
Immunopeptidomic Analysis of BoLA-I and BoLA-DR Presented Peptides from Theileria parva Infected Cells
 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. BoLA-Defined Cell Lines
2.2. Peptide-BoLA-I and Peptide-BoLA-DR Complex Purification
2.3. High Performance Liquid Chromatography (HPLC) Fractionation
2.4. LC-MS2 Analysis
2.5. MS Data Analysis
2.6. IFNG ELISPOT
2.7. In Vitro Measurement of Peptide-BoLA-I Binding
3. Results
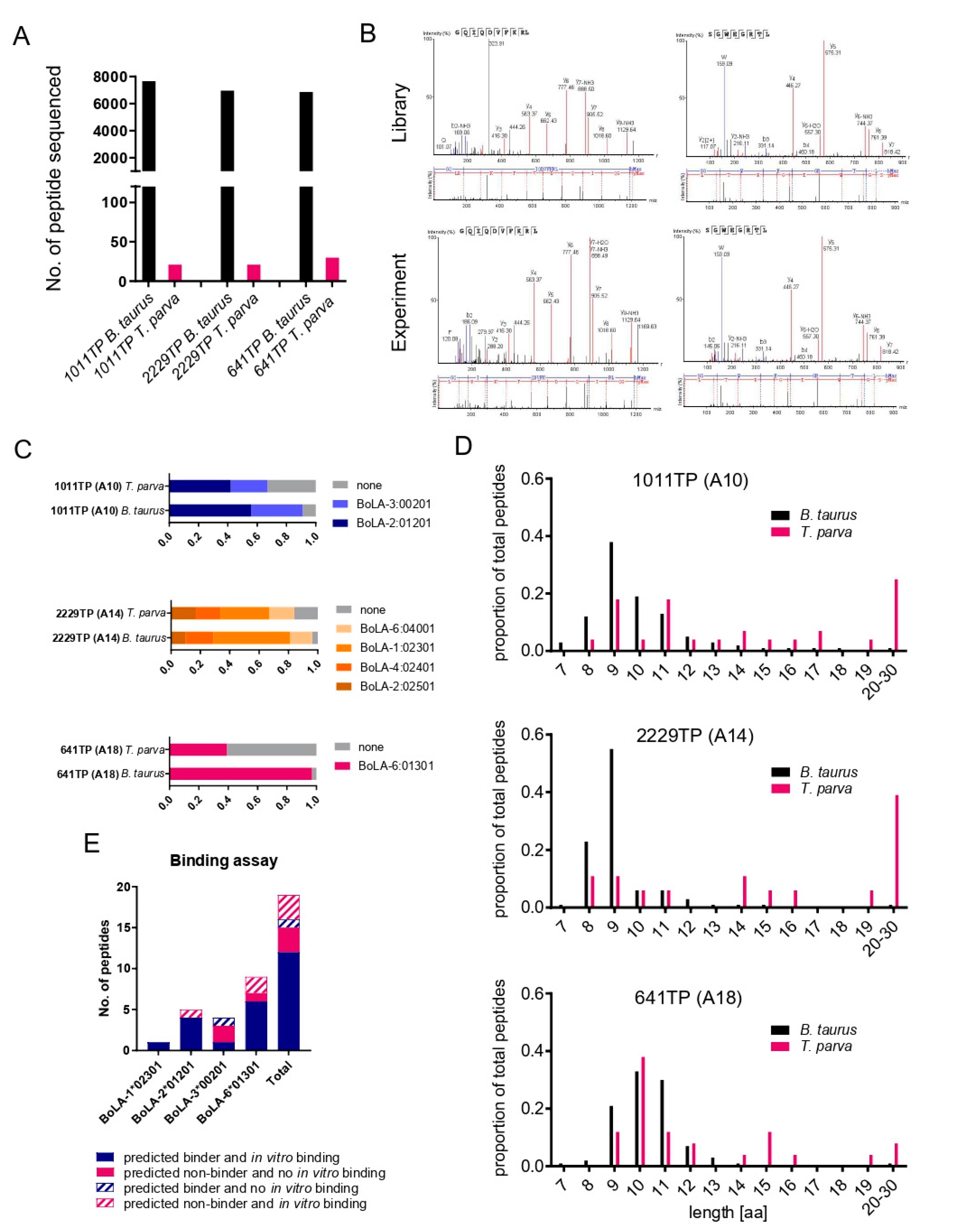
