1. Introduction
The immune system, known as the biological defense of the body, is made up of two parts: the innate immune system or non-specific immune system and the adaptive immune system. The adaptive immune system, also referred to as the acquired immune system, is characterized by the remarkable specificity and the extreme diversity of their antigen receptors [
1]. These antigen receptors of the adaptive immune response are the immunoglobulins (IG) or antibodies of the B cells [
2] and the T cell receptors (TR) of the T cells [
3]. IG are proteins that have a dual role in immunity: they recognize antigens on the surface of foreign bodies such as bacteria and viruses and trigger elimination mechanisms such as cell lysis and phagocytosis, to rid the body of these cells and particles [
2]. An IG comprises two identical heavy chains (IGH), associated with two identical light chains, kappa (IGK) or lambda (IGL). The synthesis of the immunoglobulin heavy and light chains requires gene rearrangements at the DNA level in the IGH, IGK, and IGL loci during the B cell differentiation [
4]. The IGH locus comprises four types of genes, variable (V), diversity (D), joining (J), and constant (C) [
5] whereas the IGK [
6] and IGL [
7] loci lack the D genes. In human, the immunoglobulins comprise a variable domain and a constant region which is composed of one constant domain (IG light chains) or three or four constant domains for IG heavy chain [
8]. The variable domain is the result of one rearrangement between variable (V) and joining (J) genes for IGL and IGK and two consecutive rearrangements between diversity (D) and J genes, then between V and partially rearranged D-J genes for IGH [
8]. After transcription, the V-(D)-J sequence is spliced to the constant (C) gene to give the final transcript. These rearrangement mechanisms involve a considerable repertoire of genes, which, combined with junctional N-diversity occurring during V-(D)-J gene recombination and with somatic mutations in B cell differentiation, result in a huge IG diversity [
1,
2,
8].
The anatomical and physiological similarities between humans and animals have led researchers to investigate a large range of mechanisms and evaluate new therapies in animal models before applying their discoveries to humans [
8]. Closely related to humans with 93% of its genome, the rhesus monkey (
Macaca mulatta) is therefore one of the most widely used nonhuman primate species in biomedical research [
9]. The rhesus monkey has played a key role in the development of vaccines for decades, such as hepatitis [
10], tuberculosis [
11,
12,
13], and most recently in the development of vaccines against human immunodeficiency virus-1 (HIV-1) [
14,
15,
16,
17,
18,
19,
20] and against SARS-CoV-2 [
21,
22].
IMGT
®, the international ImMunoGeneTics information system
®,
http://www.imgt.org, (accessed on 11 November 2021) [
23], is the global reference in immunogenetics and immunoinformatics [
1]. IMGT
® is a high-quality integrated knowledge resource specialized in IG, TR, major histocompatibility (MH) of human and other vertebrate species, and in the immunoglobulin superfamily (IgSF), MH superfamily (MhSF), and related proteins of the immune system (RPI) of vertebrates and invertebrates. Thanks to the currently “representative genome” of the rhesus monkey published on NCBI database: Mmul_10 [
24], IMGT
® performed the complete annotation of the three IGH, IGK, and IGL loci of this genome, and the corresponding IG germline data were integrated within IMGT
® databases, tools, and web resources, improving the amount of rhesus monkey IG germline data which until then were based on Mmul_051212 [
25] and individual gene sequences. It should be noticed that structural variations have been shown, in particular in the IGH locus of human [
26] and in rhesus monkey [
14,
27,
28]. The Mmul_10 annotation will be used as model and reference in IMGT
® for the characterization and the description of the IG genetic diversity in other
Macaca mulatta genomes assemblies. During this study, a comprehensive analysis of the V, D (for IGH), J, and C genes was performed to determine their position in each IG locus, to characterize the allele functionality and to display the V, D, and J genes conventional recombination signal (RS) sequences. Furthermore, considering the important role of non-coding DNA regions in eukaryotic gene transcription as well as the variety and range of polymorphisms that have already been observed and studied in the 5′ untranslated region (5′UTR) of IG [
29,
30], a comprehensive in silico analysis of the 5′UTR of
Macaca mulatta was carried out. Although the reasons for these changes have not yet been clearly revealed, these polymorphisms certainly modify the secondary structure of the promoter, affecting the stability, localization, transcription, and interaction of UTR with RNA-binding proteins [
29,
31]. Thus, identifying the regulatory elements of the promoter provides a basis for comparative analyses among the genes intra as well as inter species.
2. Materials and Methods
The biocuration was performed manually assisted by internally developed tools IMGT/LIGMotif [
32], NtiToVald [
33] and IMGT/Automat [
33] based on the IMGT-ONTOLOGY axioms and concepts: “IDENTIFICATION”, “DESCRIPTION”, “CLASSIFICATION”, “NUMEROTATION”, “LOCALIZATION”, “ORIENTATION”, and “OBTENTION” [
34]. IMGT-ONTOLOGY includes the controlled vocabulary and annotation rules which are indispensable to ensure accuracy and consistency.
