
Design of a Multi-Epitopes Vaccine against Hantaviruses: An Immunoinformatics and Molecular Modelling Approach



 and
and 
Abstract
1. Introduction
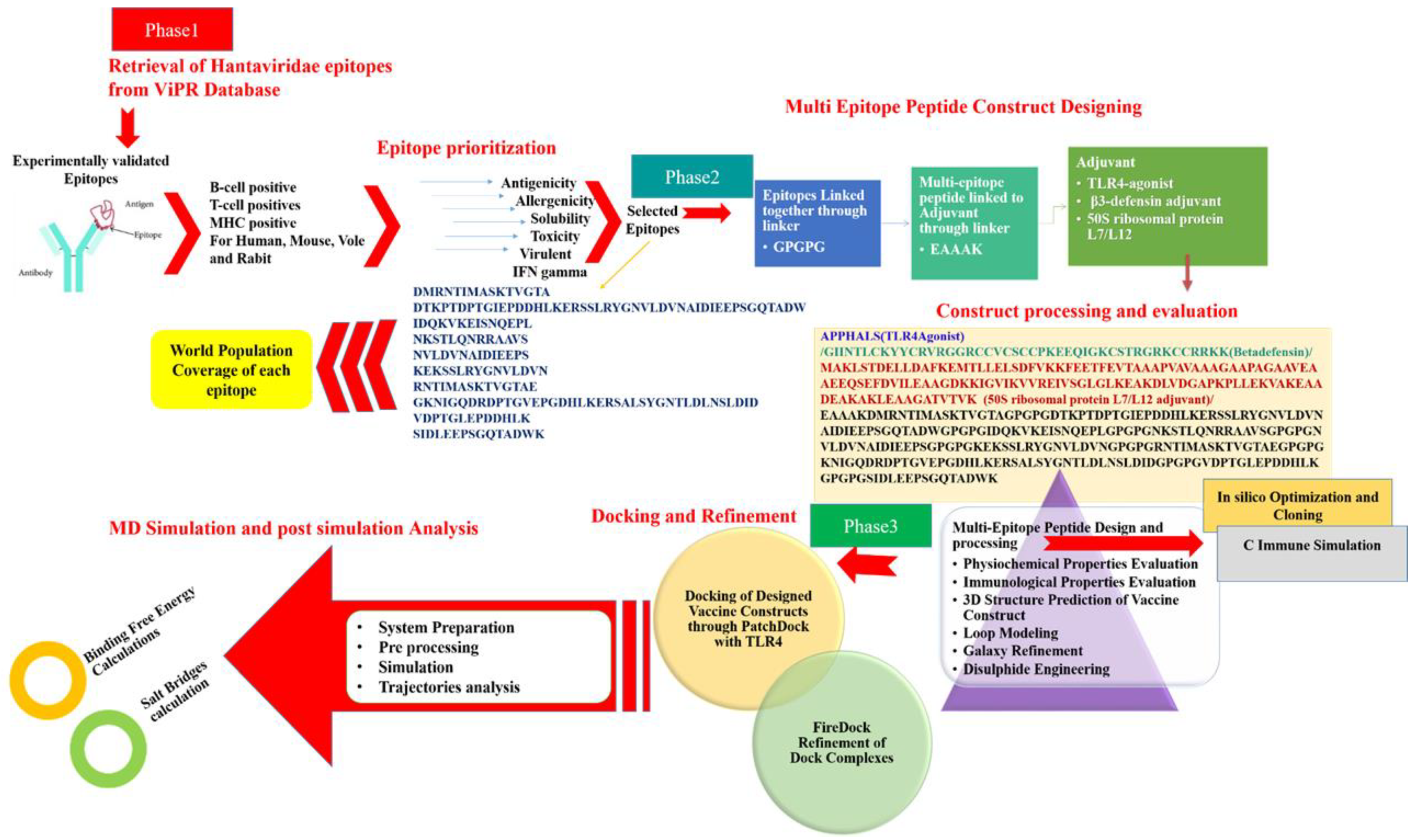
2. Materials and Methods
2.1. Target Epitopes Retrieval
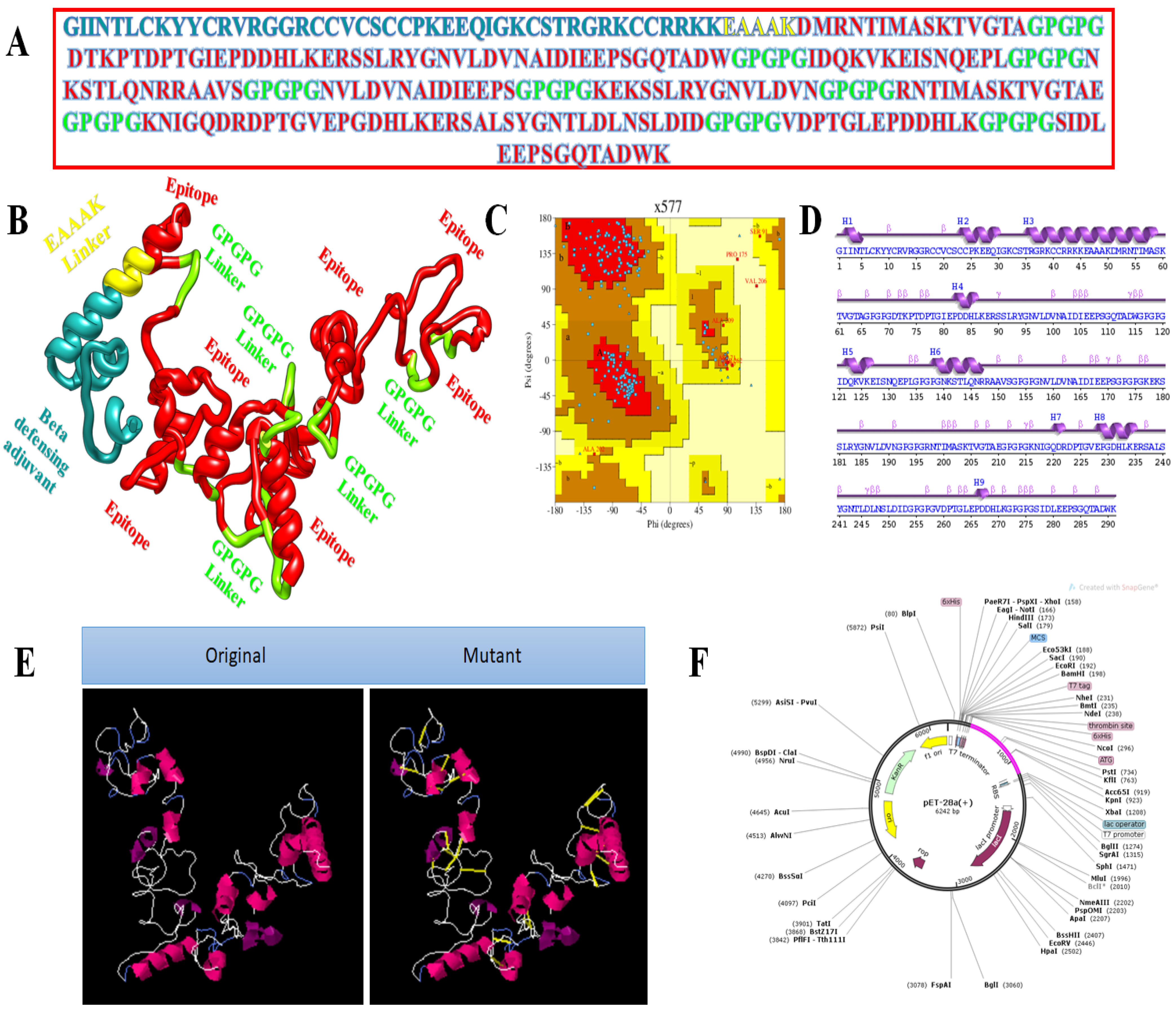
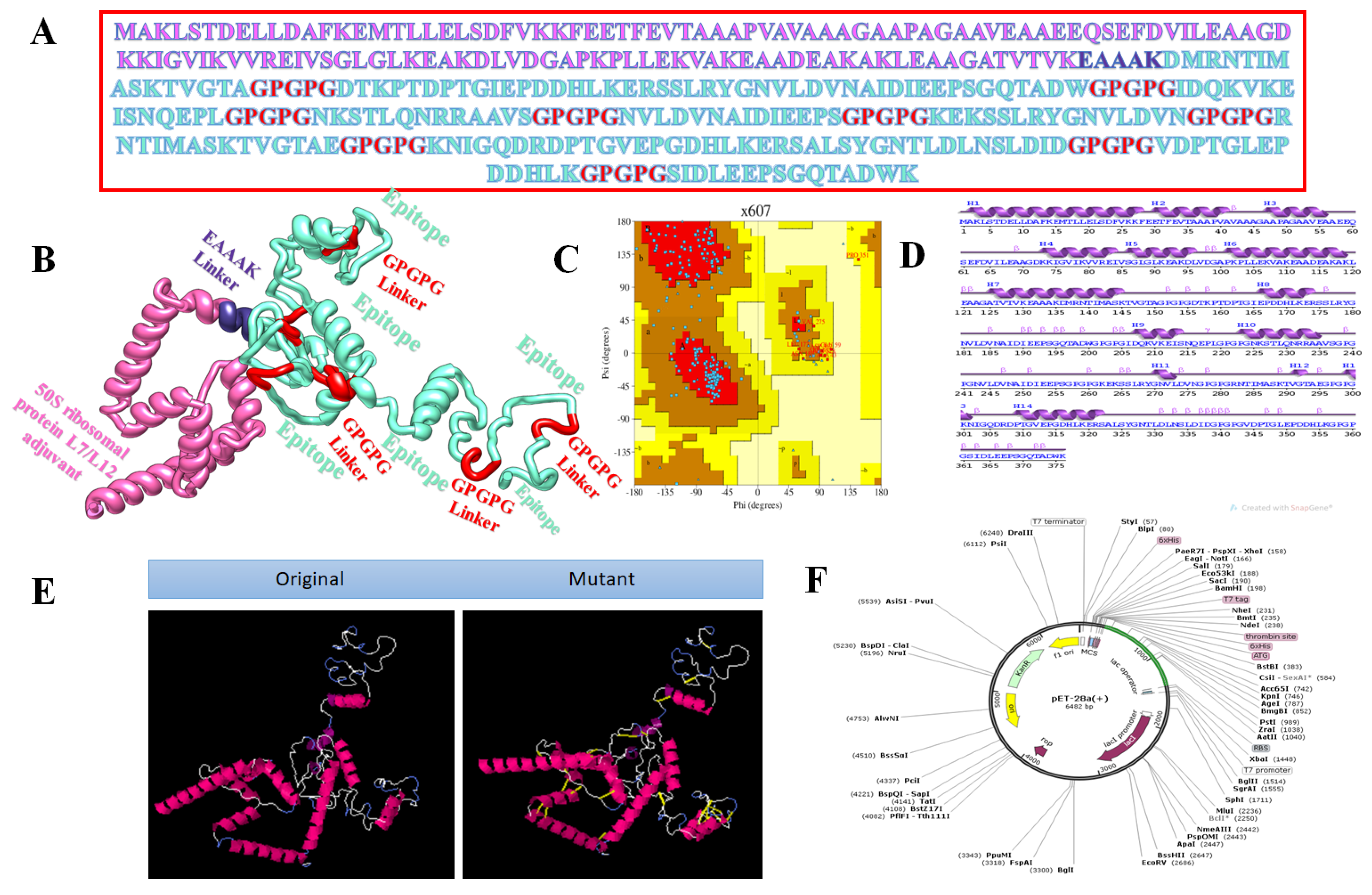
2.2. Chimeric Vaccine Designing
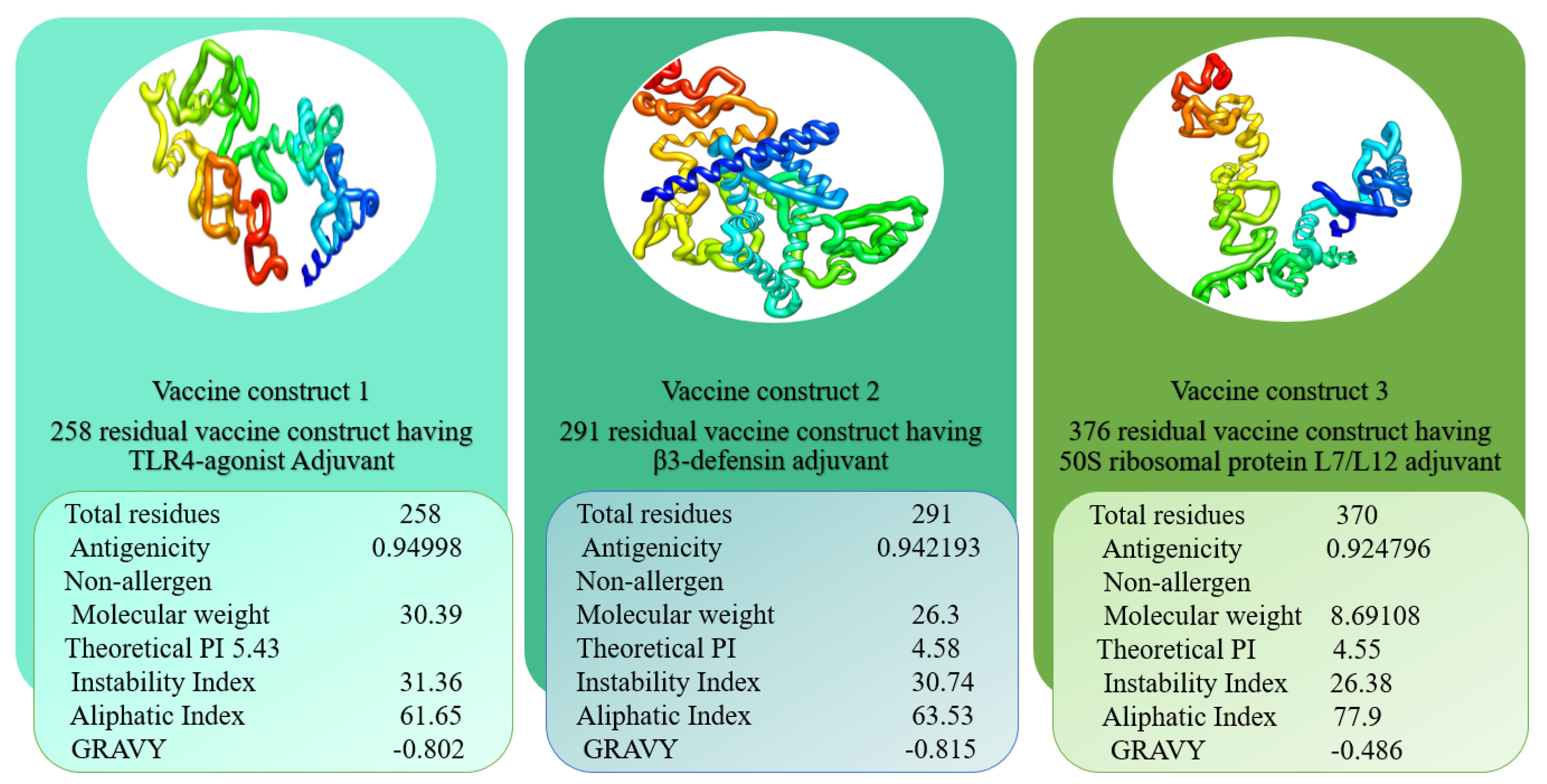
2.3. Physicochemical and Immunological Properties
2.4. Molecular Docking
2.5. Molecular Dynamics Simulations
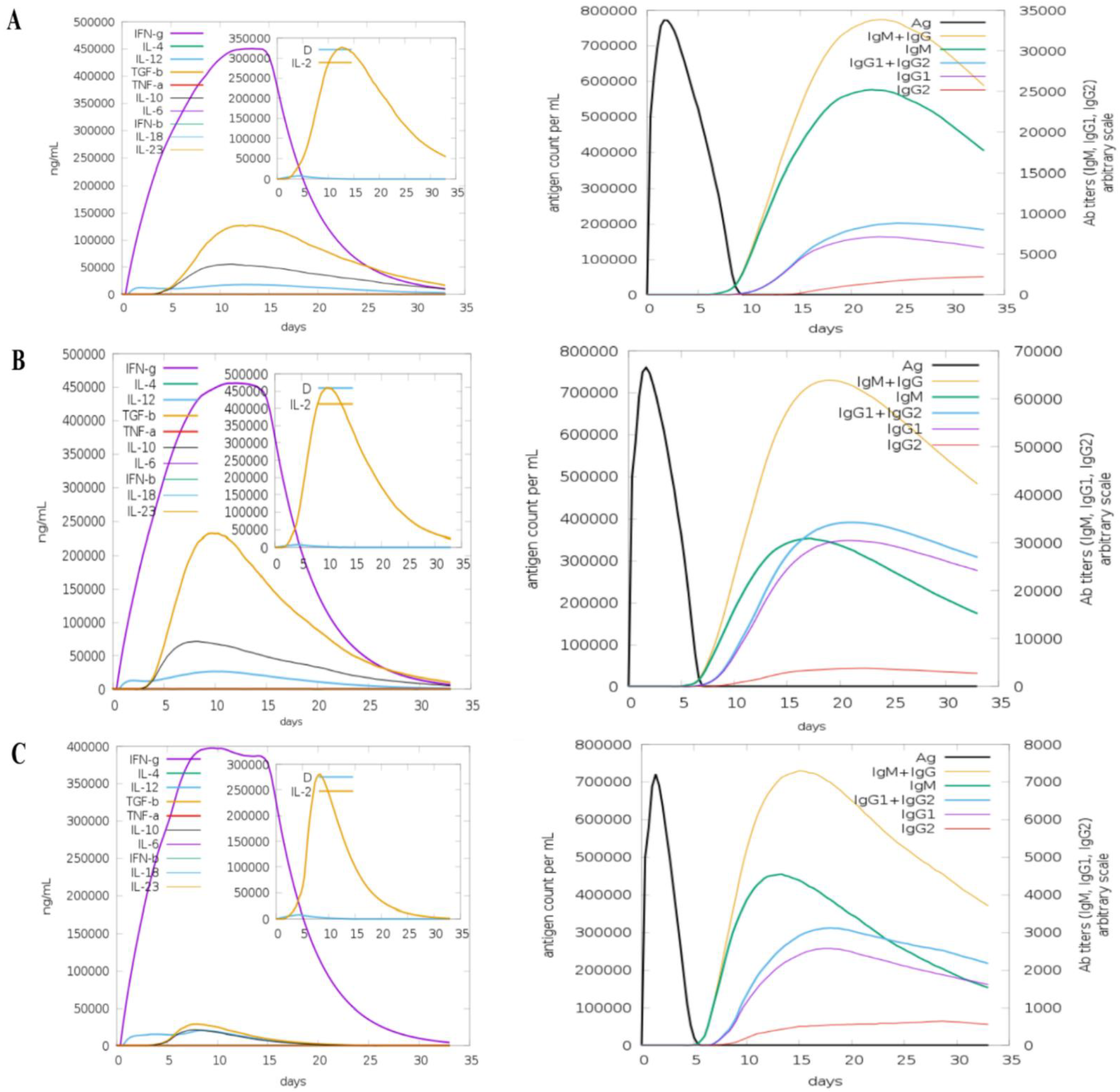
2.6. Computational Immune Simulation
2.7. Disulphide Engineering and Codon Optimisation
3. Results and Discussion
3.1. Epitopes Analysis
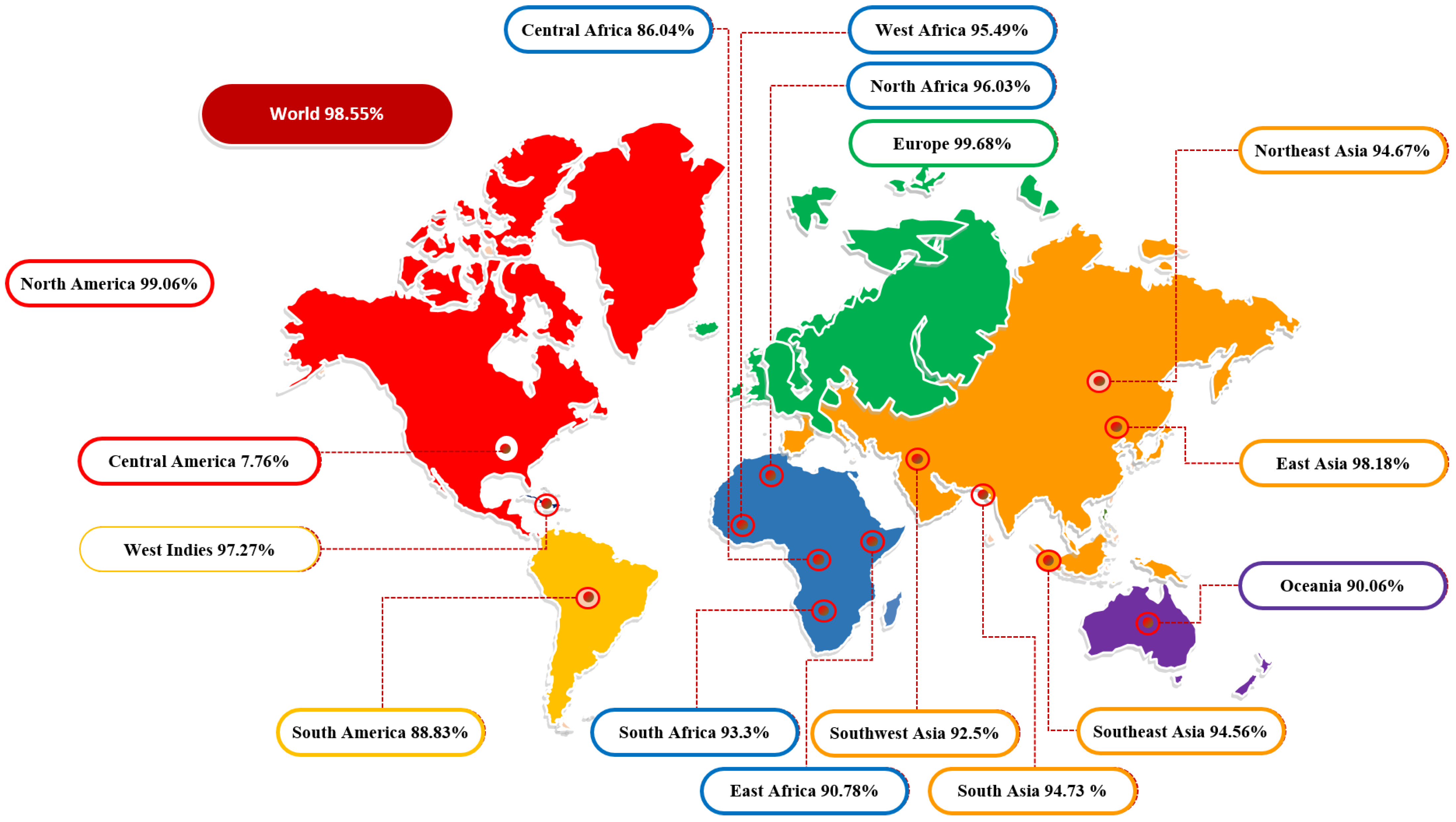
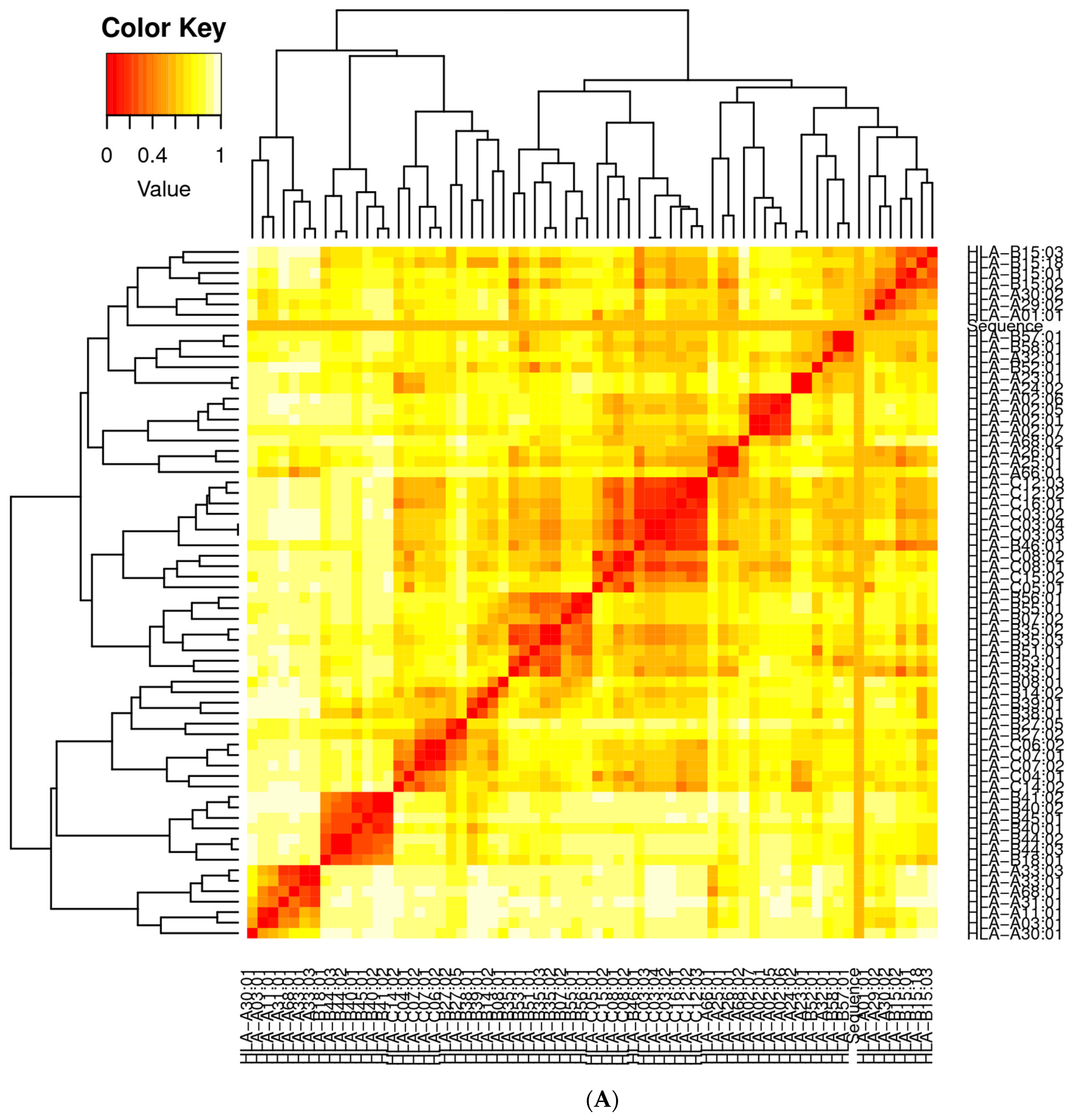
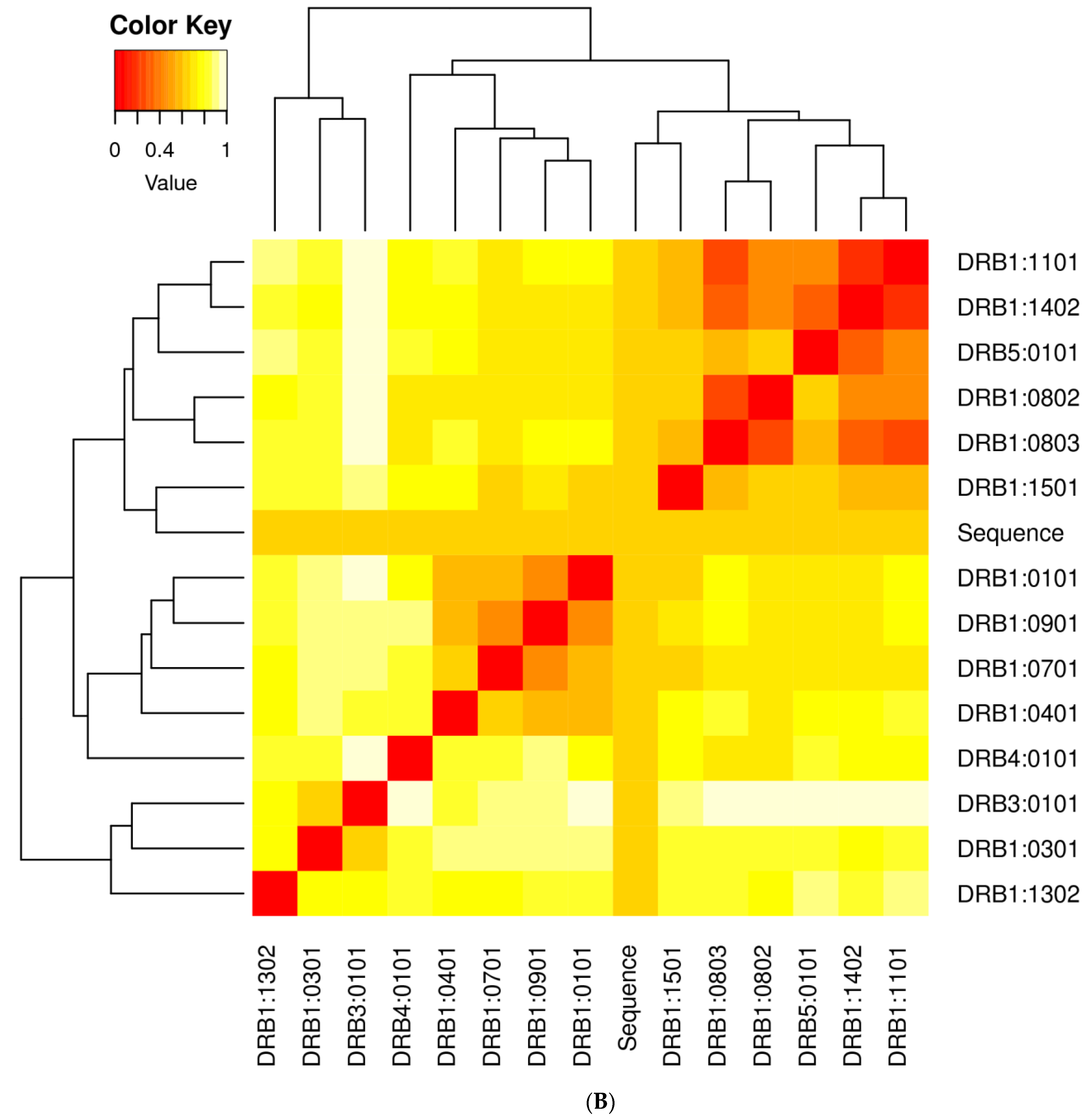
3.2. Population Coverage Analysis
3.3. Vaccine with Different Adjuvants
3.4. Multi-Epitope Vaccine Design
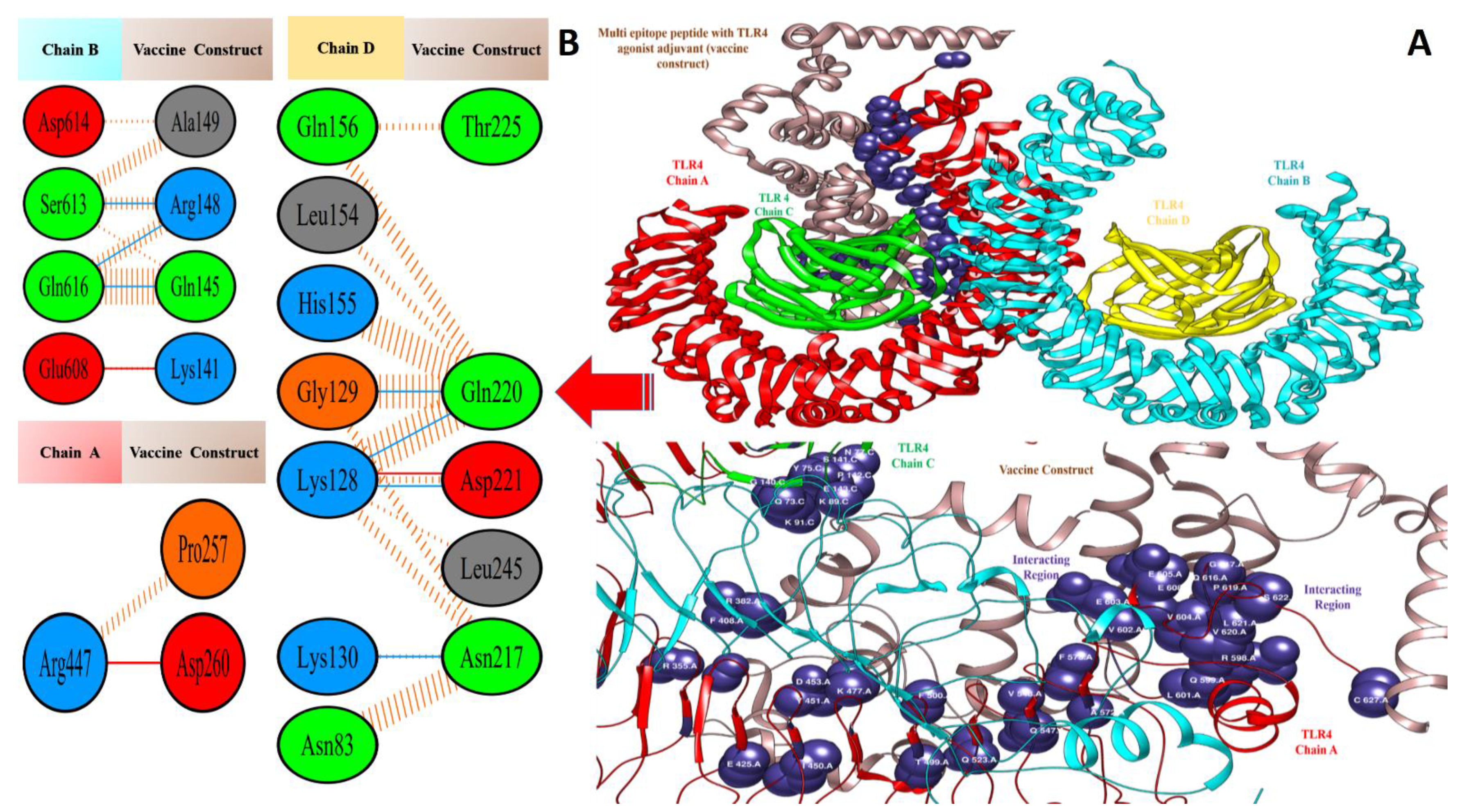
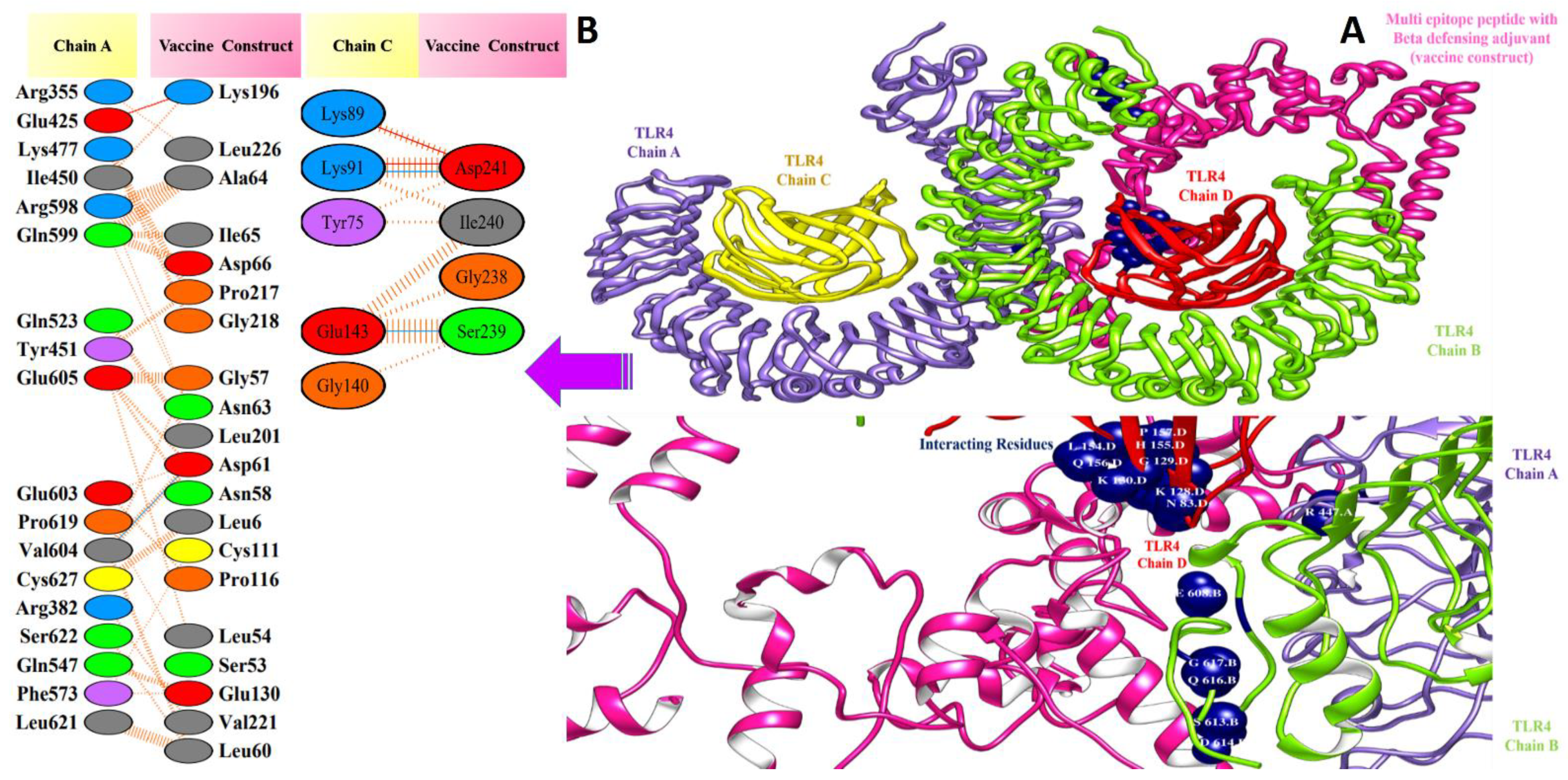
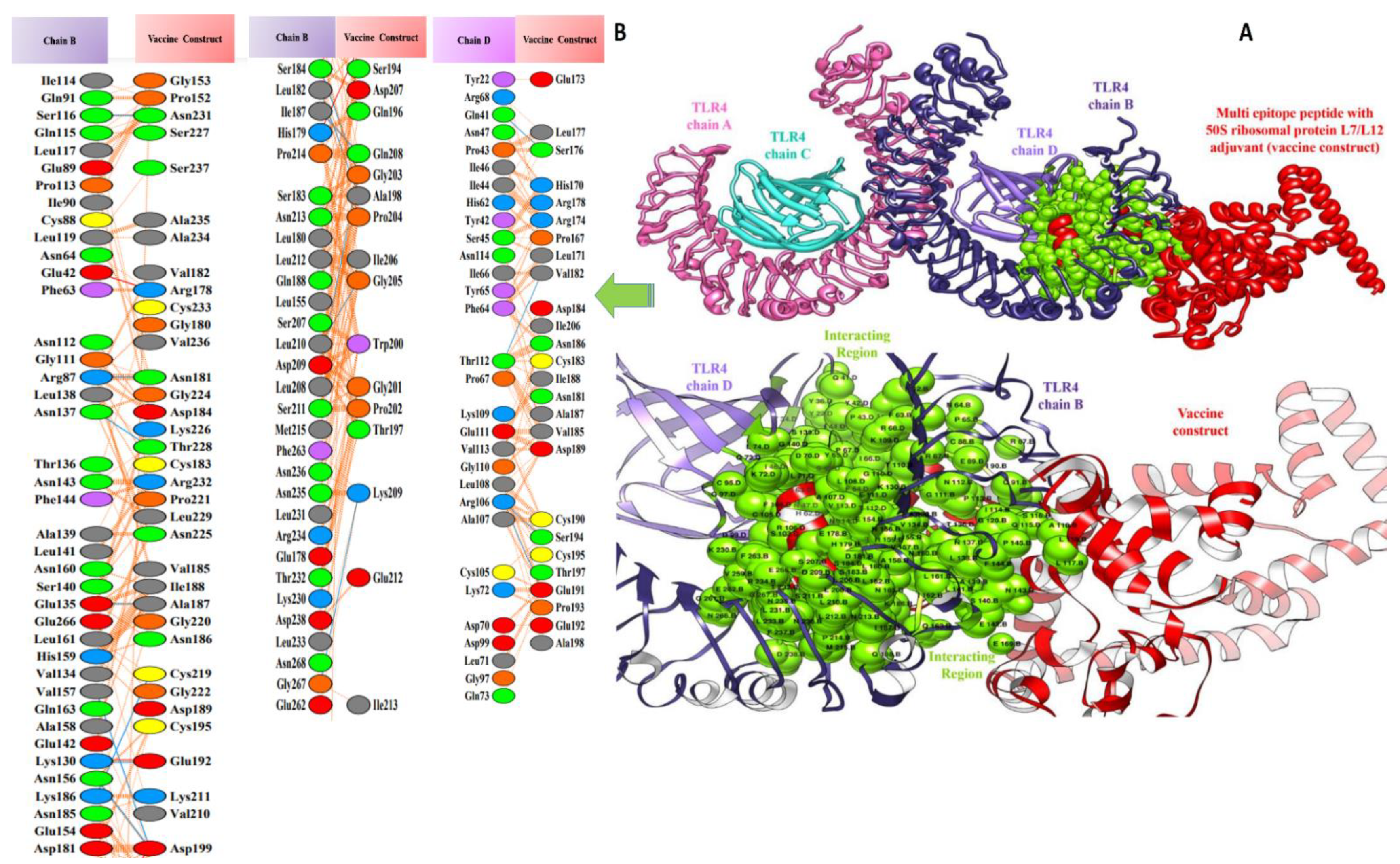
3.5. Molecular Docking of Vaccine Constructs with TLR4
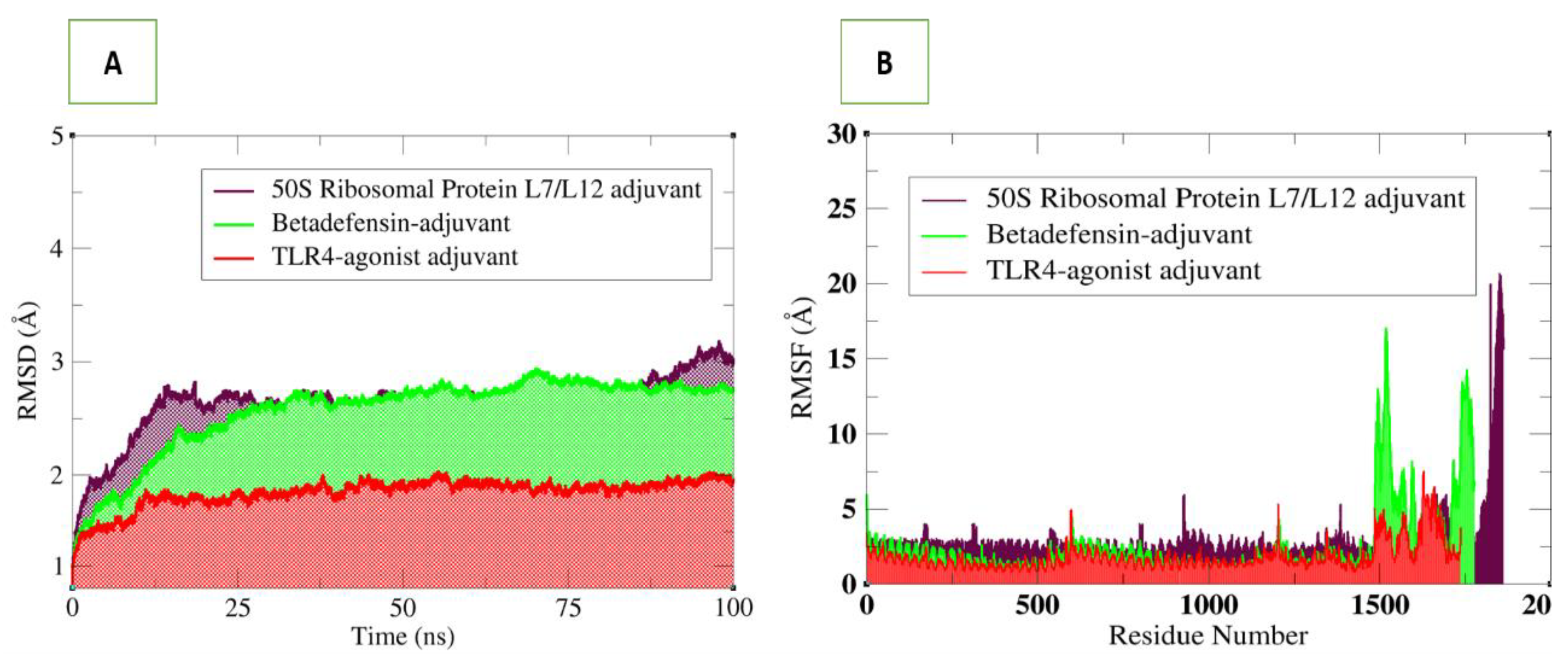
3.6. Molecular Dynamics Simulations Analysis
3.7. Salt Bridges between TLR4 and Vaccine Constructs
3.8. Binding Free Energies Calculation
3.9. In Silico Cloning
3.10. Clustering Analysis of Vaccine Constructs
3.11. Computational Immune Simulation
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, R.; Ma, H.; Shu, J.; Zhang, Q.; Han, M.; Liu, Z.; Jin, X.; Zhang, F.; Wu, X. Vaccines and Therapeutics Against Hantaviruses. Front. Microbiol. 2020, 10, 2989. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.-K.; Cho, S.; Lee, S.-H.; No, J.S.; Lee, G.-Y.; Park, K.; Lee, D.; Jeong, S.T.; Song, J.-W. Genomic Epidemiology and Active Surveillance to Investigate Outbreaks of Hantaviruses. Front. Cell. Infect. Microbiol. 2021, 10, 803. [Google Scholar] [CrossRef] [PubMed]
- Brocato, R.L.; Hooper, J.W. Progress on the Prevention and Treatment of Hantavirus Disease. Viruses 2019, 11, 610. [Google Scholar] [CrossRef]