3.1. Identification of BoLA-I Associated Peptides Derived from T. parva
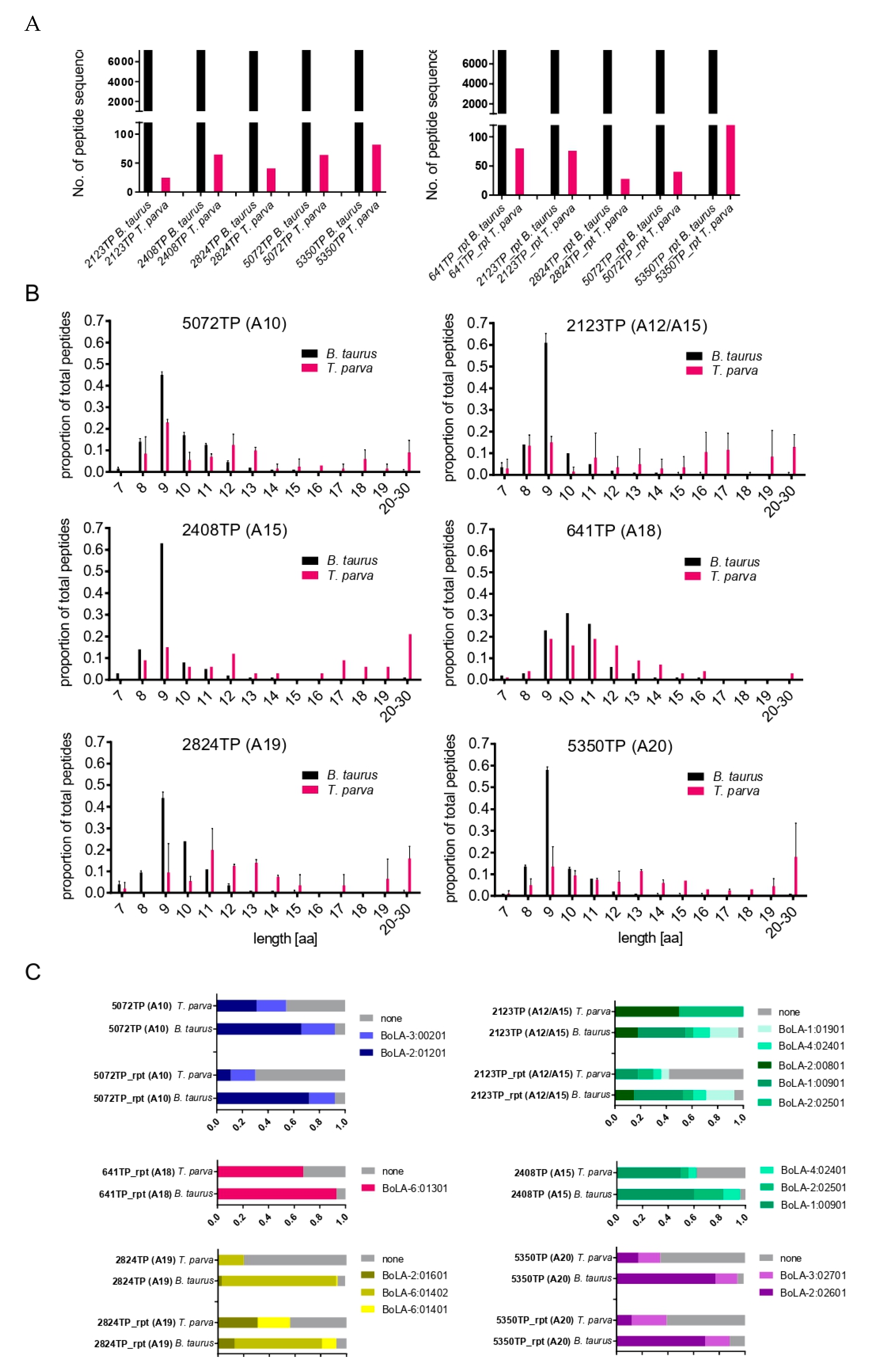
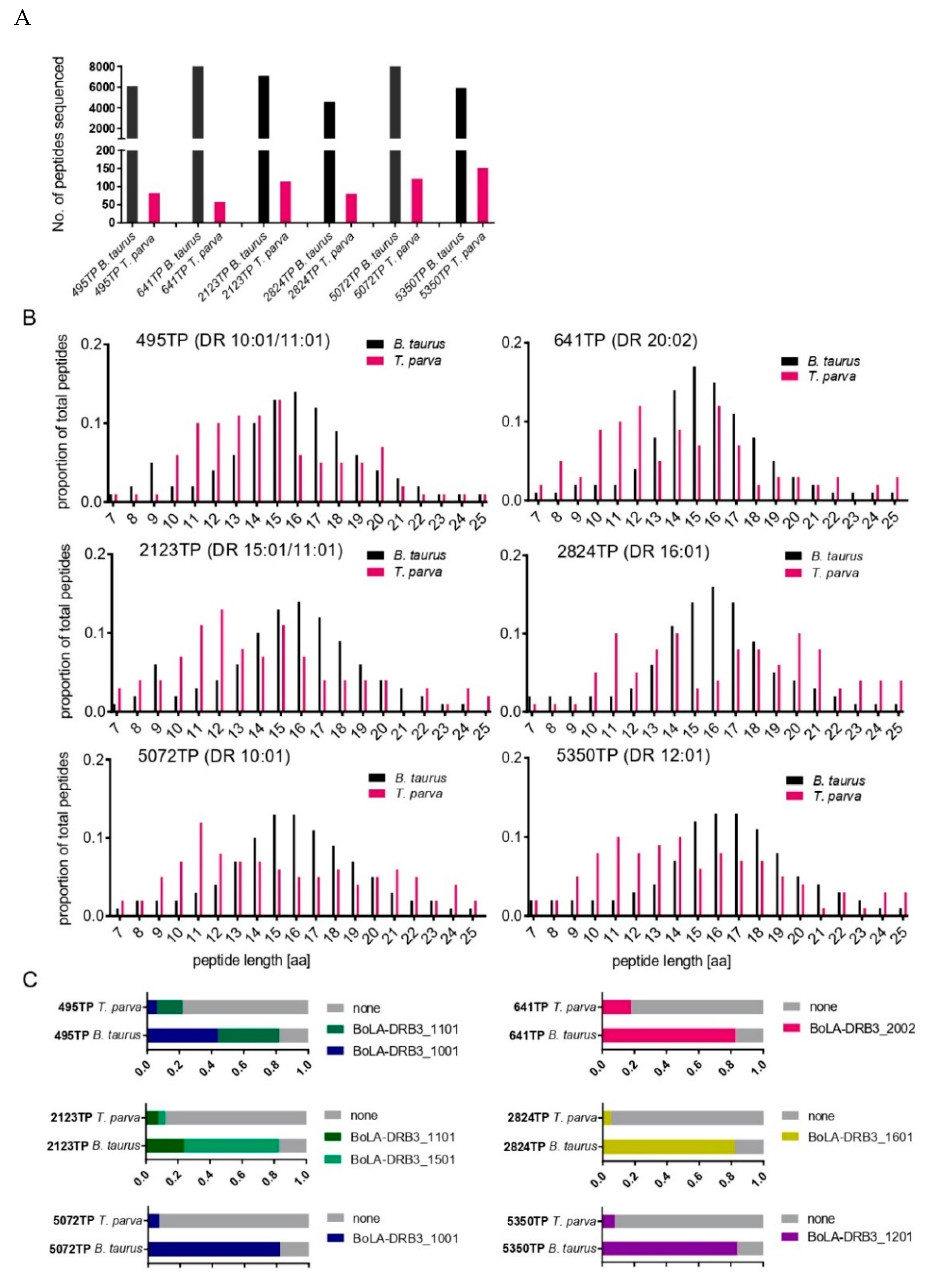
3.2. Immunopeptidome Analysis of Additional T. parva-Infected Cell Lines
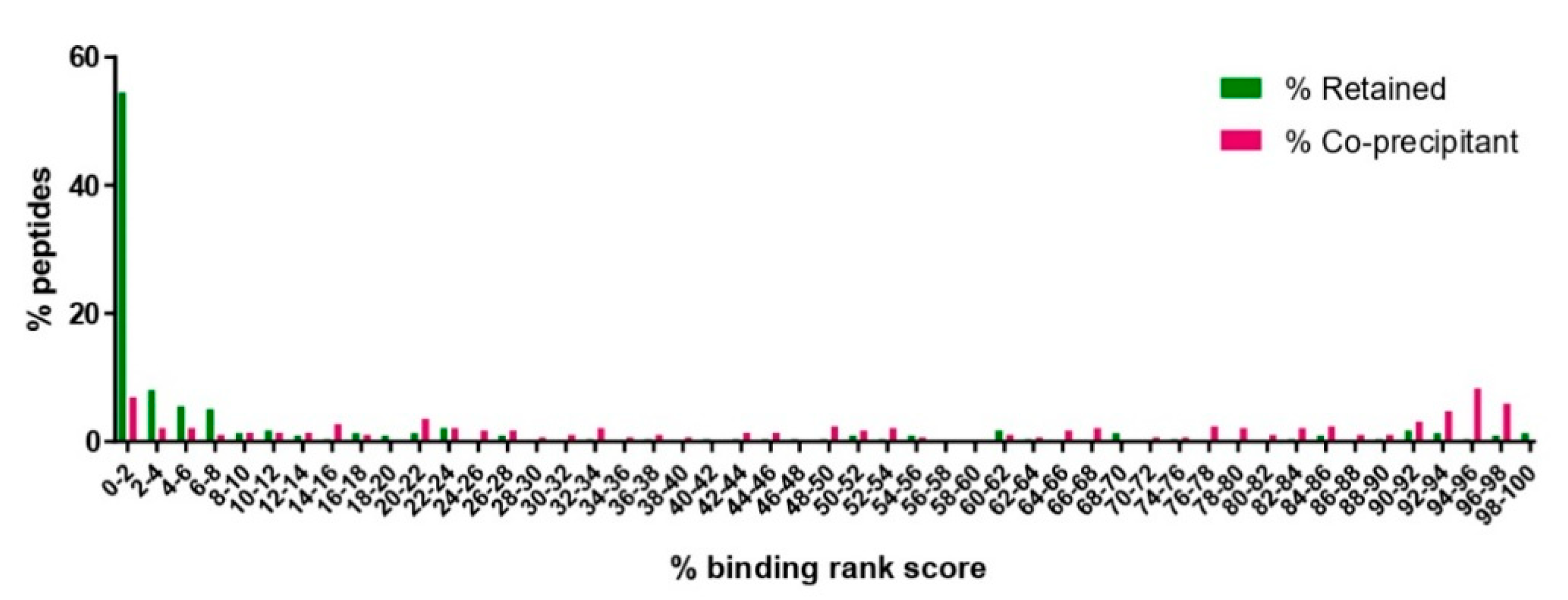
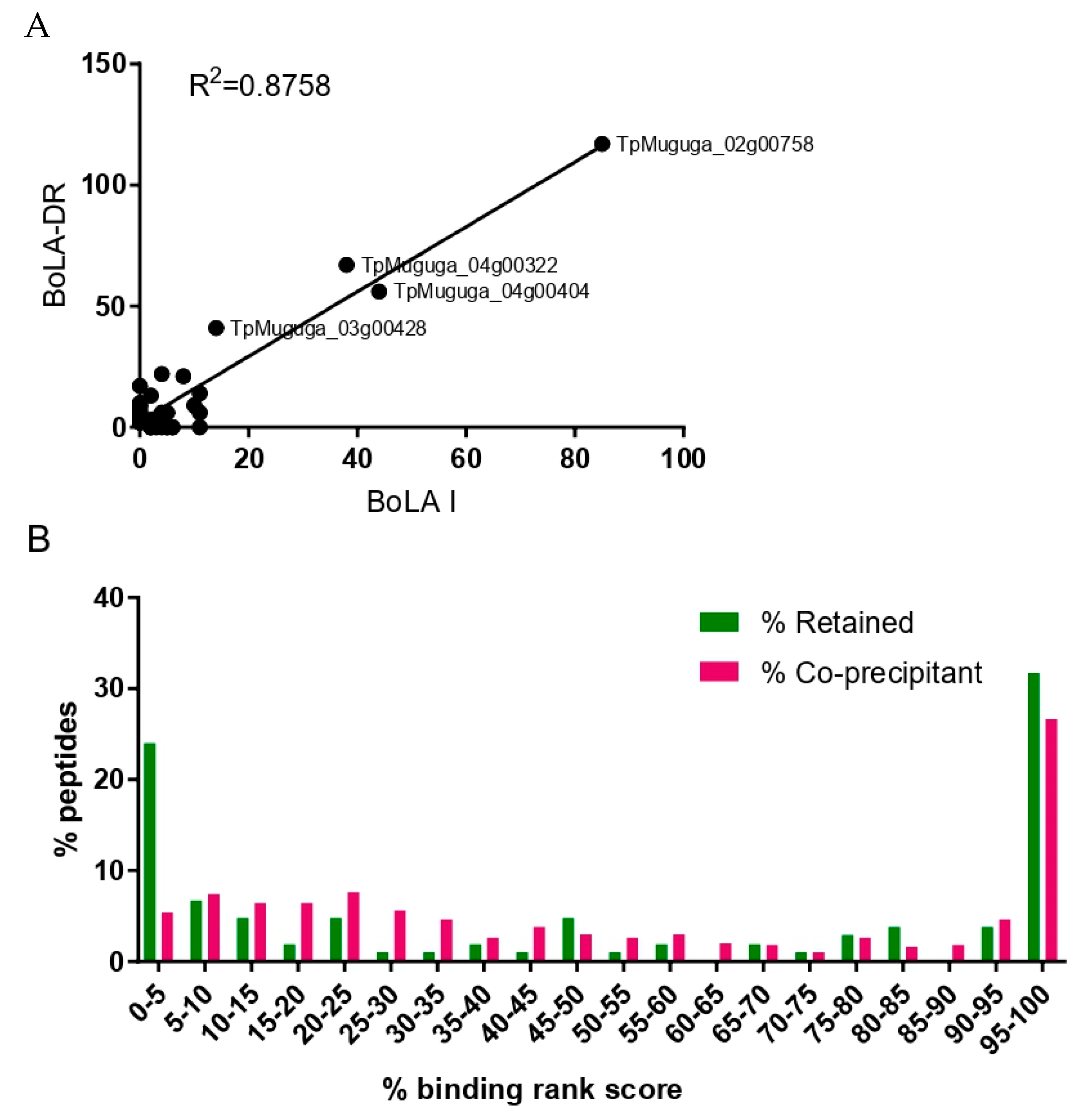
3.3. Exclusion of Putative Coprecipitating Parasite Proteins and Application of Immunoinformatics Provides a Refined List of Putative BoLA-I-eluted T. parva Peptides