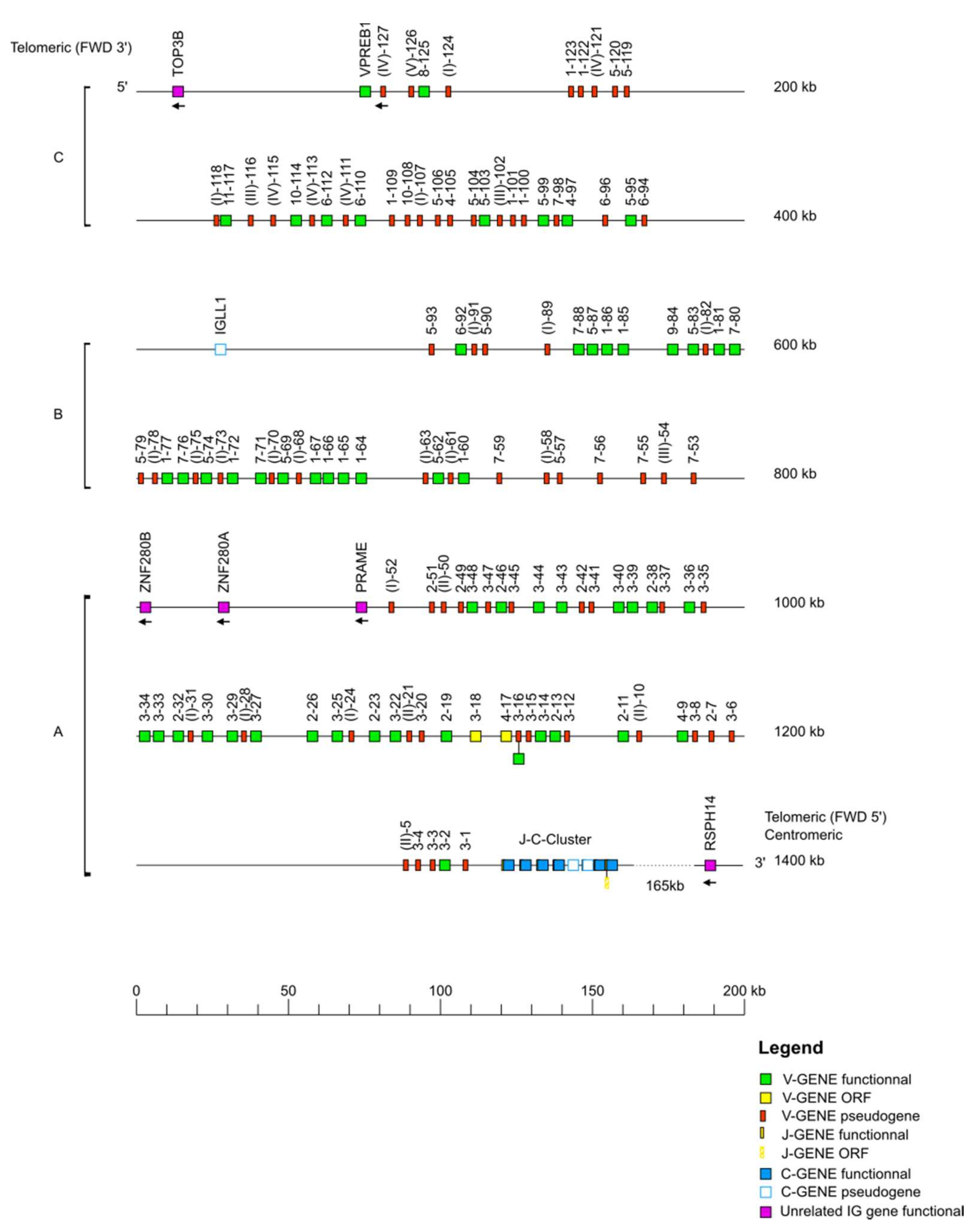
The IMGT
® biocuration pipeline for locus annotation has been described previously [
35]. Each locus sequence was localized on the corresponding chromosome and subsequently extracted from NCBI assembly [
36] Mmul_10 in GenBank format. The delimitation of the locus was performed through research of the “IMGT bornes”, which are coding genes (other than IG or TR) conserved among species, located upstream of the first or downstream of the last gene of an IG or TR locus (
http://www.imgt.org/IMGTindex/IMGTborne.php, (accessed on 11 November 2021)). The IMGT 5′ borne of the IGK locus is the paired box 8 (PAX8, Gene ID: 701906) gene and the IMGT 3′ borne of the locus is the ribose 5-phosphate isomerase A (RPIA, Gene ID: 699694) gene. The IMGT 5′ borne of the IGL locus is the DNA topoisomerase III (TOP3B, Gene ID: 698317) gene and the IMGT 3′ borne of the locus is the radial spoke head 14 homolog (RSPH14, Gene ID: 706814) gene. Similar to the
Homo sapiens locus, the IMGT 5′ and IMGT 3′ bornes of the
Macaca mulatta IGH locus could not be identified, therefore, the sequences of the V genes and C genes of the
Homo sapiens IGH locus were used to localize the V-D-J-C-CLUSTER on the rhesus monkey genome assembly. The locus orientation on a chromosome can be either forward (FWD) or reverse (REV). Therefore, the REV locus sequences were placed in the 5′ to 3′ locus orientation. Each locus sequence thus obtained was assigned an IMGT
® accession number (IGL: IMGT000062, IGK: IMGT000063, IGH: IMGT000064).
