Abstract
Antibiotic resistance (AR) is the resistance mechanism pattern in bacteria that evolves over some time, thus protecting the bacteria against antibiotics. AR is due to bacterial evolution to make itself fit to changing environmental conditions in a quest for survival of the fittest. AR has emerged due to the misuse and overuse of antimicrobial drugs, and few antibiotics are now left to deal with these superbug infections. To combat AR, vaccination is an effective method, used either therapeutically or prophylactically. In the current study, an in silico approach was applied for the design of multi-epitope-based vaccines against Providencia rettgeri, a major cause of traveler’s diarrhea. A total of six proteins: fimbrial protein, flagellar hook protein (FlgE), flagellar basal body L-ring protein (FlgH), flagellar hook-basal body complex protein (FliE), flagellar basal body P-ring formation protein (FlgA), and Gram-negative pili assembly chaperone domain proteins, were considered as vaccine targets and were utilized for B- and T-cell epitope prediction. The predicted epitopes were assessed for allergenicity, antigenicity, virulence, toxicity, and solubility. Moreover, filtered epitopes were utilized in multi-epitope vaccine construction. The predicted epitopes were joined with each other through specific GPGPG linkers and were joined with cholera toxin B subunit adjuvant via another EAAAK linker in order to enhance the efficacy of the designed vaccine. Docking studies of the designed vaccine construct were performed with MHC-I (PDB ID: 1I1Y), MHC-II (1KG0), and TLR-4 (4G8A). Findings of the docking study were validated through molecular dynamic simulations, which confirmed that the designed vaccine showed strong interactions with the immune receptors, and that the epitopes were exposed to the host immune system for proper recognition and processing. Additionally, binding free energies were estimated, which highlighted both electrostatic energy and van der Waals forces to make the complexes stable. Briefly, findings of the current study are promising and may help experimental vaccinologists to formulate a novel multi-epitope vaccine against P. rettgeri.
1. Introduction
Antibiotic resistance (AR) is the defense mechanism pattern in microbes, and it occurs when bacteria, fungi, or viruses evolve over some time, such that it protects the microorganism against antibiotics adapts itself to the environmental conditions [1]. Antibiotics are medicines used to treat bacterial infections. All commercially available antibiotics are becoming ineffective, as multi-drug resistant strains of microbes are spreading worldwide, leading to bacterial and fungal diseases with less-effective treatments [2]. This phenomenon has mainly sped up due to misuse and overuse of antibiotics [3]. AR is an alarming global challenge associated with a high mortality and morbidity rate in humans and animals [4]. Antimicrobial resistance (AMR) is becoming difficult to treat with currently available antibiotics due to the high level of genetic diversity in microbial species [5]. The excessive use of antibiotics in humans and animal medicines, agriculture, and the environment has led to AR in bacteria, which has significantly contributed to high hospital and community mortality and mobility [6]. AR is the outcome of the bacterial evolution process, making itself fit to changing environmental milieu in the quest for survival of the fittest [7]. The AR is increasing and together with poor infection-controlled clinical practices, the resistant genetic determinants are spreading fast to non-AR microbes as well as to the environment [8]. Therefore, there is a dire need to devise new strategies for effective management of AR pathogens [3,9,10,11].
Vaccines are a biological preparation that help in generating acquired immunity against any given pathogen or pathological condition [12]. Vaccines consist of either the whole microorganism or a byproduct of the microbes, e.g., toxins, etc. [13]. A vaccine is typically made from the inactivated, killed, or weakened toxins or surface proteins of the infectious agent that have only the ability to provoke the immune response of the host [14]. A potential vaccine candidate taken for the preparation of an efficient vaccine must fulfill the criteria of certain parameters, e.g., the vaccine candidates should be highly antigenic, conserved, and non-homologous to the host proteome and its normal flora (which are essential for microbe survival) and should it be easily recognizable by the immune cells of the host [15]. An effective and safe vaccine discovery against poliovirus by Salk and Sabin was carried out by Pasteur’s vaccinology concept [16]. Pasteur’s rule of vaccines led to the development of the BCG vaccine against Mycobacterium tuberculosis [17], along with the vaccine against mumps, measles, and rubella [18]. However, such vaccinology is a failure for pathogens that are unable to be cultured or grown in the laboratory [19]. In addition, culture-based developed vaccines exhibit antigenic variability [20]. Particularly, Pasteur vaccinology limitations have surfaced in the cases of Neisseria meningitides and Mycobacterium leprae. Subunit vaccines that target cellular components contain virulence factors [21], as can be exemplified by the pertussis vaccine [22], outer membrane Meningococcal Vesicle (OMV)-containing protein, PorA/porin, and pneumococcal polysaccharide vaccine (PPSV23) [23]. A potential vaccine candidate taken for the preparation of an efficient vaccine must fulfill the criteria of certain parameters, e.g., the vaccine candidates should be highly antigenic, conserved, and non-homologous to the host normal flora and proteome (which are essential for microbe survival) and should be easily recognizable by the immune cells of the host [15].
To combat AR and to eliminate this alarming global health issue, both therapeutically and prophylactically, the introduction of novel approaches remains vital [24]. Boosting host immunity via immunotherapeutic and immunological interventions among various approaches proposed by the National Institute of Allergy and Infectious Diseases (NIAID) is an attractive solution to fight the challenges of AR [25]. Reverse vaccinology (RV) is an up-to-date approach and is used to identify putative surface-associated proteins without the need to culture the microorganisms [26]. Through immunotherapy, pathogen-specific antibodies can be designed and produced; meanwhile, immunological interventions in the human immune system are trained to tackle bacterial infections by acquiring adaptive immunity [27]. The latter strategy, using vaccines in particular, holds great importance in lowering the burden of AR [28]. Conventional vaccine development is expensive, time consuming, and needs many human resources [21]. The genomic revolution will greatly aid in disclosing new vaccine candidates that, by traditional vaccine development, are hard to detect [29]. Next-generation sequencing of bacterial pathogens and advanced bioinformatics practices in vaccinology are now commonly employed for the identification of putative surface-associated antigens [30]. The meningococcal serogroup B (4CMenB) vaccine was effectively developed using the RV approach [31]. Pan-genomic reverse vaccinology (PGRV), specifically, is more effective compared to conventional RV, as it screens highly conserved targets that are strain specific [32]. For example, the genome of Streptococcus agalactiae revealed four protective antigens identified via the PGRV approach [6].
Providencia is a group of opportunistic urease-producing Gram-negative, motile, and rod-shaped bacteria belonging to the Enterobacteriaceae family. P. rettgeri can cause a variety of hospital-acquired infections, i.e., urinary tract infections, wounds, human blood infections, gastroenteritis, and bacteremia [33] P. rettgeri is ubiquitous in the environment. It may exist in both land and water habitats and may therefore be present in water, soil, and land, most necessarily in hospital and nursing vicinities [34]. Providencia shows resistance to several antibiotics and can cause several types of hospital-associated infections with high mortality and morbidity rates [35]. P. rettgeri is resistant to commercially available antibiotics such as ampicillin, polymyxins, first-generation cephalosporins [36], and gentamicin, along with tobramycin, ciprofloxacin, levofloxacin [37], carbapenem, polymyxins, and tigecycline [38]. Additionally, it is resistant to amikacin [39]. Compared to other bacterial species such as Klebsiella pneumonia and Acinetobacter baumannii, P. rettgeri is naturally resistant to tigecycline and colistin [39]. According to previous literature, about 86% of the isolates of P. rettgeri were found to be resistant to amikacin antibiotics; in addition, 71% of the clinical isolates were resistant to gentamycin drugs [34]. Furthermore, metallo-β-lactamase-1 was noted as NDM-1. Extended-spectrum beta-lactamases and amoxicillin-clavulanate resistivity patterns have also been reported [40]. The outcome of this study will provide a ready-to-use multi-epitope peptide vaccine for experimentalists to evaluate its real immune protection ability in animal models. This will not only enrich the vaccine antigens against P. rettgeri but will also speed up the vaccine development process without consuming much cost and technical expertise. From a clinical usefulness perspective, the vaccine will likely provide protection against all sequenced strains of the pathogen, as it is based on the core genome. The vaccine is composed of safe antigens and will be less risky compared to other types of vaccine. Furthermore, such vaccines are easy to produce and can be inserted into multiple carrier systems. These findings will also lead in the development of economically cheaper vaccines, will aid in stopping the spread of AR bacterial strains and will thus be useful in saving millions of lives.
2. Research Methodology
The methodology flow that is used in the design of the multi-epitope vaccine against P. rettgeri is mentioned in Figure 1.
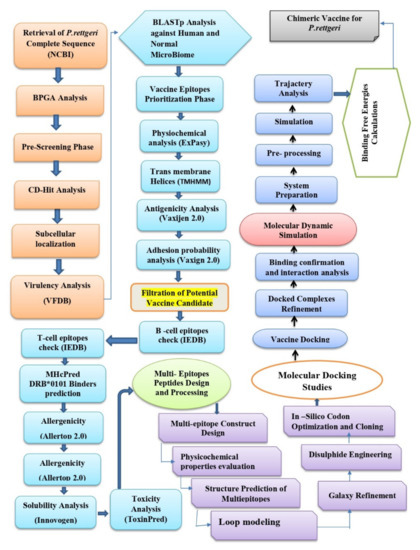
Figure 1.
Overall flow of methodology that was used in the design of the multi-epitope vaccine against P. rettgeri.
2.1. P. rettgeri Complete Proteome Retrieval
This research commenced with the retrieval of complete proteomes of P. rettgeri. The bacteria have a total of 14 strains, and their genomic/proteomic data were retrieved from the genome database of the National Center For Biotechnology Information (NCBI) database [41]. All the respective proteomes and genomes were retrieved in FASTA format [42].
2.2. BPGA Analysis
Bacterial pan-genome analysis is a bioinformatics approach mainly used for the retrieval of core, unique and accessory genes. Herein, the bacterial pan-genome analysis was considered to investigate the pan-genome of the pathogen [19]. Through the one-click analysis technique of the BPGA tool [43], all 14 strains of P. rettgeri were analyzed for core genome (conserved), along with accessory genes (dispensable) and unique genes (strain-specific genes) [44].
2.3. Pre-Screening Phase
The pre-screening phase was considered as the filtering phase/subtractive proteomic phase in which several potential antigenic targets were determined in the core genome of the pathogen and were prioritized as the potential vaccine candidates. This phase consisted of CD-HIT analysis [45], homology check, essential check, and surface localization check [23].
2.4. CD-Hit Analysis (Cluster Data with High Identity and Tolerance)
The bacterial core genome mostly consists of redundant and non-redundant proteins. Redundant proteins have double representation in the core proteome, which is not important for further processing in the design of the vaccine candidate. To remove all the redundant proteins, the CD-Hit-h analysis approach was utilized. Furthermore, the non-redundant proteins were considered for further processing [46].
2.5. Subcellular Localization Analysis
Surface-localized proteins can be easily recognized by the host immune system, and these surface-localized proteins are mainly involved in the pathogenicity of infection. Hence, due to being pathogenic in nature and surface localization, these proteins were prioritized as vaccine candidates. This task was achieved through PSORTb analysis [47].
2.6. Virulent Protein Analysis
The virulent factor database (VFDB) is a dataset for bacterial and fungal virulence factors. In virulent factor analysis, the prioritized subcellular localized proteins were examined for virulence through Basic Local Alignment Search (BLASTp) against the VFDB full proteomic dataset [48]. The cut-off values kept for prioritization were: bit score ≥ 100 and sequence identity ≥30 percent. The proteins that failed to fulfill the noted criteria were discarded [49]
2.7. BLASTp Analysis against Humans and MicroBiome
The BLASTp tool was used to check the homology of filtered virulent proteins against human normal flora and the proteome. The homology check was performed to prevent the chances of autoimmune responses by the host against self-antigens. In this analysis, similar to the selected proteins, it was checked with humans and three normal intestinal flora; Lactobacillus rhamnosus (taxid: 47715), L. johnsonii (taxid: 33959), and L. casei (taxid: 1582) [50]. The cut-off sequence identity and E values used were at ≤30% and 10−4, respectively.
2.8. Vaccine Epitopes Prioritization Phase
Vaccine candidate prioritization is the proceeding step for designing a multi-epitope vaccine. In this phase, the shortlisted proteins were further subjected to physicochemical analysis, transmembrane helices analysis, antigenicity, allergenicity, adhesion probability, and B-cell and T-cell epitopes prediction [51].
2.9. Physiochemical Analysis
Physiochemical analysis of the shortlisted proteins was performed for properties such as instability index, atomic composition, amino acid composition, theoretical PI, molecular weight, aliphatic index, estimated half-life, and grand average of hydropath city (GRAVY). This task was achieved with the help of an online web server, ProtParam [52]. The protein was identified as a good vaccine target when the cut-off value of the protein molecular weight was <110 kDa and when the instability index was <40 [53]. All the unstable proteins were excluded from the study, and stable proteins were subjected to further analysis.
2.10. Transmembrane Helices
HMMTOP 2.0 and TMHMM 2.0 tools were used to check for transmembrane helices [54]. The cut-off values for the number of transmembrane helices were 0 or 1, and proteins exceeding these values were discarded. The proteins were regarded as good vaccine candidates only when the transmembrane helices were less than the threshold, as such proteins can be easily handled in experimental studies. The selected proteins were further processed in downward analysis [55].
2.11. Antigenicity Prediction
Antigenicity is the capability of foreign antigens to bind immune cells and generate proper immune responses. Antigenicity prediction was performed through an online Vaxijen 2.0 webserver (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html, accessed on 18 June 2021) [16]. To prioritize potential vaccine candidates, ≥0.4 cut-off value was considered [56]. The higher the antigenicity of the vaccine construct is, the stronger the chances of host–immune system provocation [26].
2.12. Adhesion Probability Analysis
The adhesion probability was the next step followed for selecting and prioritizing proteins that had the ability of adhesion. The threshold value to prioritize good adhesive candidates was >0.5 [57]. This ensures active attachment of the designed vaccine with the host immune cells and ensures robust host immunity. The adhesion and binding with the host immune system developed adaptive immunity, inducing both antibodies and TCR (T-cell receptors). This task was achieved using an online Vaxign webserver [58].
2.13. Epitopes Prediction Phase
In the epitopes prediction phase, both B- and T-cell epitopes were predicted through the online immune–epitope database (IEDB) tool [59]. The IEDB covers a wide-ranging collection of immune epitopes and can be easily accessed. First, B-cell epitopes were predicted and B-cell epitopes were further used for T-cell epitope prediction [60]. The predicted epitopes were prioritized based on low percentile rank. Furthermore, through the MHcPred tool, binding potency of the predicted epitopes with DRB*10101 alleles was analyzed [61,62]. Finally, in epitope prediction, shortlisted epitopes were subjected again to antigenicity, virulence, toxicity, and water solubility analysis through Vaxijen 2.0, Virulentpred, ToxinPred, and ProteinSol, respectively [63].
2.14. Multi-Epitope Peptide Designing and Processing
Multi-epitope-based vaccines consist of several antigenic epitopes [64]. In the multi-epitope design phase, all the screened epitopes were linked via GPGPG linkers to construct a multi-epitope-based vaccine. The designed vaccine construct was further linked to cholera toxin B subunit adjuvant to enhance immunogenicity of the vaccine.