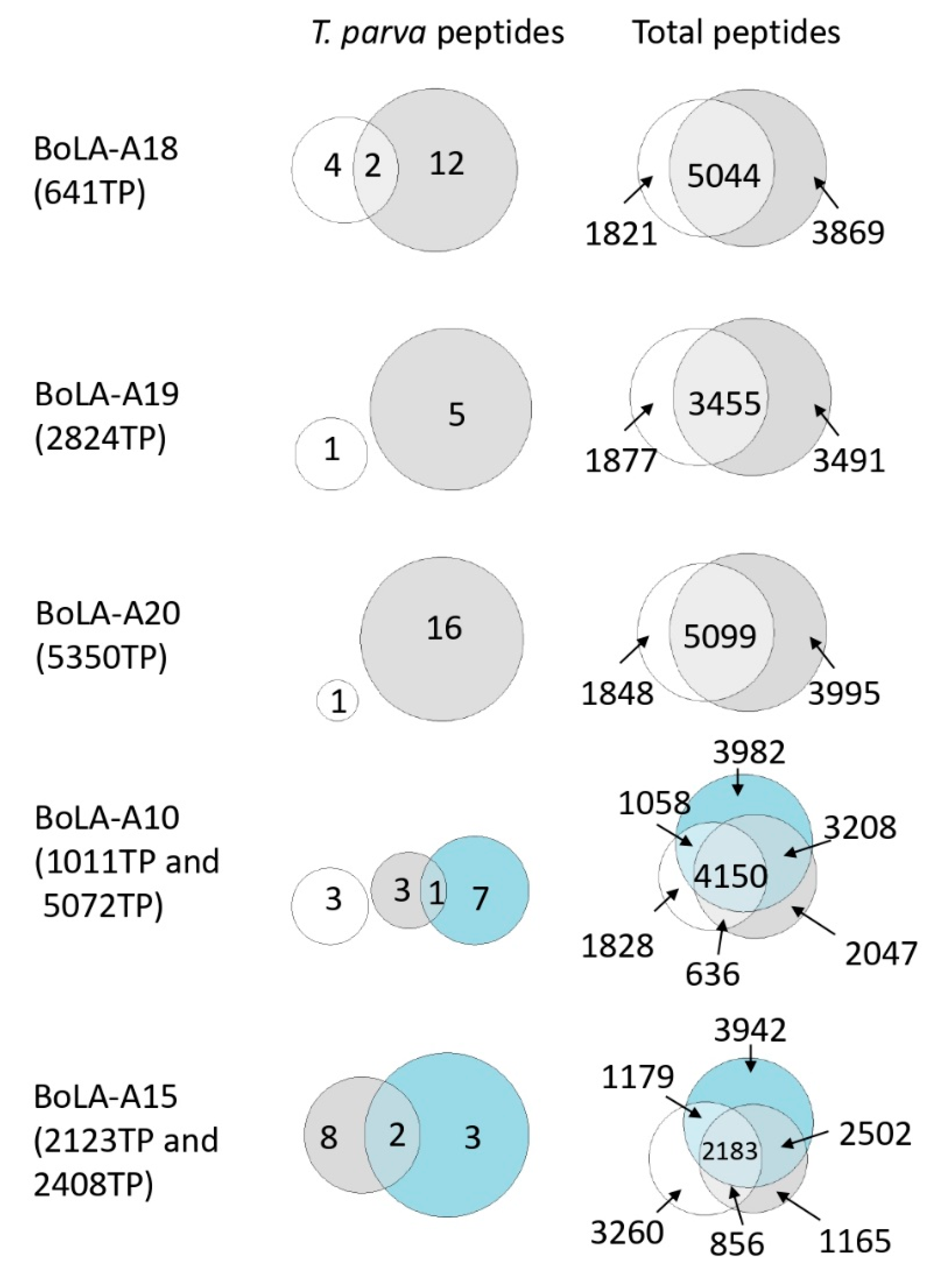
3.4. Analysis of the Reproducibility of the Identified T. parva BoLA-I Immunopeptidomes
3.5. Analysis of T. parva Peptides Presented by BoLA-DR
3.6. Comparison of T. parva Peptidome Data with Previously Identified T. parva Antigens
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- East Coast Fever. Available online: https://www.galvmed.org/livestock-and-diseases/livestock-diseases/east-coast-fever/ (accessed on 30 September 2022).
