. Virol. 1985, 56, 651–654. [Google Scholar] [CrossRef] [PubMed]
- McLaren, L.C.; Holland, J.J. Defective interfering particles from poliovirus vaccine and vaccine reference strains. Virology 1974, 60, 579–583. [Google Scholar] [CrossRef]
- Martín, J. Vaccine-derived poliovirus from long term excretors and the end game of polio eradication. Biologicals 2006, 34, 117–122. [Google Scholar] [CrossRef]
- Montgomery, J.; Gear, J.; Prinsloo, F.R.; Kahn, M.; Kirsch, Z.G. Myocarditis of the newborn; an outbreak in a maternity home in Southern Rhodesia associated with Coxsackie group-B virus infection. S. Afr. Med. J. 1955, 29, 608–612. [Google Scholar]
- Woodruff, J.F. Viral myocarditis. A review. Am. J. Pathol. 1980, 101, 425–484. [Google Scholar]
- Sandoni, M.; Ciardo, L.; Tamburini, C.; Boncompagni, A.; Rossi, C.; Guidotti, I.; Garetti, E.; Lugli, L.; Iughetti, L.; Berardi, A. Enteroviral Infections in the First Three Months of Life. Pathogens 2022, 11, 60. [Google Scholar] [CrossRef]
- Bouin, A.; Gretteau, P.-A.; Wehbe, M.; Renois, F.; N’Guyen, Y.; Lévêque, N.; Vu, M.N.; Tracy, S.; Chapman, N.M.; Bruneval, P.; et al. Enterovirus Persistence in Cardiac Cells of Patients With Idiopathic Dilated Cardiomyopathy Is Linked to 5′ Terminal Genomic RNA-Deleted Viral Populations With Viral-Encoded Proteinase Activities. Circulation 2019, 139, 2326–2338. [Google Scholar] [CrossRef]
- Yeung, W.-C.G.; Rawlinson, W.; Craig, M.E. Enterovirus infection and type 1 diabetes mellitus: Systematic review and meta-analysis of observational molecular studies. BMJ 2011, 342, d35. [Google Scholar] [CrossRef]
- Bowles, N.E.; Rose, M.L.; Taylor, P.; Banner, N.R.; Morgan-Capner, P.; Cunningham, L.; Archard, L.C.; Yacoub, M.H. End-stage dilated cardiomyopathy. Persistence of enterovirus RNA in myocardium at cardiac transplantation and lack of immune response. Circulation 1989, 80, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Tracy, S. A molecular and serologic evaluation of enteroviral involvement in human myocarditis. J. Mol. Cell. Cardiol. 1990, 22, 403–414. [Google Scholar] [CrossRef]
- Wiegand, V.; Tracy, S.; Chapman, N.; Wucherpfennig, C. Enteroviral infection in end stage dilated cardiomyopathy. Klin. Wochenschr. 1990, 68, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.J.; Willcox, A.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia 2009, 52, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Kandolf, R.; Klingel, K.; Zell, R.; Selink, H.-C.; Raab, U.; Schneider-Brachert, W.; Bültmann, B. Molecular Pathogenesis of Enterovirus-Induced Myocarditis: Virus Persistence and Chronic Inflammation. Intervirology 1993, 35, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Kandolf, R.; Klingel, K.; Mertsching, H.; Canu, A.; Hohenadl, C.; Zell, R.; Reimann, B.Y.; Heim, A.; McManus, B.M.; Foulis, A.K.; et al. Molecular studies on enteroviral heart disease: Patterns of acute and persistent infections. Eur. Heart J. 1991, 12, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Andreoletti, L.; Hober, D.; Decoene, C.; Copin, M.-C.; Lobert, P.-E.; Dewilde, A.; Stankowiac, C.; Wattre, P. Detection of enteroviral RNA by polymerase chain reaction in endomyocardial tissue of patients with chronic cardiac diseases. J. Med. Virol. 1996, 48, 53–59. [Google Scholar] [CrossRef]
- Kühl, U.; Pauschinger, M.; Seeberg, B.; Lassner, D.; Noutsias, M.; Poller, W.; Schultheiss, H.-P. Viral Persistence in the Myocardium Is Associated With Progressive Cardiac Dysfunction. Circulation 2005, 112, 1965–1970. [Google Scholar] [CrossRef]
- Lévêque, N.; Renois, F.; Talmud, D.; Nguyen, Y.; Lesaffre, F.; Boulagnon, C.; Bruneval, P.; Fornes, P.; Andréoletti, L. Quantitative Genomic and Antigenomic Enterovirus RNA Detection in Explanted Heart Tissue Samples from Patients with End-Stage Idiopathic Dilated Cardiomyopathy. J. Clin. Microbiol. 2012, 50, 3378–3380. [Google Scholar] [CrossRef]
- Rey, L.; Lambert, V.; Wattré, P.; Andréoletti, L. Detection of enteroviruses ribonucleic acid sequences in endomyocardial tissue from adult patients with chronic dilated cardiomyopathy by a rapid RT-PCR and hybridization assay. J. Med. Virol. 2001, 64, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Reetoo, K.N.; Osman, S.A.; Illavia, S.J.; Cameron-Wilson, C.L.; Banatvala, J.E.; Muir, P. Quantitative analysis of viral RNA kinetics in coxsackievirus B3-induced murine myocarditis: Biphasic pattern of clearance following acute infection, with persistence of residual viral RNA throughout and beyond the inflammatory phase of disease. J. Gen. Virol. 2000, 81, 2755–2762. [Google Scholar] [CrossRef] [PubMed]