According to the “CLASSIFICATION” axiom of IMGT-ONTOLOGY, the nomenclature of all V genes of each IG locus, was characterized based on the human V genes by using IMGT/V-QUEST [
37] and NGPhylogeny.fr [
38] to define the subgroups. All the V genes are designated by a number for the subgroup, followed by a hyphen and a number for their localization from 3′ to 5′ in the locus [
2]. Two genes are assigned to the same subgroup if their V-REGION show a percentage of identity greater than 75% at the nucleotide level. The V genes which were pseudogenes and did not match in any subgroup were assigned to a clan also characterized according to the human clans (
http://www.imgt.org/IMGTindex/Clan.php, (accessed on 11 November 2021)). Duplicated genes share the same name with an additional ‘D’ at the end of the second occurring gene. The nomenclature of the J genes and of the IGHD genes comprises a number for the sets defined according to the human sets [
2], while the number corresponding to the localization is increased from 5′ to 3′ within the locus. Finally, C genes are designated according to their isotype, followed by a number if there was more than one gene, a number which also increased from 5′ to 3′ in the locus. An allele is a polymorphic variant of a gene which is characterized by mutations at the nucleotide level, in its core sequence (V-REGION, D-REGION, J-REGION, and C-REGION for V, D, J, and C genes, respectively). Alleles are designated by the gene name followed by an asterisk and a number with two digits starting from 01. The identification of an allelic polymorphism is performed by comparison of the IMGT reference sequence with a newly annotated genomic sequence and relies on the following rule: for a given mapped gene with the same IMGT position, the same IMGT allele name is assigned if the two core nucleotide sequences have 100% identity and the new genomic sequence is qualified as “sequence from the literature” for that allele. In case of less than 100% identity in the core, a new IMGT allele name is assigned, and the new genomic sequence becomes the “IMGT reference sequence”. The IMGT
® reference directories comprise the reference sequences of all gene alleles.
The functionality of the genes and alleles was defined according to the IMGT ‘functionality’ concept, more precisely the ‘IDENTIFICATION’ axiom of IMGT-ONTOLOGY, described in
http://www.imgt.org/IMGTScientificChart/SequenceDescription/IMGTfunctionality.html (accessed on 11 November 2021). An allele is considered as functional (F) if its coding region has an open reading frame without stop codons, no defect in the splicing sites, recombination signals and/or regulatory elements. An allele is considered as open reading frame (ORF) if its coding region has an open reading frame without stop codons but shows alterations in the splicing sites, recombination signals, regulatory elements, and/or changes of conserved amino acids. A gene allele is considered as pseudogene (P) if the coding region has stop codon(s) and/or frameshift mutation(s).
The main concepts of the ‘DESCRIPTION’ axiom of IMGT-ONTOLOGY correspond to IMGT
® standardized labels in the tools and databases used to describe the organization of the IG genes in the IGH, IGL, and IGK loci [
34]. The standardized annotation of nucleotide sequences (IMGT reference sequences and sequences from literature) is performed using IMGT
®labels (
http://www.imgt.org/ligmdb/label#, (accessed on 11 November 2021)) and integrated in IMGT/LIGM-DB [
39]. This allows data entry of genes and alleles in IMGT/GENE-DB [
40] and in the IMGT
® reference directory; and the entry of amino acid sequences in IMGT/3Dstructure-DB and IMGT/2Dstructure-DB [
41]. IMGT
® reference directories are also used in the sequence analysis tools (IMGT/V-QUEST [
37], IMGT/HighV-QUEST [
42], and IMGT/DomainGapAlign [
43]). The synthesis of the annotation of genomic data is integrated in dedicated sections of IMGT
® web resources: Locus representation, Locus description, Locus in genome assembly, Locus gene order, Locus Borne, Gene tables, Potential germline repertoire, Protein displays, Alignments of alleles, Colliers de Perles [
44,
45], and germline [CDR1-IMGT.CDR2-IMGT.CDR3-IMGT] lengths (
http://imgt.org/IMGTrepertoire/, accessed on 11 November 2021).
The standardized annotation of nucleotide sequences (IMGT reference sequence and sequence from literature) is performed using IMGT
®labels (
http://www.imgt.org/ligmdb/label#, accessed on 11 November 2021) and integrated in IMGT/LIGM-DB [
39]. This allows data entry of genes and alleles in IMGT/GENE-DB [
40] and in the IMGT
® reference directory, and of amino acid sequences in IMGT/3Dstructure-DB and IMGT/2Dstructure-DB [
41].
After integration of data in IMGT/GENE-DB, the 5′ UTR of the IG V genes and alleles were extracted. Those sequences were trimmed up to ~500 bp upstream the initiation codon (atg), which include all of the core promoter elements, as reported in the literature regarding
Homo sapiens [
46,
47,
48,
49,
50,
51,
52,
53]. Then, a progressive multiple sequence alignment (MSA) was performed for the V genes 5′UTR of each locus and each subgroup separately. The MSA analysis was performed using MATLAB bioinformatics toolbox [
54], as previously described in several studies [
55,
56], along with Clustal Omega tool/EMBL-EBI [
57], MAFFT version 7 [
58] and NGPhylogeny.fr [
59]. The results of the MSA analysis were visualized through the Jalview platform [
60]. Thus, guided by the IMGT
® reference of the
Homo sapiens promoter sequences and distances between elements, the motif identification and extraction, along with the calculation of the distance between elements were carried out in
Macaca mulatta loci. For further investigation and element validation, the study was assisted by bioinformatics tools and biological databases concerning eukaryotic promoter elements, including PROMO [
61], gene-regulation [
62], TRANSFAC database [
63], Sequence Manipulation Suite [
64], Transcription factor Affinity Prediction (TRAP) Web Tools [
65], GPMiner [
66], and SoftBerry/Nsite [
67].