- D’Souza, M.H.; Patel, T.R. Biodefense Implications of New-World Hantaviruses. Front. Bioeng. Biotechnol. 2020, 8, 925. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, P.; Tia, M.; Alabi, A.; Anon, J.-C.; Auste, B.; Essbauer, S.; Gnionsahe, A.; Kigninlman, H.; Klempa, B.; Kraef, C.; et al. Human Infections by Non–Rodent-Associated Hantaviruses in Africa. J. Infect. Dis. 2016, 214, 1507–1511. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zheng, X.; Wang, L.; Du, H.; Wang, P.; Bai, X. Hantavirus infection: A global zoonotic challenge. Virol. Sin. 2017, 32, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Schmaljohn, C.S.; Nichol, S.T. Bunyaviridae. In Fields’ Virology, 5th ed.; Lippincott Williams Wilkins: Philadelphia, PA, USA, 2007; pp. 1741–1789. [Google Scholar]
- Meyer, B.J.; Schmaljohn, C.S. Persistent hantavirus infections: Characteristics and mechanisms. Trends Microbiol. 2000, 8, 61–67. [Google Scholar] [CrossRef]
- Witkowski, P.T.; Drexler, J.F.; Kallies, R.; Ličková, M.; Bokorová, S.; Mananga, G.D.; Szemes, T.; Leroy, E.M.; Krüger, D.H.; Drosten, C. Phylogenetic Analysis of a Newfound Bat-Borne Hantavirus Supports a Laurasiatherian Host Association for Ancestral Mammalian Hantaviruses. Infect. Genet. Evol. 2016, 41, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Yanagihara, R.; Gu, S.H.; Arai, S.; Kang, H.J.; Song, J.-W. Hantaviruses: Rediscovery and new beginnings. Virus Res. 2014, 187, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Z. Discovery of hantaviruses in bats and insectivores and the evolution of the genus Hantavirus. Virus Res. 2014, 187, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Forbes, K.M.; Sironen, T.; Plyusnin, A. Hantavirus maintenance and transmission in reservoir host populations. Curr. Opin. Virol. 2018, 28, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Nuzum, E.O.; Rossi, C.A.; Stephenson, E.H.; LeDuc, J.W. Aerosol Transmission of Hantaan and Related Viruses to Laboratory Rats. Am. J. Trop. Med. Hyg. 1988, 38, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Voutilainen, L.; Sironen, T.; Tonteri, E.; Bäck, A.T.; Razzauti, M.; Karlsson, M.; Wahlström, M.; Niemimaa, J.; Henttonen, H.; Lundkvist, Å. Life-Long Shedding of Puumala Hantavirus in Wild Bank Voles (Myodes Glareolus). J. Gen. Virol. 2015, 96, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Sargianou, M.; Watson, D.C.; Chra, P.; Papa, A.; Starakis, I.; Gogos, C.; Panos, G. Hantavirus Infections for the Clinician: From Case Presentation to Diagnosis and Treatment. Crit. Rev. Microbiol. 