- Goddeeris, B.M.; Morrison, W.I.; Toye, P.G.; Bishop, R. Strain specificity of bovine Theileria parva-specific cytotoxic T cells is determined by the phenotype of the restricting class I MHC. Immunology 1990, 69, 38–44. [Google Scholar] [PubMed]
- Goddeeris, B.M.; Morrison, W.I.; Teale, A.J. Generation of bovine cytotoxic cell lines, specific for cells infected with the protozoan parasite Theileria parva and restricted by products of the major histocompatibility complex. Eur. J. Immunol. 1986, 16, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- McKeever, D.J.; Taracha, E.L.; Innes, E.L.; MacHugh, N.D.; Awino, E.; Goddeeris, B.M.; Morrison, W.I. Adoptive transfer of immunity to Theileria parva in the CD8+ fraction of responding efferent lymph. Proc. Natl. Acad. Sci. USA 1994, 91, 1959–1963. [Google Scholar] [CrossRef] [PubMed]
- Taracha, E.L.; Goddeeris, B.; Teale, A.J.; Kemp, S.J.; Morrison, W.I. Parasite strain specificity of bovine cytotoxic T cell responses to Theileria parva is determined primarily by immunodominance. J. Immunol. 1995, 155, 4854–4860. [Google Scholar]
- Christensen, J.P.; Marker, O.; Thomsen, A.R. The role of CD4+ T cells in cell-mediated immunity to LCMV: Studies in MHC class I and class II deficient mice. Scand. J. Immunol. 1994, 40, 373–382. [Google Scholar] [CrossRef]
- Thomsen, A.R.; Johansen, J.; Marker, O.; Christensen, J. Exhaustion of CTL memory and recrudescence of viremia in lymphocytic choriomeningitis virus-infected MHC class II-deficient mice and B cell-deficient mice. J. Immunol. 1996, 157, 3074–3080. [Google Scholar]
- Palmateer, N.C.; Tretina, K.; Orvis, J.; Ifeonu, O.O.; Crabtree, J.; Drabék, E.; Pelle, R.; Awino, E.; Gotia, H.T.; Munro, J.B.; et al. Capture-based enrichment of Theileria parva DNA enables full genome assembly of first buffalo-derived strain and reveals exceptional intra-specific genetic diversity. PLoS Negl. Trop. Dis. 2020, 14, e0008781. [Google Scholar] [CrossRef]
- Atuhaire, D.K.; Muleya, W.; Mbao, V.; Bazarusanga, T.; Gafarasi, I.; Salt, J.; Namangala, B.; Musoke, A.J. Sequence diversity of cytotoxic T cell antigens and satellite marker analysis of Theileria parva informs the immunization against East Coast fever in Rwanda. Parasites Vectors 2020, 13, 452. [Google Scholar] [CrossRef]
- Hemmink, J.D.; Sitt, T.; Pelle, R.; De Klerk-Lorist, L.-M.; Shiels, B.; Toye, P.G.; Morrison, W.I.; Weir, W. Ancient diversity and geographical sub-structuring in African buffalo Theileria parva populations revealed through metagenetic analysis of antigen-encoding loci. Int. J. Parasitol. 2018, 48, 287–296. [Google Scholar] [CrossRef]
- Pelle, R.; Graham, S.P.; Njahira, M.N.; Osaso, J.; Saya, R.M.; Odongo, D.O.; Toye, P.G.; Spooner, P.R.; Musoke, A.J.; Mwangi, D.M.; et al. Two Theileria parva CD8 T cell antigen genes are more variable in buffalo than cattle parasites, but differ in pattern of sequence diversity. PLoS ONE 2011, 6, e19015. [Google Scholar] [CrossRef]
- Graham, S.P.; Pellé, R.; Honda, Y.; Mwangi, D.M.; Tonukari, N.J.; Yamage, M.; Glew, E.J.; de Villiers, E.P.; Shah, T.; Bishop, R.; et al. Theileria parva candidate vaccine antigens recognized by immune bovine cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 3286–3291. [Google Scholar] [CrossRef] [PubMed]
- Morrison, W.I.; Aguado, A.; Sheldrake, T.A.; Palmateer, N.C.; Ifeonu, O.O.; Tretina, K.; Parsons, K.; Fenoy, E.; Connelley, T.; Nielsen, M.; et al. CD4 T Cell Responses to Theileria parva in Immune Cattle Recognize a Diverse Set of Parasite Antigens Presented on the Surface of Infected Lymphoblasts. J. Immunol. 2021, 207, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Tretina, K.; Pelle, R.; Orvis, J.; Gotia, H.T.; Ifeonu, O.O.; Kumari, P.; Palmateer, N.C.; Iqbal, S.B.A.; Fry, L.M.; Nene, V.M.; et al. Re-annotation of the Theileria parva genome refines 53% of the proteome and uncovers essential components of N-glycosylation, a conserved pathway in many organisms. BMC Genom. 2020, 21, 279. [Google Scholar] [CrossRef] [PubMed]
- MacHugh, N.D.; Connelley, T.; Graham, S.; Pelle, R.; Formisano, P.; Taracha, E.L.; Ellis, S.A.; McKeever, D.J.; Burrells, A.; Morrison, W.I. CD8+ T-cell responses to Theileria parva are preferentially directed to a single dominant antigen: Implications for parasite strain-specific immunity. Eur. J. Immunol. 2009, 39, 2459–2469. [Google Scholar] [CrossRef]
- Han, K.-C.; Park, D.; Ju, S.; Lee, Y.E.; Heo, S.-H.; Kim, Y.-A.; Lee, J.E.; Lee, Y.; Park, K.H.; Park, S.-H.; et al. Streamlined selection of cancer antigens for vaccine development through integrative multi-omics and high-content cell imaging. Sci. Rep. 2020, 10, 5885. [Google Scholar] [CrossRef]
- Newey, A.; Griffiths, B.; Michaux, J.; Pak, H.S.; Stevenson, B.J.; Woolston, A.; Semiannikova, M.; Spain, G.; Barber, L.J.; Matthews, N.; et al. Immunopeptidomics of colorectal cancer organoids reveals a sparse HLA class I neoantigen landscape and no increase in neoantigens with interferon or MEK-inhibitor treatment. J. Immunother. Cancer 2019, 7, 309. [Google Scholar] [CrossRef]
- Teck, A.T.; Urban, S.; Quass, P.; Nelde, A.; Schuster, H.; Letsch, A.; Busse, A.; Walz, J.S.; Keilholz, U.; Ochsenreither, S. Cancer testis antigen Cyclin A1 harbors several HLA-A*02:01-restricted T cell epitopes, which are presented and recognized in vivo. Cancer Immunol. Immunother. 2020, 69, 1217–1227. [Google Scholar] [CrossRef]
- Mou, Z.; Li, J.; Boussoffara, T.; Kishi, H.; Hamana, H.; Ezzati, P.; Hu, C.; Yi, W.; Liu, D.; Khadem, F.; et al. Identification of broadly conserved cross-species protective Leishmania antigen and its responding CD4+ T cells. Sci. Transl. Med. 2015, 7, 310ra167. [Google Scholar] [CrossRef]
- McMurtrey, C.; Trolle, T.; Sansom, T.; Remesh, S.G.; Kaever, T.; Bardet, W.; Jackson, K.; McLeod, R.; Sette, A.; Nielsen, M.; et al. Toxoplasma gondii peptide ligands open the gate of the HLA class I binding groove. eLife 2016, 5, e12556. [Google Scholar] [CrossRef]