4. Discussion
The
Macaca mulatta (rhesus monkey) is one of the most widely used primate species in biomedical research and is used extensively as a model for studying human diseases, as it is evolutionarily close to humans [
74,
75,
76]. In this study, an in silico research of the heavy and light chain IG genes was conducted based on the IMGT biocuration pipeline by using the “representative genome” (assembly Mmul_10) of the rhesus monkey from NCBI. Moreover, after a benchmarking of all the rhesus monkey assemblies available on NCBI, the Mmul_10 assembly [
24] was found to be of better quality in terms of number of gaps and correct order of clusters for each IG locus. For example, within the assembly Mmul_8, the order of clusters from 5′ to 3′ for the IGH locus is: V-CLUSTER -> D-CLUSTER -> J-CLUSTER -> C-CLUSTER -> V-CLUSTER instead of V-CLUSTER -> D-CLUSTER -> J-CLUSTER -> C-CLUSTER. As another example for the IGH locus, the Mmul_8 assembly has 92 gaps, the Mmul_051212 assembly has 65 gaps, and the rheMacS_1.0 assembly has six gaps, while the Mmul_10 has only two gaps. It is worth noting that the current study focuses on the analysis of the Mmul_10 assembly, including the previously available rhesus monkey data, within IMGT
®. However, a considerable IG genetic diversity in the form of allelic polymorphism and structural variation has been shown in previous genomic and germline gene inference studies [
27,
28], the results of which need to be taken into account for an improved overview of the IG gene repertoires of
Macaca mulatta. Future work, within IMGT
®, aims at analyzing the additional assemblies (for example RheMacS assembly [
27] and the ASM545330 contigs [
77]) based on the same model as well as incorporating the inferred alleles validated by the community. This will lead to the description of haplotypes taking into account the localization and the relative order of the genes in each locus in order to highlight new allelic polymorphisms and/or structural variations. This will also lead to more accurate gene assignments and calculations of somatic hypermutation (SHM) in this species. Extreme caution should be taken to correctly identify this genetic diversity.
Taking advantage of the IMGT biocuration pipeline, the IG germline repertoire and the IMGT® reference directories were established according to IMGT® nomenclature. The annotation of sequences, genes and structural data were integrated in the IMGT® databases, tools and web resources. As a result of this effort, 597 IG genes and 908 IG alleles have been integrated into IMGT®. Despite the high similarity of the human and rhesus monkey genome, some differences have also been observed. Indeed, the genomic organization and characterization of IG genes highlighted that, based on the assembly Mmul_10 of the rhesus monkey and the data available on the IMGT/GENE-DB, the rhesus monkey has more V genes than human (excluding orphons). For example, the IMGT/GENE-DB contains 228 IGHV genes for rhesus monkeys versus 162 IGHV genes for humans; 149 IGLV genes for rhesus monkeys versus 79 IGLV genes for humans; 138 IGKV genes for rhesus monkeys versus 77 IGKV genes for humans. Consequently, this difference has an impact on the number of functional genes found in these species, 86 F IGHV genes for rhesus monkeys versus 57 F IGHV genes for humans; 73 F IGLV genes for rhesus monkeys versus 33 F IGLV genes for humans; 85 F IGKV genes for rhesus monkeys versus 41 F IGKV genes for humans. It would be interesting to study the genomic organization and characterization of IG genes in the other available Macaca mulatta assemblies, as well as in the genome of additional nonhuman primates to determine the proportion of genes for each species and track their evolution.
It was noted that, even though the germline CDR-IMGT lengths of the rhesus monkey vary between subgroups, and in some cases within a subgroup, certain CDR-IMGT lengths are much more frequent and are found in several subgroups. For example, the CDR-IMGT lengths [8.8.2] are frequently found in subgroups IGHV1, IGHV3, IGHV4, IGHV5, and IGHV7.
