
Cytotoxic T-Cell-Based Vaccine against SARS-CoV-2: A Hybrid Immunoinformatic Approach

Abstract
:1. Introduction
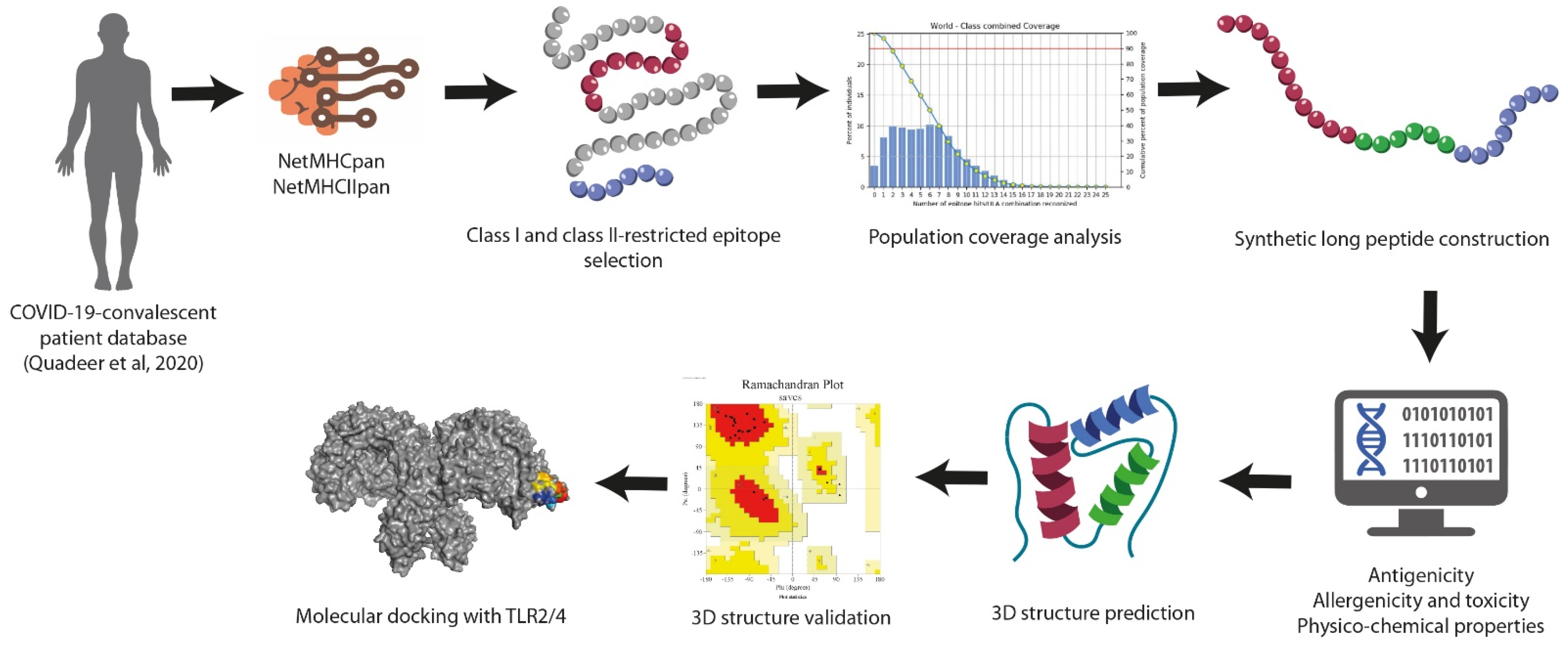
2. Materials and Methods
2.1. Epitope Screening
- Degree of conservation (so that mutations identified in various SARS-CoV-2 will not influence the antigen processing and presentation significantly);
- Cross-specificity—multiple HLA allele coverage.
2.2. Population Coverage Analysis
2.3. Synthetic Long Peptide Construction
- The HLA class II molecule is much more permissive in terms of epitope sequence length compared to the HLA class I molecule.
- Class I-restricted epitope could undergo further cleavage by ERAP (endoplasmic reticulum aminopeptidase) in the presence of HLA class I molecule inside the endoplasmic reticulum, cleaving the remaining amino acids originating from the linker. As a result, peptides with 9-11 amino acids can fit perfectly to the HLA class I binding groove, as stated by the “molecular ruler” hypothesis [27].