2012, 38, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Du, H.; Wang, L.M.; Wang, P.Z.; Bai, X.F. Hemorrhagic Fever with Renal Syndrome: Pathogenesis and Clinical Picture. Front. Cell. Infect. Microbiol. 2016, 6, 1. [Google Scholar] [CrossRef]
- Munir, N.; Jahangeer, M.; Hussain, S.; Mahmood, Z.; Ashiq, M.; Ehsan, F.; Akram, M.; Shah, S.M.A.; Riaz, M.; Sana, A. Hantavirus diseases pathophysiology, their diagnostic strategies and therapeutic approaches: A review. Clin. Exp. Pharmacol. Physiol. 2020, 48, 20–34. [Google Scholar] [CrossRef]
- Muranyi, W.; Bahr, U.; Zeier, M.; van der Woude, F.J. Hantavirus Infection. J. Am. Soc. Nephrol. 2005, 16, 3669–3679. [Google Scholar] [CrossRef]
- Zhang, L. Multi-epitope vaccines: A promising strategy against tumors and viral infections. Cell. Mol. Immunol. 2017, 15, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Abdulla, F.; Nain, Z.; Hossain, M.M.; Sayed, S.B.; Khan, M.S.A.; Adhikari, U.K. Computational Approach for Screening the Whole Proteome of Hantavirus and Designing a Multi-Epitope Subunit Vaccine. bioRxiv 2019, 832980. [Google Scholar] [CrossRef]
- Brennick, C.; George, M.M.; Corwin, W.L.; Srivastava, P.; Ebrahimi-Nik, H. Neoepitopes as cancer immunotherapy targets: Key challenges and opportunities. Immunotherapy 2017, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, L.; Consortium, H. Developments in Cancer Vaccines for Hepatocellular Carcinoma. Cancer Immunol. Immunother. 2016, 65, 93–99. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Yang, X.; Liu, C.; Chen, X.; Wang, L.; Xiao, M.; Ye, J.; Wu, Y.; Ye, L. Efficient Control of Chronic LCMV Infection by a CD4 T Cell Epitope-Based Heterologous Prime-Boost Vaccination in a Murine Model. Cell. Mol. Immunol. 2017, 15, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.; Wang, C.; Badakhshan, T.; Chilukuri, S.; BenMohamed, L. The Challenges and Opportunities for the Development of a T-Cell Epitope-Based Herpes Simplex Vaccine. Vaccine 2014, 32, 6733–6745. [Google Scholar] [CrossRef] [PubMed]