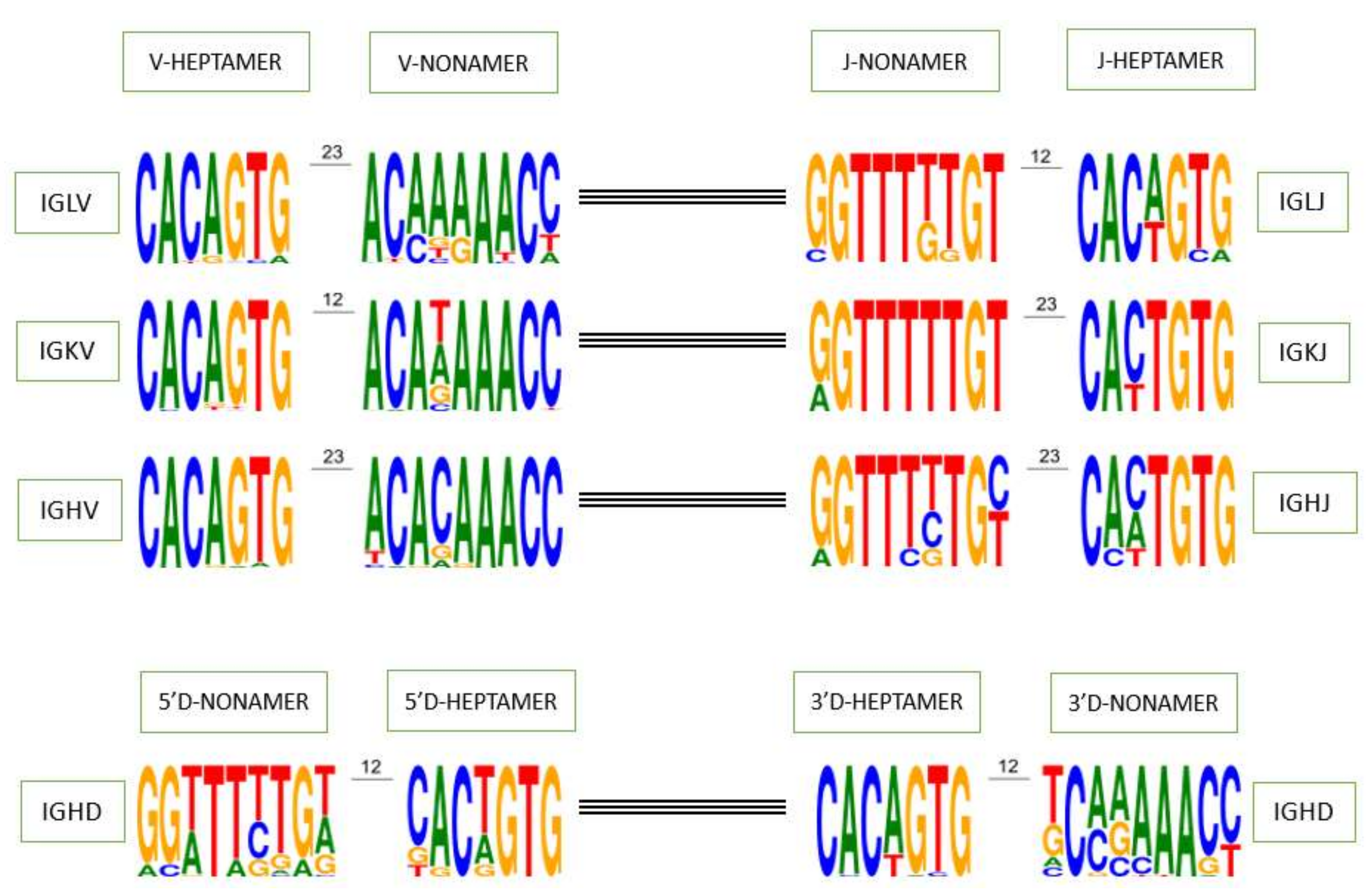
The heptamer and nonamer of the V, D and J genes recombination signals (RS) play a crucial role in the recombination process, their first three and last three nucleotides as well as the poly-A or poly-T tract of the nonamer are the specific characteristics used to identify them on genomic sequence (
Figure 3). In order to determine whether a heptamer or nonamer is canonical and if it could be useful during the rearrangement process or not, the IMGT
® annotation rule for RS is as follows: if a heptamer or a nonamer is found in more than one functional gene in IMGT/GENE-DB for a given locus whatever the species, it is considered as canonical. If not, heptamers with at most one mutation and nonamers with at most two mutations compared with the corresponding consensus sequences in the locus are also considered as canonical. However, there are also exceptions where the heptamer and nonamer do not obey this rule and are found rearranged in cDNAs available in IMGT/LIGM-DB and generalist nucleotide databases (GenBank and ENA). For example, the J-HEPTAMER of the gene IGHJ2 (‘ggctgtg’ instead of ‘cactgtg’ or ‘caatgtg’) has been found rearranged in 159 cDNAs in IMGT/LIGM-DB [
39] and the V-HEPTAMER of the allele IGLV8-125*01 (‘cacggcg’ instead of ‘cacagtg’) has also been found in cDNAs. Interestingly, the allele IGLV3-16*01, which is a pseudogene because there is no INIT-CODON (
https://www.imgt.org/ligmdb/view?id=IMGT000062, accessed on 11 November 2021), is found rearranged in the cDNA accession number KCWN01002936.1 (
https://www.ncbi.nlm.nih.gov/nuccore/KCWN01002936.1, accessed on 11 November 2021).