2.4. Allergenicity Screening
2.5. Toxicity Screening
2.6. Physico-Chemical Properties and Antigenicity
2.7. Three-Dimensional Structure Prediction
2.8. Three-Dimensional Structure Validation
2.9. Molecular Docking Studies
3. Results
3.1. 19 Peptides from Convalescent Patients Express High Degree of Conservation, Cross-Specificity and Bind Strongly to HLA Molecules
3.2. 90% Probability That 2 Peptides Will Be Recognized by Any Individual
3.3. SLP Constructs Express High In Silico Immunogenicity and Are Stable under Laboratory Conditions
3.4. Peptide Constructs Did Not Express Allergenicity nor Toxicity
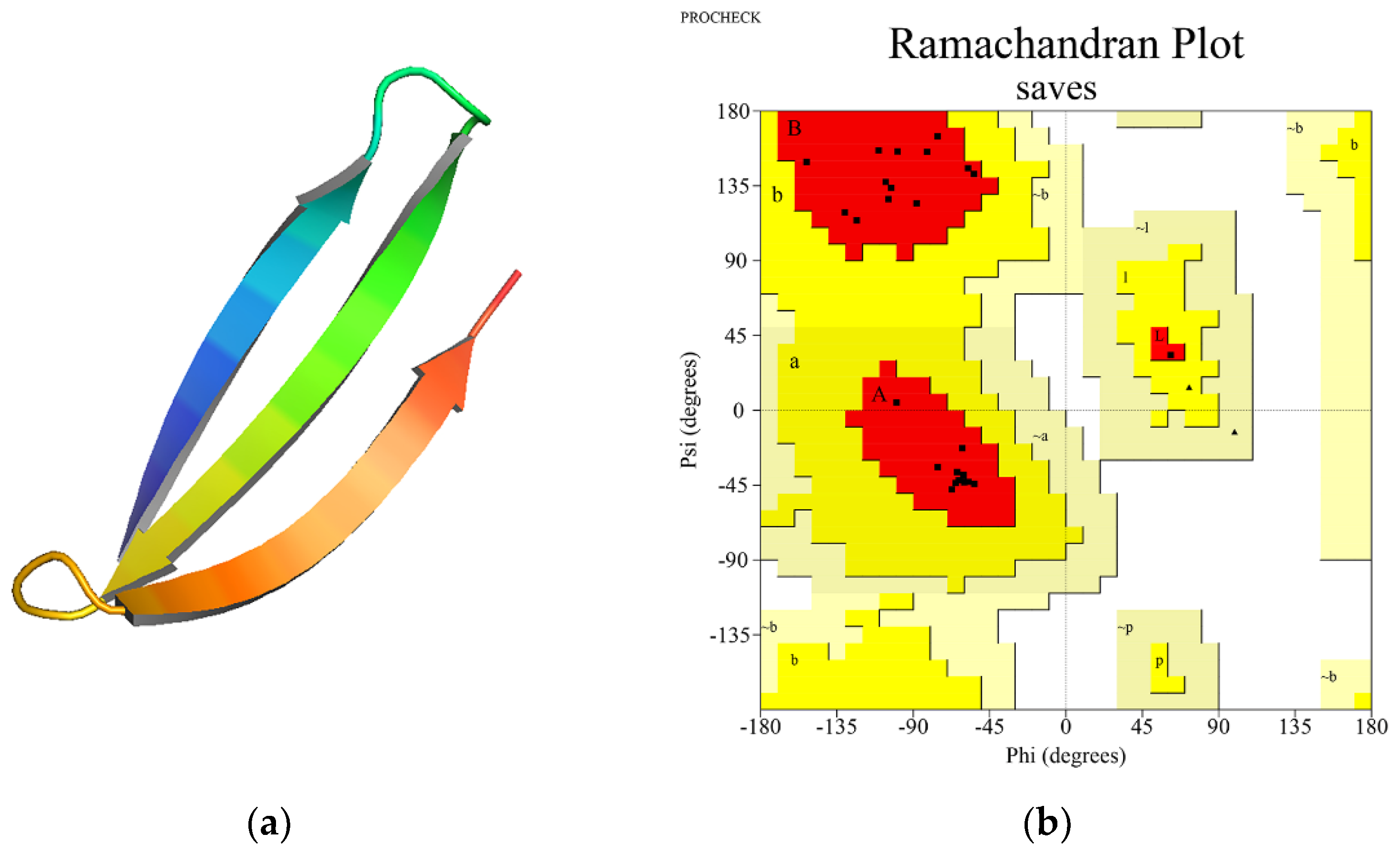
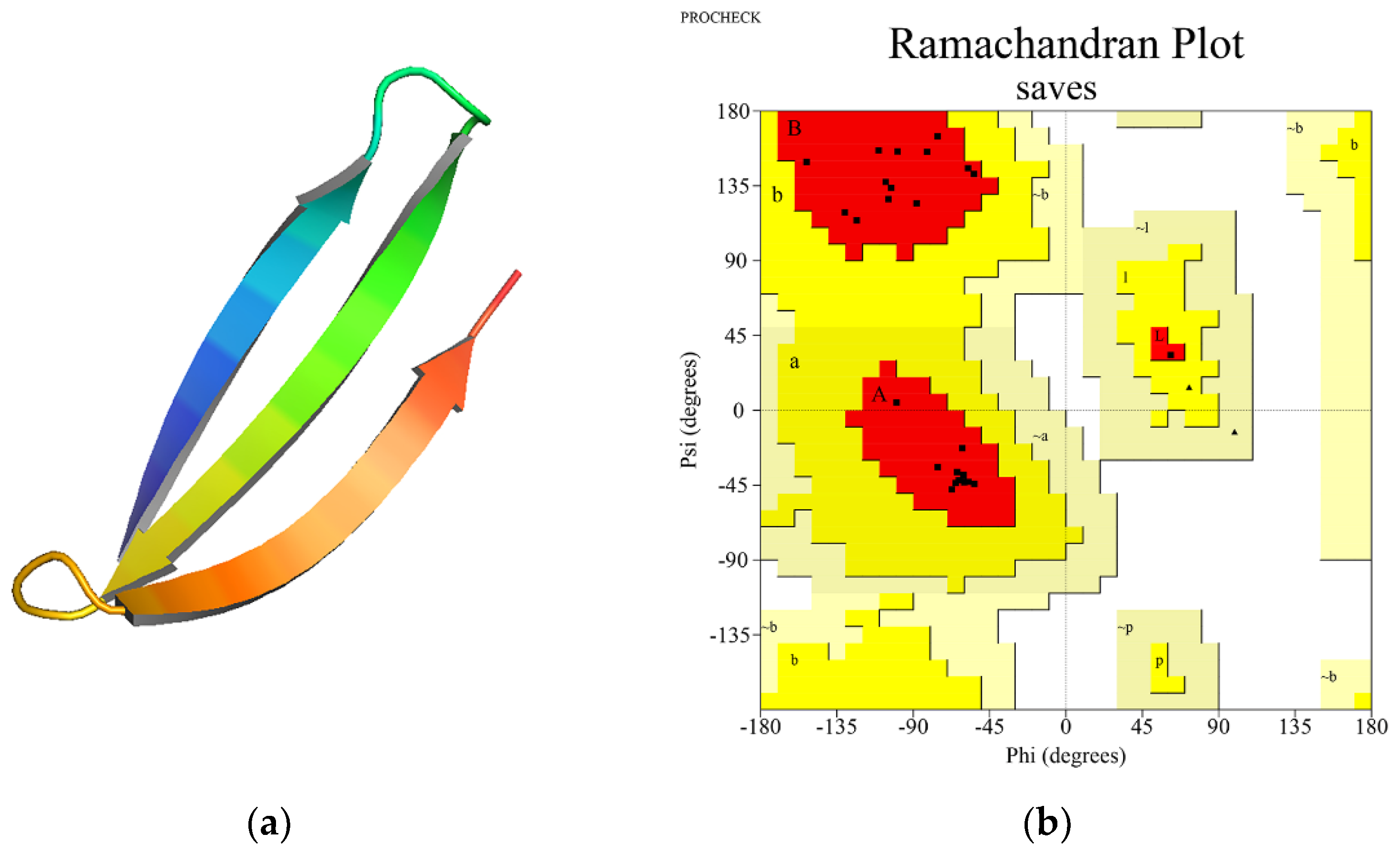
3.5. SLP Three-Dimensional Structure Prediction and Validation
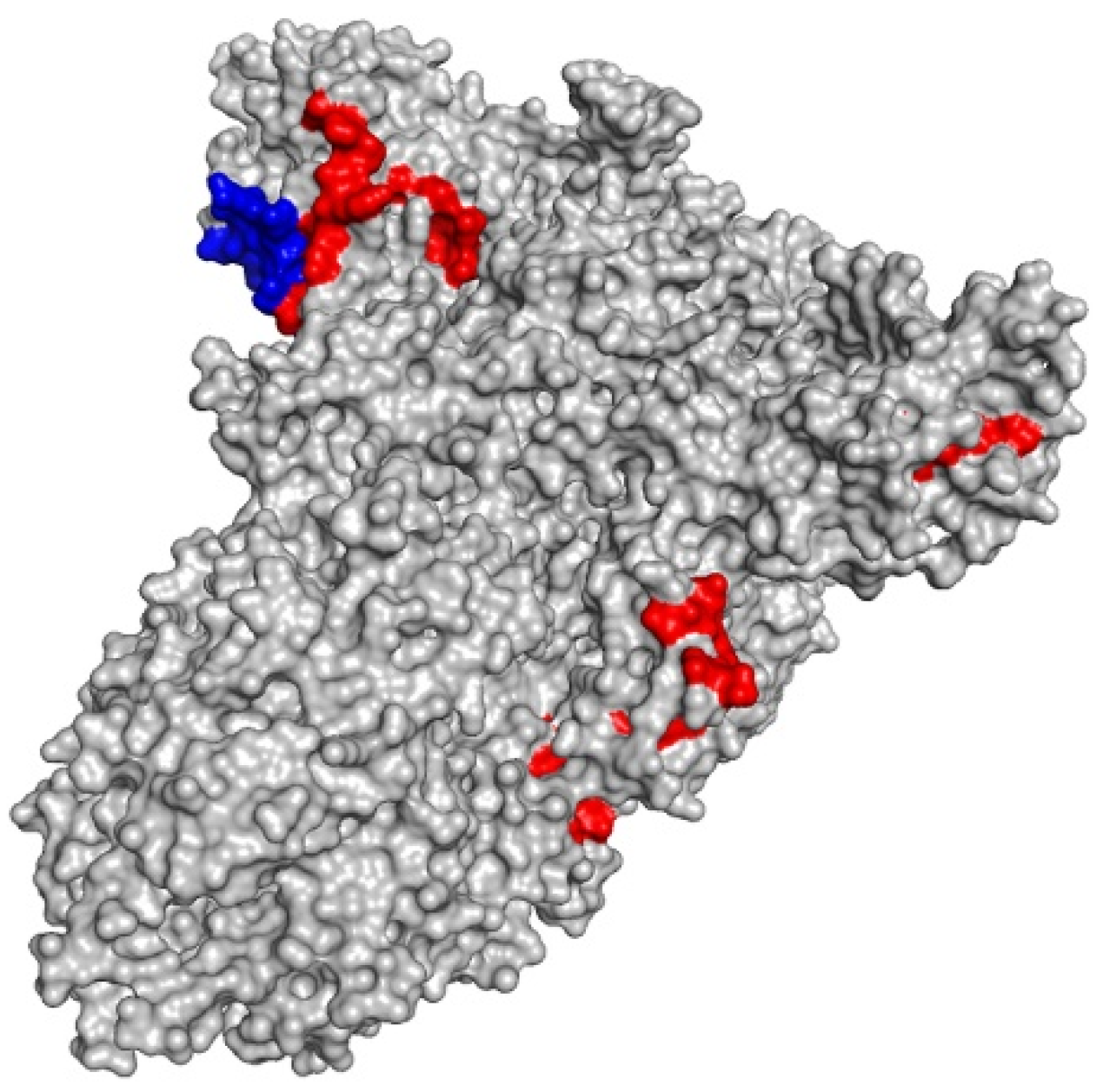
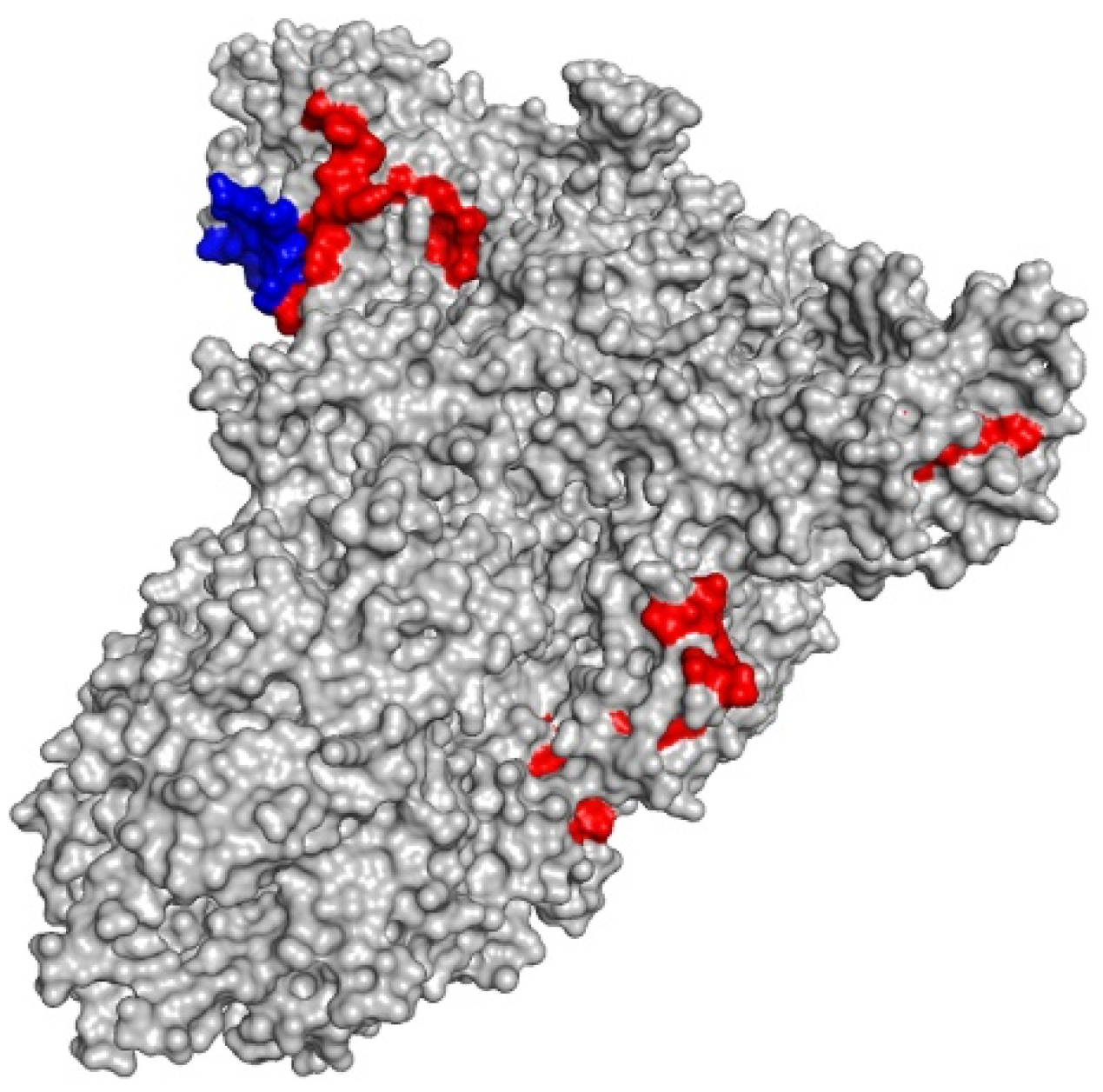
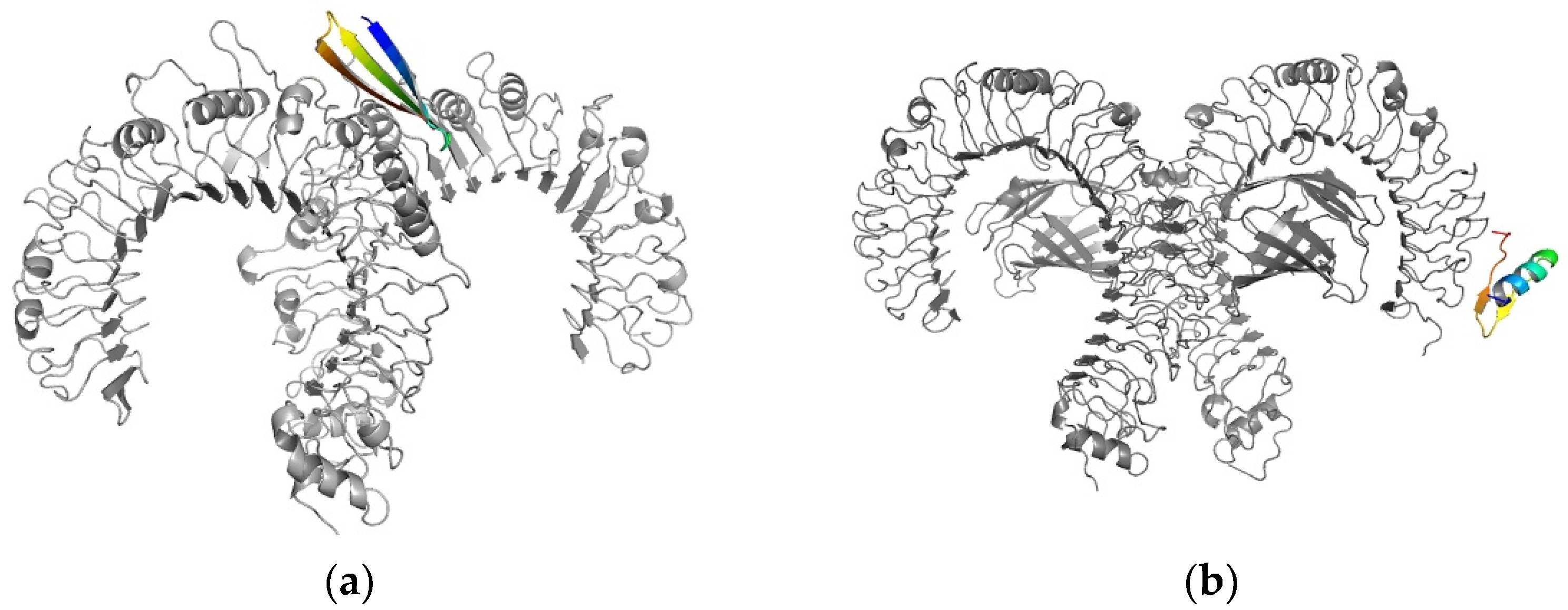
3.6. Syntethic Long Peptides Present Favourable Interaction with Toll-Like Receptors 2 and 4
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- le Bert, N.; Tan, A.T.; Kunasegaran, K.; Tham, C.Y.L.; Hafezi, M.; Chia, A.; Chng, M.H.Y.; Lin, M.; Tan, N.; Linster, M.; et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 2020, 584, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Stadnytskyi, V.; Bax, C.E.; Bax, A.; Anfinrud, P. The airborne lifetime of small speech droplets and