- Lu, I.-N.; Farinelle, S.; Sausy, A.; Muller, C.P. Identification of a CD4 T-Cell Epitope in the Hemagglutinin Stalk Domain of Pandemic H1N1 Influenza Virus and Its Antigen-Driven TCR Usage Signature in BALB/c Mice. Cell. Mol. Immunol. 2016, 14, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Shin, O.S.; Na, J.; Kim, J.K.; Seong, R.-K.; Park, M.-S.; Noh, J.Y.; Song, J.Y.; Cheong, H.J.; Park, Y.H. A Systems Vaccinology Approach Reveals the Mechanisms of Immunogenic Responses to Hantavax Vaccination in Humans. Sci. Rep. 2019, 9, 4760. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Park, H.; Jung, J. Effectiveness of Inactivated Hantavirus Vaccine on the Disease Severity of Hemorrhagic Fever with Renal Syndrome. Kidney Res. Clin. Pract. 2018, 37, 366. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Ma, T.; Zhang, X.; Ying, Q.; Han, M.; Zhang, M.; Yang, R.; Li, Y.; Wang, F.; Liu, R. Incorporation of CD40 Ligand or Granulocyte-Macrophage Colony Stimulating Factor into Hantaan Virus (HTNV) Virus-like Particles Significantly Enhances the Long-Term Immunity Potency against HTNV Infection. J. Med. Microbiol. 2019, 68, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Nicacio, C.D.C.; Della Valle, M.G.; Padula, P.; Bjorling, E.; Plyusnin, A.; Lundkvist, A. Cross-Protection against Challenge with Puumala Virus after Immunization with Nucleocapsid Proteins from Different Hantaviruses. J. Virol. 2002, 76, 6669–6677. [Google Scholar] [CrossRef]
- Schmaljohn, C.S.; Hasty, E.S.; Dalrymple, J.M. Preparation of candidate vaccinia-vectored vaccines for haemorrhagic fever with renal syndrome. Vaccine 1992, 10, 10–13. [Google Scholar] [CrossRef]
- Safronetz, D.; Hegde, N.R.; Ebihara, H.; Denton, M.; Kobinger, G.P.; Jeor, S.S.; Feldmann, H.; Johnson, D.C. Adenovirus Vectors Expressing Hantavirus Proteins Protect Hamsters against Lethal Challenge with Andes Virus. J. Virol. 2009, 83, 7285–7295. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.S.; Safronetz, D.; Marzi, A.; Ebihara, H.; Feldmann, H. Vesicular Stomatitis Virus-Based Vaccine Protects Hamsters against Lethal Challenge with Andes Virus. J. Virol. 2011, 85, 12781–12791. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, E.F.; Josleyn, M.; Ullman, D.; Fisher, D.; Dalrymple, L.; Sellers-Myers, K.; Loudon, P.; Rusnak, J.; Rivard, R.; Schmaljohn, C.; et al. A Phase 1 clinical trial of Hantaan virus and Puumala virus M-segment DNA vaccines for hemorrhagic fever with renal syndrome. Vaccine 2012, 30, 1951–1958. [Google Scholar] [CrossRef]