2.15. Physiochemical Properties of the Final Vaccine Construct
The physiochemical properties such as number of amino acids, molecular weight, theoretical PI value, instability index, aliphatic index, and grand average of hydropathicity (GRAVY) were analyzed [53]. This task was achieved through an online webserver, ProtParam [65].
2.16. Structure Modeling of the Final Vaccine Construct
The 3D structure of the vaccine construct was modeled using the 3Dpro tool to predict the most stable vaccine structure, as it is essential for molecular recognition. The 3D structure was checked to ensure the construct’s stability and durability [19].
2.17. Loops Modeling
The vaccine was passed through Galaxy Loop of the GalaxyWeb server to remove the unnecessary loops from the vaccine construct [66]. This was vital to obtain the most suitable vaccine 3D structure.
2.18. Galaxy Refinement
The final loop-modeled vaccine construct was further analyzed in GalaxyRefine of GalaxyWeb server. The vaccine was reconstructed for its side chains, and steric clashes were removed. The refined vaccine construct was considered as a good and potential vaccine candidate [67].
2.19. Disulfide Engineering
To enhance stability of the vaccine, disulfide engineering was performed. The stability of the vaccine was enhanced by reducing the conformational energy of the twisted and folded structures. For enhanced and improved stability, both inner and outer chain bonding was examined computationally. The Design 2.0 webserver was used for disulfide engineering [68].
2.20. Codon Optimization
The Java Codon Adaptation Tool (JCat) server was used to convert the multi-epitope vaccine sequence into DNA and was then cloned in the Escherichia coli expression system. The vaccine maximum expression is in the E. coli expression system and was calculated with the codon adaptation index (CAI) and its GC percentage value. The value of 1 was estimated as an ideal value for the said expression system [69].
2.21. Molecular Docking
Molecular docking studies were applied in order to check the binding efficacy of the designed vaccine construct with different types of immune cell receptors. A blind docking approach was used to predict the binding affinity of the vaccine to TLR-4 (4G8A), MHC-I (1I1Y), and MHC-II (1KG0) receptors retrieved from PDB. The docking study was performed through an online web server of PatchDock [70]. The docking works on the following principles: sample conformation and function scoring. The docked complexes were then refined via the FireDock server [71].
2.22. Molecular Dynamic Simulation
Molecular dynamics simulation (MDS) is a computational approach to predict the movement of molecules and atoms. In MDS, the molecules and atoms interact for a specific period. MDS of the vaccine with innate immune receptors was carried out using AMBER 20 [72]. The FF14SB was used as a force field, while TIP3P water box (12 Angstrom is size) was used for vaccine–receptor complex submersion. The SHAKE algorithm was used for constraining hydrogen bonds. The complexes were heated and equilibrated, and then a production run was performed for 250 ns. CCPTRAJ was used for trajectory analysis.
2.23. Binding Free Energies Calculation
Additionally, the intermolecular affinity of the vaccine–receptor complex was validated through the AMBER MMPBSA.py module [73]. The binding free energies demonstrate and ensure the system stability, and the lesser the energy is, the more stable the vaccine construct. [74]. In total, 100 frames were analyzed during this analysis.
2.24. C-Immune Simulation
C-ImmSim server was used to decipher host–immune responses to the designed vaccine [75]. This server was used for antigen characterization and its profiling to calculate immune responses of a host, i.e., human. The server was used to run the process of simulation for three separate mammalian organs, i.e., lymph nodes, thymus, and bone marrow, while the parameters used for the process of the simulation were by default [76].
3. Results
AR in bacterial species is becoming a worldwide public health concern [77]. One example is the rapid emergence of AR in P. rettgeri, resulting in pathogenic hospital-associated infections [78]. As no licensed vaccine is available against the pathogen, computational vaccine design is an alternative approach to speed up vaccine development [79]. In the current research study, we have designed a computational-based multi-epitope recombinant vaccine against P. rettgeri.
3.1. Providencia rettgeri Complete Proteome Retrieval
In the current research, the complete proteome of 14 strains of P. rettgeri was retrieved from the NCBI databases (https://www.ncbi.nlm.nih.gov/) (accessed on 4 April 2021), followed by a pan-genome analysis.
3.2. Bacterial Pan Genome Analysis
The total proteome size of P. rettgeri is 13,580 proteins, as mentioned in Figure S1, while all 14 bacterial strains and their genome size are figuratively represented and presented in Figure 1. Due to genome plasticity, there are high chances of gaining new genes over time because the pathogen genomes are open, which is indicated in the core-pan plot. Moreover, the core proteins are typically involved in metabolic regulation in addition to metabolic biogenesis, which has been tested by the COG distribution analysis [80]. The storage and processing of information genes are present, mainly in unique proteins set. The processing of RNA and the process of replication, transcription and translation of recombination genes are all part of the core genome. Likewise, the pan-phylogeny tree of all 14 P. rettgeri is shown in Figure S1B.
3.3. CD-HIT Analysis
The CD-hit analysis was accomplished for the retrieval of core proteomes without duplicate sequences [81]. The core proteome of P. rettgeri comprised 926 non-redundant proteins, while 12,654 were found to be redundant proteins, as shown in Figure 2. The redundant protein sequences were discarded from the study because they were not required in the vaccine development process due to repeating copies of the same proteins, while non-redundant core proteins were further used in the subcellular localization phase and virulent analysis [82].
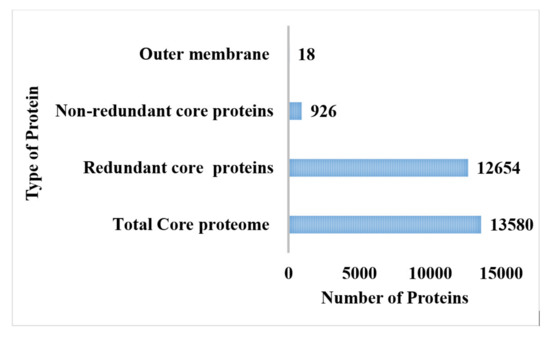
Figure 2.
Number of the total proteome, core proteome, and redundant, non-redundant, and virulent proteins.
3.4. Subcellular Localization
The surface or membrane proteins are easily recognized by the host immune system; hence, potential immune responses are generated when these proteins are used in the design of a vaccine. Subcellular localization analysis was performed as an essential check for surface proteins [83]. In the non-redundant core proteome, seven were found to be extracellular, 18 were in the outer membrane, and 25 periplasmic membrane proteins were identified in the subcellular localization analysis.
3.5. VFDB Analysis
Virulent proteins are mainly involved in the pathogenesis of infectious pathogens [48]. In VFDB analysis, these protein sequences were considered as virulent, which fulfilled the bit score >100 percent with the sequence identity of higher than 30% [23]. Among the 50 subcellular localized proteins, only 13 protein sequences were found as virulent.
3.6. Human and Normal Flora, Adhesion Probability, Physiochemical Property Analysis
To avoid the autoimmune response, all the virulent protein sequences were further checked for homology analysis against human and normal flora proteomes [84], and here in this study, only two proteins have shown homology with humans, and two protein sequences have shown similarities with normal flora of host and were hence discarded. The remaining non-similar proteins were subjected to transmembrane helices check, which is essential for feasibility in experimental evaluation [85]. All the sequences that had more than one transmembrane helix were removed, while those that had 0 or 1 transmembrane helices were considered for further analysis [86]. In this step, only one sequence was discarded due to not fulfilling the above criteria. Proteins with more than one transmembrane that showed similarity to humans and their microbiota and that were non-adhesive were discarded. Furthermore, the proteins were predicted to be stable when their instability index was less than 40 with a molecular weight of less than 100 kDa. Physiochemical properties of the filtered protein sequences are mentioned in Table 1. Finally, among the eight filtered proteins, only two proteins were discarded in the adhesion probability check. Concluding, among the total 13 virulent sequences, seven were discarded in the homology check with the host and normal flora proteome, the transmembrane helices check, and the adhesion probability check, as shown in Figure 3.

Table 1.
Physiochemical properties of shortlisted proteins. Molecular weight (MW), isoelectric point (PI).
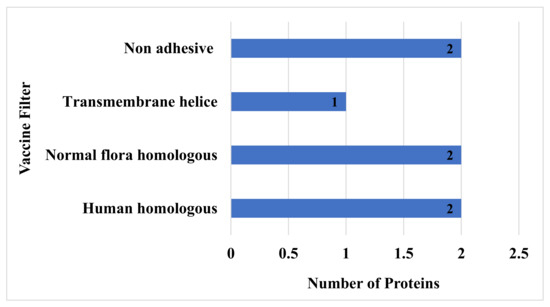
Figure 3.
Number of non-adhesive normal flora and homologous proteins that have more than one transmembrane helix.
3.7. Vaccine Epitopes Prioritization Phase
In the current study, all six prioritized proteins, which were filtered from the above steps, and checks were subjected to the epitope prioritization phase. In the epitope prioritization phase, both B- and T-cell epitopes were predicted to generate B- and T-cell immune responses [87].
3.8. B-Cell Epitopes Prediction
An immune response that is dependent on antibodies may also be referred to as humoral immunity; after stimulation, B-cells convert into plasma cells [88]. From six shortlisted proteins: fimbrial protein, flagellar hook protein (FlgE), flagellar basal body L-ring protein (FlgH), flagellar hook-basal body complex protein (FliE), flagellar basal body P-ring formation protein (FlgA) and Gram-negative pili assembly chaperone domain protein, B-cell epitopes were predicted, as tabulated in Table S1.
3.9. T-Cell Epitopes Prediction
The function of the T-cell epitopes is mainly to generate a cellular immune response. T-cell-dependent immunity is also known as cellular immunity. The resultant multiplication and differentiation of T-cell lymphocytes as an outcome of recognizing peptide antigens is to develop the primary immune response [89]. B-cell-derived T-cell epitopes that have the ability to activate the cellular immune response were predicted using B-cell epitopes generated from T-cell epitopes, and therefore MHC-I and MHC-II epitopes were recognized on the basis of lowest percentile scores [90]. The following MHC-I subset alleles are: HLA-A*01:01, HLA-A*01:01, HLA-A*02:01, HLA-A*02:01, HLA-A*02:03, LA-A*02:03, HLA-A*02:06, HLA-A*02:06, HLA-A*03:01, HLA-A*03:01, HLA-A*11:01, HLA-A*11:01, HLA-A*23:01, HLA-A*23:01, HLA-A*24:02, HLA-A*24:02, HLA-A*26:01, HLA-A*26:01, HLA-A*30:01, HLA-A*30:01, HLA-A*30:02, HLA-A*30:02, HLA-A*31:01, HLA-A*31:01, HLA-A*32:01, HLA-A*32:01, HLA-A*33:01, HLA-A*33:01, HLA-A*68:01, HLA-A*68:01, HLA-A*68:02, HLA-A*68:02, HLA-B*07:02, HLA-B*07:02, HLA-B*08:01, HLA-B*08:01, HLA-B*15:01, HLA-B*15:01, HLA-B*35:01, HLA-B*35:01, HLA-B*40:01, HLA-B*40:01, HLA-B*44:02, HLA-B*44:02, HLA-B*44:03, HLA-B*44:03, HLA-B*51:01, HLA-B*51:01, HLA-B*53:01, HLA-B*53:01, HLA-B*57:01, HLA-B*57:01, HLA-B*58:01, HLA-B*58:01; and MHC-II alleles are: HLA-DRB1*01:01, HLA-DRB1*03: *04:01, HLA-DRB101,HLA-DRB1*04:05, HLA-DRB1*07:01, HLA-DQA1*03:01/DQB1*03:02, HLADQA1*03:01/DQB1*03:02, HLADQA1*01:02/DQB1*06:02, HLADPA1*02:01/DPB1*01:01, HLADPA1*01:03/DPB1*04:01, HLADPA1*03:01/DPB1*04:02, HLADPA1*02:01/DPB1*05:01, HLADPA1*02:01/DPB1*14:01. MHC-I and MHC-II molecules of the T-cell are tabulated in following Table S2.
3.10. Epitope Prioritization Phase
In the epitope analysis and prioritization phase, all the predicted B- and T-cell shortlisted epitopes were next subjected to further analyses, such as DRB*0101 binding affinity [91] followed by allergenicity, solubility, and toxicity analyses [92].
3.11. DRB*0101 Binding Analysis
Vaccine binding affinity to host immune cell receptors is important for a proper immune response. In the DRB*0101 analysis, all selected epitopes were further checked for the potential of binding with the HLA DRB*0101 allele [93,94]. Only those epitopes of IC50 values <100 nM for DRB*0101 alleles were selected, as they represent strong binding [95]. The shortlisted epitopes whose values are less than the above-mentioned threshold are listed in Table 2.
Table 2.
List of probable antigenic, good water-soluble, non-toxigenic, and non-allergic DRB*0101 binding affinity epitopes.
3.12. Antigenicity, Allergenicity, Solubility, and Toxicity Analysis of Selected Epitopes
Only antigenic proteins can stimulate host immune responses [73]. To achieve this task, only antigenic proteins were included in the study, and all probable non-antigenic protein sequences were excluded. To avoid allergic and toxic responses, allergenicity and toxicity analyses were performed for removal of all toxic and allergic protein sequences, as well as poor water soluble epitopes [92,96]. To achieve solubility prediction, an online webserver InvivoGen was used, which is available at https://www.invivogen.com/ova-peptide and accessed on 7 June 2021. All shortlisted probable antigenic, non-allergic, nontoxic, and good water-soluble peptides are mentioned in Table 2.
3.13. Multi-Epitope Vaccine Construction and Processing
A multi-epitope-based vaccine construct consists of different types of epitopes, rather than a single epitope. The vaccine construct is designed by linking screened epitopes with each other through specific linkers, i.e., GPGPG linkers for the purpose of overcoming the limitations of single-peptide-based vaccines that are unable to generate effective immune responses against variants of the same pathogen [97]. Another linker, i.e., EAAAK, is used to link the adjuvant CTBS, to enhance the immune efficacy of the vaccine construct [98]. These specific linkers are used because they are rigid and allow for the separation of epitopes that have efficiently been recognized by the immune system [99]. Consequently, safe, robust, and efficient immune responses are generated against the designed vaccine [64]. The designed multi-epitope vaccine construct is mentioned in Figure 4. The amino acid length of the designed vaccine is 264.
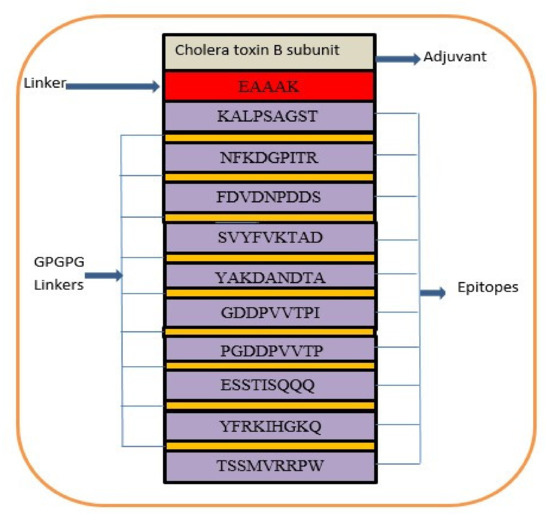
Figure 4.