their potential importance in SARS-CoV-2 transmission. Proc. Natl. Acad. Sci. USA 2020, 117, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. COVID-19: Pfizer’s paxlovid is 89% effective in patients at risk of serious illness, company reports. BMJ 2021, 375, n2713. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.A.; Eron, J., Jr.; Holman, W.; Cohen, M.S.; Fang, L.; Szewczyk, L.J.; Sheahan, T.P.; Baric, R.; Mollan, K.R.; Wolfe, C.R.; et al. CORONAVIRUS A Phase 2a clinical trial of Molnupiravir in patients with COVID-19 shows accelerated SARS-CoV-2 RNA clearance and elimination of infectious virus. Sci. Transl. Med. 2021, 14, eabl7430. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.-C.; Liu, W.; Zhang, P.-H.; Zhang, F.; Richardus, J.H. Disappearance of Antibodies to SARS-Associated Coronavirus after Recovery. N. Engl. J. Med. 2007, 357, 1162–1163. [Google Scholar] [CrossRef]
- Ng, O.; Chia, A.; Tan, A.T.; Jadi, R.S.; Nam, H.; Bertoletti, A.; Leong, H.N.; Tan, Y.-J. Memory T cell responses targeting the SARS coronavirus persist up to 11 years post-infection. Vaccine 2016, 34, 2008–2014. [Google Scholar] [CrossRef]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Strålin, K.; Gorin, J.B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. bioRxiv 2020, 183, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Morgulchik, N.; Athanasopoulou, F.; Chu, E.; Lam, Y.; Kamaly, N. Potential