- Draheim, M.; Wlodarczyk, M.F.; Crozat, K.; Saliou, J.-M.; Alayi, T.D.; Tomavo, S.; Hassan, A.; Salvioni, A.; Demarta-Gatsi, C.; Sidney, J.; et al. Profiling MHC II immunopeptidome of blood-stage malaria reveals that cDC1 control the functionality of parasite-specific CD4 T cells. EMBO Mol. Med. 2017, 9, 1605–1621. [Google Scholar] [CrossRef]
- De Wit, J.; Emmelot, M.E.; Meiring, H.; Brink, J.A.M.V.G.-V.D.; Els, C.A.C.M.V.; Kaaijk, P. Identification of Naturally Processed Mumps Virus Epitopes by Mass Spectrometry: Confirmation of Multiple CD8+ T-Cell Responses in Mumps Patients. J. Infect. Dis. 2020, 221, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Ramarathinam, S.H.; Gras, S.; Alcantara, S.; Yeung, A.W.; Mifsud, N.A.; Sonza, S.; Illing, P.T.; Glaros, E.N.; Center, R.J.; Thomas, S.R.; et al. Identification of Native and Posttranslationally Modified HLA-B*57:01-Restricted HIV Envelope Derived Epitopes Using Immunoproteomics. Proteomics 2018, 18, 1700253. [Google Scholar] [CrossRef]
- Ternette, N.; Yang, H.; Partridge, T.; Llano, A.; Cedeño, S.; Fischer, R.; Charles, P.D.; Dudek, N.L.; Mothe, B.; Crespo, M.; et al. Defining the HLA class I-associated viral antigen repertoire from HIV-1-infected human cells. Eur. J. Immunol. 2016, 46, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Bettencourt, P.; Müller, J.; Nicastri, A.; Cantillon, D.; Madhavan, M.; Charles, P.D.; Fotso, C.B.; Wittenberg, R.; Bull, N.; Pinpathomrat, N.; et al. Identification of antigens presented by MHC for vaccines against tuberculosis. NPJ Vaccines 2020, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Ellis, S.A.; Staines, K.A.; Stear, M.J.; Hensen, E.J.; Morrison, W.I. DNA typing for BoLA class I using sequence-specific primers (PCR-SSP). Eur. J. Immunogenet. 1998, 25, 365–370. [Google Scholar] [CrossRef]
- Baxter, R.; Hastings, N.; Law, A.; Glass, E.J. A rapid and robust sequence-based genotyping method for BoLA-DRB3 alleles in large numbers of heterozygous cattle. Anim. Genet. 2008, 39, 561–563. [Google Scholar] [CrossRef]
- Vasoya, D.; Law, A.; Motta, P.; Yu, M.; Muwonge, A.; Cook, E.; Li, X.; Bryson, K.; MacCallam, A.; Sitt, T.; et al. Rapid identification of bovine MHCI haplotypes in genetically divergent cattle populations using next-generation sequencing. Immunogenetics 2016, 68, 765–781. [Google Scholar] [CrossRef]
- Goddeeris, B.M.; Morrison, W.I. Techniques for the generation, cloning, and characterization of bovine cytotoxic T cells specific for the protozoan Theileria parva. J. Tissue Cult. Methods 1988, 11, 101–110. [Google Scholar] [CrossRef]
- Purcell, A.W.; Ramarathinam, S.H.; Ternette, N. Mass spectrometry–based identification of MHC-bound peptides for immunopeptidomics. Nat. Protoc. 2019, 14, 1687–1707. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Fisch, A.; Reynisson, B.; Benedictus, L.; Nicastri, A.; Vasoya, D.; Morrison, W.I.; Buus, S.; Ferreira, B.R.; de Miranda Santos, I.K.; Ternette, N.; et al. Integral Use of Immunopeptidomics and Immunoinformatics for the Characterization of Antigen Presentation and Rational Identification of BoLA-DR-Presented Peptides and Epitopes. J. Immunol. 2021, 206, 2489–2497. [Google Scholar] [CrossRef] [PubMed]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Svitek, N.; Hansen, A.M.; Steinaa, L.; Saya, R.; Awino, E.; Nielsen, M.; Buus, S.; Nene, V. Use of “one-pot, mix-and-read” peptide-MHC class I tetramers and predictive algorithms to improve detection of cytotoxic T lymphocyte responses in cattle. Vet. Res. 2014, 45, 50. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.M.; Rasmussen, M.; Svitek, N.; Harndahl, M.; Golde, W.T.; Barlow, J.; Nene, V.; Buus, S.; Nielsen, M. Characterization of binding specificities of bovine leucocyte class I molecules: Impacts for rational epitope discovery. Immunogenetics 2014, 66, 705–718. [Google Scholar] [CrossRef]
- Harndahl, M.; Rasmussen, M.; Roder, G.; Buus, S. Real-time, high-throughput measurements of peptide–MHC-I dissociation using a scintillation proximity assay. J. Immunol. Methods 2011, 374, 5–12. [Google Scholar] [CrossRef][Green Version]
- Codner, G.F.; Stear, M.J.; Reeve, R.; Matthews, L.; Ellis, S.A. Selective forces shaping diversity in the class I region of the major histocompatibility complex in dairy cattle. Anim. Genet. 2012, 43, 239–249. [Google Scholar] [CrossRef]
- Nielsen, M.; Connelley, T.; Ternette, N. Improved Prediction of Bovine Leucocyte Antigens (BoLA) Presented Ligands by Use of Mass-Spectrometry-Determined Ligand and in Vitro Binding Data. J. Proteome Res. 2018, 17, 559–567. [Google Scholar] [CrossRef]
- Andersson, L.; Rask, L. Characterization of the MHC class II region in cattle. The number of DQ genes varies between haplotypes. Immunogenetics 1988, 27, 110–120. [Google Scholar] [CrossRef]
- Zhou, H.; Hickford, J.; Fang, Q.; Byun, S. Short Communication: Identification of Allelic Variation at the Bovine DRA Locus by Polymerase Chain Reaction-Single Strand Conformational Polymorphism. J. Dairy Sci. 2007, 90, 1943–1946. [Google Scholar] [CrossRef]
- Burke, M.G.; Stone, R.T.; Muggli-Cockett, N.E. Nucleotide sequence and northern analysis of a bovine major histocompatibility class II DR beta-like cDNA. Anim. Genet. 1991, 22, 343–352. [Google Scholar] [CrossRef]
- Mayer, R.L.; Impens, F. Immunopeptidomics for next-generation bacterial vaccine development. Trends Microbiol. 2021, 29, 1034–1045. [Google Scholar] [CrossRef]
- Bettencourt, P. Current Challenges in the Identification of Pre-Erythrocytic Malaria Vaccine Candidate Antigens. Front. Immunol. 2020, 11, 190. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.P.; Pellé, R.; Yamage, M.; Mwangi, D.M.; Honda, Y.; Mwakubambanya, R.S.; de Villiers, E.P.; Abuya, E.; Awino, E.; Gachanja, J.; et al. Characterization of the fine specificity of bovine CD8 T-cell responses to defined antigens from the protozoan parasite Theileria parva. Infect. Immun. 2008, 76, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.P.; Srivastava, A. Immuno-informatics Analysis to Identify Novel Vaccine Candidates and Design of a Multi-Epitope Based Vaccine Candidate Against Theileria parasites. Front. Immunol. 2018, 9, 2213. [Google Scholar] [CrossRef]