Schematic presentation of final vaccine epitope construct.
3.14. Structure Modeling
The three-dimensional structure of the vaccine was modeled using 3Dpro [100], as shown in Figure 5. The structure modeling was performed ab initio rather than by homology-based or threading because no appropriate template structure was available.
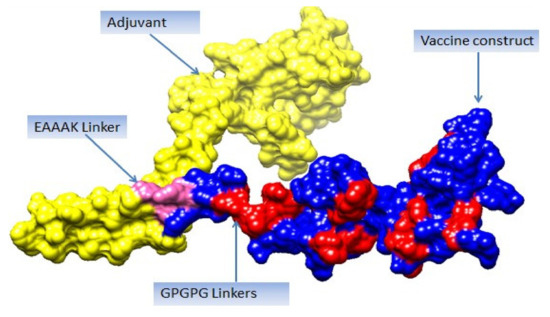
Figure 5.
Vaccine 3D structure. Yellow color represents adjuvant (cholera toxin B subunit), blue color shows vaccine construct, and pink color represents EAAAK linker, while red color represents GPGPG linkers.
3.15. Loops Modeling and Refinement
Structure stability is necessary for a good vaccine candidate [101]. To avoid structure instability, all the present loops in the vaccine candidate were modeled for the following residues: Met1-Leu4, Ala19-Gly21, Csy30-Ile38, Ser51-Asn65, Gly66-Val73, Glu100-Asn111, Leu132-Thr151, Arg152-Gly171, Ser172-Asn191, Asp192-Gly211, Pro212-Pro226 and Ser230-Gln235, Gln236-Gly255, and Thr256-Trp264.
3.16. Disulfide Engineering
For structure stability, disulfide engineering was performed [102]. The covalent bonds ensure protein structure stability, and therefore, the geometry of the construct remains intact [103]. Moreover, some amino acid residues are sensitive to enzyme degradation. Hence, all the enzyme-degradable amino acid residues were replaced with cysteine residues, which are shown as yellow sticks in Figure 6B [104].
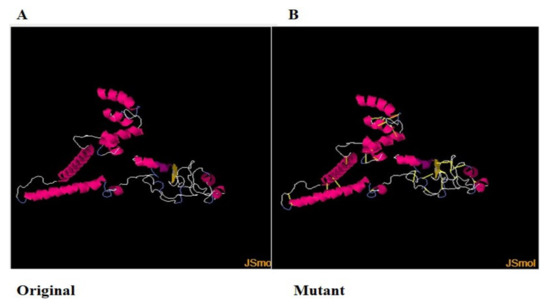
Figure 6.
(A). Original wild structure of the vaccine construct and (B) mutated structure of the vaccine. The yellow sticks are the disulfide bonds introduced via disulfide engineering.
3.17. Codon Optimization
Codon optimization is a specific genetic engineering technique that makes sure the codon optimization of the construct is consistent with the host immune usage pattern for maximum production and expression of proteins. The codon adaptation index threshold value is 0.92 and GC content is 57.08%. The values of CAI are estimated to be ideal for maximum expression and production of proteins [105,106,107]. Finally, pET-28a (+) expression vector was used to optimize the expression of the vaccine. The in silico cloned vaccine construct is shown in Figure 7.
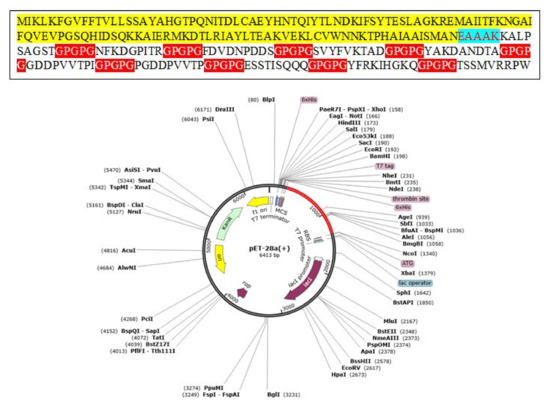
Figure 7.
Cloning of multi-epitope vaccine constructs computationally into pET28a (+) vector. The vaccine is shown in red color.
3.18. Molecular Docking
Interaction of the designed vaccine construct with both innate and adaptive immune cells of the host is imperative for activation of efficient cellular and humoral immunity. Therefore, a docking study was performed to predict the binding affinity of the vaccine with host immune receptors [108]. A blind docking approach was carried out to evaluate the binding affinities with MHC-I (PDB ID:1L1Y) and MHC-II (1KG0) and TLR-4 (PDB:4G8A), which are receptors of the host [109]. Results were obtained from PatchDock server in which 20 resultant solutions were produced, as tabulated in Tables S3–S5 [110].
3.19. Refinement of Docked Complexes
Results of PatchDock were additionally subjected to refinement. The complexes with the lowermost global energy were ranked top and selected further for binding mode and interaction studies through UCSF Chimera 1.13.1 [111]. For each receptor, the top-docked solution was selected. In the case of MHC-I, solution 8 was selected, as it has the lowest global energy of −12.72 kJ.mol−1 with good contribution from attractive van der Waals (−6.67 kJ.mol−1), repulsive van der Waals (0.73 kJ.mol−1), ACE (1.39 kJ.mol−1) and hydrogen bond (−0.43 kJ.mol−1) energy. Similarly, for MHC-II and TLR4, solutions 2 and 5 were selected based on the above criteria. [112]. The FireDock refinement results for MHC-I, MHC-II and TLR4 are shown in Tables S6–S8, respectively. The docked intermolecular conformation of the vaccine with MHC-I, MHC-II, and TLR4 is shown in Figure 8.
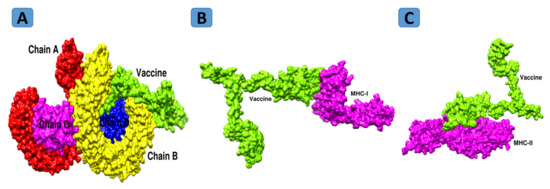
Figure 8.
Docked conformation of vaccine with TLR4 (A), MHC-I (B), and MHC-II (C).
3.20. Chemical Interactions of Vaccine to Immune Cells Receptors
Interaction between vaccine and host immune cell receptors is crucial in order to generate proper immune responses [113]. The chemical interactions between vaccine construct and TLR-4, MHC-I and MHC-II immune receptors were determined using the protein–peptide molecular docking approach [114], and specific residue-wise interactions of MHC-I, MHC-II and TLR 4 were checked using the UCSF chimera tool [111]. The model vaccine construct showed interactions with different residues of MHC-I within 3 Å. These interactions are both hydrophobic and hydrophilic. The interactions are shown in Table 3. Similarly, the vaccine also produced a strong interaction network with the MHC-II molecule. All the interactions are within close distance and are of different types, including hydrogen bonding, salt-bridges and van der Waals interactions [115]. The interacting residue network of the vaccine to MHC-II is presented in Table 3. Toll-like receptors are a class of several proteins that initiate acquired and adaptive immune response; among them, TLR-4 is one of the members of the TLR family, usually expressed on dendritic and macrophage cells [116]. The interacting residues of the model vaccine to TLR-4 are mentioned in Table 3.
Table 3.
Residue-wise interactions of vaccine to MHC-I, MHC-II, and TLR-4.
3.21. Molecular Dynamic Simulation
The molecular dynamics simulation is an in silico simulation for analyzing the dynamic behavior of macromolecules. In this system, the atoms and molecules are allowed to interact for a specific time period, giving a view of the dynamic “evolution” of the system. In the most common version, the trajectories of atoms and molecules are determined through numerically solving Newton’s equations of motion [117]. The docked complexes were analyzed in 250 nanosecond periods, whereas it is an important step to obtain and to ensure the binding affinity of the vaccine construct with dock receptors, i.e., MHC-I, MHC-II, and TLR-4 in a specific time [118]. However, it is mandatory to ensure that the vaccine antigens are efficiently exposed and recognizable by the host immune system to develop a robust immune response. No drastic changes were observed throughout the simulation period, as shown in Figure 9. The first analysis performed was root mean square deviation (RMSD) based on carbon alpha atoms. All three systems revealed increasing RMSD. The TLR4 and MHC-I system with the vaccine showed good binding stability compared to the MHC-II vaccine complex (Figure 9A). The mean TLR4-vaccine and MHC-I-vaccine RMSD values are ~4 and 4.2 angstrom, respectively. The MHC-II–vaccine complex reported a mean RMSD of 5.8 angstrom. The deviation in the systems is because of a larger size and a high number of flexible loops. This is evident in the root mean square fluctuation (RMSF) analysis (Figure 9B). The system’s intermolecular stability can also be witnessed by the radius of gyration (RoG), which reflected the highly compact nature of the vaccine–immune receptor complexes (Figure 9C).
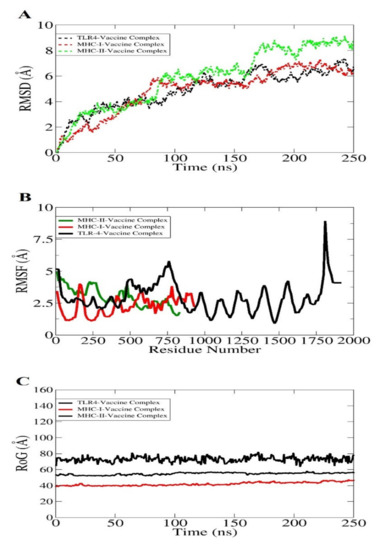
Figure 9.
Different statistical analyses of the simulation trajectories. RMSD (A), RMSF (B), and RoG (C).
3.22. Hydrogen Bonding
Hydrogen bonds are formed between electronegative charged atoms of hydrogen and other electronegative charged particle atoms. Hydrogen bonds are non-covalent forces among electronegative atoms [119]. These bonds are formed between electronegative acceptors and donors. The VMD plugin was used to identify and count the hydrogen bonds between the vaccine and receptors formed in the simulation process [120], which is shown in Figure 10. The cut-off distance value is 3 Å. In each case, a strong and high number of hydrogen bonds (>35) are revealed between the vaccine and its relevant immune receptor.
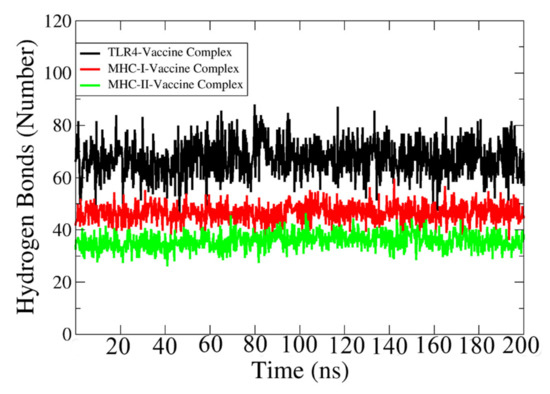
Figure 10.
Number of hydrogen bonds between TLR-4, MHC-I, and MHC-II with designed vaccine construct.
3.23. Binding Free Energies Calculation
The MM-GB/PBSA approach was used to calculate dock complexes binding free energies [73]. The total free binding affinities for the receptor TLR4 with vaccine construct was −90.9 kcal/mol, while with MHC-I and the vaccine construct was −75.07.44 kcal/mol and the MHC-II with its vaccine construct was estimated to be −77.82 kcal/mol. In the MM-GB/PBSA, the net electrostatic and van der Waal energies are the most favorable in the complex formation. The overall gas phase energy dominates all three complexes. On the other side, polar energy is non-favorable and is non-polar in the complex formation. The different binding energy terms are tabulated in Table 4.
Table 4.
MMGBSA/PBSA binding free energies results from the vaccine construct with MHC- I, MHC-II, and TLR4 complexes. The energy values are reported in kcal/mol.
3.24. In Silico Immune Simulation
For in silico immune simulation, the C-ImmSim server was used to evaluate the immune protective potency of the designed vaccine construct stimulating the host immune system using in silico approaches [121]. The position-specific score matrix (PSSM) is used by this approach, along with the other machining learning techniques to study and prioritize epitopes and their immune interactions [122]. In this analysis, the maximum level of exposure to the vaccine antigen was made certain for about 350 days regarding the human immune system. Hence, there was an enhancement in the provocation of adaptive immunity, which can be detected by high IgG and IgM antibody production. The IgM antibody level was also detected to be high. Secondary immune responses subsequently followed by tertiary immune responses led to the maximum production of B-cells and the high levels of IgM + IgG, IgM, IgG1 + IgG2, and IgG1 and IgG2, as shown in Figure S2A. Similarly, the production of interferon–gamma that was greater than 400,000 counts per ml for almost 350 days was observed, as shown in Figure S2B. The humoral and cellular immune responses to the vaccine are presented in Figure S3. Similarly, the response of different immune cells to the vaccine is shown in Figure S4.
4. Discussion
The multi-drug resistant microbes are evolving rapidly and are causing fatal infections not only in humans but in other organisms as well. When bacteria evolve, the efficacy of the drugs is reduced, and hence, AMR develops [123]. Vaccination is considered one of the most important alternatives and effective methods for the prevention of bacterial infections. It is of dire need to develop alternative methods to counter multi-drug resistant bacteria because the presently available antibiotics have become inefficient [124]. In the past decade, almost half of the antibiotics have become ineffective against the present evolving pathogens. Experimental vaccination is time consuming and requires a longer period. Conversely, computational methods along with the advancement in genomic sciences has brought many-fold progress in developing broad spectrum vaccines against AMR pathogens in considerably less time and using less resources [125].
P. rettgeri is resistant to many antibiotics such as ampicillin, polymyxin, first-generation cephalosporins, gentamicin, tobramycin, carbapenem, and tigecycline [36,37,38]. Additionally, it is susceptible to amikacin, according to one study [34]. Furthermore, metallo-β-lactamase-1 and amoxicillin-clavulanate resistivity patterns are noted [40]. The MDR P. rettgeri genome sequence revealed that the pathogen harbors 17 resistance genes and various virulence and heavy metal resistance genes [126]. The high resistance spectrum of P. rettgeri has prompted research to use genomic information of the pathogen to devise new therapeutic strategies, in particular, a vaccine to stop the spread of MDR strains and effectively manage its infections [127].
Vaccine preparation is effective and is the most successful method to prevent infection in a host against pathogenic microbes [128]. The bioinformatics approach is an easier method to design an in silico vaccine. The step-wise rationale of a computational vaccine design approach consists of the following steps: (i) target antigen identification, (ii) select vaccine platform, (iii) optimize gene expression factors, (IV) construct a vaccine candidate, and (v) check immunogenicity and efficacy of vaccine construct and ensure its biological safety, which must be evaluated by using cellular models in vitro and animal models [129].