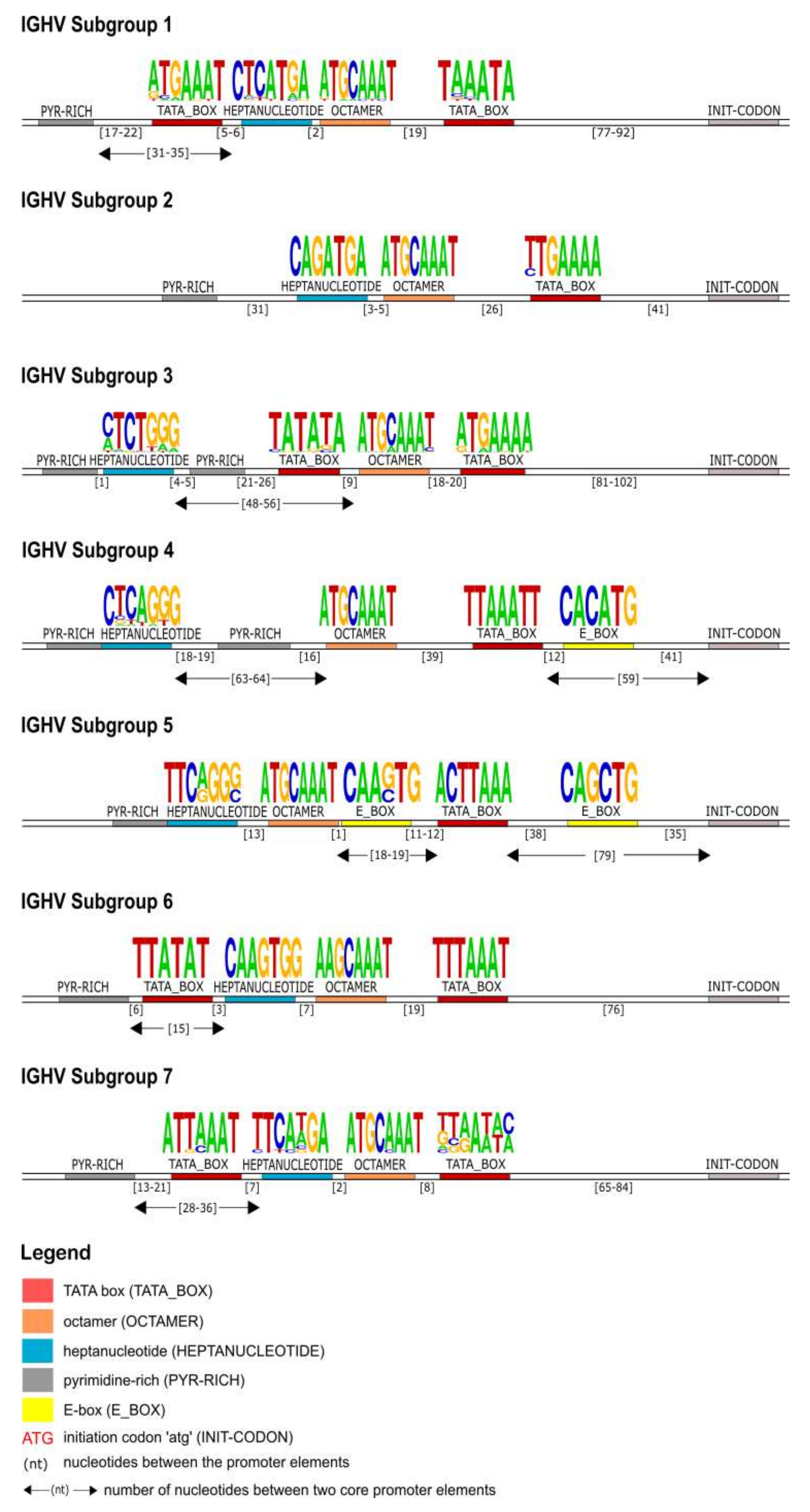
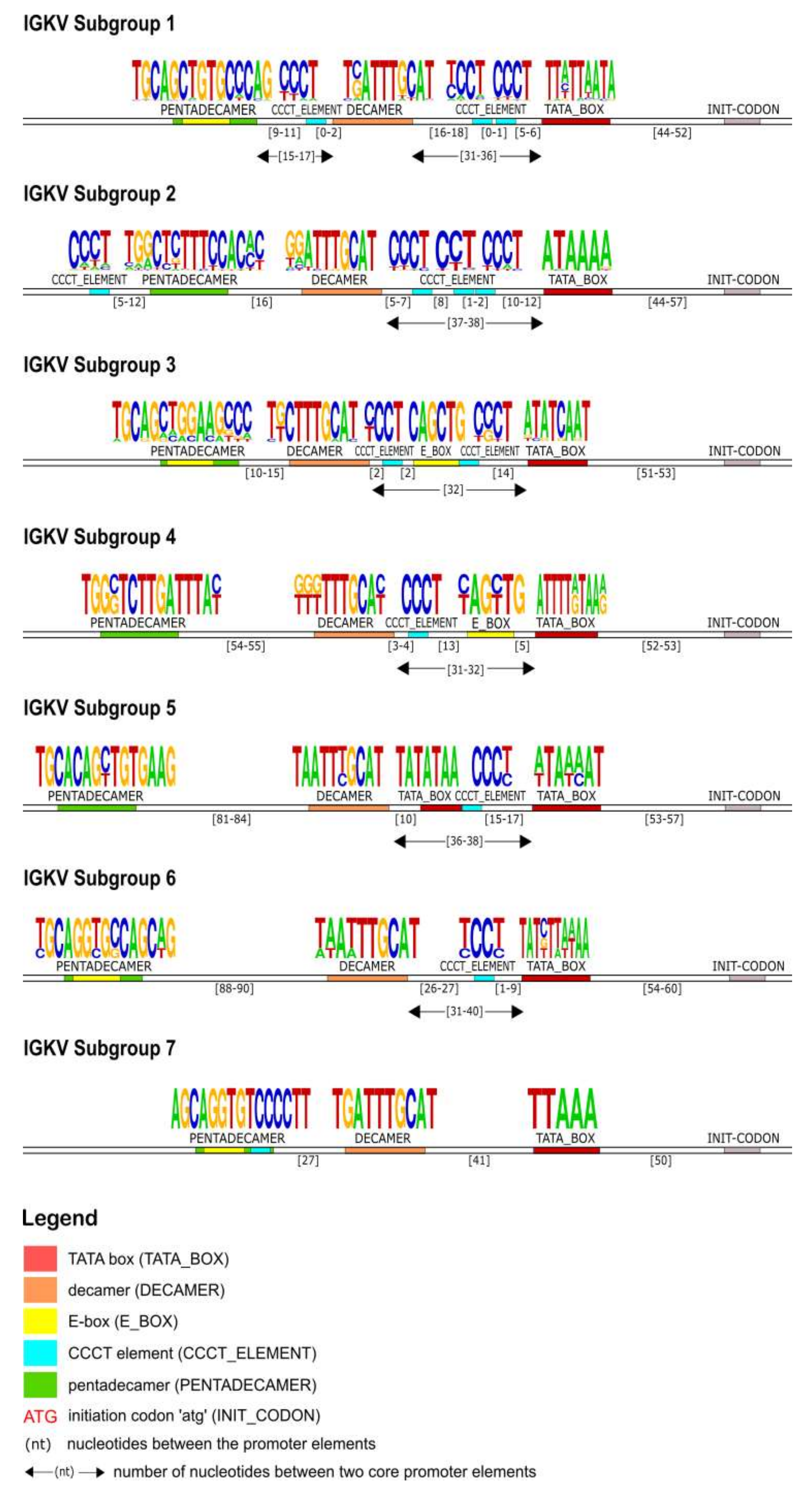
The identification of the core regulatory elements, located in the 5′UTR of the IG, reveals their conservation among eukaryotic species and would suggest their essential role in the transcription activity of the promoter. In this IG 5′UTR analysis of the rhesus monkey, all the core regulatory elements of the promoter region, that have been experimentally described in other eukaryotic species [
29,
30], have also been identified. Each IG chain, heavy and light, is characterized by different elements, however, a TATA box is always present (
Figure 4,
Figure 7 and
Figure 10). This conserved A/T rich region upstream of the initiation codon could indicate and validate its important role in the transcriptional promoter activity. The rhesus monkey’s heavy chain promoters contain the octamer motif upstream the TATA box, whereas light chain promoters contain the identical sequence in an inverted and complementary form at the same position, with two additional conserved nucleotides in 5′ end (decamer). This element (octamer/decamer) is highly conserved in all of the subgroups in both heavy and light chain promoters, and its presence seems to be sufficient and necessary for the promoter activation [
78,
79]. Besides, experimental data in eukaryotes have shown that the octamer/decamer interacts and binds to central regulatory sequence-specific factors (Oct family proteins) in order to achieve a high transcriptional stimulation [
78,
79].
Heptanucleotide and pentadecamer are two core elements, characteristic for heavy chain and light chain promoters, respectively. It is observed that their sequences, in our dataset, are highly similar to the consensus sequences which were previously identified in experimental studies of eukaryotic species [
80,
81]. Although they are well aligned with these consensuses, in some cases, nucleotide alterations appear in their motifs, which provide uniqueness to the subgroup they are identified in. These two elements seem to remain conserved, to ensure a high rate of transcriptional activation, acting synergistically to the octamer or decamer elements [
78,
79]. However, the precise mechanism underlying this synergism remains to be elucidated for the
Macaca mulatta.