- Karunakaran, K.P.; Yu, H.; Jiang, X.; Chan, Q.W.T.; Foster, L.J.; Johnson, R.M.; Brunham, R.C. Discordance in the Epithelial Cell-Dendritic Cell Major Histocompatibility Complex Class II Immunoproteome: Implications for Chlamydia Vaccine Development. J. Infect. Dis. 2020, 221, 841–850. [Google Scholar] [CrossRef]
- Alvarez, B.; Reynisson, B.; Barra, C.; Buus, S.; Ternette, N.; Connelley, T.; Andreatta, M.; Nielsen, M. NNAlign_MA.; MHC Peptidome Deconvolution for Accurate MHC Binding Motif Characterization and Improved T-cell Epitope Predictions. Mol. Cell. Proteom. 2019, 18, 2459–2477. [Google Scholar] [CrossRef] [PubMed]
- Obara, I.; Nielsen, M.; Jeschek, M.; Nijhof, A.; Mazzoni, C.J.; Svitek, N.; Steinaa, L.; Awino, E.; Olds, C.; Jabbar, A.; et al. Sequence diversity between class I MHC loci of African native and introduced Bos taurus cattle in Theileria parva endemic regions: In silico peptide binding prediction identifies distinct functional clusters. Immunogenetics 2016, 68, 339–352. [Google Scholar] [CrossRef]
- Vasoya, D.; Oliveira, P.S.; Muriel, L.A.; Tzelos, T.; Vrettou, C.; Morrison, W.I.; Santos, I.K.F.M.; Connelley, T. High throughput analysis of MHC-I and MHC-DR diversity of Brazilian cattle populations. HLA 2021, 98, 93–113. [Google Scholar] [CrossRef] [PubMed]
- Partridge, T.; Nicastri, A.; Kliszczak, A.E.; Yindom, L.-M.; Kessler, B.; Ternette, N.; Borrow, P. Discrimination Between Human Leukocyte Antigen Class I-Bound and Co-Purified HIV-Derived Peptides in Immunopeptidomics Workflows. Front. Immunol. 2018, 9, 912. [Google Scholar] [CrossRef]
- Schneider, I.; Haller, D.; Kullmann, B.; Beyer, D.; Ahmed, J.S.; Seitzer, U. Identification, molecular characterization and subcellular localization of a Theileria annulata parasite protein secreted into the host cell cytoplasm. Parasitol. Res. 2007, 101, 1471–1482. [Google Scholar] [CrossRef]
- Tajeri, S.; Langsley, G. Theileria secretes proteins to subvert its host leukocyte. Biol. Cell 2021, 113, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Woods, K.; Perry, C.; Brühlmann, F.; Olias, P. Theileria’s Strategies and Effector Mechanisms for Host Cell Transformation: From Invasion to Immortalization. Front. Cell Dev. Biol. 2021, 9, 662805. [Google Scholar] [CrossRef]
- Von Schubert, C.; Xue, G.; Schmuckli-Maurer, J.; Woods, K.L.; Nigg, E.A.; Dobbelaere, D.A.E. The Transforming Parasite Theileria Co-opts Host Cell Mitotic and Central Spindles to Persist in Continuously Dividing Cells. PLoS Biol. 2010, 8, e1000499. [Google Scholar] [CrossRef]
- Bishop, R.; Shah, T.; Pelle, R.; Hoyle, D.; Pearson, T.; Haines, L.; Brass, A.; Hulme, H.; Graham, S.P.; Taracha, E.L.; et al. Analysis of the transcriptome of the protozoan Theileria parva using MPSS reveals that the majority of genes are transcriptionally active in the schizont stage. Nucleic Acids Res. 2005, 33, 5503–5511. [Google Scholar] [CrossRef] [PubMed]
- Witschi, M.; Xia, D.; Sanderson, S.; Baumgartner, M.; Wastling, J.; Dobbelaere, D. Proteomic analysis of the Theileria annulata schizont. Int. J. Parasitol. 2013, 43, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Wickström, S.L.; Lövgren, T.; Volkmar, M.; Reinhold, B.; Duke-Cohan, J.S.; Hartmann, L.; Rebmann, J.; Mueller, A.; Melief, J.; Maas, R.; et al. Cancer Neoepitopes for Immunotherapy: Discordance Between Tumor-Infiltrating T Cell Reactivity and Tumor MHC Peptidome Display. Front. Immunol. 2019, 10, 2766. [Google Scholar] [CrossRef]
- Bosch-Camós, L.; López, E.; Navas, M.J.; Pina-Pedrero, S.; Accensi, F.; Correa-Fiz, F.; Park, C.; Carrascal, M.; Domínguez, J.; Salas, M.L. Identification of Promiscuous African Swine Fever Virus T-Cell Determinants Using a Multiple Technical Approach. Vaccines 2021, 9, 29. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Bennink, J.R. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu. Rev. Immunol. 1999, 17, 51–88. [Google Scholar] [CrossRef]
- Kastenmuller, W.; Gasteiger, G.; Gronau, J.H.; Baier, R.; Ljapoci, R.; Busch, D.H.; Drexler, I. Cross-competition of CD8+ T cells shapes the immunodominance hierarchy during boost vaccination. J. Exp. Med. 2007, 204, 2187–2198. [Google Scholar] [CrossRef]
- Dominguez, M.; Silveira, E.L.V.; De Vasconcelos, J.R.C.; De Alencar, B.C.G.; Machado, A.V.; Bruña-Romero, O.; Gazzinelli, R.T.; Rodrigues, M.M. Subdominant/Cryptic CD8 T Cell Epitopes Contribute to Resistance against Experimental Infection with a Human Protozoan Parasite. PLoS ONE 2011, 6, e22011. [Google Scholar] [CrossRef]
- Aagaard, C.S.; Hoang, T.T.K.T.; Vingsbo-Lundberg, C.; Dietrich, J.; Andersen, P. Quality and Vaccine Efficacy of CD4+T Cell Responses Directed to Dominant and Subdominant Epitopes in ESAT-6 from Mycobacterium tuberculosis. J. Immunol. 2009, 183, 2659–2668. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.W.; Hansen, P.R.; Holm, A.; Andersen, P. Efficient protection against Mycobacterium tuberculosis by vaccination with a single subdominant epitope from the ESAT-6 antigen. Eur. J. Immunol. 2000, 30, 1724–1732. [Google Scholar] [CrossRef]
- Frahm, N.; Kiepiela, P.; Adams, S.; Linde, C.H.; Hewitt, H.S.; Sango, K.; E Feeney, M.; Addo, M.; Lichterfeld, M.; Lahaie, M.P.; et al. Control of human immunodeficiency virus replication by cytotoxic T lymphocytes targeting subdominant epitopes. Nat. Immunol. 2006, 7, 173–178. [Google Scholar] [CrossRef]
- Holtappels, R.; Simon, C.O.; Munks, M.W.; Thomas, D.; Deegen, P.; Kuhnapfel, B.; Daubner, T.; Emde, S.F.; Podlech, J.; Grzimek, N.K.A.; et al. Subdominant CD8 T-Cell Epitopes Account for Protection against Cytomegalovirus Independent of Immunodomination. J. Virol. 2008, 82, 5781–5796. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Haddad, E.K.; Marceau, J.; Morabito, K.M.; Rao, S.S.; Filali-Mouhim, A.; Sekaly, R.P.; Graham, B.S. A Numerically Subdominant CD8 T Cell Response to Matrix Protein of Respiratory Syncytial Virus Controls Infection with Limited Immunopathology. PLoS Pathog. 2016, 12, e1005486. [Google Scholar] [CrossRef] [PubMed]
- Im, E.-J.; Hong, J.P.; Roshorm, Y.; Bridgeman, A.; Létourneau, S.; Liljeström, P.; Potash, M.J.; Volsky, D.J.; McMichael, A.J.; Hanke, T. Protective Efficacy of Serially Up-Ranked Subdominant CD8+ T Cell Epitopes against Virus Challenges. PLoS Pathog. 2011, 7, e1002041. [Google Scholar] [CrossRef]