- Klingel, K.; Stephan, S.; Sauter, M.; Zell, R.; McManus, B.M.; Bültmann, B.; Kandolf, R. Pathogenesis of murine enterovirus myocarditis: Virus dissemination and immune cell targets. J. Virol. 1996, 70, 8888–8895. [Google Scholar] [CrossRef] [PubMed]
- Destombes, J.; Couderc, T.; Thiesson, D.; Girard, S.; Wilt, S.G.; Blondel, B. Persistent poliovirus infection in mouse motoneurons. J. Virol. 1997, 71, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Kyu, B.; Matsumori, A.; Sato, Y.; Okada, I.; Chapman, N.M.; Tracy, S. Cardiac persistence of cardioviral RNA detected by polymerase chain reaction in a murine model of dilated cardiomyopathy. Circulation 1992, 86, 522–530. [Google Scholar] [CrossRef]
- See, D.M.; Tilles, J.G. Pathogenesis of Virus-Induced Diabetes in Mice. J. Infect. Dis. 1995, 171, 1131–1138. [Google Scholar] [CrossRef]
- Tam, P.E.; Schmidt, A.M.; Ytterberg, S.R.; Messner, R.P. Duration of virus persistence and its relationship to inflammation in the chronic phase of coxsackievirus B1-induced murine polymyositis. J. Lab. Clin. Med. 1994, 123, 346–356. [Google Scholar]
- Jaïdane, H.; Gharbi, J.; Lobert, P.-E.; Lucas, B.; Hiar, R.; Ben M’Hadheb, M.; Brilot, F.; Geenen, V.; Aouni, M.; Hober, D. Prolonged Viral RNA Detection in Blood and Lymphoid Tissues from Coxsackievirus B4 E2 Orally-Inoculated Swiss Mice. Microbiol. Immunol. 2006, 50, 971–974. [Google Scholar] [CrossRef]
- Caforio, A.L.P.; Baboonian, C.; McKenna, W.J. Postviral autoimmune heart disease--fact or fiction? Eur. Heart J. 1997, 18, 1051–1055. [Google Scholar] [CrossRef][Green Version]
- Fairweather, D.; Frisancho-Kiss, S.; Rose, N.R. Viruses as adjuvants for autoimmunity: Evidence from Coxsackievirus-induced myocarditis. Rev. Med. Virol. 2004, 15, 17–27. [Google Scholar] [CrossRef]
- Bearden, D.; Collett, M.; Quan, P.L.; Costa-Carvalho, B.T.; Sullivan, K.E. Enteroviruses in X-Linked Agammaglobulinemia: Update on Epidemiology and Therapy. J. Allergy Clin. Immunol. Pract. 2016, 4, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Lwoff, A. LYSOGENY. Bacteriol. Rev. 1953, 17, 269–337. [Google Scholar] [CrossRef] [PubMed]
- Carson, S.D.; Chapman, N.; Hafenstein, S.; Tracy, S. Variations of Coxsackievirus B3 Capsid Primary Structure, Ligands, and Stability Are Selected for in a Coxsackievirus and Adenovirus Receptor-Limited Environment. J. Virol. 2011, 85, 3306–3314. [Google Scholar] [CrossRef] [PubMed]
- Nekoua, M.; Bertin, A.; Sane, F.; Gimeno, J.-P.; Fournier, I.; Salzet, M.; Engelmann, I.; Alidjinou, E.; Hober, D. Persistence of Coxsackievirus B4 in Pancreatic β Cells Disturbs Insulin Maturation, Pattern of Cellular Proteins, and DNA Methylation. Microorganisms 2021, 9, 1125. [Google Scholar] [CrossRef] [PubMed]
- Pinkert, S.; Klingel, K.; Lindig, V.; Dörner, A.; Zeichhardt, H.; Spiller, O.B.; Fechner, H. Virus-Host Coevolution in a Persistently Coxsackievirus B3-Infected Cardiomyocyte Cell Line. J. Virol. 2011, 85, 13409–13419. [Google Scholar] [CrossRef] [PubMed]
- Klingel, K.; Kandolf, R. The role of enterovirus replication in the development of acute and chronic heart muscle disease indifferent immunocompetent mouse strains.Scand. J. Infect. Dis. Suppl. 1993, 88, 79–85. [Google Scholar]
- Huang, A.S.; Baltimore, D. Defective Viral Particles and Viral Disease Processes. Nature 1970, 226, 325–327. [Google Scholar] [CrossRef]
- Roux, L.; Simon, A.E.; Holland, J.J. Effects of Defective Interfering Viruses on Virus Replication and Pathogenesis In Vitro and In Vivo. Adv. Virus Res. 1991, 40, 181–211. [Google Scholar] [CrossRef]
- Li, D.; Lott, W.; Lowry, K.; Jones, A.; Thu, H.M.; Aaskov, J. Defective Interfering Viral Particles in Acute Dengue Infections. PLoS ONE 2011, 6, e19447. [Google Scholar] [CrossRef]
- Lui, W.-Y.; Yuen, C.-K.; Li, C.; Wong, W.M.; Lui, P.-Y.; Lin, C.-H.; Chan, K.-H.; Zhao, H.; Chen, H.; To, K.K.W.; et al. SMRT sequencing revealed the diversity and characteristics of defective interfering RNAs in influenza A (H7N9) virus infection. Emerg. Microbes Infect. 2019, 8, 662–674. [Google Scholar] [CrossRef]
- Pesko, K.N.; Fitzpatrick, K.A.; Ryan, E.M.; Shi, P.-Y.; Zhang, B.; Lennon, N.J.; Newman, R.M.; Henn, M.R.; Ebel, G.D. Internally deleted WNV genomes isolated from exotic birds in New Mexico: Function in cells, mosquitoes, and mice. Virology 2012, 427, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Vasilijevic, J.; Zamarreño, N.; Oliveros, J.C.; Rodriguez-Frandsen, A.; Gómez, G.; Rodriguez, G.; Pérez-Ruiz, M.; Rey, S.; Barba, I.; Pozo, F.; et al. Reduced accumulation of defective viral genomes contributes to severe outcome in influenza virus infected patients. PLoS Pathog. 2017, 13, e1006650. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.N.; Smoler, D.; Wimmer, E.; Baltimore, D. Defective Interfering Particles of Poliovirus I. Isolation and Physical Properties. J. Virol. 1971, 7, 478–485. [Google Scholar] [CrossRef]