therapeutic approaches for targeted inhibition of inflammatory cytokines following COVID-19 infection-induced cytokine storm. Interface Focus 2021, 12, 20210006. [Google Scholar] [CrossRef] [PubMed]
- Savage, H.R.; Santos, V.S.; Edwards, T.; Giorgi, E.; Krishna, S.; Planche, T.D.; Staines, H.M.; Fitchett, J.R.; Kirwan, D.E.; Atienzar, A.I.C.; et al. Prevalence of neutralising antibodies against SARS-CoV-2 in acute infection and convalescence: A systematic review and meta-analysis. PLoS Negl. Trop. Dis. 2021, 15, e0009551. [Google Scholar] [CrossRef]
- Farahani, M.; Niknam, Z.; Mohammadi Amirabad, L.; Amiri-Dashatan, N.; Koushki, M.; Nemati, M.; Pouya, F.D.; Rezaei-Tavirani, M.; Rasmi, Y.; Tayebi, L. Molecular pathways involved in COVID-19 and potential pathway-based therapeutic targets. Biomed. Pharmacother. 2022, 145, 112420. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Toth, I. Non-invasive mucosal vaccine delivery: Advantages, challenges and the future. Expert Opin. Drug Deliv. 2020, 17, 435–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Jang, Y.S. The development of mucosal vaccines for both mucosal and systemic immune induction and the roles played by adjuvants. Clin. Exp. Vaccine Res. 2017, 6, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Yang, J.; Wang, L.; Liu, B. Personalized neoantigen vaccination with synthetic long peptides: Recent advances and future perspectives. Theranostics 2020, 10, 6011–6023. [Google Scholar] [CrossRef] [PubMed]