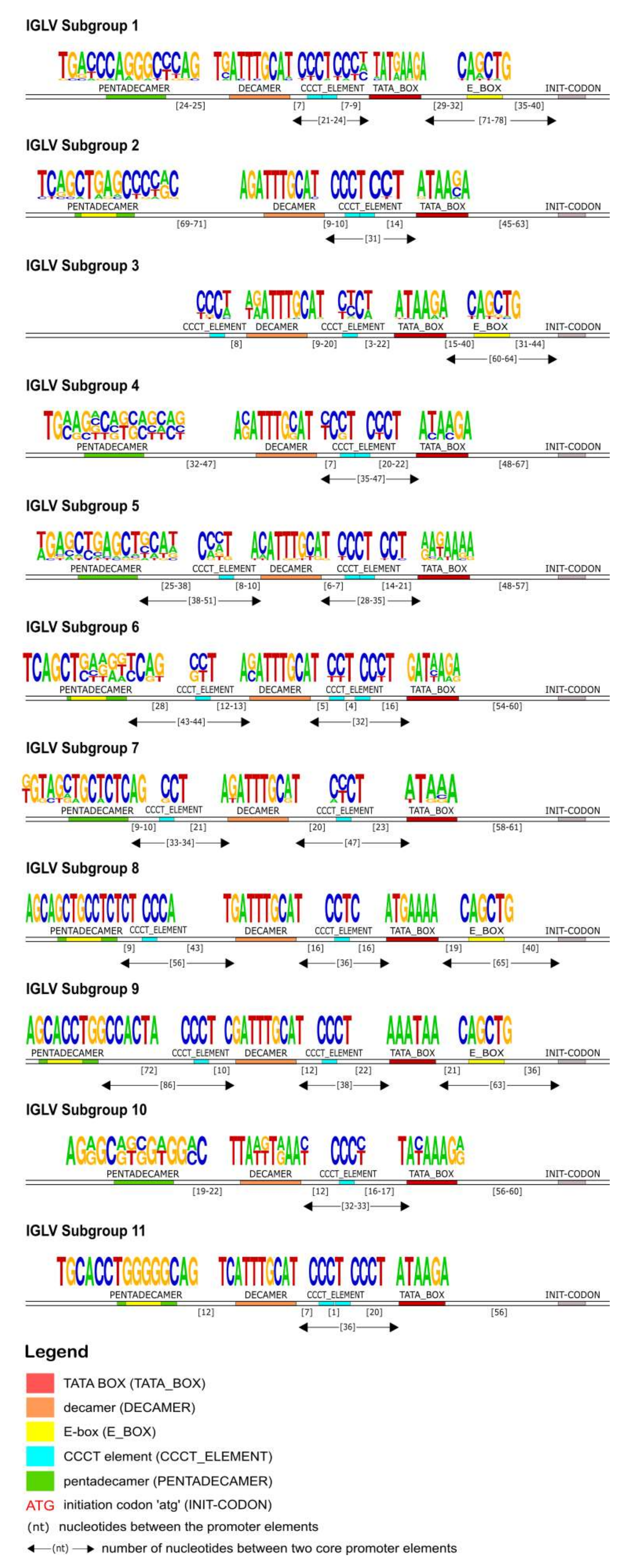
Our IG promoter analysis revealed additional regulatory elements in the 5′ flanking sequences of immunoglobulin V genes, which could increase the rate of transcription [
78]. These elements include a pyrimidine-rich region in heavy chain promoters (
Figure 4), a CCCT element in light chain promoters (
Figure 7 and
Figure 10), and additional E-boxes and TATA boxes in both of them (heavy and light chains). More specifically, a CCCT element occurs in one or two repeats in the decamer flanking sequence of kappa and lambda chain UTRs. It has been shown experimentally in mouse cells that this element is very important for IG expression, and even in cases where the pentadecamer is absent it acts along with the decamer to stimulate transcription [
79]. Moreover, a previous study [
78] justifies that in the promoters where the CCCT element is missing, an E-box is detected and could fulfill the transcriptional activity, an observation that we also make in our analysis. For example, in the IGLV3 subgroup (
Figure 7), the promoter region is lacking the pentadecamer, but the presence of two repeats of the CCCT element in the 5′ and 3′ flanking decamer and the E-box could probably activate the transcription.
Overall, our findings support the high degree of conservation of the fundamental regulatory promoter elements upstream the initiation codon of a V gene (ATG), as well as the existence of a distinct IG promoter organization. Despite this conservation, the consensus sequences and distances of the core elements of each subgroup vary significantly, indicating an auxiliary method for subgroup characterization and gene classification within subgroups. Notably, this observation along with the results of recent polymorphism studies in the human antibody upstream sequences could be valuable for the IG variable genes annotation process [
29,
30]. The elements identified and revealed in our study were previously described in the literature in other eukaryotic species and showed their necessary role in IG transcription, as well as the compensatory factor between elements, as their presence or absence could increase or decrease transcriptional activity, respectively [
78,
79]. Therefore, based on the Evo-devo scenarios and conservation, we could hypothesize that
Macaca mulatta elements could have a similar fundamental role in IG regulation as in other eukaryotic species, however further in-depth experimental research needs to be done to validate this hypothesis. There is increasing evidence that 5′UTRs have a fundamental role in gene expression and further genomic studies could be performed to clarify and even explain these IG expression differences among individuals.
The study focuses on Mmul_10 assembly of rhesus monkey in order to provide to the scientific community rich and precise details about genes and alleles in this genome. For a given gene, up to 200 different information fields may be available for its annotation (IMGT labels of description, IMGT nomenclature, IMGT numbering, functionalities, isolate, reference sequence and sequence of the literature, description of alleles, protein display, colliers de perles, FR and CDR-length, etc.). This includes detailed characterisation of the regulatory elements of their promoters and their conventional recombination signals (RS), an overview of which is presented in this manuscript.
Taking into account the IG genetic diversity in the form of allelic and structural variations previously described [
27,
28], the approach described in this manuscript, and the resulting characterization of IG genes in Mmul_10, will be used as reference and model for the annotation of other published assemblies. The identification of position and order of genes in the IG loci of Mmul_10 provides a starting point for the characterization of allelic polymorphisms and of structural variations.
A comprehensive understanding of both innate and adaptive immune responses is essential for vaccine design and development [
82,
83]. Characterization of the
Macaca mulatta IG loci in genomic assemblies, as shown in this study, provides important baseline information for this model species. However, the characterization of IG loci of additional assemblies and different animals will be required to generate a more complete IGH reference directory within IMGT, work that is currently underway.


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}