- Lundquist, R.E.; Sullivan, M.; Maizel, J.V. Characterization of a new isolate of polioviru defective interfering particles. Cell 1979, 18, 759–769. [Google Scholar] [CrossRef]
- Ogram, S.; Flanegan, J.B. Non-template functions of viral RNA in picornavirus replication. Curr. Opin. Virol. 2011, 1, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.K.; Kräusslich, H.G.; Nicklin, M.J.; Duke, G.M.; Palmenberg, A.C.; Wimmer, E. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol. 1988, 62, 2636–2643. [Google Scholar] [CrossRef]
- Paul, A.V.; Wimmer, E. Initiation of protein-primed picornavirus RNA synthesis. Virus Res. 2015, 206, 12–26. [Google Scholar] [CrossRef]
- Kim, K.-S.; Tracy, S.; Tapprich, W.; Bailey, J.; Lee, C.-K.; Barry, W.H.; Chapman, N.M. 5′-Terminal Deletions Occur in Coxsackievirus B3 during Replication in Murine Hearts and Cardiac Myocyte Cultures and Correlate with Encapsidation of Negative-Strand Viral RNA. J. Virol. 2005, 79, 7024–7041. [Google Scholar] [CrossRef]
- Herold, J.; Andino, R. Poliovirus Requires a Precise 5′ End for Efficient Positive-Strand RNA Synthesis. J. Virol. 2000, 74, 6394–6400. [Google Scholar] [CrossRef]
- Frohman, M.A.; Dush, M.K.; Martin, G.R. Rapid production of full-length cDNAs from rare transcripts: Amplification using a single gene-specific oligonucleotide primer. Proc. Natl. Acad. Sci. USA 1988, 85, 8998–9002. [Google Scholar] [CrossRef]
- Bouin, A.; Nguyen, Y.; Wehbe, M.; Renois, F.; Fornes, P.; Bani-Sadr, F.; Metz, D.; Andreoletti, L. Major Persistent 5′ Terminally Deleted Coxsackievirus B3 Populations in Human Endomyocardial Tissues. Emerg. Infect. Dis. 2016, 22, 1488–1490. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.M.; Kim, K.-S.; Drescher, K.M.; Oka, K.; Tracy, S. 5′ terminal deletions in the genome of a coxsackievirus B2 strain occurred naturally in human heart. Virology 2008, 375, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Glenet, M.; N’Guyen, Y.; Mirand, A.; Henquell, C.; Lebreil, A.L.; Berri, F.; Bani-Sadr, F.; Lina, B.; Schuffenecker, I.; Andreoletti, L.; et al. Major 5′terminally deleted enterovirus populations modulate type I IFN response in acute myocarditis patients and in human cultured cardiomyocytes. Sci. Rep. 2020, 10, 11947. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, B.; Horn, C.; Tapprich, W.E. Structure of the 5′ Untranslated Region of Enteroviral Genomic RNA. J. Virol. 2019, 93, e01288-19. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Salas, E.; Francisco-Velilla, R.; Fernandez, J.; Lozano, G.; Diaz-Toledano, R. Picornavirus IRES elements: RNA structure and host protein interactions. Virus Res. 2015, 206, 62–73. [Google Scholar] [CrossRef]
- Kloc, A.; Rai, D.K.; Rieder, E. The Roles of Picornavirus Untranslated Regions in Infection and Innate Immunity. Front. Microbiol. 2018, 9, 485. [Google Scholar] [CrossRef]
- Hunziker, I.P.; Cornell, C.T.; Whitton, J.L. Deletions within the 5′UTR of coxsackievirus B3: Consequences for virus translation and replication. Virology 2007, 360, 120–128. [Google Scholar] [CrossRef]
- Liu, Z.; Carthy, C.M.; Cheung, P.; Bohunek, L.; Wilson, J.E.; McManus, B.M.; Yang, D. Structural and Functional Analysis of the 5′ Untranslated Region of Coxsackievirus B3 RNA: In Vivo Translational and Infectivity Studies of Full-Length Mutants. Virology 1999, 265, 206–217. [Google Scholar] [CrossRef]
- Kim, K.-S.; Chapman, N.; Tracy, S. Replication of Coxsackievirus B3 in Primary Cell Cultures Generates Novel Viral Genome Deletions. J. Virol. 2008, 82, 2033–2037. [Google Scholar] [CrossRef]
- Andino, R.; Rieckhof, G.E.; Baltimore, D. A functional ribonucleoprotein complex forms around the 5′ end of poliovirus RNA. Cell 1990, 63, 369–380. [Google Scholar] [CrossRef]
- Tracy, S.; Smithee, S.; Alhazmi, A.; Chapman, N. Coxsackievirus can persist in murine pancreas by deletion of 5′ terminal genomic sequences. J. Med. Virol. 2015, 87, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Andino, R.; Rieckhof, G.E.; Trono, D.; Baltimore, D. Substitutions in the protease (3Cpro) gene of poliovirus can suppress a mutation in the 5′ noncoding region. J. Virol. 1990, 64, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Ohlenschläger, O.; Wöhnert, J.; Bucci, E.; Seitz, S.; Häfner, S.; Ramachandran, R.; Zell, R.; Görlach, M. The Structure of the Stemloop D Subdomain of Coxsackievirus B3 Cloverleaf RNA and Its Interaction with the Proteinase 3C. Structure 2004, 12, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Sidigi, K.; Bucci, E.; Stelzner, A.; Görlach, M. Determinants of the recognition of enteroviral cloverleaf RNA by coxsackievirus B3 proteinase 3C. RNA 2002, 8, 188–201. [Google Scholar] [CrossRef][Green Version]