Traditional and conventional vaccine development is based on Pasteur principles that consist of isolation, inactivation, and injection of non-virulent pathogenic microbes or part of the microbe into the host [130]. Conventional vaccines are vital pharmacological products, but production and development are expensive, not only economically; they also require much time. It takes years to prioritize a potential vaccine candidate against particular infectious microbes [131]. Particularly, the traditional vaccine development phase requires time and grants for making good antigenic candidates. Besides, there are chances of adverse autoimmune side effects. Moreover, the culturing of pathogenic fatal microbes is risky to deal with in laboratory conditions. Therefore, to reduce cost and time, along with eliminating the risk of spreading infections, vaccinomic applications and tools have been established for the development of a vaccine [132]. Potential vaccine candidates are rapidly developed with the aid of computer-aided software/prescreening servers. Various types of vaccines have been designed through in silico approaches, i.e., COVID-19, Dengue, Cancer, Salmonella typhi, and Meningitis [133,134].
The current work is based on a multi-epitope vaccine against P. rettgeri. The potential surface membrane and secretory proteins were prioritized using bioinformatics tools. The complete proteome of P. rettgeri was retrieved from the NCBI databases. BPGA tool core genes from the genomic pool of P. rettgeri were extracted, followed by various types of analyses to prioritize potential vaccine candidates: (1) CD-hit analysis, (2) homology check, (3) essential check, and (4) localization check. Furthermore, the virulence factors were checked via VFDB, followed by transmembrane helices checked with HMMTOP 2.0 and TMHMM 2.0 web servers. Blastp server was used to check for homology with the host genes and normal flora of the host. The physicochemical properties calculated were computationally evaluated with the ProtParam server and ExPasy tool. Vaxign 2.0 webserver was used to check adhesion probability. An antigenicity check was performed to prioritize highly potential vaccine candidates with the aid of Vaxijen 2.0. Moreover, allergenic sequences were found through Allertop 2.0 web server and excluded from the study. B-cell and B-cell-derived T-cell epitopes were predicted from shortlisted filtered proteins. All the predicted epitopes were linked with each other to design multi-epitope vaccine, as highlighted by Ismail et al., 2020. Multi-epitope-based vaccines can combat a wide range of infectious caused by P. rettgeri strains. Multi-epitope vaccines are consistent of several different types of epitopes versus single epitopes, and they have the capacity to generate both humoral and cellular immunity. The designed multi-epitope vaccine was docked with different immune cell receptors. The interaction of the vaccine with immune cells is important in generating a proper immune response; hence, these interactions were analyzed through molecular docking. The docking results were validated through molecular dynamics simulation and binding free energies calculations. Results of the molecular dynamic simulation revealed no drastic changes throughout the simulation time, which is important for the recognition of peptides by the immune system in order to provoke an immune response. Overall, this study concluded that the designed vaccine construct can tackle infectious caused by P. rettgeri. However, in vivo and in vitro experiments will further support the outcomes of this study.
5. Conclusions and Limitations
The excessive use of antibiotics in humans and animal medicine, agriculture, and the environment has led to AR in bacteria, which has significantly contributed to high hospital and community mortality and mobility. AR in P. rettgeri leas to life-threatening health issues around the globe and is becoming difficult to treat. To tackle infections of the pathogen, no significant work of vaccinology is under process. The use of bioinformatics webservers will not only lessen the cost of vaccine research work but will also reduce the time to identify vaccine targets. In this study, a pan-secretome and pan-exoproteome-based recombinant vaccine candidate was designed using immunoinformatics and reverse vaccinology approaches that will induce robust immune responses against P. rettgeri. For the above purpose, potential antigenic and highly non-allergenic surface membrane peptides epitopes were selected. With the aid of various immunoinformatics web tools, it is suggested that this vaccine candidate might provoke strong and active immunologic immune responses in the host. Furthermore, wet laboratory experimental applications are recommended to validate these in silico predictions. It is hoped that this research might help to develop the interest of scientists and researchers in the bioinformatics field on the above subject. Despite the promising results of the study, several methodological limitations can be overcome in the future. For example, the use of more refined tools/servers in terms of algorithms to validate the predictions. Similarly, the optimal ordering of epitopes in the vaccine construct needs strong experimental proof. Lastly, experimental validation of in vivo and in vitro vaccine models is a must.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/vaccines10020189/s1, Figure S1. (A) Core pan plot of 14 P. rettgeri genomes. (B) Pan-phylogeny tree of 14 complete genomes of P. rettgeri. Figure S2. (A) Antibody titer as shown in different color peaks in response to the vaccine injection (black color peak). (B) Simulation of interleukins and interferon level after injection of vaccine. Figure S3. Different B- and T-cell responses against the vaccine. Figure S4. Different immune cell responses are generated in response to the chimeric vaccine construct. Tc (cytotoxic killer T-cell), macrophages (Mφ), natural killer cells, dendritic and epithelial cells. Table S1. Predicted B-cell epitopes. Table S2. MHC-I and MHC-II predicted epitopes with a percentile score. Table S3. Docking score of top 20 docked vaccine complex with MHC-I molecule. Table S4. Docking score of top 20 docked vaccine complex to MHC-II molecule. Table S5. Docking score of top 20 docked vaccine complex to TLR4 molecule. Table S6. Top 10 refined docked complexes of vaccine–MHC-1 complexes. Table S7. Top 10 refined docked complexes of vaccine–MHC-II. Table S8. Top 10 refined docked complexes of vaccine-TLR4.
Author Contributions
Conceptualization, S.A., M.K., M.T.u.Q. and K.S.A.; Data curation, S.G., A.U., S.I., A.R.H., A.G.A. and F.A.; Formal analysis, S.G. and S.A.; Funding acquisition, K.S.A.; Methodology, S.A., A.U., S.I., M.K., M.T.u.Q., A.R.H., A.G.A., F.A. and K.S.A.; Project administration, K.S.A.; Resources, M.T.u.Q., A.R.H., A.G.A. and F.A.; Software, S.A.; Supervision, S.A. and K.S.A.; Validation, A.U., S.I., M.K., A.R.H., A.G.A., F.A. and K.S.A.; Visualization, S.G., S.A. and M.T.u.Q.; Writing—original draft, S.G. and S.A.; Writing—review and editing, A.U., S.I., M.K., M.T.u.Q., A.R.H., A.G.A., F.A. and K.S.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available within the article.
Acknowledgments
The researchers would like to thank the Deanship of Scientific Research, Qassim University for funding the publication of this project.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bowler, P.; Murphy, C.; Wolcott, R. Biofilm exacerbates antibiotic resistance: Is this a current oversight in antimicrobial stewardship? Antimicrob. Resist. Infect. Control 2020, 9, 162. [Google Scholar] [CrossRef] [PubMed]
- Roth, N.; Käsbohrer, A.; Mayrhofer, S.; Zitz, U.; Hofacre, C.; Domig, K.J. The application of antibiotics in broiler production and the resulting antibiotic resistance in Escherichia coli: A global overview. Poult. Sci. 2019, 98, 1791–1804. [Google Scholar] [CrossRef] [PubMed]
- Van Houten, C.B.; Cohen, A.; Engelhard, D.; Hays, J.P.; Karlsson, R.; Moore, E.; Fernández, D.; Kreisberg, R.; Collins, L.V.; de Waal, W. Antibiotic misuse in respiratory tract infections in children and adults—A prospective, multicentre study (TAILORED Treatment). Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frieri, M.; Kumar, K.; Boutin, A. Antibiotic resistance. J. Infect. Public Health 2017, 10, 369–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, R.; Halder, U.; Kabiraj, A.; Mondal, A.; Bandopadhyay, R. Overview on the role of heavy metals tolerance on developing antibiotic resistance in both Gram-negative and Gram-positive bacteria. Arch. Microbiol. 2021, 203, 2761–2770. Available online: https://link.springer.com/article/10.1007/s00203-021-02275-w (accessed on 18 June 2021). [CrossRef]
- Caniça, M.; Manageiro, V.; Abriouel, H.; Moran-Gilad, J.; Franz, C.M.A.P. Antibiotic resistance in foodborne bacteria. Trends Food Sci. Technol. 2019, 84, 41–44. [Google Scholar] [CrossRef]
- MacLean, R.C.; San Millan, A. The evolution of antibiotic resistance. Science 2019, 365, 1082–1083. [Google Scholar] [CrossRef]
- Koch, B.J.; Hungate, B.A.; Price, L.B. Food-animal production and the spread of antibiotic resistance: The role of ecology. Front. Ecol. Environ. 2017, 15, 309–318. [Google Scholar] [CrossRef]
- Wang, J.; Seebacher, N.; Shi, H.; Kan, Q.; Duan, Z. Novel strategies to prevent the development of multidrug resistance (MDR) in cancer. Oncotarget 2017, 8, 84559. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Singh, A.P.; Kumar, S.; Giri, B.S.; Kim, K.-H. Antibiotic resistance in major rivers in the world: A systematic review on occurrence, emergence, and management strategies. J. Clean. Prod. 2019, 234, 1484–1505. [Google Scholar] [CrossRef]
- Chin, W.; Zhong, G.; Pu, Q.; Yang, C.; Lou, W.; De Sessions, P.F.; Periaswamy, B.; Lee, A.; Liang, Z.C.; Ding, X. A macromolecular approach to eradicate multidrug resistant bacterial infections while mitigating drug resistance onset. Nat. Commun. 2018, 9, 917. [Google Scholar] [CrossRef] [PubMed]
- Covián, C.; Fernández-Fierro, A.; Retamal-Díaz, A.; Díaz, F.E.; Vasquez, A.E.; Lay, M.K.; Riedel, C.A.; González, P.A.; Bueno, S.M.; Kalergis, A.M. BCG-induced cross-protection and development of trained immunity: Implication for vaccine design. Front. Immunol. 2019, 10, 2806. [Google Scholar] [CrossRef] [PubMed]
- Halloran, M.E.; Longini, I.M.; Struchiner, C.J.; Longini, I.M. Design and Analysis of Vaccine Studies; Springer: Berlin/Heidelberg, Germany, 2010; Volume 18. [Google Scholar]
- Ansari, A.; Madan, A.; Prakash, D. Vaccine development—A complex science. EPRA Int. J. Multidiscip. Res. 2021, 7, 34–37. [Google Scholar] [CrossRef]
- Gasperini, G.; Alfini, R.; Arato, V.; Mancini, F.; Aruta, M.G.; Kanvatirth, P.; Pickard, D.; Necchi, F.; Saul, A.; Rossi, O. Salmonella Paratyphi A Outer Membrane Vesicles Displaying Vi Polysaccharide as a Multivalent Vaccine against Enteric Fever. Infect. Immun. 2021, 89, e00699-20. [Google Scholar] [CrossRef]
- Chen, H.; Gao, Z.; Bai, S.; Liu, X.; Han, S.; Xiao, Y.; Liu, F.; Yu, Y.; Sun, H.; Yang, X. Immunogenicity and safety of sabin-strain based inactivated poliovirus vaccine replacing salk-strain based inactivated poliovirus vaccine: An innovative application of different strain-IPVs replacement. Vaccine 2021, 39, 2467–2474. [Google Scholar] [CrossRef]
- Kaufmann, S.H.E. The Tuberculosis Vaccine Development Pipeline: Present and Future Priorities and Challenges for Research and Innovation. In Essential Tuberculosis; Springer: Berlin/Heidelberg, Germany, 2021; pp. 395–405. [Google Scholar]
- Gong, W.; Aspatwar, A.; Wang, S.; Parkkila, S.; Wu, X. COVID-19 pandemic: SARS-CoV-2 specific vaccines and challenges, protection via BCG trained immunity, and clinical trials. Expert Rev. Vaccines 2021, 20, 857–880. Available online: https://www.tandfonline.com/doi/full/10.1080/14760584.2021.1938550 (accessed on 18 June 2021). [CrossRef]
- Dar, H.A.; Ismail, S.; Waheed, Y.; Ahmad, S.; Jamil, Z.; Aziz, H.; Hetta, H.F.; Muhammad, K. Designing a multi-epitope vaccine against Mycobacteroides abscessus by pangenome-reverse vaccinology. Sci. Rep. 2021, 11, 11197. [Google Scholar] [CrossRef]
- Thompson, M.G.; Burgess, J.L.; Naleway, A.L.; Tyner, H.L.; Yoon, S.K.; Meece, J.; Olsho, L.E.W.; Caban-Martinez, A.J.; Fowlkes, A.; Lutrick, K. Interim estimates of vaccine effectiveness of BNT162b2 and mRNA-1273 COVID-19 vaccines in preventing SARS-CoV-2 infection among health care personnel, first responders, and other essential and frontline workers—Eight US locations, December 2020–March 2021. Morb. Mortal. Wkly. Rep. 2021, 70, 495. [Google Scholar] [CrossRef]
- Bidmos, F.A.; Siris, S.; Gladstone, C.A.; Langford, P.R. Bacterial Vaccine Antigen Discovery in the Reverse Vaccinology 2.0 Era: Progress and Challenges. Front. Immunol. 2018, 9, 2315. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Huo, C.; Lang, S.; Caution, K.; Nick, S.T.; Dubey, P.; Deora, R.; Huang, X. Chemical Synthesis and Immunological Evaluation of a Pentasaccharide Bearing Multiple Rare Sugars as a Potential Anti-pertussis Vaccine. Angew. Chem. 2020, 132, 6513–6520. [Google Scholar] [CrossRef]
- Ismail, S.; Ahmad, S.; Azam, S.S. Vaccinomics to design a novel single chimeric subunit vaccine for broad-spectrum immunological applications targeting nosocomial Enterobacteriaceae pathogens. Eur. J. Pharm. Sci. 2020, 146, 105258. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R. What are the most powerful immunogen design vaccine strategies? Reverse vaccinology 2.0 shows great promise. Cold Spring Harb. Perspect. Biol. 2017, 9, a030262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wetzler, L.M.; Feavers, I.M.; Gray-Owen, S.D.; Jerse, A.E.; Rice, P.A.; Deal, C.D. Summary and recommendations from the National Institute of Allergy and Infectious Diseases (NIAID) workshop “Gonorrhea Vaccines: The Way forward. ” Clin. Vaccine Immunol. 2016, 23, 656–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalsass, M.; Brozzi, A.; Medini, D.; Rappuoli, R. Comparison of open-source Reverse Vaccinology programs for bacterial vaccine antigen discovery. Front. Immunol. 2019, 10, 113. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Yang, Q.; Chan, L.W.; Bhatia, S.N.; Ruoslahti, E.; Sailor, M.J. Fusogenic porous silicon nanoparticles as a broad-spectrum immunotherapy against bacterial infections. Nanoscale Horiz. 2021, 6, 330–340. [Google Scholar] [CrossRef]
- Gagneux-Brunon, A.; Lucht, F.; Launay, O.; Berthelot, P.; Botelho-Nevers, E. Vaccines for healthcare-associated infections: Present, future, and expectations. Expert Rev. Vaccines 2018, 17, 421–433. [Google Scholar] [CrossRef]
- Ahmad, S.; Waheed, Y.; Ismail, S.; Abbasi, S.W.; Najmi, M.H. A computational study to disclose potential drugs and vaccine ensemble for COVID-19 conundrum. J. Mol. Liq. 2021, 324, 114734. [Google Scholar] [CrossRef]
- Yero, D.; Conchillo-Solé, O.; Daura, X. Antigen Discovery in Bacterial Panproteomes. In Vaccine Delivery Technology; Springer: Berlin/Heidelberg, Germany, 2021; pp. 43–62. [Google Scholar]
- Abdullah, M.; Kadivella, M.; Sharma, R.; Faisal, S.M.; Azam, S. Designing of multiepitope-based vaccine against Leptospirosis using Immuno-Informatics approaches. bioRxiv 2021. Available online: https://www.biorxiv.org/content/10.1101/2021.02.22.431920v1 (accessed on 18 June 2021).