- Rabu, C.; Rangan, L.; Florenceau, L.; Fortun, A.; Charpentier, M.; Dupré, E.; Paolini, L.; Beauvillain, C.; Dupel, E.; Latouche, J.-B.; et al. Cancer vaccines: Designing artificial synthetic long peptides to improve presentation of class I and class II T cell epitopes by dendritic cells. OncoImmunology 2019, 8, e1560919. [Google Scholar] [CrossRef]
- Coppola, M.; van den Eeden, S.J.F.; Wilson, L.; Franken, K.L.M.C.; Ottenhoff, T.H.M.; Geluk, A. Synthetic long peptide derived from Mycobacterium tuberculosis latency antigen Rv1733c protects against tuberculosis. Clin. Vaccine Immunol. 2015, 22, 1060–1069. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Khan, S.; Ali, A.; Akbar, H.; Sayaf, A.M.; Khan, A.; Wei, D.-Q. Immunoinformatics approaches to explore Helicobacter Pylori proteome (Virulence Factors) to design B and T cell multi-epitope subunit vaccine. Sci. Rep. 2019, 9, 13321. [Google Scholar] [CrossRef]
- Nezafat, N.; Karimi, Z.; Eslami, M.; Mohkam, M.; Zandian, S.; Ghasemi, Y. Designing an efficient multi-epitope peptide vaccine against Vibrio cholerae via combined immunoinformatics and protein interaction based approaches. Comput. Biol. Chem. 2016, 62, 82–95. [Google Scholar] [CrossRef]