- Mok, H.; Lee, S.; Wright, D.W.; Crowe, J.E., Jr. Enhancement of the CD8+ T cell response to a subdominant epitope of respiratory syncytial virus by deletion of an immunodominant epitope. Vaccine 2008, 26, 4775–4782. [Google Scholar] [CrossRef]
- Riedl, P.; Wieland, A.; Lamberth, K.; Buus, S.; Lemonnier, F.; Reifenberg, K.; Reimann, J.; Schirmbeck, R. Elimination of immunodominant epitopes from multispecific DNA-based vaccines allows induction of CD8 T cells that have a striking antiviral potential. J. Immunol. 2009, 183, 370–380. [Google Scholar] [CrossRef]
- Wieland, A.; Riedl, P.; Reimann, J.; Schirmbeck, R. Silencing an immunodominant epitope of hepatitis B surface antigen reveals an alternative repertoire of CD8 T cell epitopes of this viral antigen. Vaccine 2009, 28, 114–119. [Google Scholar] [CrossRef]
- Woodworth, J.S.; Aagaard, C.S.; Hansen, P.R.; Cassidy, J.P.; Agger, E.M.; Andersen, P. Protective CD4 T Cells Targeting Cryptic Epitopes of Mycobacterium tuberculosis Resist Infection-Driven Terminal Differentiation. J. Immunol. 2014, 192, 3247–3258. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | −10lgP | Length | Sample | #Spec | Allele | Icore | %Rank | Accession | Protein Description | Peptide Location Start | Peptide Location Stop | Spectral Match |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VANTKIEFPEI | 31.7 | 11 | 641TP | 4 | 6:01301 | VANTKIEFPEI | 7.15 | TpMuguga_01g00263 | hypothetical protein | 2371 | 2381 | ND |
| FSVPNQVKAAKVEATIPSHLEKKVITNKKN | 73.1 | 30 | 1011TP | 1 | 3:00201 | FSVPNQVKAAK | 92.00 | TpMuguga_01g00293 | 60S ribosomal protein L38, putative | 50 | 79 | ND |
| QAYQQKVDL | 21.3 | 9 | 2229TP | 1 | 2:02501 | QAYQQKVDL | 0.45 | TpMuguga_01g00386 | hypothetical protein | 1588 | 1596 | ND |
| SSISSSLLSVK | 23.7 | 11 | 1011TP | 2 | 2:01201 | SSISSSLLSVK | 0.05 | TpMuguga_01g00421 | hypothetical protein, conserved | 365 | 375 | ND |
| RMYGKGKGISSSSIP | 46.8 | 15 | 641TP | 3 | 6:01301 | RMYGKGKGISSSSI | 19.53 | TpMuguga_01g00502 | 40S ribosomal protein S13, putative | 3 | 17 | ND |
| SEKDSYLSSIKKLNL | 59.1 | 15 | 641TP | 1 | 6:01301 | SEKDSYLSSIKKLNL | 3.93 | TpMuguga_01g00946 | ribosome biogenesis regulatory protein, putative | 221 | 235 | ND |
| GKFKNPTKTHKMDEVSESSLQ | 28.6 | 21 | 2229TP | 1 | 1:02301 | GKFKNPTKTH | 90.00 | TpMuguga_01g01108 | hypothetical protein | 2 | 22 | ND |
| GKFKNPTKTHKMDEVSESSLQQK | 54.9 | 23 | 2229TP | 1 | 1:02301 | GKFKNPTKTH | 91.67 | TpMuguga_01g01108 | hypothetical protein | 2 | 24 | ND |
| LNIGRIELIDYI | 27.8 | 12 | 641TP | 6 | 6:01301 | LNIGRIELIDYI | 26.37 | TpMuguga_01g01149 | hypothetical protein | 914 | 925 | ND |
| RALFDDLHRY | 22.9 | 10 | 2229TP | 1 | 4:02401 | RALFDDLHRY | 0.28 | TpMuguga_01g01198 | hypothetical protein | 46 | 55 | ND |
| ILGFDFNKLGKI | 28 | 12 | 641TP | 2 | 6:01301 | ILGFDFNKLGKI | 7.89 | TpMuguga_02g00219 | hypothetical protein | 495 | 506 | ND |
| SLQLKFAQGSDLPNL | 51 | 15 | 641TP | 1 | 6:01301 | SLQLKFAQGSDLPNL | 5.77 | TpMuguga_02g00321 | hypothetical protein | 188 | 202 | ND |
| RVRKLCEYAI | 22.2 | 10 | 641TP | 3 | 6:01301 | RVRKLCEYAI | 2.40 | TpMuguga_02g00476 | crooked neck protein, putative | 499 | 508 | ND |
| ERLAALALYGDYGEFDRKTKEDSK | 64.6 | 24 | 2229TP | 1 | 6:04001 | ERLAALALY | 95.00 | TpMuguga_02g00758 | hypothetical protein | 552 | 575 | ND |
| ERLAALALYGDYGEFD | 61 | 16 | 641TP | 1 | 6:01301 | ERLAALALYGDYGEF | 100.0 | TpMuguga_02g00758 | hypothetical protein | 552 | 567 | ND |
| DRKLFSTKRPSLSL | 20.4 | 14 | 2229TP | 1 | 6:04001 | RKLFSTKRPSLSL | 24.46 | TpMuguga_02g00803 | 60S ribosomal protein L18, putative | 178 | 191 | ND |
| QPSYLSQAL | 26.7 | 9 | 1011TP | 1 | 3:00201 | QPSYLSQAL | 7.04 | TpMuguga_03g00202 | DNA polymerase alpha, putative | 159 | 167 | ND |
| SKVDRVSL | 23.6 | 8 | 2229TP | 6 | 1:02301 | SKVDRVSL | 0.09 | TpMuguga_03g00507 | tRNA nucleotidyltransferase (putative) | 495 | 502 | ND |
| SMKGKHELTL | 22.1 | 10 | 641TP | 3 | 6:01301 | SMKGKHELTL | 0.01 | TpMuguga_03g00747 | ATP-dependent RNA helicase, putative | 2223 | 2232 | ND |
| SSIDVNVKL | 21.1 | 9 | 1011TP | 1 | 3:00201 | SSIDVNVKL | 0.09 | TpMuguga_03g02030 | N-terminal region of Chorein, a TM vesicle-mediated sorter family protein | 3510 | 3518 | ND |
| KQVVRDAMVEQDML | 32 | 14 | 641TP | 1 | 6:01301 | KQVVRDAMVEQDML | 1.58 | TpMuguga_03g02350 | RF-1 domain protein | 534 | 547 | ND |
| LELIRARNEI | 23.6 | 10 | 641TP | 1 | 6:01301 | LELIRARNEI | 6.82 | TpMuguga_04g00031 | 26S proteasome aaa-ATPase subunit Rpt3, putative | 47 | 56 | ND |
| SPDQPDQHHQPTPAAQP | 43.3 | 17 | 1011TP | 1 | 3:00201 | SPDQPDQHHQPTPAAQP | 61.79 | TpMuguga_04g00051 | polymorphic immunodominant molecule | 228 | 244 | ND |
| FRNEKDLGF | 29.1 | 9 | 641TP | 7 | 6:01301 | FRNEKDLGF | 7.44 | TpMuguga_04g00227 | hypothetical protein | 1403 | 1411 | ND |
| VKKRVHKGKKKARSETYSTYIF | 28.5 | 22 | 1011TP | 1 | 3:00201 | KARSETYSTYIF | 96.88 | TpMuguga_04g00404 | histone H2B-III, putative | 2 | 23 | ND |
| ENKLVEEALK | 34.4 | 10 | 641TP | 2 | 6:01301 | ENKLVEEAL | 50.94 | TpMuguga_04g00505 | hypothetical protein | 99 | 108 | ND |
| KLLYVLKPFI | 22.1 | 10 | 641TP | 2 | 6:01301 | KLLYVLKPFI | 4.10 | TpMuguga_04g00611 | hypothetical protein | 234 | 243 | ND |
| LSPIDILDVAGLVT | 21.1 | 14 | 1011TP | 2 | 3:00201 | LSPIDILDVAGLVT | 61.30 | TpMuguga_04g00621 | DNA repair exonuclease, putative | 401 | 414 | ND |
| IGSAIKDNPAFITL | 46.3 | 14 | 2229TP | 2 | 6:04001 | IGSAIKDNPAFITL | 1.66 | TpMuguga_01g00188 | prohibitin, putative | 231 | 244 | Positive |
| RMDDKSGGLL | 42.7 | 10 | 641TP | 1 | 6:01301 | RMDDKSGGLL | 1.20 | TpMuguga_01g00736 | hypothetical protein | 20 | 29 | Positive |
| GEFEKKYIPTL | 50.7 | 11 | 641TP | 1 | 6:01301 | GEFEKKYIPTL | 0.15 | TpMuguga_01g00757 | GTP-binding nuclear protein ran, putative | 30 | 40 | Positive |
| VQHIPVDDFSGLQTEVVANE | 63.5 | 20 | 1011TP | 6 | 3:00201 | VQHIPVDDFSGLQTEV | 97.35 | TpMuguga_01g00924 | 60S ribosomal protein L31, putative | 99 | 118 | Positive |