- Santos, A.; Ali, A.; Barbosa, E.; Silva, A.; Miyoshi, A.; Barh, D.; Azevedo, V. The reverse vaccinology—A contextual overview. IIOABJ 2011, 2, 8–15. [Google Scholar]
- Yuan, C.; Wei, Y.; Zhang, S.; Cheng, J.; Cheng, X.; Qian, C.; Wang, Y.; Zhang, Y.; Yin, Z.; Chen, H. Comparative Genomic Analysis Reveals Genetic Mechanisms of the Variety of Pathogenicity, Antibiotic Resistance, and Environmental Adaptation of Providencia Genus. Front. Microbiol. 2020, 11, 572642. [Google Scholar] [CrossRef]
- Sagar, S.; Narasimhaswamy, N.; d’Souza, J. Providencia rettgeri: An emerging nosocomial uropathogen in an indwelling urinary catheterised patient. J. Clin. Diagn. Res. JCDR 2017, 11, DD01–DD02. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Sharma, P.; Soni, P. First case report of Providencia rettgeri neonatal sepsis. BMC Res. Notes 2017, 10, 17–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.; Jeong, S.H.; Lee, H.; Hong, J.S.; Park, M.-J.; Song, W. Emergence of multidrug-resistant Providencia rettgeri isolates co-producing NDM-1 carbapenemase and PER-1 extended-spectrum β-lactamase causing a first outbreak in Korea. Ann. Clin. Microbiol. Antimicrob. 2018, 17, 20. [Google Scholar] [CrossRef] [PubMed]
- Koreishi, A.F.; Schechter, B.A.; Karp, C.L. Ocular Infections Caused by Providencia rettgeri. Ophthalmology 2006, 113, 1463–1466. [Google Scholar] [CrossRef] [PubMed]
- Tshisevhe, V.S.; Lekalakala, M.R.; Tshuma, N.; Janse Van Rensburg, S.; Mbelle, N. Outbreak of carbapenem-resistant Providencia rettgeri in a tertiary hospital. S. Afr. Med. J. 2017, 107, 31–33. [Google Scholar] [CrossRef] [Green Version]
- Iwata, S.; Tada, T.; Hishinuma, T.; Tohya, M.; Oshiro, S.; Kuwahara-Arai, K.; Ogawa, M.; Shimojima, M.; Kirikae, T. Emergence of carbapenem-resistant Providencia rettgeri and Providencia stuartii producing IMP-type metallo-β-lactamase in Japan. Antimicrob. Agents Chemother. 2020, 64, e00382-20. [Google Scholar] [CrossRef] [PubMed]
- Carvalho-Assef, A.P.D.; Pereira, P.S.; Albano, R.M.; Berião, G.C.; Chagas, T.P.G.; Timm, L.N.; Da Silva, R.C.F.; Falci, D.R.; Asensi, M.D. Isolation of NDM-producing Providencia rettgeri in Brazil. J. Antimicrob. Chemother. 2013, 68, 2956–2957. [Google Scholar] [CrossRef] [Green Version]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33, D501–D504. [Google Scholar] [CrossRef] [Green Version]
- Pérez de la Lastra, J.M.; Asensio-Calavia, P.; González-Acosta, S.; Baca-González, V.; Morales-delaNuez, A. Bioinformatic Analysis of Genome-Predicted Bat Cathelicidins. Molecules 2021, 26, 1811. [Google Scholar] [CrossRef]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA-an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [Green Version]
- Zeb, S.; Gulfam, S.M.; Bokhari, H. Comparative core/pan genome analysis of Vibrio cholerae isolates from Pakistan. Infect. Genet. Evol. 2020, 82, 104316. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, H.; Dyer, R.; Severin, A.J.; Rajan, H. Comprehensive Analysis of Non Redundant Protein Database. Res. Sq. 2020. Available online: https://www.researchgate.net/publication/343753529_Comprehensive_Analysis_of_Non_Redundant_Protein_Database (accessed on 18 June 2021).
- Wheeler, C.R.; Scarbrough, D. Implementing Bioinformatic Tools to Predict Vaccine Potential from Prioritized Staphylococcus aureus Antigens. 2020. Available online: https://scholarworks.boisestate.edu/icur/2020/Poster_Session/119/ (accessed on 18 June 2021).
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Abbas, G.; Zafar, I.; Ahmad, S.; Azam, S.S. Immunoinformatics design of a novel multi-epitope peptide vaccine to combat multi-drug resistant infections caused by Vibrio vulnificus. Eur. J. Pharm. Sci. 2020, 142, 105160. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Ranaghan, K.E.; Azam, S.S. Combating tigecycline resistant Acinetobacter baumannii: A leap forward towards multi-epitope based vaccine discovery. Eur. J. Pharm. Sci. 2019, 132, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chand, Y.; Singh, S. Prioritization of potential vaccine candidates and designing a multiepitope-based subunit vaccine against multidrug-resistant Salmonella Typhi str. CT18: A subtractive proteomics and immunoinformatics approach. Microb. Pathog. 2021, 159, 105150. [Google Scholar] [CrossRef]
- ProtParam—Expasy. ExPASy-ProtParam Tool. 2017. Available online: https://web.expasy.org/protparam/ (accessed on 2 May 2021).
- Sahoo, P.R.; Sahoo, G.; Behera, P.C. Comparative Insilco physiochemical and phylogenetic analysis of insulin like growth factor 1 receptor (IGF-1R) in domestic animals. Indian J. Anim. Res. 2019, 53, 1033–1035. [Google Scholar] [CrossRef]
- Lüthje, S.; Ramanathan, K. In Silico Analysis of Class III Peroxidases: Hypothetical Structure, Ligand Binding Sites, Posttranslational Modifications, and Interaction with Substrates. In Plant Proteomics; Springer: Berlin/Heidelberg, Germany, 2020; pp. 325–339. [Google Scholar]
- Ferdous, N.; Reza, M.N.; Emon, M.T.H.; Islam, M.S.; Mohiuddin, A.K.M.; Hossain, M.U. Molecular characterization and functional annotation of a hypothetical protein (SCO0618) of Streptomyces coelicolor A3 (2). Genom. Inform. 2020, 18, e28. [Google Scholar] [CrossRef]
- Ahmad, S.; Navid, A.; Farid, R.; Abbas, G.; Ahmad, F.; Zaman, N.; Parvaiz, N.; Azam, S.S. Design of a novel multi epitope-based vaccine for pandemic coronavirus disease (COVID-19) by vaccinomics and probable prevention strategy against avenging zoonotics. Eur. J. Pharm. Sci. 2020, 151, 105387. [Google Scholar] [CrossRef]
- Ong, E.; Cooke, M.F.; Huffman, A.; Xiang, Z.; Wong, M.U.; Wang, H.; Seetharaman, M.; Valdez, N.; He, Y. Vaxign2: The second generation of the first Web-based vaccine design program using reverse vaccinology and machine learning. Nucleic Acids Res. 2021, 49, W671–W678. [Google Scholar] [CrossRef]
- Adeoti, O.M. Prediction of multi-epitopic domains of a putative oral vaccine against hepatitis C virus. Int. J. Immunol. Microbiol. 2021, 1, 16–22. [Google Scholar]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; Greenbaum, J.A.; Marcatili, P. IEDB-AR: Immune epitope database—Analysis resource in 2019. Nucleic Acids Res. 2019, 47, W502–W506. [Google Scholar] [CrossRef] [Green Version]
- Girija, A.S.S.; Shoba, G.; Priyadharsini, J.V. Accessing the T-Cell and B-Cell immuno-dominant peptides from A. baumannii biofilm associated protein (bap) as vaccine candidates: A computational approach. Int. J. Pept. Res. Ther. 2021, 27, 37–45. [Google Scholar] [CrossRef]
- Guan, P.; Doytchinova, I.A.; Zygouri, C.; Flower, D.R. MHCPred: A server for quantitative prediction of peptide–MHC binding. Nucleic Acids Res. 2003, 31, 3621–3624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taiwo, A.A.; Falilat, A.J.; Ezemuel, Y.S. Computational design of peptide vaccine against Acinetobacter baumannii infection using comparative genomic approach. Comput. Biol. Bioinform. 2014, 2, 13–18. [Google Scholar] [CrossRef]
- Ahmad, S.; Azam, S.S. A novel approach of virulome based reverse vaccinology for exploring and validating peptide-based vaccine candidates against the most troublesome nosocomial pathogen: Acinetobacter baumannii. J. Mol. Graph. Model. 2018, 83, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Saadi, M.; Karkhah, A.; Nouri, H.R. Development of a multi-epitope peptide vaccine inducing robust T cell responses against brucellosis using immunoinformatics based approaches. Infect. Genet. Evol. 2017, 51, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Jerin, T.; Sharmin, N.; Raihan, S. Silico Functional Annotation and Molecular Characterization of an Uncharacterized Protein MBO_502153 of Mycobacterium Tuberculosis Variant Bovis. Int. J. Sci. Res. Dent. Med. Sci. 2021, 3, 78–85. [Google Scholar]
- Sarkar, M.; Saha, S. Structural insight into the role of novel SARS-CoV-2 E protein: A potential target for vaccine development and other therapeutic strategies. PLoS ONE 2020, 15, e0237300. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Saba Ismail, S.A.; Mirza, M.U.; Abbasi, S.W.; Ashfaq, U.A.; Chen, L.-L. Development of a Novel Multi-Epitope Vaccine against Crimean-Congo Hemorrhagic Fever Virus: An Integrated Reverse Vaccinology, Vaccine Informatics and Biophysics Approach. Front. Immunol. 2021, 12, 669812. [Google Scholar] [CrossRef]
- Devi, A.; Chaitanya, N.S.N. In silico designing of multi-epitope vaccine construct against human coronavirus infections. J. Biomol. Struct. Dyn. 2021, 39, 6903–6917. [Google Scholar] [CrossRef]
- Samad, A.; Ahammad, F.; Nain, Z.; Alam, R.; Imon, R.R.; Hasan, M.; Rahman, M.S. Designing a multi-epitope vaccine against SARS-CoV-2: An immunoinformatics approach. J. Biomol. Struct. Dyn. 2020, 17, 1–17. [Google Scholar] [CrossRef]
- Krishnan, S.; Joshi, A.; Akhtar, N.; Kaushik, V. Immunoinformatics designed T cell multi epitope dengue peptide vaccine derived from non structural proteome. Microb. Pathog. 2021, 150, 104728. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, T.; Rizarullah, R. Predicting Multi-Epitope Peptide Cancer Vaccine from Novel TAA Topo48. J. Sci. Appl. Technol. 2021, 5, 171–178. [Google Scholar] [CrossRef]
- Debnath, S.; Sen, D. Mushrooms are potential foods against cancer: Identified by molecular docking and molecular dynamics simulation. Nat. Prod. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Shahid, F.; Tahir ul Qamar, M.; Abbasi, S.W.; Sajjad, W.; Ismail, S.; Alrumaihi, F.; Allemailem, K.S.; Almatroudi, A.; Ullah Saeed, H.F. Immuno-Informatics Analysis of Pakistan-Based HCV Subtype-3a for Chimeric Polypeptide Vaccine Design. Vaccines 2021, 9, 293. [Google Scholar] [CrossRef]
- Huang, K.; Luo, S.; Cong, Y.; Zhong, S.; Zhang, J.Z.H.; Duan, L. An accurate free energy estimator: Based on MM/PBSA combined with interaction entropy for protein–ligand binding affinity. Nanoscale 2020, 12, 10737–10750. [Google Scholar] [CrossRef]
- Bhardwaj, A. In silico Multi Subunit Vaccine Design Referring Spike Glycoprotein of SARS-COV-2 (COVID-19): The World Pandemic. Indian J. Pharm. Sci. 2021, 83, 21–31. [Google Scholar] [CrossRef]
- Omoniyi, A.A.; Adebisi, S.S.; Musa, S.A.; Nzalak, J.O.; Danborno, B.; Bauchi, Z.M.; Badmus, I.T.; Olatomide, O.D.; Oladimeji, O.J.; Nyengaard, J.R. Immunoinformatics Analysis and In-Silico Design of Multi-Epitopes Vaccine against Lassa Virus. Available online: https://www.researchgate.net/publication/350394848_Immunoinformatics_Analysis_and_In-silico_Design_of_Multi-_Epitopes_Vaccine_Against_Lassa_Virus (accessed on 16 August 2021).