- Jiang, D.-B.; Zhang, J.-P.; Cheng, L.-F.; Zhang, G.-W.; Li, Y.; Li, Z.-C.; Lu, Z.-H.; Zhang, Z.-X.; Lu, Y.-C.; Zheng, L.-H.; et al. Hantavirus Gc Induces Long-Term Immune Protection via LAMP-Targeting DNA Vaccine Strategy. Antiviral Res. 2018, 150, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An Open Bioinformatics Database and Analysis Resource for Virology Research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP—A server for in silico prediction of allergens. BMC Bioinform. 2013, 14, S4. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P. Peptide Toxicity Prediction. In Computational Peptidology; Zhou, P., Huang, J., Eds.; Springer: Chandigarh, India, 2015; pp. 143–157. [Google Scholar]
- Garg, A.; Gupta, D. VirulentPred: A SVM Based Prediction Method for Virulent Proteins in Bacterial Pathogens. BMC Bioinform. 2008, 9, 62. [Google Scholar] [CrossRef]
- Rost, B.; Sander, C.; Casadio, R.; Fariselli, P. Transmembrane Helices Predicted at 95% Accuracy. Protein Sci. 1995, 4, 521–533. [Google Scholar] [CrossRef]
- Li, W.; Joshi, M.D.; Singhania, S.; Ramsey, K.H.; Murthy, A.K. Peptide Vaccine: Progress and Challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef]
- Abbas, G.; Zafar, I.; Ahmad, S.; Azam, S.S. Immunoinformatics Design of a Novel Multi-Epitope Peptide Vaccine to Combat Multi-Drug Resistant Infections Caused by Vibrio Vulnificus. Eur. J. Pharm. Sci. 2019, 142, 105160. [Google Scholar] [CrossRef]
- Khan, S.; Khan, A.; Rehman, A.U.; Ahmad, I.; Ullah, S.; Khan, A.A.; Ali, S.S.; Afridi, S.G.; Wei, D.-Q. Immunoinformatics and Structural Vaccinology Driven Prediction of Multi-Epitope Vaccine against Mayaro Virus and Validation through in-Silico Expression. Infect. Genet. Evol. 2019, 73, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Nezafat, N.; Karimi, Z.; Eslami, M.; Mohkam, M.; Zandian, S.; Ghasemi, Y. Designing an Efficient Multi-Epitope Peptide Vaccine against Vibrio Cholerae via Combined Immunoinformatics and Protein Interaction Based Approaches. Comput. Biol. Chem. 2016, 62, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Motamedi, M.J.; Amani, J.; Shahsavandi, S.; Salmanian, A.H. In Silico Design of Multimeric HN-F Antigen as a Highly Immunogenic Peptide Vaccine against Newcastle Disease Virus. Int. J. Pept. Res. Ther. 2014, 20, 179–194. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. Proteom. Protoc. Handb. 2005, 71–607. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A Protein Structure and Structural Feature Prediction Server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A. PDBsum: Summaries and Analyses of PDB Structures. Nucleic Acids Res. 2001, 29, 221–222. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein Structure Refinement Driven by Side-Chain Repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef]
- Ismail, S.; Ahmad, S.; Azam, S.S. Vaccinomics to Design a Novel Single Chimeric Subunit Vaccine for Broad-Spectrum Immunological Applications Targeting Nosocomial Enterobacteriaceae Pathogens. Eur. J. Pharm. Sci. 2020, 146, 105258. [Google Scholar] [CrossRef]
- Yu, H.-T.; Jiang, H.; Zhang, Y.; Nan, X.-P.; Li, Y.; Wang, W.; Jiang, W.; Yang, D.-Q.; Su, W.-J.; Wang, J.-P.; et al. Hantaan Virus Triggers TLR4-Dependent Innate Immune Responses. Viral Immunol. 2012, 25, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for Rigid and Symmetric Docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast interaction refinement in molecular docking. Proteins Struct. Funct. Bioinform. 2007, 69, 139–159. [Google Scholar] [CrossRef] [PubMed]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A Web Server for Fast Interaction Refinement in Molecular Docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A Program to Generate Schematic Diagrams of Protein-Ligand Interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Case, D.; Cerutti, D.; Cheateham, T.; Darden, T.; Duke, R.; Giese, T.; Gohlke, H.; Goetz, A.; Greene, D.; Homeyer, N.; et al. AMBER16 Package. Univ. Calif. San. Fr. 2016. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham III, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- AMBER 10.0 Introductory Tutorial. Available online: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.497.2524&rep=rep1&type=pdf (accessed on 12 February 2021).