- Klump, W.M.; Bergmann, I.; Müller, B.C.; Ameis, D.; Kandolf, R. Complete nucleotide sequence of infectious Coxsackievirus B3 cDNA: Two initial 5′ uridine residues are regained during plus-strand RNA synthesis. J. Virol. 1990, 64, 1573–1583. [Google Scholar] [CrossRef]
- Vogt, D.A.; Andino, R. An RNA Element at the 5′-End of the Poliovirus Genome Functions as a General Promoter for RNA Synthesis. PLoS Pathog. 2010, 6, e1000936. [Google Scholar] [CrossRef]
- Jaramillo, L.; Smithee, S.; Tracy, S.; Chapman, N. Domain I of the 5′ non-translated genomic region in coxsackievirus B3 RNA is not required for productive replication. Virology 2016, 496, 127–130. [Google Scholar] [CrossRef][Green Version]
- Lloyd, R.E. Nuclear proteins hijacked by mammalian cytoplasmic plus strand RNA viruses. Virology 2015, 479–480, 457–474. [Google Scholar] [CrossRef]
- Sharma, N.; Ogram, S.A.; Morasco, B.J.; Spear, A.; Chapman, N.M.; Flanegan, J.B. Functional role of the 5′ terminal cloverleaf in Coxsackievirus RNA replication. Virology 2009, 393, 238–249. [Google Scholar] [CrossRef]
- Toyoda, H.; Franco, D.; Fujita, K.; Paul, A.V.; Wimmer, E. Replication of Poliovirus Requires Binding of the Poly(rC) Binding Protein to the Cloverleaf as Well as to the Adjacent C-Rich Spacer Sequence between the Cloverleaf and the Internal Ribosomal Entry Site. J. Virol. 2007, 81, 10017–10028. [Google Scholar] [CrossRef]
- Zell, R.; Ihle, Y.; Seitz, S.; Gündel, U.; Wutzler, P.; Görlach, M. Poly(rC)-binding protein 2 interacts with the oligo(rC) tract of coxsackievirus B3. Biochem. Biophys. Res. Commun. 2008, 366, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Lévêque, N.; Garcia, M.; Bouin, A.; Nguyen, J.H.C.; Tran, G.P.; Andreoletti, L.; Semler, B.L. Functional Consequences of RNA 5′-Terminal Deletions on Coxsackievirus B3 RNA Replication and Ribonucleoprotein Complex Formation. J. Virol. 2017, 91, e00423-17. [Google Scholar] [CrossRef] [PubMed]
- Spear, A.; Ogram, S.A.; Morasco, B.J.; Smerage, L.E.; Flanegan, J.B. Viral precursor protein P3 and its processed products perform discrete and essential functions in the poliovirus RNA replication complex. Virology 2015, 485, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Smithee, S.; Tracy, S.; Chapman, N.M. Mutational Disruption of cis-Acting Replication Element 2C in Coxsackievirus B3 Leads to 5′-Terminal Genomic Deletions. J. Virol. 2015, 89, 11761–11772. [Google Scholar] [CrossRef] [PubMed]
- Smithee, S.; Tracy, S.; Chapman, N.M. Reversion to wildtype of a mutated and nonfunctional coxsackievirus B3CRE(2C). Virus Res. 2016, 220, 136–149. [Google Scholar] [CrossRef]
- Nugent, C.I.; Johnson, K.L.; Sarnow, P.; Kirkegaard, K. Functional Coupling between Replication and Packaging of Poliovirus Replicon RNA. J. Virol. 1999, 73, 427–435. [Google Scholar] [CrossRef]
- Novak, J.E.; Kirkegaard, K. Improved method for detecting poliovirus negative strands used to demonstrate specificity of positive-strand encapsidation and the ratio of positive to negative strands in infected cells. J. Virol. 1991, 65, 3384–3387. [Google Scholar] [CrossRef]
- Schulte, M.B.; Andino, R. Single-Cell Analysis Uncovers Extensive Biological Noise in Poliovirus Replication. J. Virol. 2014, 88, 6205–6212. [Google Scholar] [CrossRef]
- Klingel, K.; Hohenadl, C.; Canu, A.; Albrecht, M.; Seemann, M.; Mall, G.; Kandolf, R. Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: Quantitative analysis of virus replication, tissue damage, and inflammation. Proc. Natl. Acad. Sci. USA 1992, 89, 314–318. [Google Scholar] [CrossRef]
- Tam, P.E.; Messner, R.P. Molecular Mechanisms of Coxsackievirus Persistence in Chronic Inflammatory Myopathy: Viral RNA Persists through Formation of a Double-Stranded Complex without Associated Genomic Mutations or Evolution. J. Virol. 1999, 73, 10113–10121. [Google Scholar] [CrossRef]