- Yadav, S.; Kapley, A. Antibiotic resistance: Global health crisis and metagenomics. Biotechnol. Rep. 2021, 29, e00604. [Google Scholar] [CrossRef]
- Idomir, M.E. providencia species-involvement in pathology and multidrug resistance in a Romanian county hospital. Bull. Transilv. Univ. Brasov Med. Sci. Ser. VI 2021, 14, 43–50. [Google Scholar] [CrossRef]
- Micoli, F.; Bagnoli, F.; Rappuoli, R.; Serruto, D. The role of vaccines in combatting antimicrobial resistance. Nat. Rev. Microbiol. 2021, 19, 287–302. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Kristensen, D.M.; Makarova, K.S.; Wolf, Y.I.; Koonin, E. V Microbial genome analysis: The COG approach. Brief. Bioinform. 2019, 20, 1063–1070. [Google Scholar] [CrossRef]
- Ehsan, N.; Ahmad, S.; Navid, A.; Azam, S.S. Identification of potential antibiotic targets in the proteome of multi-drug resistant Proteus mirabilis. Meta Gene 2018, 18, 167–173. [Google Scholar] [CrossRef]
- Uddin, R.; Jamil, F. Prioritization of potential drug targets against P. aeruginosa by core proteomic analysis using computational subtractive genomics and Protein-Protein interaction network. Comput. Biol. Chem. 2018, 74, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Da Fiúza, T.S.; de Souza, G.A. Identification of core immunogenic peptides of Shigella sonnei for a Peptide-Based Vaccine. Bio-Manguinhos 2021, 5, 102. [Google Scholar]
- Li, J.; Qiu, J.; Huang, Z.; Liu, T.; Pan, J.; Zhang, Q.; Liu, Q. Reverse vaccinology approach for the identifications of potential vaccine candidates against Salmonella. Int. J. Med. Microbiol. 2021, 311, 151508. [Google Scholar] [CrossRef] [PubMed]
- Wadood, A.; Ghufran, M.; Khan, A.; Azam, S.S.; Uddin, R.; Waqas, M.; Saleem, S. The methicillin-resistant S. epidermidis strain RP62A genome mining for potential novel drug targets identification. Gene Rep. 2017, 8, 88–93. [Google Scholar] [CrossRef]
- Paramasivam, N.; Linke, D. ClubSub-P: Cluster-based subcellular localization prediction for Gram-negative bacteria and archaea. Front. Microbiol. 2011, 2, 218. [Google Scholar] [CrossRef] [Green Version]
- Raoufi, E.; Hemmati, M.; Eftekhari, S.; Khaksaran, K.; Mahmodi, Z.; Farajollahi, M.M.; Mohsenzadegan, M. Epitope prediction by novel immunoinformatics approach: A state-of-the-art review. Int. J. Pept. Res. Ther. 2020, 26, 1155–1163. [Google Scholar] [CrossRef]
- Yasser, E.-M.; Dobbs, D.; Honavar, V.G. In silico prediction of linear B-cell epitopes on proteins. In Prediction of Protein Secondary Structure; Springer: Berlin/Heidelberg, Germany, 2017; pp. 255–264. [Google Scholar]
- Kollmann, T.R. Variation between populations in the innate immune response to vaccine adjuvants. Front. Immunol. 2013, 4, 81. [Google Scholar] [CrossRef] [Green Version]
- Albagi, S.; Ahmed, O.H.; Gumaa, M.A.; Abd_elrahman, K.A.; Abu-Haraz, A.H. Immunoinformatics-peptide driven vaccine and in silico modeling for Duvenhage rabies virus glycoprotein G. J Clin Cell Immunol 2017, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.; Lundegaard, C.; Lund, O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinform. 2007, 8, 238. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Kumar, A. Designing an efficient multi-epitope vaccine against Campylobacter jejuni using immunoinformatics and reverse vaccinology approach. Microb. Pathog. 2020, 147, 104398. [Google Scholar] [CrossRef] [PubMed]
- Bibi, N.; Zaidi, N.-S.S.; Tahir, M.; Babar, M.M. Vaccinomics driven proteome-wide screening of Haemophilus influenzae for the prediction of common putative vaccine candidates. Can. J. Microbiol. 2021, 67, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Naz, A.; Obaid, A.; Paracha, R.Z.; Naz, K.; Awan, F.M.; Muhmmad, S.A.; Janjua, H.A.; Ahmad, J.; Ali, A. Pangenome and immuno-proteomics analysis of Acinetobacter baumannii strains revealed the core peptide vaccine targets. BMC Genom. 2016, 17, 732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naz, A.; Awan, F.M.; Obaid, A.; Muhammad, S.A.; Paracha, R.Z.; Ahmad, J.; Ali, A. Identification of putative vaccine candidates against Helicobacter pylori exploiting exoproteome and secretome: A reverse vaccinology based approach. Infect. Genet. Evol. 2015, 32, 280–291. [Google Scholar] [CrossRef]