- Brice, A.R.; Dominy, B.N. Examining Electrostatic Influences on Base-Flipping: A Comparison of TIP3P and GB Solvent Models. Commun. Comput. Phys. 2013, 13, 223–237. [Google Scholar] [CrossRef]
- Case, D.; Babin, V.; Berryman, J.; Betz, R.; Cai, Q.; Cerutti, D.; Cheatham, T., III; Darden, T.; Duke, R.; Gohlke, H. The FF14SB Force Field. Amber 2014, 14, 29–31. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A Fast SHAKE Algorithm to Solve Distance Constraint Equations for Small Molecules in Molecular Dynamics Simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Rapin, N.; Lund, O.; Castiglione, F. C-Immsim 10.1 Server. Available online: http://www.cbs.dtu.dk/services/C-ImmSim-10.1/ (accessed on 12 February 2021).
- Creighton, T.E. Disulphide Bonds and Protein Stability. BioEssays 1988, 8, 57–63. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A Novel Tool to Adapt Codon Usage of a Target Gene to Its Potential Expression Host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Pandey, R.K.; Khatoon, N.; Narula, A.; Mishra, A.; Prajapati, V.K. Exploring Dengue Genome to Construct a Multi-Epitope Based Subunit Vaccine by Utilizing Immunoinformatics Approach to Battle against Dengue Infection. Sci. Rep. 2017, 7, 9232. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.K.; Bhatt, T.K.; Prajapati, V.K. Novel Immunoinformatics Approaches to Design Multi-Epitope Subunit Vaccine for Malaria by Investigating Anopheles Salivary Protein. Sci. Rep. 2018, 8, 1125. [Google Scholar] [CrossRef] [PubMed]
- Shey, R.A.; Ghogomu, S.M.; Esoh, K.K.; Nebangwa, N.D.; Shintouo, C.M.; Nongley, N.F.; Asa, B.F.; Ngale, F.N.; Vanhamme, L.; Souopgui, J. In-Silico Design of a Multi-Epitope Vaccine Candidate against Onchocerciasis and Related Filarial Diseases. Sci. Rep. 2019, 9, 4409. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wu, B.; Zhang, T.; Jia, J.; Lu, J.; Chen, Z.; Ni, Z.; Tan, T. Effect of Linker Length and Flexibility on the Clostridium Thermocellum Esterase Displayed on Bacillus Subtilis Spores. Appl. Biochem. Biotechnol. 2017, 182, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, A.; Kaushik, A.C.; Wei, D. Prediction and Validation of Potent Peptides against Herpes Simplex Virus Type 1 via Immunoinformatic and Systems Biology Approach. Chem. Biol. Drug Des. 2019, 94, 1868–1883. [Google Scholar] [CrossRef] [PubMed]
- Qamar, M.T.U.; Saba Ismail, S.A.; Mirza, M.U.; Abbasi, S.W.; Ashfaq, U.A.; Chen, L.-L. Development of a Novel Multi-Epitope Vaccine Against Crimean-Congo Hemorrhagic Fever Virus: An Integrated Reverse Vaccinology, vaccine Informatics and Biophysics Approach. Front Immunol. 2021, 12, 669812. [Google Scholar] [CrossRef]
- Mehmood, A.; Khan, M.T.; Kaushik, A.C.; Khan, A.S.; Irfan, M.; Wei, D.-Q. Structural Dynamics Behind Clinical Mutants of PncA-Asp12Ala, Pro54Leu, and His57Pro of Mycobacterium Tuberculosis Associated with Pyrazinamide Resistance. Front. Bioeng. Biotechnol. 2019, 7, 404. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, A.; Kaushik, A.C.; Wang, Q.; Li, C.-D.; Wei, D.-Q. Bringing Structural Implications and Deep Learning-Based Drug Identification for KRAS Mutants. J. Chem. Inf. Model. 2021, 61, 571–586. [Google Scholar] [CrossRef]
- Bosshard, H.R.; Marti, D.N.; Jelesarov, I. Protein Stabilization by Salt Bridges: Concepts, Experimental Approaches and Clarification of Some Misunderstandings. J. Mol. Recognit. 2004, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.; Lundegaard, C.; Buus, S.; Lund, O.; Nielsen, M. MHCcluster, a Method for Functional Clustering of MHC Molecules. Immunogenetics 2013, 65, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Vaheri, A.; LeDuc, J.W.; Kruger, D.H.; Avsic-Zupanc, T.; Arikawa, J.; Song, J.-W.; Markotić, A.; Clement, J.; Liang, M.; et al. Meeting report: Tenth international conference on hantaviruses. Antivir. Res. 2016, 133, 234–241. [Google Scholar] [CrossRef] [PubMed][Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Epitopes | Host (s) | Allergenicity | Antigenicity | Solubility | IFN | Toxicity | Virulence | |
|---|---|---|---|---|---|---|---|---|
| DMRNTIMASKTVGTA | Human | Nonallergen | 0.95 | Soluble | Positive | Nontoxic | Virulent | 1.04 |
| DTKPTDPTGIEPDDHLKERSSLRYGNVLDVNAIDIEEPSGQTADW | Vole | Nonallergen | 0.88 | Soluble | Positive | Nontoxic | Virulent | 0.99 |
| IDQKVKEISNQEPL | Human, rabbit, vole | Nonallergen | 0.84 | Soluble | Positive | Nontoxic | Virulent | 1.02 |
| NKSTLQNRRAAVS | Human Mouse | Nonallergen | 0.88 | soluble | Positive | Nontoxic | Virulent | 1.05 |
| NVLDVNAIDIEEPS | Human, Rabbit, Vole | Nonallergen | 0.75 | soluble | Positive | Nontoxic | Virulent | 1.05 |
| KEKSSLRYGNVLDVN | Mouse, | Nonallergen | 1.06 | Soluble | Positive | Nontoxic | Virulent | 1.02 |
| RNTIMASKTVGTAE | Human, rabbit, vole | Nonallergen | 0.73 | soluble | Positive | Nontoxic | Virulent | 1.03 |
| GKNIGQDRDPTGVEPGDHLKERSALSYGNTLDLNSLDID | Mouse | Nonallergen | 0.80 | soluble | Positive | Nontoxic | Virulent | 0.99 |
| VDPTGLEPDDHLK | Human Mouse | Nonallergen | 0.94 | soluble | Positive | Nontoxic | Virulent | 1.05 |
| SIDLEEPSGQTADWK | Human | Nonallergen | 0.70 | soluble | Positive | Nontoxic | Virulent | 1.05 |
| TLR4-Agonist | ||||||
| Model | GDT-HA | RMSD | MolProbity | Clash Score | Poor Rotamers | Rama Favoured |
| Initial | 1 | 0 | 3.692 | 104.6 | 4.9 | 83.6 |
| MODEL 1 | 0.9225 | 0.499 | 2.123 | 15.4 | 0.5 | 93.4 |
| MODEL 2 | 0.9254 | 0.494 | 2.184 | 15.4 | 0.5 | 91.8 |
| MODEL 3 | 0.9254 | 0.491 | 2.163 | 15.7 | 0.5 | 92.6 |
| MODEL 4 | 0.9283 | 0.471 | 2.134 | 14.1 | 0.5 | 92.2 |
| MODEL 5 | 0.9176 | 0.507 | 2.061 | 13.8 | 0.5 | 93.8 |