- Pandey, R.K.; Bhatt, T.K.; Prajapati, V.K. Novel Immunoinformatics Approaches to Design Multi-epitope Subunit Vaccine for Malaria by Investigating Anopheles Salivary Protein. Sci. Rep. 2018, 8, 1125. [Google Scholar] [CrossRef] [Green Version]
- Tosta, S.F.D.O.; Passos, M.S.; Kato, R.; Salgado, Á.; Xavier, J.; Jaiswal, A.K.; Soares, S.C.; Azevedo, V.; Giovanetti, M.; Tiwari, S.; et al. Multi-epitope based vaccine against yellow fever virus applying immunoinformatics approaches. J. Biomol. Struct. Dyn. 2020, 39, 219–235. [Google Scholar] [CrossRef]
- Abraham Peele, K.; Srihansa, T.; Krupanidhi, S.; Vijaya Sai, A.; Venkateswarulu, T.C. Design of multi-epitope vaccine candidate against SARS-CoV-2: A in-silico study. J. Biomol. Struct. Dyn. 2020, 39, 3793–3801. [Google Scholar] [CrossRef] [PubMed]
- Naz, A.; Shahid, F.; Butt, T.T.; Awan, F.M.; Ali, A.; Malik, A. Designing Multi-Epitope Vaccines to Combat Emerging Coronavirus Disease 2019 (COVID-19) by Employing Immuno-Informatics Approach. Front. Immunol. 2020, 11, 1663. [Google Scholar] [CrossRef] [PubMed]
- Bojin, F.; Gavriliuc, O.; Mărgineanu, M.-B.; Păunescu, V. Design of an Epitope-Based Synthetic Long Peptide Vaccine to Counteract the Novel China Coronavirus (2019-nCoV). 2020. Available online: https://www.preprints.org/manuscript/202002.0102/v1 (accessed on 4 January 2022).