- Sanami, S.; Zandi, M.; Pourhossein, B.; Mobini, G.-R.; Safaei, M.; Abed, A.; Arvejeh, P.M.; Chermahini, F.A.; Alizadeh, M. Design of a multi-epitope vaccine against SARS-CoV-2 using immunoinformatics approach. Int. J. Biol. Macromol. 2020, 164, 871–883. [Google Scholar] [CrossRef]
- Jafari, E.; Mahmoodi, S. Design, expression, and purification of a multi-epitope vaccine against Helicobacter pylori based on Melittin as an adjuvant. Microb. Pathog. 2021, 157, 104970. [Google Scholar] [CrossRef]
- Javadi, M.; Oloomi, M.; Bouzari, S. In Silico Design of a Poly-epitope Vaccine for Urinary Tract Infection Based on Conserved Antigens by Modern Vaccinology. Int. J. Pept. Res. Ther. 2021, 27, 909–921. [Google Scholar] [CrossRef]
- Ismail, M.; Sajid, Z.; Ali, A.; Wu, X.; Muhammad, S.A.; Shaikh, R.S. Prediction of Prophylactic Peptide Vaccine Candidates for Human Papillomavirus (HPV): Immunoinformatics and Reverse Vaccinology Approaches. Curr. Proteom. 2021, 18, 178–192. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [Green Version]
- Gupta, K.; Varadarajan, R. Insights into protein structure, stability and function from saturation mutagenesis. Curr. Opin. Struct. Biol. 2018, 50, 117–125. [Google Scholar] [CrossRef]
- Sayed, S.B.; Nain, Z.; Khan, M.S.A.; Abdulla, F.; Tasmin, R.; Adhikari, U.K. Exploring lassa virus proteome to design a multi-epitope vaccine through immunoinformatics and immune simulation analyses. Int. J. Pept. Res. Ther. 2020, 26, 2089–2107. [Google Scholar] [CrossRef] [PubMed]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombkowski, A.A.; Sultana, K.Z.; Craig, D.B. Protein disulfide engineering. FEBS Lett. 2014, 588, 206–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, R.K.; Bhatt, T.K.; Prajapati, V.K. Novel immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating anopheles salivary protein. Sci. Rep. 2018, 8, 1125. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.; Pandey, R.K.; Khatoon, N.; Narula, A.; Mishra, A.; Prajapati, V.K. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci. Rep. 2017, 7, 9232. [Google Scholar] [CrossRef] [PubMed]
- Shey, R.A.; Ghogomu, S.M.; Esoh, K.K.; Nebangwa, N.D.; Shintouo, C.M.; Nongley, N.F.; Asa, B.F.; Ngale, F.N.; Vanhamme, L.; Souopgui, J. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci. Rep. 2019, 9, 4409. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Fu, A.; Zhang, L. Progress in molecular docking. Quant. Biol. 2019, 7, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Rehman, A.; Ahmad, S.; Shahid, F.; Albutti, A.; Alwashmi, A.S.S.; Aljasir, M.A.; Alhumeed, N.; Qasim, M.; Ashfaq, U.A.; Tahir ul Qamar, M. Integrated Core Proteomics, Subtractive Proteomics, and Immunoinformatics Investigation to Unveil a Potential Multi-Epitope Vaccine against Schistosomiasis. Vaccines 2021, 9, 658. [Google Scholar] [CrossRef]
- Jyotisha, S.S.; Qureshi, I.A. Multi-epitope vaccine against SARS-CoV-2 applying immunoinformatics and molecular dynamics simulation approaches. J. Biomol. Struct. Dyn. 2020. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7682209/ (accessed on 18 June 2021).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Nemati, A.S.; Tafrihi, M.; Sheikhi, F.; Tabari, A.R.; Haditabar, A. Designing a New Multi Epitope-Based Vaccine against COVID-19 Disease: An Immunoinformatic Study Based on Reverse Vaccinology Approach. 2021. Available online: https://assets.researchsquare.com/files/rs-206270/v1/17abe418-b7e9-4ff5-84da-eaae0965ca5d.pdf?c=1631875889 (accessed on 18 June 2021).
- Rizvi, S.M.; Raghavan, M. Direct peptide-regulatable interactions between MHC class I molecules and tapasin. Proc. Natl. Acad. Sci. USA 2006, 103, 18220–18225. [Google Scholar] [CrossRef] [Green Version]
- Bonvin, A.M.J.J. Flexible protein–protein docking. Curr. Opin. Struct. Biol. 2006, 16, 194–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofving, T. Small Intestinal Neuroendocrine Tumours—Disease Models, Tumour Development, and Remedy. Ph.D. Thesis, University of Gothenburg, Gothenburg, Sweden, 2019. Available online: https://gupea.ub.gu.se/bitstream/2077/58488/5/gupea_2077_58488_5.pdf (accessed on 18 June 2021).