| β-Defensin | ||||||
| Model | GDT-HA | RMSD | MolProbity | Clash Score | Poor Rotamers | Rama Favoured |
| Initial | 1 | 0 | 3.707 | 101.6 | 5.1 | 82.7 |
| MODEL 1 | 0.8978 | 0.55 | 2.164 | 17.9 | 0 | 93.8 |
| MODEL 2 | 0.8995 | 0.54 | 2.153 | 16.7 | 0.9 | 93.4 |
| MODEL 3 | 0.8952 | 0.559 | 2.169 | 18.1 | 0.4 | 93.8 |
| MODEL 4 | 0.8918 | 0.557 | 2.164 | 17.9 | 0.9 | 93.8 |
| MODEL 5 | 0.9012 | 0.545 | 2.18 | 17.9 | 0.4 | 93.4 |
| 50S Ribosomal Protein L7/L12 | ||||||
| Model | GDT-HA | RMSD | MolProbity | Clash Score | Poor Rotamers | Rama Favoured |
| Initial | 1 | 0 | 3.449 | 98.8 | 3.7 | 89.8 |
| MODEL 1 | 0.9162 | 0.495 | 1.99 | 14.8 | 0.7 | 95.5 |
| MODEL 2 | 0.9182 | 0.492 | 2.1 | 14.8 | 1.4 | 95.5 |
| MODEL 3 | 0.9162 | 0.493 | 1.965 | 13.9 | 0.7 | 95.5 |
| MODEL 4 | 0.9195 | 0.487 | 1.96 | 13.7 | 0.3 | 95.5 |
| MODEL 5 | 0.9182 | 0.498 | 1.909 | 12 | 0.3 | 95.5 |
| TLR4−Agonist | ||||||
| Rank | Solution Number | Global Energy | Attractive VdW | Repulsive VdW | Atomic Contact Energy | Hydrogen Bond Energy |
| ↓ | ||||||
| 1 | 4 | −29.63 | −44.67 | 41.32 | 4.84 | −5.12 |
| 2 | 7 | 6.71 | −15.14 | 12.21 | −1.23 | −1.20 |
| 3 | 3 | 16.63 | −23.10 | 24.78 | 8.02 | −2.98 |
| 4 | 8 | 30.40 | −17.64 | 22.04 | 12.26 | −2.67 |
| 5 | 2 | 79.31 | −8.74 | 81.87 | 2.58 | −0.84 |
| 6 | 10 | 83.47 | −49.20 | 155.60 | 15.30 | −4.87 |
| 7 | 9 | 107.83 | −42.49 | 146.03 | 17.89 | −5.37 |
| 8 | 5 | 121.17 | −32.38 | 193.07 | 6.66 | −2.80 |
| 9 | 1 | 432.16 | −30.72 | 570.71 | 4.75 | −2.82 |
| 10 | 6 | 4149.68 | −63.85 | 5310.77 | 8.60 | −4.70 |
| β−Defensin | ||||||
| Rank | Solution Number | Global Energy | Attractive VdW | Repulsive VdW | Atomic Contact Energy | Hydrogen Bond Energy |
| ↓ | ||||||
| 1 | 9 | −3.41 | −5.43 | 1.69 | 2.98 | −2.97 |
| 2 | 7 | 10.42 | −3.55 | 0.40 | 1.50 | 0.00 |
| 3 | 2 | 10.51 | −58.47 | 80.50 | 20.95 | −10.00 |
| 4 | 4 | 31.11 | −3.73 | 0.00 | 4.15 | 0.00 |
| 5 | 6 | 364.44 | −51.20 | 547.20 | 5.50 | −10.07 |
| 6 | 1 | 615.86 | −53.65 | 872.84 | 4.94 | −1.23 |
| 7 | 10 | 918.64 | −57.19 | 1206.60 | 8.00 | −9.37 |
| 8 | 3 | 1371.21 | −72.88 | 1860.69 | 4.42 | −14.33 |
| 9 | 5 | 1617.02 | −83.97 | 2179.88 | 17.45 | −18.53 |
| 10 | 8 | 4445.60 | −95.90 | 5699.42 | 15.36 | −13.35 |
| 50S Ribosomal Protein L7/L12 | ||||||
| Rank | Solution Number | Global Energy | Attractive VdW | Repulsive VdW | Atomic Contact Energy | Hydrogen Bond Energy |
| ↓ | ||||||
| 1 | 9 | −11.03 | −9.67 | 7.15 | 0.93 | −1.31 |
| 2 | 4 | −0.43 | −3.27 | 1.17 | 2.48 | −1.33 |
| 3 | 5 | 2.61 | −4.54 | 6.50 | 2.57 | −0.73 |
| 4 | 7 | 13.94 | −10.63 | 5.23 | 2.94 | −0.45 |
| 5 | 10 | 18.08 | −1.80 | 0.00 | 3.89 | −0.95 |
| 6 | 2 | 54.13 | −39.14 | 116.61 | 8.41 | −2.65 |
| 7 | 8 | 108.38 | −40.95 | 195.97 | 11.18 | −6.06 |
| 8 | 3 | 192.03 | −16.85 | 216.91 | 14.88 | −2.43 |
| 9 | 1 | 3011.31 | −37.19 | 3794.70 | 6.44 | −3.85 |
| 10 | 6 | 6854.97 | −119.49 | 8726.07 | 23.31 | −21.54 |
| MMGBSA | MMPBSA | ||||||
|---|---|---|---|---|---|---|---|
| TLR4-Agonist Vaccine with TLR4 | |||||||
| Energy Component | Average | Std Dev | Err. of Mean | Energy Component | Average | Std Dev | Err. of Mean |
| Van der Waals Energy | −220.7 | 35.5 | 3.5 | Van der Waals Energy | −220.7 | 35.5 | 3.5 |
| Electrostatic Energy | 1798.2 | 82.3 | 8.2 | Electrostatic Energy | 1798.2 | 82.3 | 8.2 |
| Gas-Phase Energy | 1577.5 | 55.4 | 5.5 | Gas-Phase Energy | 1577.5 | 55.4 | 5.5 |
| Solvation Energy | −1628.4 | 56.6 | 5.66 | Solvation Energy | −1615.3 | 54.0 | 5.4 |
| Total | −1628.4 | 13.3 | 1.33 | Total | −37.7 | 15.9 | 1.5 |
| β-Defensin Vaccine with TLR4 | |||||||
| Van der Waals Energy | −90.2 | 17.1 | 1.7 | Van der Waals Energy | −90.2 | 17.1 | 1.7 |
| Electrostatic Energy | 790.7 | 74.6 | 7.4 | Electrostatic Energy | 790.7 | 74.6 | 7.4 |
| Gas-Phase Energy | 700.4 | 59.4 | 5.9 | Gas-Phase Energy | 700.4 | 59.4 | 5.9 |
| Solvation Energy | −710.29 | 53.6 | 5. | Solvation Energy | −742.8 | 52.5 | 5.2 |
| Total | −9.8 | 8.1 | 0.8 | Total | −42.3 | 10.3 | 1.0 |
| 50S Ribosomal Protein L7/L12 Vaccine with TLR4 | |||||||
| Van der Waals Energy | −361.1 | 35.6 | 3.5 | Van der Waals Energy | −361.1 | 35.6 | 3.5 |
| Electrostatic Energy | 1723.3 | 52.5 | 5.2 | Electrostatic Energy | 1723.3 | 52.5 | 5.2 |
| Gas-Phase Energy | 1362.1 | 45.7 | 4.5 | Gas-Phase Energy | 1362.1 | 45.7 | 4.5 |
| Solvation Energy | −1556.8 | 48.6 | 4.8 | Solvation Energy | −1512.8 | 72.3 | 7.2 |
| Total | −194.6 | 40.9 | 4.0 | Total | −150.6 | 65.6 | 6.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ismail, S.; Abbasi, S.W.; Yousaf, M.; Ahmad, S.; Muhammad, K.; Waheed, Y. Design of a Multi-Epitopes Vaccine against Hantaviruses: An Immunoinformatics and Molecular Modelling Approach. Vaccines 2022, 10, 378. https://doi.org/10.3390/vaccines10030378
Ismail S, Abbasi SW, Yousaf M, Ahmad S, Muhammad K, Waheed Y. Design of a Multi-Epitopes Vaccine against Hantaviruses: An Immunoinformatics and Molecular Modelling Approach. Vaccines. 2022; 10(3):378. https://doi.org/10.3390/vaccines10030378
Chicago/Turabian StyleIsmail, Saba, Sumra Wajid Abbasi, Maha Yousaf, Sajjad Ahmad, Khalid Muhammad, and Yasir Waheed. 2022. "Design of a Multi-Epitopes Vaccine against Hantaviruses: An Immunoinformatics and Molecular Modelling Approach" Vaccines 10, no. 3: 378. https://doi.org/10.3390/vaccines10030378
APA StyleIsmail, S., Abbasi, S. W., Yousaf, M., Ahmad, S., Muhammad, K., & Waheed, Y. (2022). Design of a Multi-Epitopes Vaccine against Hantaviruses: An Immunoinformatics and Molecular Modelling Approach. Vaccines, 10(3), 378. https://doi.org/10.3390/vaccines10030378