- Feuer, R.; Ruller, C.M.; An, N.; Tabor-Godwin, J.M.; Rhoades, R.E.; Maciejewski, S.; Pagarigan, R.R.; Cornell, C.T.; Crocker, S.J.; Kiosses, W.B.; et al. Viral Persistence and Chronic Immunopathology in the Adult Central Nervous System following Coxsackievirus Infection during the Neonatal Period. J. Virol. 2009, 83, 9356–9369. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.M.; Koch, G. Purification and Characterization of Poliovirus-induced Infectious Double-stranded Ribonucleic Acid. J. Biol. Chem. 1967, 242, 1736–1743. [Google Scholar] [CrossRef]
- Kruse, I.; Peyret, H.; Saxena, P.; Lomonossoff, G.P. Encapsidation of Viral RNA in Picornavirales: Studies on Cowpea Mosaic Virus Demonstrate Dependence on Viral Replication. J. Virol. 2019, 93, e01520-18. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Liu, Y.; Ma, H.-C.; Paul, A.V.; Wimmer, E. Picornavirus Morphogenesis. Microbiol. Mol. Biol. Rev. 2014, 78, 418–437. [Google Scholar] [CrossRef]
- Chandler-Bostock, R.; Mata, C.P.; Bingham, R.J.; Dykeman, E.C.; Meng, B.; Tuthill, T.J.; Rowlands, D.J.; Ranson, N.A.; Twarock, R.; Stockley, P.G. Assembly of infectious enteroviruses depends on multiple, conserved genomic RNA-coat protein contacts. PLoS Pathog. 2020, 16, e1009146. [Google Scholar] [CrossRef]
- Glenet, M.; Heng, L.; Callon, D.; Lebreil, A.-L.; Gretteau, P.-A.; Nguyen, Y.; Berri, F.; Andreoletti, L. Structures and Functions of Viral 5′ Non-Coding Genomic RNA Domain-I in Group-B Enterovirus Infections. Viruses 2020, 12, 919. [Google Scholar] [CrossRef]
- Pathak, H.B.; Arnold, J.J.; Wiegand, P.N.; Hargittai, M.R.S.; Cameron, C.E. Picornavirus Genome Replication. J. Biol. Chem. 2007, 282, 16202–16213. [Google Scholar] [CrossRef]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; Dos Remedios, C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef]
- Willcox, A.; Richardson, S.J.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. Immunohistochemical analysis of the relationship between islet cell proliferation and the production of the enteroviral capsid protein, VP1, in the islets of patients with recent-onset type 1 diabetes. Diabetologia 2011, 54, 2417–2420. [Google Scholar] [CrossRef]
- Feuer, R.; Mena, I.; Pagarigan, R.; Slifka, M.K.; Whitton, J.L. Cell Cycle Status Affects Coxsackievirus Replication, Persistence, and Reactivation In Vitro. J. Virol. 2002, 76, 4430–4440. [Google Scholar] [CrossRef]
- Belov, G.A.; Lidsky, P.V.; Mikitas, O.V.; Egger, D.; Lukyanov, K.A.; Bienz, K.; Agol, V.I. Bidirectional Increase in Permeability of Nuclear Envelope upon Poliovirus Infection and Accompanying Alterations of Nuclear Pores. J. Virol. 2004, 78, 10166–10177. [Google Scholar] [CrossRef] [PubMed]
- Brunner, J.E.; Ertel, K.J.; Rozovics, J.M.; Semler, B.L. Delayed kinetics of poliovirus RNA synthesis in a human cell line with reduced levels of hnRNP C proteins. Virology 2010, 400, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.D.; Chase, A.J.; Cathcart, A.L.; Tran, G.P.; Semler, B.L. Viral Proteinase Requirements for the Nucleocytoplasmic Relocalization of Cellular Splicing Factor SRp20 during Picornavirus Infections. J. Virol. 2013, 87, 2390–2400. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Knoch, K.-P.; Nath-Sain, S.; Petzold, A.; Schneider, H.; Beck, M.; Wegbrod, C.; Sönmez, A.; Münster, C.; Friedrich, A.; Roivainen, M.; et al. PTBP1 is required for glucose-stimulated cap-independent translation of insulin granule proteins and Coxsackieviruses in beta cells. Mol. Metab. 2014, 3, 518–530. [Google Scholar] [CrossRef]
- Lin, J.-Y.; Shih, S.-R.; Pan, M.; Li, C.; Lue, C.-F.; Stollar, V.; Li, M.-L. hnRNP A1 Interacts with the 5′ Untranslated Regions of Enterovirus 71 and Sindbis Virus RNA and Is Required for Viral Replication. J. Virol. 2009, 83, 6106–6114. [Google Scholar] [CrossRef]
- Lizcano-Perret, B.; Michiels, T. Nucleocytoplasmic Trafficking Perturbation Induced by Picornaviruses. Viruses 2021, 13, 1210. [Google Scholar] [CrossRef]
- Ullmer, W.; Semler, B.L. Direct and Indirect Effects on Viral Translation and RNA Replication Are Required for AUF1 Restriction of Enterovirus Infections in Human Cells. mBio 2018, 9, e01669-18. [Google Scholar] [CrossRef]
- Holmes, A.C.; Semler, B.L. Picornaviruses and RNA Metabolism: Local and Global Effects of Infection. J. Virol. 2019, 93, e02088-17. [Google Scholar] [CrossRef]
- Schepens, B.; Tinton, S.A.; Bruynooghe, Y.; Parthoens, E.; Haegman, M.; Beyaert, R.; Cornelis, S. A role for hnRNP C1/C2 and Unr in internal initiation of translation during mitosis. EMBO J. 2006, 26, 158–169. [Google Scholar] [CrossRef]
- Piñol-Roma, S.; Dreyfuss, G. Cell cycle-regulated phosphorylation of the pre-mRNA-binding (heterogeneous nuclear ribonucleoprotein) C proteins. Mol. Cell. Biol. 1993, 13, 5762–5770. [Google Scholar] [CrossRef]