- Quadeer, A.A.; Ahmed, S.F.; McKay, M.R. Landscape of epitopes targeted by T cells in 852 individuals recovered from COVID-19: Meta-analysis, immunoprevalence, and web platform. Cell Rep. Med. 2021, 2, 100312. [Google Scholar] [CrossRef] [PubMed]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Bui, H.H.; Sidney, J.; Dinh, K.; Southwood, S.; Newman, M.J.; Sette, A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006, 7, 153. [Google Scholar] [CrossRef] [Green Version]
- Evnouchidou, I.; van Endert, P. Peptide trimming by endoplasmic reticulum aminopeptidases: Role of MHC class I binding and ERAP dimerization. Hum. Immunol. 2019, 80, 290–295. [Google Scholar] [CrossRef]
- Maurer-Stroh, S.; Krutz, N.L.; Kern, P.S.; Gunalan, V.; Nguyen, M.N.; Limviphuvadh, V.; Gerberick, G. AllerCatPro-prediction of protein allergenicity potential from the protein sequence. Bioinformatics 2019, 35, 3020–3027. [Google Scholar] [CrossRef] [Green Version]
- Guruprasad, K.; Reddy, B.V.B.; Pandit, M.W. Correlation between stability of a protein and its dipeptide composition: A novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. Des. Sel. 1990, 4, 155–161. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, 526–531. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Bradley, P.; Greisen, P., Jr.; Liu, Y.; Mulligan, V.K.; Kim, D.E.; Baker, D.; DiMaio, F. Simultaneous optimization of biomolecular energy function on features from small molecules and macromolecules. J. Chem. Theory Comput. 2016, 12, 6201–6212. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Wu, S.; Zhang, Y. Ab initio protein structure prediction. In From Protein Structure to Function with Bioinformatics; Springer: Dordrecht, the Netherlands, 2009; pp. 3–25. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.; van Dijk, M.; de Vries, S.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Wang, Y.; Wang, J.; Mifune, Y.; Morin, M.D.; Jones, B.T.; Moresco, E.M.Y.; Boger, D.L.; Beutler, B.; Zhang, H. Structural Basis of TLR2/TLR1 Activation by the Synthetic Agonist Diprovocim. J. Med. Chem. 2019, 62, 2938–2949. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.S.; Lee, H.; Lee, J.O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein-protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [PubMed]
- Vangone, A.; Bonvin, A.M.J.J. Contacts-based prediction of binding affinity in protein–protein complexes. eLife 2015, 4, e07454. [Google Scholar] [CrossRef]
- Topuzoğullari, M.; Acar, T.; Pelit Arayici, P.; Uçar, B.; Uğurel, E.; Abamor, E.Ş.; Arasoğlu, T.; Balik-Turgut, D.; Derman, S. An insight into the epitope-based peptide vaccine design strategy and studies against COVID-19. Turk. J. Biol. 2020, 44, 215–227. [Google Scholar] [CrossRef]
- Raies, A.B.; Bajic, V.B. In silico toxicology: Computational methods for the prediction of chemical toxicity. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2016, 6, 147–172. [Google Scholar] [CrossRef] [Green Version]
- Alshammary, A.F.; Al-Sulaiman, A.M. The journey of SARS-CoV-2 in human hosts: A review of immune responses, immunosuppression, and their consequences. Virulence 2021, 12, 1771–1794. [Google Scholar] [CrossRef] [PubMed]
- Dong Kim, K.; Zhao, J.; Auh, S.; Yang, X.; Du, P.; Tang, H.; Fu, Y.-X. Adaptive immune cells temper initial innate responses. Nat. Med. 2007, 13, 1248–1252. [Google Scholar] [CrossRef]