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharadwaj, K.K.; Srivastava, A.; Panda, M.K.; Singh, Y.D.; Maharana, R.; Mandal, K.; Singh, B.S.M.; Singh, D.; Das, M.; Murmu, D. Computational intelligence in vaccine design against COVID-19. In Computational Intelligence Methods in COVID-19: Surveillance, Prevention, Prediction and Diagnosis; Springer: Berlin/Heidelberg, Germany, 2021; pp. 311–329. [Google Scholar]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alagumuthu, M.; Rajpoot, S.; Baig, M.S. Structure-based design of novel peptidomimetics targeting the SARS-CoV-2 spike protein. Cell. Mol. Bioeng. 2021, 14, 177–185. [Google Scholar] [CrossRef]
- Rafi, S.; Yasmin, S.; Uddin, R. A molecular dynamic simulation approach: Development of dengue virus vaccine by affinity improvement techniques. J. Biomol. Struct. Dyn. 2022, 40, 61–76. [Google Scholar] [CrossRef]
- Abraham Peele, K.; Srihansa, T.; Krupanidhi, S.; Ayyagari, V.S.; Venkateswarulu, T.C. Design of multi-epitope vaccine candidate against SARS-CoV-2: A in-silico study. J. Biomol. Struct. Dyn. 2021, 39, 3793–3801. [Google Scholar] [CrossRef]
- Mahapatra, S.R.; Dey, J.; Kushwaha, G.S.; Puhan, P.; Mohakud, N.K.; Panda, S.K.; Lata, S.; Misra, N.; Suar, M. Immunoinformatic approach employing modeling and simulation to design a novel vaccine construct targeting MDR efflux pumps to confer wide protection against typhoidal Salmonella serovars. J. Biomol. Struct. Dyn. 2021, 1–13. Available online: https://pubmed.ncbi.nlm.nih.gov/34463211/ (accessed on 16 June 2021). [CrossRef]
- Vrancianu, C.O.; Gheorghe, I.; Czobor, I.B.; Chifiriuc, M.C. Antibiotic resistance profiles, molecular mechanisms and innovative treatment strategies of Acinetobacter baumannii. Microorganisms 2020, 8, 935. [Google Scholar] [CrossRef]
- Rosini, R.; Nicchi, S.; Pizza, M.; Rappuoli, R. Vaccines against antimicrobial resistance. Front. Immunol. 2020, 11, 1048. [Google Scholar] [CrossRef] [PubMed]
- Mat Rahim, N.; Lee, H.; Strych, U.; AbuBakar, S. Facing the challenges of multidrug-resistant Acinetobacter baumannii: Progress and prospects in the vaccine development. Hum. Vaccines Immunother. 2021, 17, 3784–3794. [Google Scholar] [CrossRef]
- Mbelle, N.M.; Osei Sekyere, J.; Amoako, D.G.; Maningi, N.E.; Modipane, L.; Essack, S.Y.; Feldman, C. Genomic analysis of a multidrug-resistant clinical Providencia rettgeri (PR002) strain with the novel integron ln1483 and an A/C plasmid replicon. Ann. N. Y. Acad. Sci. 2020, 1462, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Ntshobeni, N.B.; Allam, M.; Ismail, A.; Amoako, D.G.; Essack, S.Y.; Chenia, H.Y. Draft genome sequence of Providencia rettgeri APW139_S1, an NDM-18-producing clinical strain originating from hospital effluent in South Africa. Microbiol. Resour. Announc. 2019, 8, e00259-19. [Google Scholar] [CrossRef] [Green Version]
- Principi, N.; Esposito, S. Vaccine-preventable diseases, vaccines and Guillain-Barre’syndrome. Vaccine 2019, 37, 5544–5550. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Samuels, E.L.; Bocchini Jr, J.A. Vaccine development: From laboratory to policy. Pediatr. Ann. 2020, 49, e509–e515. [Google Scholar] [CrossRef] [PubMed]
- Palatnik-de-Sousa, C.B. What Would Jenner and Pasteur Have Done About COVID-19 Coronavirus? The Urges of a Vaccinologist. Front. Immunol. 2020, 11, 2173. [Google Scholar] [CrossRef] [PubMed]
- Rizwan, M.; Naz, A.; Ahmad, J.; Naz, K.; Obaid, A.; Parveen, T.; Ahsan, M.; Ali, A. VacSol: A high throughput in silico pipeline to predict potential therapeutic targets in prokaryotic pathogens using subtractive reverse vaccinology. BMC Bioinform. 2017, 18, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sette, A.; Rappuoli, R. Reverse vaccinology: Developing vaccines in the era of genomics. Immunity 2010, 33, 530–541. [Google Scholar] [CrossRef] [Green Version]
- Noé, A.; Cargill, T.N.; Nielsen, C.M.; Russell, A.J.C.; Barnes, E. The application of single-cell RNA sequencing in vaccinology. J. Immunol. Res. 2020, 2020, 8624963. [Google Scholar] [CrossRef]
- Goumari, M.M.; Farhani, I.; Nezafat, N.; Mahmoodi, S. Multi-epitope vaccines (MEVs), as a novel strategy against infectious diseases. Curr. Proteom. 2020, 17, 354–364. [Google Scholar] [CrossRef]
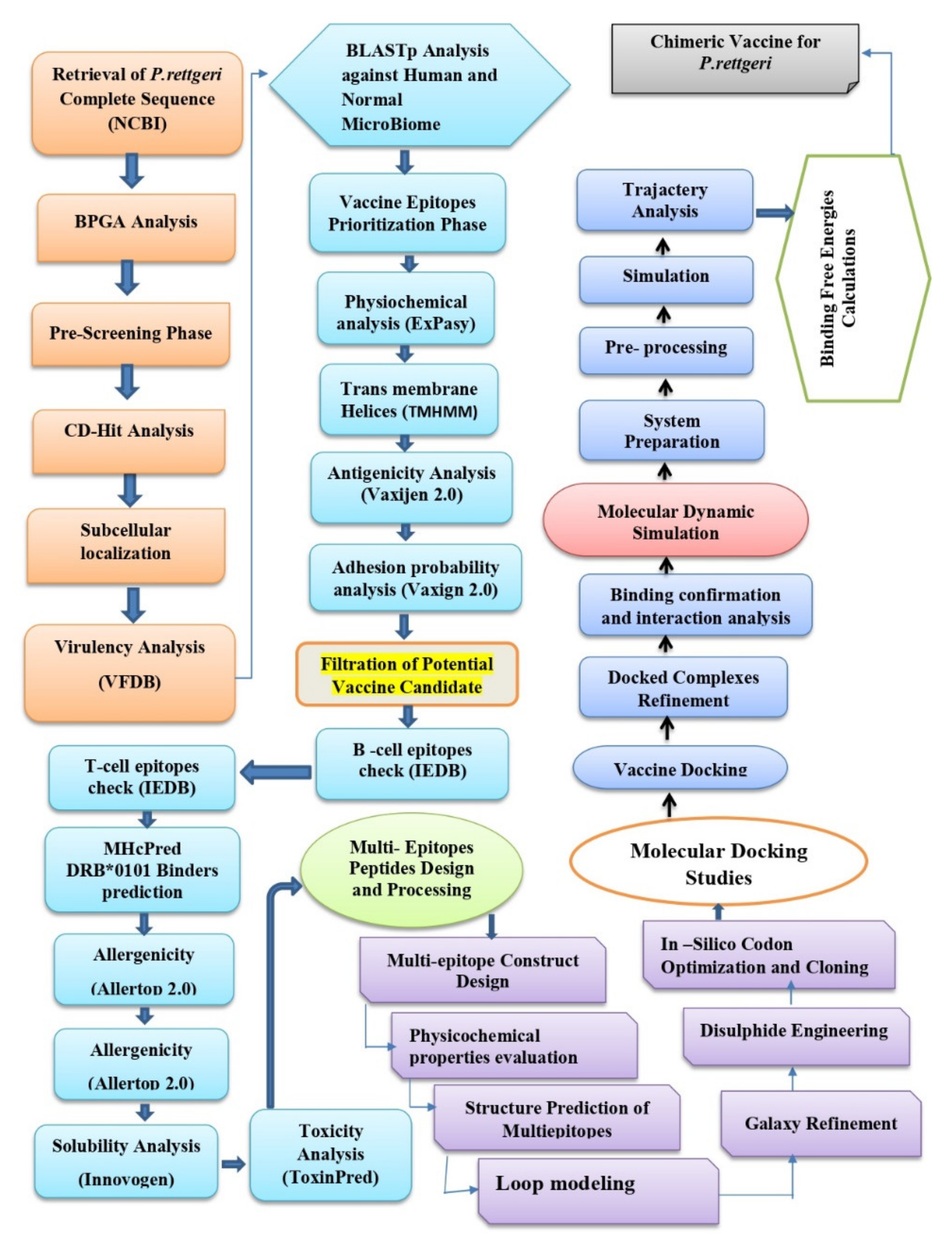

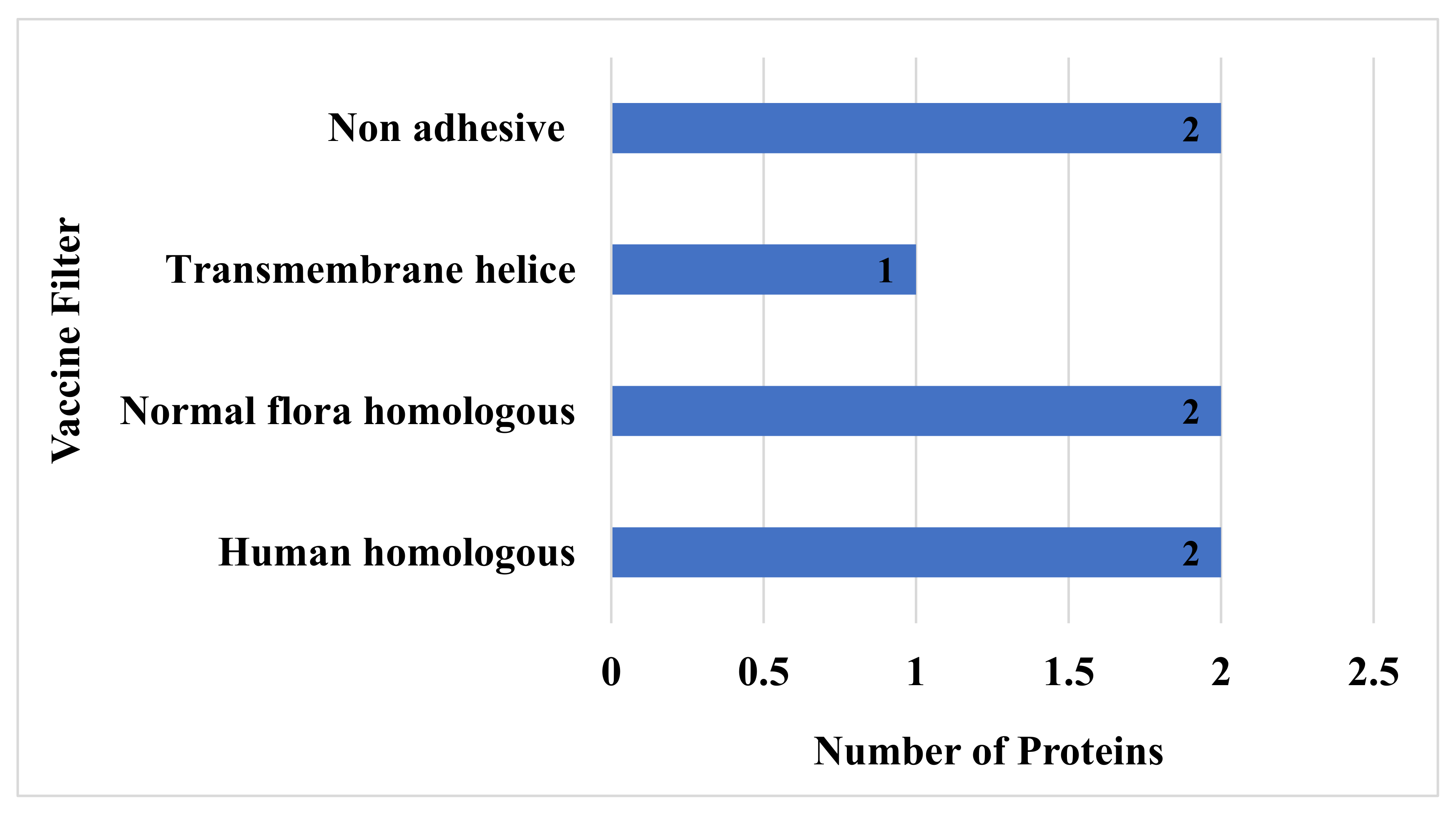
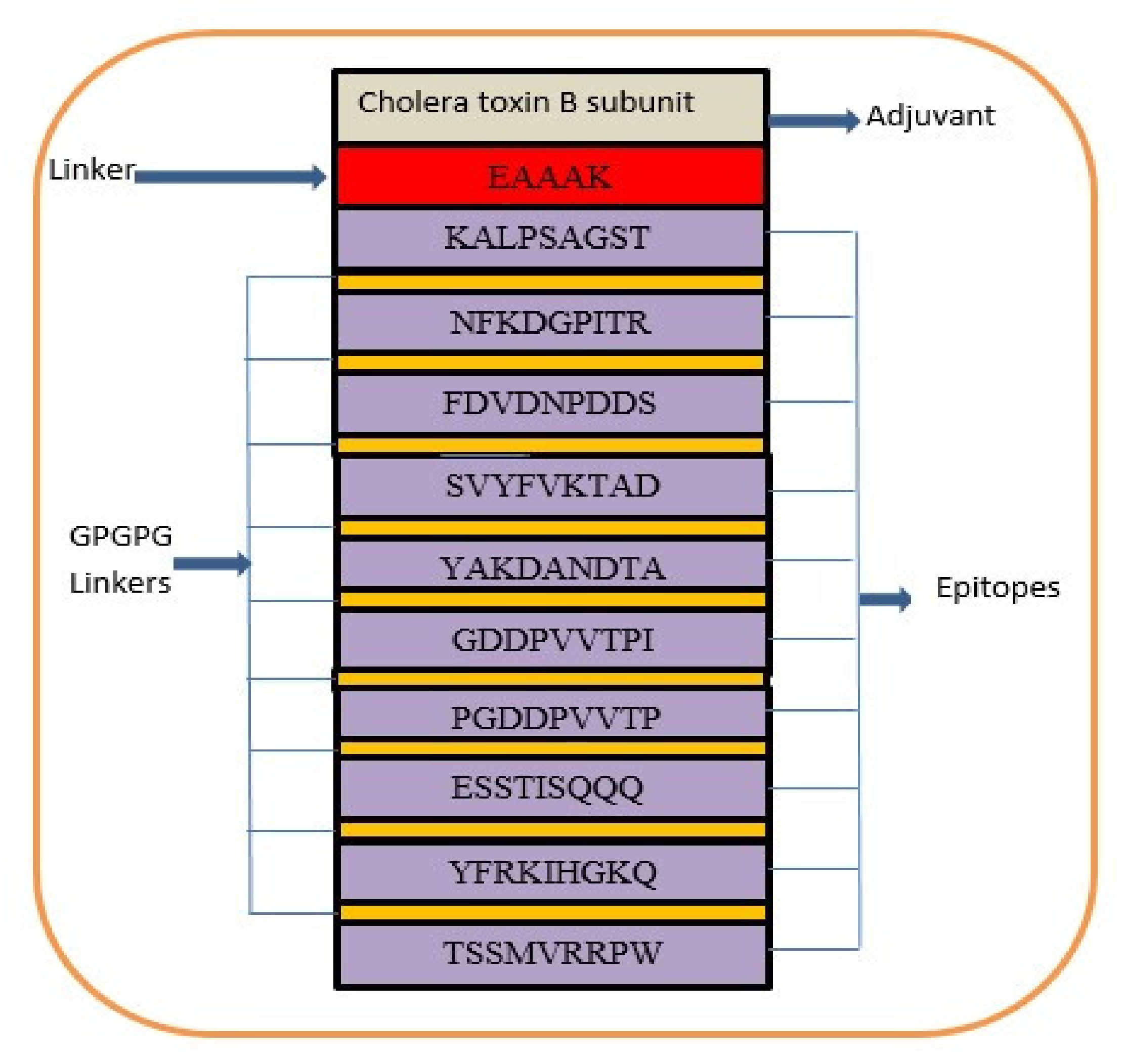
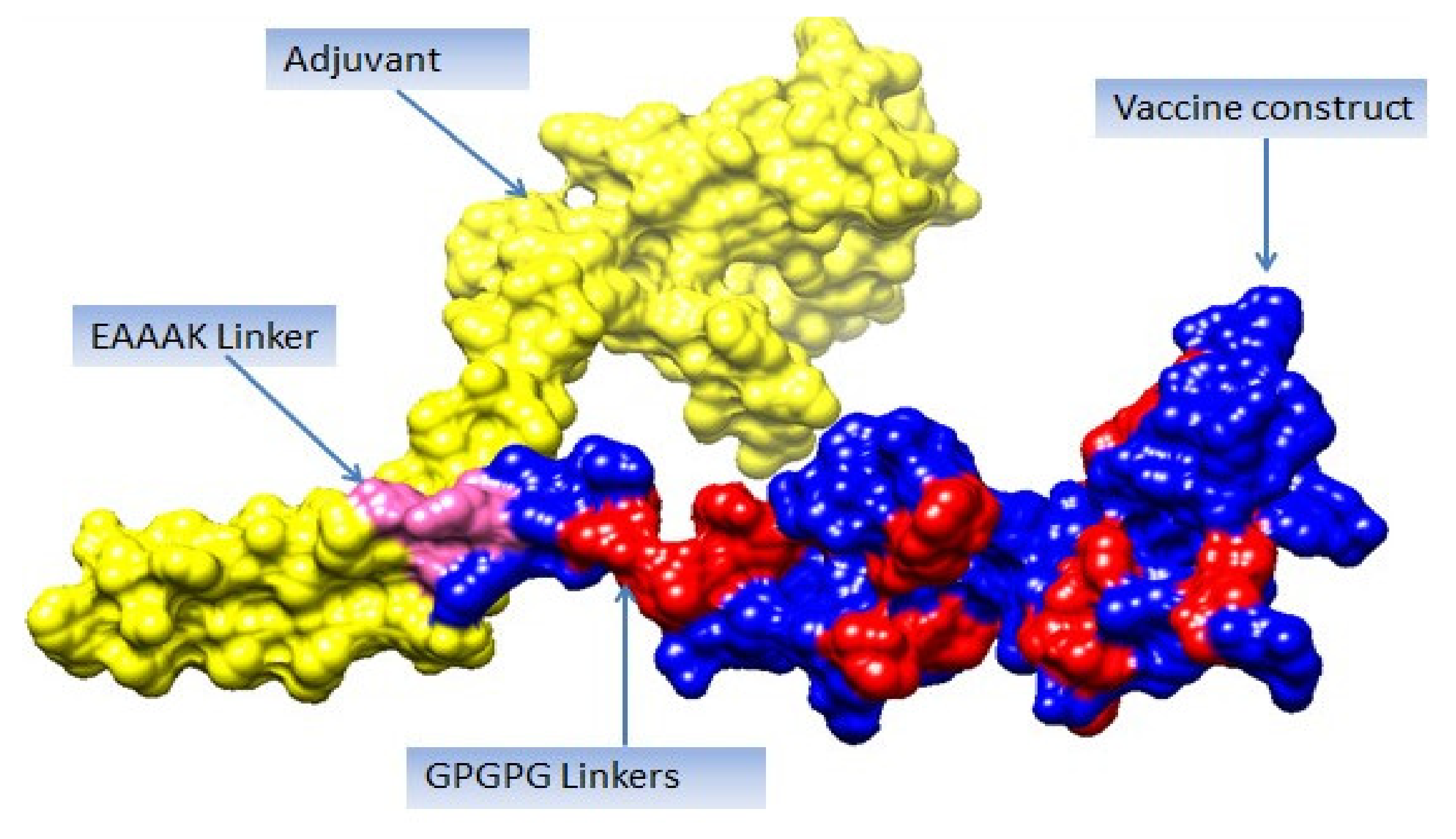
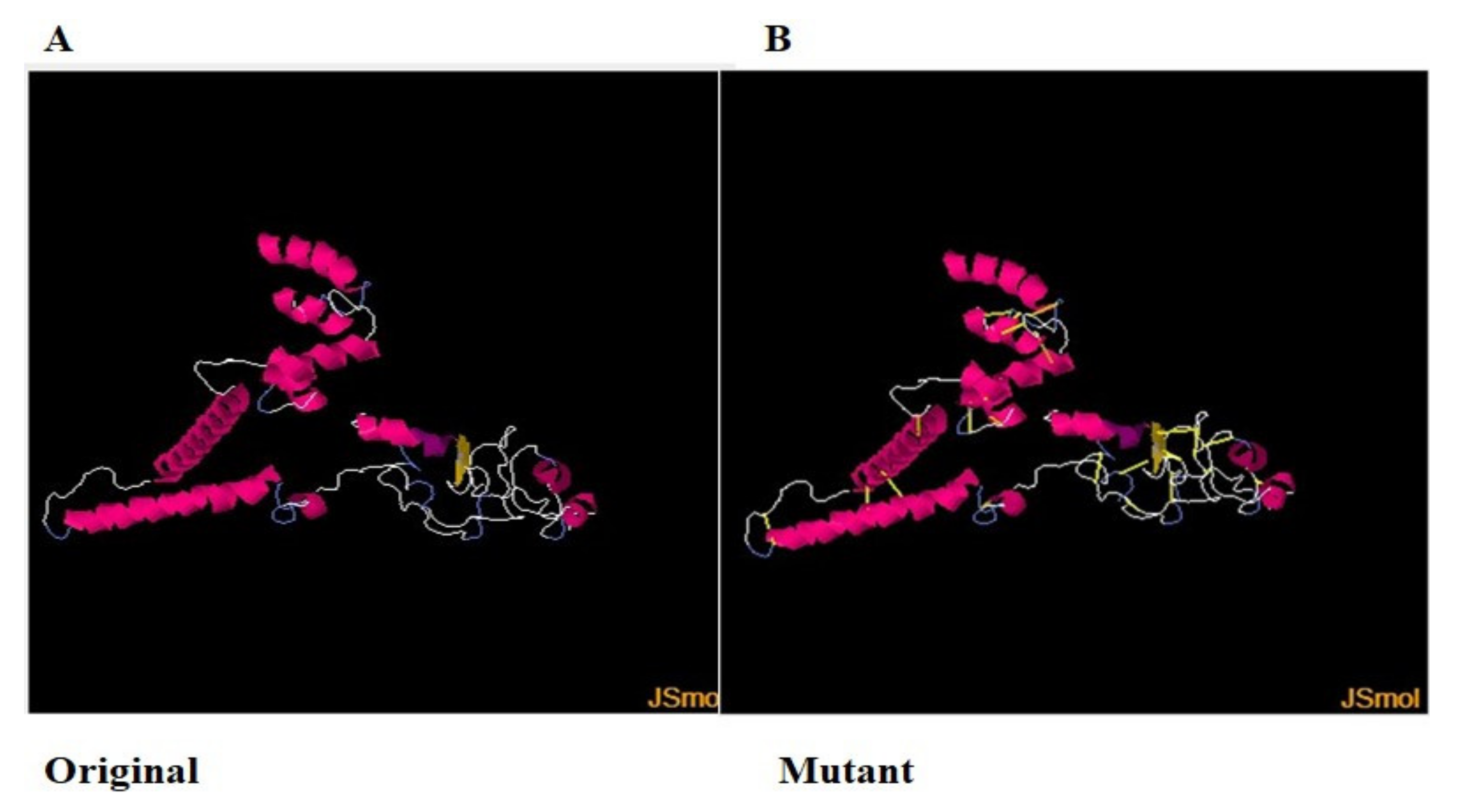
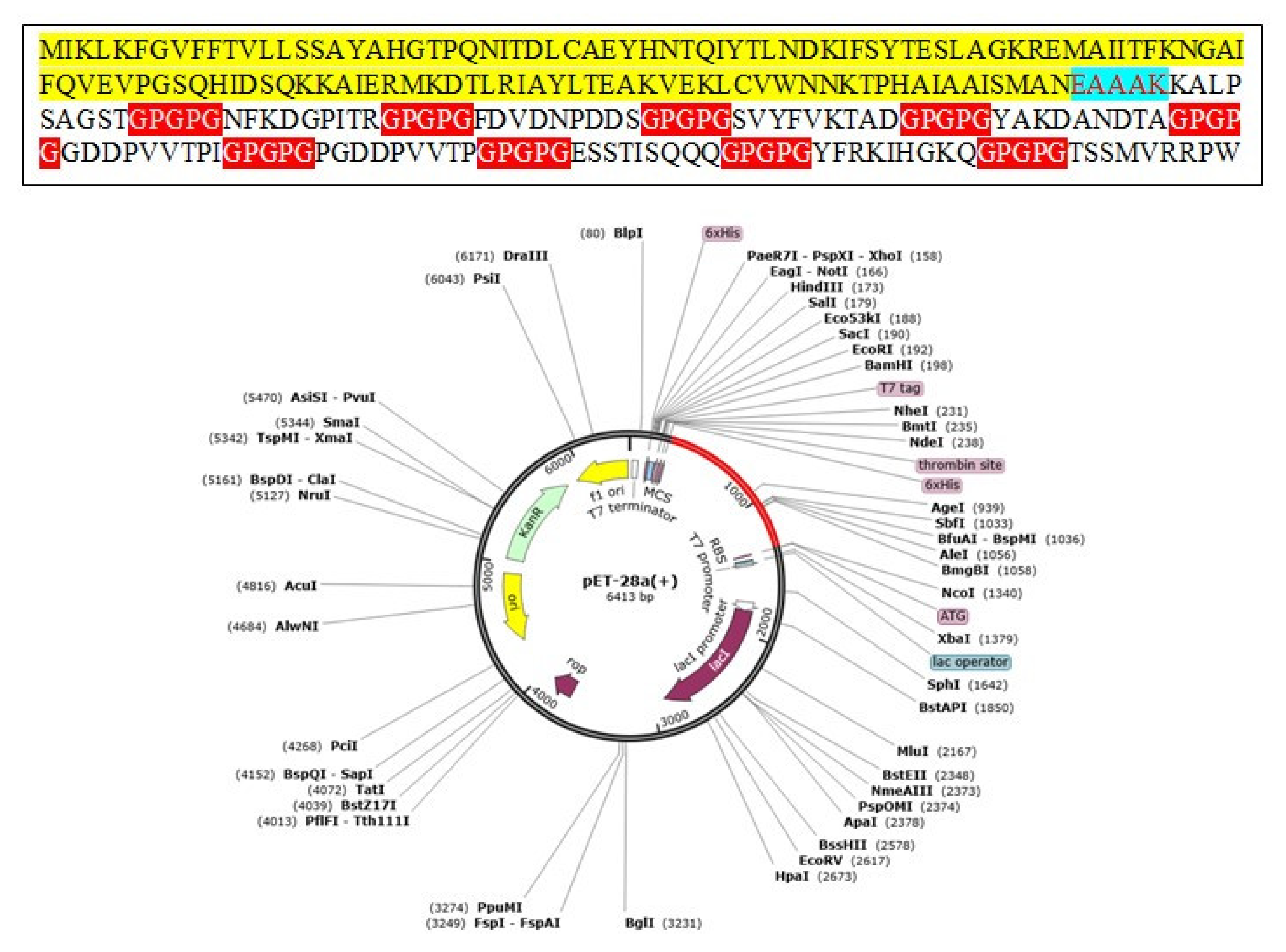
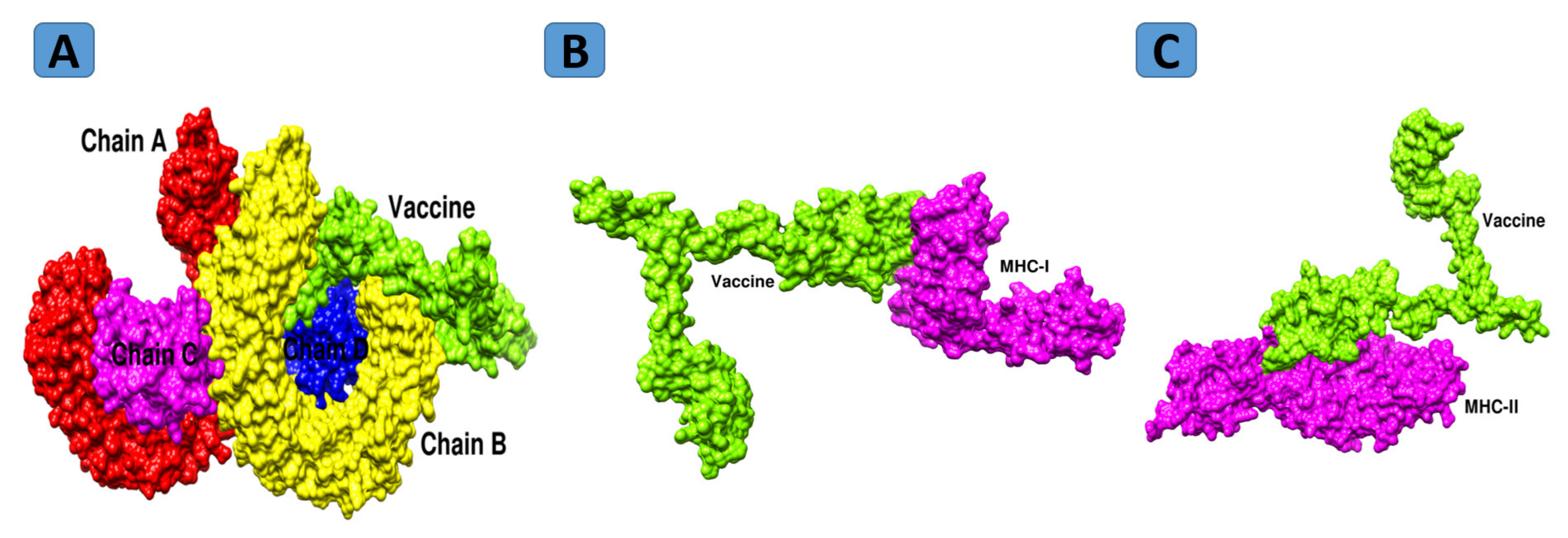
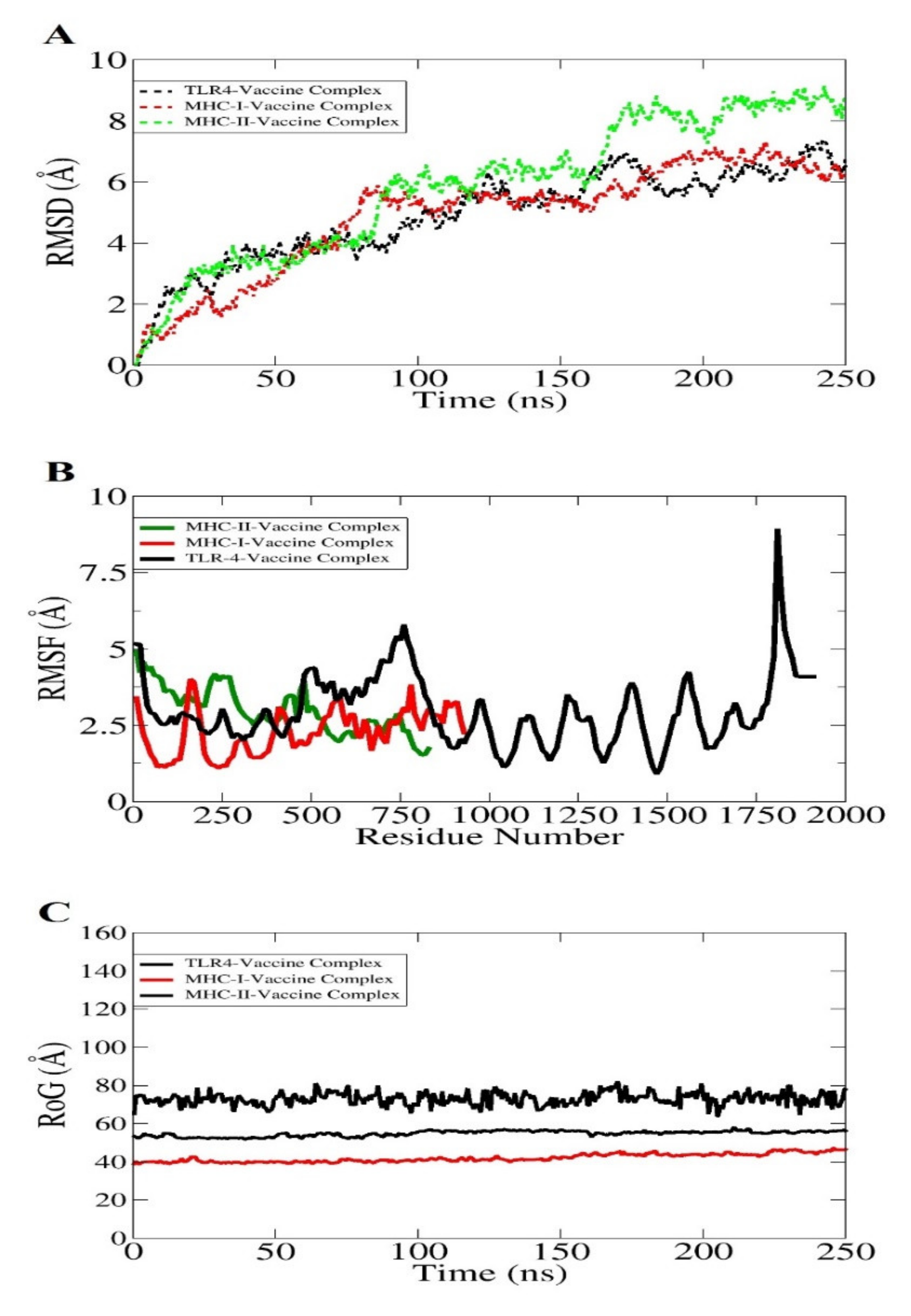
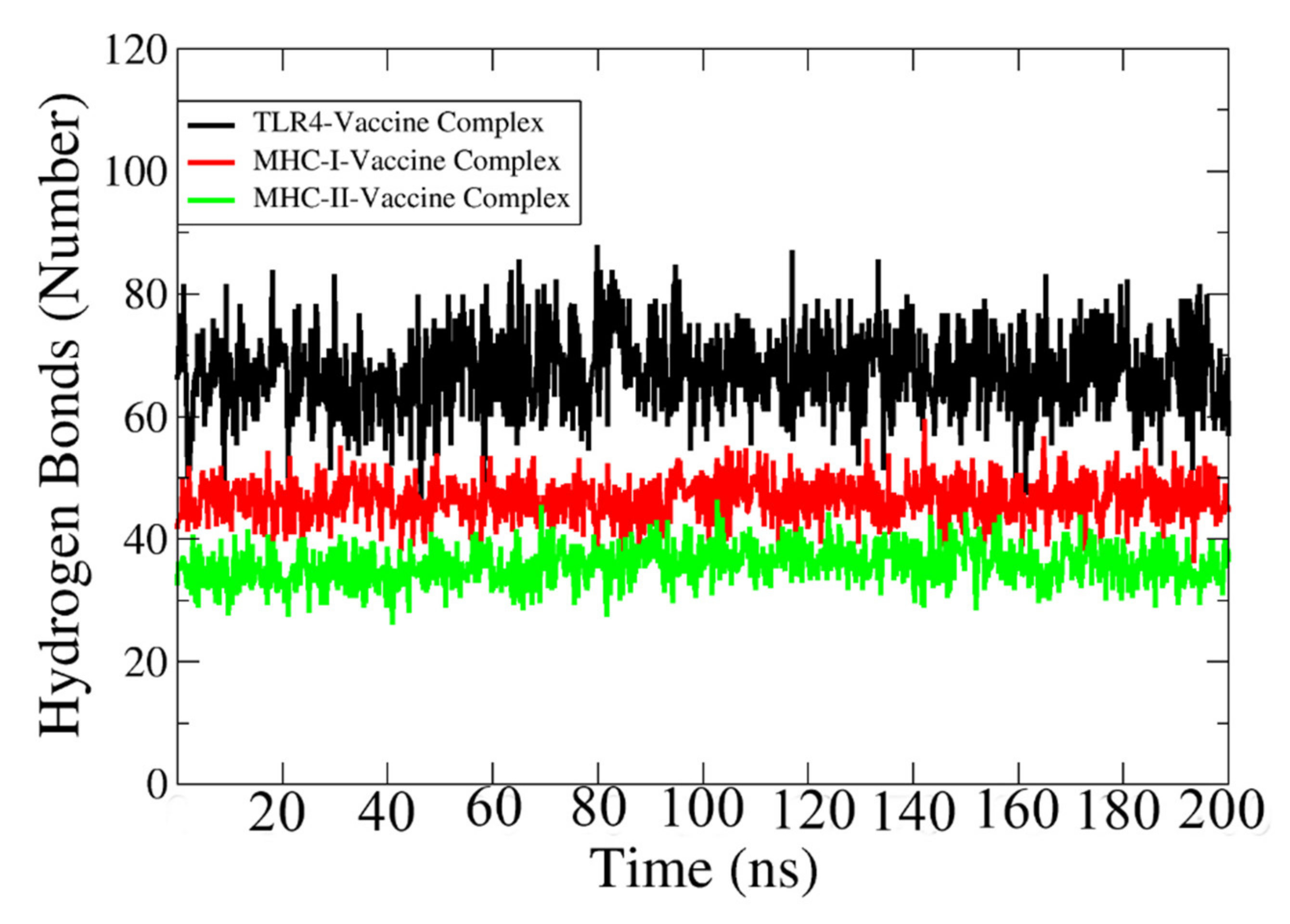
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).