- Nakielny, S.; Dreyfuss, G. The hnRNP C proteins contain a nuclear retention sequence that can override nuclear export signals. J. Cell Biol. 1996, 134, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.D.; Grabowski, P.J.; Sharp, P.A.; Dreyfuss, G. Heterogeneous Nuclear Ribonucleoproteins: Role in RNA Splicing. Science 1986, 231, 1534–1539. [Google Scholar] [CrossRef] [PubMed]
- Aviner, R.; Hofmann, S.; Elman, T.; Shenoy, A.; Geiger, T.; Elkon, R.; Ehrlich, M.; Elroy-Stein, O. Proteomic analysis of polyribosomes identifies splicing factors as potential regulators of translation during mitosis. Nucleic Acids Res. 2017, 45, 5945–5957. [Google Scholar] [CrossRef] [PubMed]
- Roehl, H.H.; Parsley, T.B.; Ho, T.V.; Semler, B.L. Processing of a cellular polypeptide by 3CD proteinase is required for poliovirus ribonucleoprotein complex formation. J. Virol. 1997, 71, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Brunner, J.E.; Nguyen, J.H.C.; Roehl, H.H.; Ho, T.V.; Swiderek, K.M.; Semler, B.L. Functional Interaction of Heterogeneous Nuclear Ribonucleoprotein C with Poliovirus RNA Synthesis Initiation Complexes. J. Virol. 2005, 79, 3254–3266. [Google Scholar] [CrossRef]
- Ertel, K.J.; Brunner, J.E.; Semler, B.L. Mechanistic Consequences of hnRNP C Binding to Both RNA Termini of Poliovirus Negative-Strand RNA Intermediates. J. Virol. 2010, 84, 4229–4242. [Google Scholar] [CrossRef]
- Wan, L.; Kim, J.-K.; Pollard, V.W.; Dreyfuss, G. Mutational Definition of RNA-binding and Protein-Protein Interaction Domains of Heterogeneous Nuclear RNP C1. J. Biol. Chem. 2001, 276, 7681–7688. [Google Scholar] [CrossRef]
- Dave, P.; George, B.; Balakrishnan, S.; Sharma, D.K.; Raheja, H.; Dixit, N.M.; Das, S. Strand-specific affinity of host factor hnRNP C1/C2 guides positive to negative-strand ratio in Coxsackievirus B3 infection. RNA Biol. 2019, 16, 1286–1299. [Google Scholar] [CrossRef]
- Shim, B.-S.; Wu, W.; Kyriakis, C.S.; Bakre, A.; Jorquera, P.A.; Perwitasari, O.; Tripp, R.A. MicroRNA-555 has potent antiviral properties against poliovirus. J. Gen. Virol. 2016, 97, 659–668. [Google Scholar] [CrossRef]
- Ogram, S.A.; Boone, C.D.; McKenna, R.; Flanegan, J.B. Amiloride inhibits the initiation of Coxsackievirus and poliovirus RNA replication by inhibiting VPg uridylylation. Virology 2014, 464–465, 87–97. [Google Scholar] [CrossRef]
- Van Ooij, M.J.M.; Vogt, D.A.; Paul, A.; Castro, C.; Kuijpers, J.; Van Kuppeveld, F.J.M.; Cameron, C.E.; Wimmer, E.; Andino, R.; Melchers, W.J.G. Structural and functional characterization of the coxsackievirus B3 CRE(2C): Role of CRE(2C) in negative- and positive-strand RNA synthesis. J. Gen. Virol. 2006, 87, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, I.; Polacek, C.; Andino, R.; Evans, D.J. The poliovirus 2C cis-acting replication element-mediated uridylylation of VPg is not required for synthesis of negative-sense genomes. J. Gen. Virol. 2003, 84, 2359–2363. [Google Scholar] [CrossRef]
- Morasco, B.J.; Sharma, N.; Parilla, J.; Flanegan, J.B. Poliovirus cre (2C)-Dependent Synthesis of VPgpUpU Is Required for Positive- but Not Negative-Strand RNA Synthesis. J. Virol. 2003, 77, 5136–5144. [Google Scholar] [CrossRef]
- Eruera, A.-R.; McSweeney, A.; McKenzie-Goldsmith, G.; Ward, V. Protein Nucleotidylylation in +ssRNA Viruses. Viruses 2021, 13, 1549. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, I.; Chaudhry, Y.; Richardson, A.; Meredith, J.; Almond, J.W.; Barclay, W.; Evans, D.J. Identification of a cis -Acting Replication Element within the Poliovirus Coding Region. J. Virol. 2000, 74, 4590–4600. [Google Scholar] [CrossRef] [PubMed]
- Rieder, E.; Paul, A.V.; Kim, D.W.; van Boom, J.H.; Wimmer, E. Genetic and Biochemical Studies of Poliovirus cis -Acting Replication Element cre in Relation to VPg Uridylylation. J. Virol. 2000, 74, 10371–10380. [Google Scholar] [CrossRef]
- Crowder, S.; Kirkegaard, K. Trans-dominant inhibition of RNA viral replication can slow growth of drug-resistant viruses. Nat. Genet. 2005, 37, 701–709. [Google Scholar] [CrossRef]
- Paul, A.V.; Van Boom, J.H.; Filippov, D.V.; Wimmer, E. Protein-primed RNA synthesis by purified poliovirus RNA polymerase. Nature 1998, 393, 280–284. [Google Scholar] [CrossRef]
- Lowry, K.; Woodman, A.; Cook, J.; Evans, D.J. Recombination in Enteroviruses Is a Biphasic Replicative Process Involving the Generation of Greater-than Genome Length ‘Imprecise’ Intermediates. PLoS Pathog. 2014, 10, e1004191. [Google Scholar] [CrossRef]
- Tu, Z.; Chapman, N.M.; Hufnagel, G.; Tracy, S.; Romero, J.R.; Barry, W.H.; Zhao, L.; Currey, K.; Shapiro, B. The cardiovirulent phenotype of coxsackievirus B3 is determined at a single site in the genomic 5′ nontranslated region. J. Virol. 1995, 69, 4607–4618. [Google Scholar] [CrossRef]