- Ascough, S.; Vlachantoni, I.; Kalyan, M.; Haijema, B.J.; Wallin-Weber, S.; Dijkstra-Tiekstra, M.; Ahmed, M.S.; Van Roosmalen, M.; Grimaldi, R.; Zhang, Q.; et al. Local and Systemic Immunity against Respiratory Syncytial Virus Induced by a Novel Intranasal Vaccine A Randomized, Double-Blind, Placebo-controlled Clinical Trial. Am. J. Respir. Crit. Care Med. 2019, 200, 481–492. [Google Scholar] [CrossRef]
- Ashraf, M.U.; Kim, Y.; Kumar, S.; Seo, D.; Ashraf, M.; Bae, Y.S. COVID-19 vaccines (Revisited) and oral-mucosal vector system as a potential vaccine platform. Vaccines 2021, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Sequence | HLA Class | Start | End | HLA Alleles | Protein | Conservation |
|---|---|---|---|---|---|---|
| WTAGAAAYY | I | 258 | 266 | A *01:01, A *26:01, A *29:02, B *35:01 | S | 0.948138 |
| LTDEMIAQY | I | 865 | 873 | A *01:01, A *29:02, B *35:01, C *07:02 | S | 0.99844 |
| ATSRTLSYY | I | 171 | 179 | A *11:01, A *01:01, B *57:01 | M | 0.998295 |
| LPPAYTNSF | I | 24 | 32 | B *53:01, B *35:01, B *07:02 | S | 0.969828 |
| LSYFIASFR | I | 93 | 101 | A *11:01, A *31:01, A *68:01 | M | 0.998614 |
| NSFTRGVYY | I | 30 | 38 | A *68:01, A *26:01, A *29:02 | S | 0.995425 |
| TSNQVAVLY | I | 604 | 612 | B *57:01, A *26:01, B *35:01 | S | 0.999212 |
| KTFPPTEPK | I | 361 | 369 | A *11:01, A *03:01, A *68:01 | N | 0.973513 |
| VASQSIIAY | I | 687 | 695 | B *35:01, B *15:01, A *29:02 | S | 0.993504 |
| CVADYSVLY | I | 361 | 369 | A *29:02, B *15:01, A *26:01 | S | 0.994539 |
| GVYFASTEK | I | 89 | 97 | A *68:01, A *11:01, A *03:01 | S | 0.957055 |
| RLFRKSNLK | I | 454 | 462 | A *31:01, A *03:01, A *11:01 | S | 0.995434 |
| TISLAGSYK | I | 1504 | 1512 | A *68:01, A *11:01, A *03:01 | ORF1a | 0.987915 |
| LPFNDGVYF | I | 84 | 92 | B *35:01, B *51:01, B *07:02 | S | 0.98813 |
| AEIRASANL | I | 1016 | 1024 | B *40:01, B *44:02, B *44:03 | S | 0.99695 |
| PINLVRDLPQGFSAL | II | 209 | 223 | DRB1 *03:01, DRB3 *01:01 | S | 0.878983 |
| SRTLSYYKLGASQRV | II | 173 | 187 | DRB5 *01:01, DRB5 *01:02 | M | 0.997732 |
| SYYKLGASQRVAGDS ITRFQTLLALHRSYL | II | 177 | 191 | DQA1 *05:01, DQB1 *03:01 DRB1 *01:01, DRB1 *07:01 | M | 0.998787 |
| II | 235 | 249 | DRB1 *01:01 | S | 0.983345 |
| Coverage | Average Hit | PC90 | |
|---|---|---|---|
| Class I | 85.94% | 4.49 | 0.71 |
| Class II | 75.42% | 1.27 | 0.41 |
| Combined | 96.54% | 5.76 | 1.81 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tirziu, A.; Paunescu, V. Cytotoxic T-Cell-Based Vaccine against SARS-CoV-2: A Hybrid Immunoinformatic Approach. Vaccines 2022, 10, 218. https://doi.org/10.3390/vaccines10020218
Tirziu A, Paunescu V. Cytotoxic T-Cell-Based Vaccine against SARS-CoV-2: A Hybrid Immunoinformatic Approach. Vaccines. 2022; 10(2):218. https://doi.org/10.3390/vaccines10020218
Chicago/Turabian StyleTirziu, Alexandru, and Virgil Paunescu. 2022. "Cytotoxic T-Cell-Based Vaccine against SARS-CoV-2: A Hybrid Immunoinformatic Approach" Vaccines 10, no. 2: 218. https://doi.org/10.3390/vaccines10020218
APA StyleTirziu, A., & Paunescu, V. (2022). Cytotoxic T-Cell-Based Vaccine against SARS-CoV-2: A Hybrid Immunoinformatic Approach. Vaccines, 10(2), 218. https://doi.org/10.3390/vaccines10020218