- Genoyer, E.; López, C.B. The Impact of Defective Viruses on Infection and Immunity. Annu. Rev. Virol. 2019, 6, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Wessely, R.; Klingel, K.; Santana, L.; Dalton, N.; Hongo, M.; Lederer, W.J.; Kandolf, R.; Knowlton, K.U. Transgenic expression of replication-restricted enteroviral genomes in heart muscle induces defective excitation-contraction coupling and dilated cardiomyopathy. J. Clin. Investig. 1998, 102, 1444–1453. [Google Scholar] [CrossRef] [PubMed]
- Xiong, D.; Yajima, T.; Lim, B.-K.; Stenbit, A.; Dublin, A.; Dalton, N.D.; Summers-Torres, D.; Molkentin, J.; Duplain, H.; Wessely, R.; et al. Inducible Cardiac-Restricted Expression of Enteroviral Protease 2A Is Sufficient to Induce Dilated Cardiomyopathy. Circulation 2007, 115, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Bouin, A.; Semler, B.L. Picornavirus Cellular Remodeling: Doubling Down in Response to Viral-Induced Inflammation. Curr. Clin. Microbiol. Rep. 2020, 7, 31–37. [Google Scholar] [CrossRef]
- Bird, S.W.; Maynard, N.D.; Covert, M.W.; Kirkegaard, K. Nonlytic viral spread enhanced by autophagy components. Proc. Natl. Acad. Sci. USA 2014, 111, 13081–13086. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A.; Stempinski, E.S.; Connelly, P.S.; Ma, H.-C.; et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef]
- Robinson, S.M.; Tsueng, G.; Sin, J.; Mangale, V.; Rahawi, S.; McIntyre, L.L.; Williams, W.; Kha, N.; Cruz, C.; Hancock, B.M.; et al. Coxsackievirus B Exits the Host Cell in Shed Microvesicles Displaying Autophagosomal Markers. PLoS Pathog. 2014, 10, e1004045. [Google Scholar] [CrossRef]
- Mutsafi, Y.; Altan-Bonnet, N. Enterovirus Transmission by Secretory Autophagy. Viruses 2018, 10, 139. [Google Scholar] [CrossRef]
- Altan-Bonnet, N.; Perales, C.; Domingo, E. Extracellular vesicles: Vehicles of en bloc viral transmission. Virus Res. 2019, 265, 143–149. [Google Scholar] [CrossRef]
- Bou, J.-V.; Geller, R.; Sanjuán, R. Membrane-Associated Enteroviruses Undergo Intercellular Transmission as Pools of Sibling Viral Genomes. Cell Rep. 2019, 29, 714–723.e4. [Google Scholar] [CrossRef]
- Boersma, S.; Rabouw, H.H.; Bruurs, L.J.; Pavlovič, T.; van Vliet, A.L.; Beumer, J.; Clevers, H.; van Kuppeveld, F.J.; Tanenbaum, M.E. Translation and Replication Dynamics of Single RNA Viruses. Cell 2020, 183, 1930–1945.e23. [Google Scholar] [CrossRef] [PubMed]
- McCune, B.T.; Lanahan, M.R.; Tenoever, B.R.; Pfeiffer, J.K. Rapid Dissemination and Monopolization of Viral Populations in Mice Revealed Using a Panel of Barcoded Viruses. J. Virol. 2020, 94, e01590-19. [Google Scholar] [CrossRef] [PubMed]
- Hur, S. Double-Stranded RNA Sensors and Modulators in Innate Immunity. Annu. Rev. Immunol. 2019, 37, 349–375. [Google Scholar] [CrossRef]
- Visser, L.J.; Langereis, M.A.; Rabouw, H.H.; Wahedi, M.; Muntjewerff, E.M.; de Groot, R.J.; van Kuppeveld, F.J.M. Essential Role of Enterovirus 2A Protease in Counteracting Stress Granule Formation and the Induction of Type I Interferon. J. Virol. 2019, 93, e00222-19. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.I.; Coyne, C.B. Enteroviruses: A Gut-Wrenching Game of Entry, Detection, and Evasion. Viruses 2019, 11, 460. [Google Scholar] [CrossRef]
- Zhang, X.; Paget, M.; Wang, C.; Zhu, Z.; Zheng, H. Innate immune evasion by picornaviruses. Eur. J. Immunol. 2020, 50, 1268–1282. [Google Scholar] [CrossRef]
- Muir, P.; Nicholson, F.; Illavia, S.J.; McNeil, T.S.; Ajetunmobi, J.F.; Dunn, H.; Starkey, W.G.; Reetoo, K.N.; Cary, N.R.; Parameshwar, J.; et al. Serological and molecular evidence of enterovirus infection in patients with end-stage dilated cardiomyopathy. Heart 1996, 76, 243–249. [Google Scholar] [CrossRef]
- Dunne, J.L.; Richardson, S.J.; Atkinson, M.A.; Craig, M.E.; Dahl-Jørgensen, K.; Flodström-Tullberg, M.; Hyöty, H.; Insel, R.A.; Lernmark, Å.; Lloyd, R.E.; et al. Rationale for enteroviral vaccination and antiviral therapies in human type 1 diabetes. Diabetologia 2019, 62, 744–753. [Google Scholar] [CrossRef]
- Wang, S.-H.; Wang, K.; Zhao, K.; Hua, S.-C.; Du, J. The Structure, Function, and Mechanisms of Action of Enterovirus Non-structural Protein 2C. Front. Microbiol. 2020, 11, 615965. [Google Scholar] [CrossRef]
- Van Der Linden, L.; Wolthers, K.C.; Van Kuppeveld, F.J.M. Replication and Inhibitors of Enteroviruses and Parechoviruses. Viruses 2015, 7, 4529–4562. [Google Scholar] [CrossRef]
- Egorova, A.; Ekins, S.; Schmidtke, M.; Makarov, V. Back to the future: Advances in development of broad-spectrum capsid-binding inhibitors of enteroviruses. Eur. J. Med. Chem. 2019, 178, 606–622. [Google Scholar] [CrossRef] [PubMed]
- Kühl, U.; Pauschinger, M.; Schwimmbeck, P.L.; Seeberg, B.; Lober, C.; Noutsias, M.; Poller, W.; Schultheiss, H.-P. Interferon-β Treatment Eliminates Cardiotropic Viruses and Improves Left Ventricular Function in Patients with Myocardial Persistence of Viral Genomes and Left Ventricular Dysfunction. Circulation 2003, 107, 2793–2798. [Google Scholar] [CrossRef] [PubMed]
- Meshram, R.J.; Kathwate, G.H.; Gacche, R.N. Progress, evolving therapeutic/diagnostic approaches, and challenges in the management of hepatitis C virus infections. Arch. Virol. 2022, 167, 717–736. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| What are the cellular conditions that generate TD genomes in differentiated cells? What difference in host environmental factors in differentiated quiescent cells prevents normal positive-strand initiation and leads to TD viral genomes? Are these conditions a result of cell cycle arrest? |
| What is the mechanism for nucleotidylation of VPg in TD initiation of positive-strand enterovirus RNA, and what does this mean for the functional assays of 3D polymerase and VPg? As multiply mutated CRE-2C reverts in the CVB3-TD-CKO, despite retention of the 5′ terminal deletion, what is the role of the CRE-2C in the replication of CVB3-TD? |
| As these TD viruses package negative- as well as positive-strand genomes, is the replication of single-stranded RNA the critical factor in packaging of enterovirus genomes, rather than an RNA-encoded signal? |
| This work has focused on a few enterovirus B genotypes, but the question arises whether other enterovirus types, indeed other picornavirus genomes, which utilize the same mechanism for efficient and specific virus positive-strand initiation, are prone to generation of TDs as well. If cell cycle arrest generates TDs from enterovirus B genotypes, will cell cycle arrest generate TDs from other picornavirus species? |
| What are the effects of TD enteroviral infection on cardiomyocytes, neuronal cells, and beta cells of TD virus infection? This question requires the ability to express TD viruses in cells with efficiency, and examine cultures with low-level infections. If host factors or cell cycle conditions are defined for the generation of TD positive-strand initiation, can these be used to generate cultures in which the majority of cells are infected with TD viruses to address the effects of TD virus infection? |
| How does the presence of a TD virus population majority prevent wildtype virus replication and translation at normal levels? Is this simply competition for host factors? If a population of cells with TD virus infection is generated by host factor/cell cycle conditions, can these be superinfected with wildtype virus to determine whether there is interference by the TD viruses? To what extent does the exosomal transmission of enteroviruses allow the minority wildtype population to persist? |
| If such a poorly replicating enterovirus variant is present in human tissues following acute infection, how common is this in enterovirus-associated human disease, given the technical requirements for detecting such a low-level infection? Given the discovery of TD enteroviruses in peripheral blood plasma of acute myocarditis patients [55], is this predictive of cardiac TD enterovirus persistence? Is the presence of TD enterovirus in plasma an indication of TD enterovirus persistence in other tissues? |
| What are the most important mechanisms for these variants to avoid immune clearance, and how might treatment for persistent enteroviruses utilize these targets? We know a great deal about immune evasion by wildtype virus. Do the same mechanisms allow persistent TD virus infections? |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chapman, N.M. Persistent Enterovirus Infection: Little Deletions, Long Infections. Vaccines 2022, 10, 770. https://doi.org/10.3390/vaccines10050770
Chapman NM. Persistent Enterovirus Infection: Little Deletions, Long Infections. Vaccines. 2022; 10(5):770. https://doi.org/10.3390/vaccines10050770
Chicago/Turabian StyleChapman, Nora M. 2022. "Persistent Enterovirus Infection: Little Deletions, Long Infections" Vaccines 10, no. 5: 770. https://doi.org/10.3390/vaccines10050770
APA StyleChapman, N. M. (2022). Persistent Enterovirus Infection: Little Deletions, Long Infections. Vaccines, 10(5), 770. https://doi.org/10.3390/vaccines10050770