| VQHIPVDDFSGLQTEVVANE | 57.3 | 20 | 2229TP | 3 | 6:04001 | VQHIPVDDFSGL | 95.77 | TpMuguga_01g00924 | 60S ribosomal protein L31, putative | 99 | 118 | Positive |
| RQMQVKLNLP | 23.8 | 10 | 641TP | 2 | 6:01301 | RQMQVKLNL | 0.79 | TpMuguga_01g00980 | 40S ribosomal protein S26e, putative | 101 | 110 | Positive |
| TQYERIKERL | 28.8 | 10 | 641TP | 1 | 6:01301 | TQYERIKERL | 0.01 | TpMuguga_01g01210 | hypothetical protein | 2 | 11 | Positive |
| GQIQDVFKRL | 33.1 | 10 | 641TP | 1 | 6:01301 | GQIQDVFKRL | 0.25 | TpMuguga_02g00123 | RNA helicase-1, putative | 189 | 198 | Positive |
| SKDEHKKLY | 39.5 | 9 | 2229TP | 3 | 1:02301 | SKDEHKKLY | 0.07 | TpMuguga_02g00142 | hypothetical protein | 91 | 99 | Positive |
| ATIIGFHK | 28 | 8 | 1011TP | 1 | 2:01201 | ATIIGFHK | 0.11 | TpMuguga_02g00222 | 40S ribosomal protein S29, putative | 45 | 52 | Positive |
| SLKSALIDT | 21.5 | 9 | 641TP | 1 | 6:01301 | SLKSALIDT | 22.80 | TpMuguga_02g00333 | translation initiation factor 6, putative | 239 | 247 | Positive |
| SLKSALIDTLI | 25.5 | 11 | 641TP | 4 | 6:01301 | SLKSALIDTLI | 2.47 | TpMuguga_02g00333 | translation initiation factor 6, putative | 239 | 249 | Positive |
| EIKERLAALAL | 35.5 | 11 | 1011TP | 5 | 3:00201 | EIKERLAALAL | 14.51 | TpMuguga_02g00758 | hypothetical protein | 549 | 559 | Positive |
| EIKERLAALAL | 27 | 11 | 2229TP | 2 | 6:04001 | EIKERLAALAL | 14.92 | TpMuguga_02g00758 | hypothetical protein | 549 | 559 | Positive |
| EIKERLAALALYGDYGEFDRKT | 66.9 | 22 | 1011TP | 3 | 3:00201 | EIKERLAALAL | 98.82 | TpMuguga_02g00758 | hypothetical protein | 549 | 570 | Positive |
| EIKERLAALALYGDYGEFDRKT | 73.5 | 22 | 2229TP | 3 | 6:04001 | EIKERLAALALALY | 95.00 | TpMuguga_02g00758 | hypothetical protein | 549 | 570 | Positive |
| EIKERLAALALYGDYGEFDRKT | 72.2 | 22 | 641TP | 4 | 6:01301 | EIKERLAALAL | 95.00 | TpMuguga_02g00758 | hypothetical protein | 549 | 570 | Positive |
| ERLAALALYGDYGEFDRKT | 71.2 | 19 | 1011TP | 3 | 3:00201 | RLAALALYGDYGEFDRK | 93.50 | TpMuguga_02g00758 | hypothetical protein | 552 | 570 | Positive |
| ERLAALALYGDYGEFDRKT | 67.9 | 19 | 2229TP | 5 | 6:04001 | ERLAALALYGDYGEFDRKT | 95.00 | TpMuguga_02g00758 | hypothetical protein | 552 | 570 | Positive |
| ERLAALALYGDYGEFDRKT | 72.2 | 19 | 641TP | 2 | 6:01301 | ERLAALALYGDYGEFDRKT | 100.00 | TpMuguga_02g00758 | hypothetical protein | 552 | 570 | Positive |
| ERLAALALYGDYGEFDRKTK | 61.9 | 20 | 2229TP | 1 | 6:04001 | RLAALALYGDYGEFDRKTK | 92.50 | TpMuguga_02g00758 | hypothetical protein | 552 | 571 | Positive |
| ERLAALALYGDYGEFDRKTKE | 58.1 | 21 | 2229TP | 1 | 6:04001 | ERLAALALY | 95.00 | TpMuguga_02g00758 | hypothetical protein | 552 | 572 | Positive |
| YGDYGEFDRKT | 52.8 | 11 | 1011TP | 3 | 3:00201 | YGDYGEFDRK | 16.16 | TpMuguga_02g00758 | hypothetical protein | 560 | 570 | Positive |
| YGDYGEFDRKTK | 47.7 | 12 | 1011TP | 2 | 2:01201 | YGDYGEFDRKTK | 13.40 | TpMuguga_02g00758 | hypothetical protein | 560 | 571 | Positive |
| YGDYGEFDRKTKEDSK | 53.7 | 16 | 1011TP | 3 | 2:01201 | YGDYGEFDRKTKEDSK | 36.34 | TpMuguga_02g00758 | hypothetical protein | 560 | 575 | Positive |
| YGDYGEFDRKTKEDSK | 37.9 | 16 | 2229TP | 1 | 6:04001 | YGDYGEFDRKTKEDSK | 81.67 | TpMuguga_02g00758 | hypothetical protein | 560 | 575 | Positive |
| YGDYGEFDRKTKEDSKN | 36.3 | 17 | 1011TP | 1 | 3:00201 | YGDYGEFDRKTKEDSK | 94.00 | TpMuguga_02g00758 | hypothetical protein | 560 | 576 | Positive |
| AKFPGMKKSKGPKDK | 54.3 | 15 | 2229TP | 3 | 1:02301 | AKFPGMKKSKGPKDK | 39.77 | TpMuguga_02g00895 | hypothetical protein | 67 | 81 | Positive |
| FRDDLGSSFTSGYTK | 59.3 | 15 | 1011TP | 2 | 2:01201 | FRDDLGSSFTSGYTK | 2.96 | TpMuguga_02g00895 | hypothetical protein | 48 | 62 | Positive |
| RDDLGSSFTSGYTK | 59.9 | 14 | 1011TP | 1 | 2:01201 | RDDLGSSFTSGYTK | 0.58 | TpMuguga_02g00895 | hypothetical protein | 49 | 62 | Positive |
| SSFTSGYTK | 38.4 | 9 | 1011TP | 1 | 2:01201 | SSFTSGYTK | 0.01 | TpMuguga_02g00895 | hypothetical protein | 54 | 62 | Positive |
| SSFTSGYTKQDLDAKFPGMK | 68.6 | 20 | 1011TP | 5 | 2:01201 | SSFTSGYTK | 25.30 | TpMuguga_02g00895 | hypothetical protein | 54 | 73 | Positive |
| KTAPVTGGVK | 20.4 | 10 | 1011TP | 1 | 2:01201 | KTAPVTGGVK | 0.46 | TpMuguga_03g00152: TpMuguga_04g00321 | histone H3, putative | 28 | 37 | Negative |
| KTAPVTGGVKK | 22.7 | 11 | 1011TP | 4 | 2:01201 | KTAPVTGGVKK | 0.16 | TpMuguga_03g00152: TpMuguga_04g00321 | histone H3, putative | 28 | 38 | Negative |
| SGWEGRTL | 32.1 | 8 | 2229TP | 1 | 6:04001 | SGWEGRTL | 0.21 | TpMuguga_03g00428 | 40S ribosomal protein S28e, putative | 33 | 40 | Positive |
| SGWEGRTLI | 30.8 | 9 | 1011TP | 1 | 3:00201 | SGWEGRTLI | 0.67 | TpMuguga_03g00428 | 40S ribosomal protein S28e, putative | 33 | 41 | Positive |
| ILRTIVQQL | 32.7 | 9 | 641TP | 3 | 6:01301 | ILRTIVQQL | 0.10 | TpMuguga_03g00716 | 40S ribosomal protein S19, putative | 121 | 129 | Positive |
| NSFVTDTFEKL | 43.5 | 11 | 1011TP | 1 | 3:00201 | NSFVTDTFEKL | 1.68 | TpMuguga_04g00404 | histone H2B-III, putative | 44 | 54 | Positive |
| SETYSTYIFKVLK | 47.8 | 13 | 1011TP | 2 | 2:01201 | SETYSTYIFKVLK | 3.65 | TpMuguga_04g00404 | histone H2B-III, putative | 15 | 27 | Positive |
| RLFNFATKRI | 37.6 | 10 | 641TP | 1 | 6:01301 | RLFNFATKRI | 2.32 | TpMuguga_04g00503 | hypothetical protein | 236 | 245 | Positive |
| Peptide | −10lgP | Peptide Length | Sample | Accession | Allele | Core | %Rank | SpectralMatch | 1*02301 | 2*01201 | 3*00201 | 6*01301 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TQYERIKERL | 28.82 | 10 | 641TP | TpMuguga_01g01210 | 6:01301 | TQYERIKERL | 0.01 | positive | 0.00 | 0.00 | 0.00 | 36.54 |
| SSFTSGYTK | 38.38 | 9 | 1011TP | TpMuguga_02g00895 | 2:01201 | SSFTSGYTK | 0.01 | positive | 0.00 | 1.43 | 0.00 | 0.22 |
| SKDEHKKLY | 39.48 | 9 | 2229TP | TpMuguga_02g00142 | 1:02301 | SKDEHKKLY | 0.07 | positive | 31.74 | 0.00 | 0.00 | 0.00 |
| ILRTIVQQL | 32.71 | 9 | 641TP | TpMuguga_03g00716 | 6:01301 | ILRTIVQQL | 0.10 | positive | 0.00 | 0.00 | 0.00 | 24.03 |
| ATIIGFHK | 27.98 | 8 | 1011TP | TpMuguga_02g00222 | 2:01201 | ATIIGFHK | 0.11 | positive | 0.00 | 0.76 | 0.00 | 0.76 |
| GEFEKKYIPTL | 50.67 | 11 | 641TP | TpMuguga_01g00757 | 6:01301 | GEFEKKYIPTL | 0.15 | positive | 0.00 | 0.00 | 0.00 | 24.56 |
| KTAPVTGGVKK | 22.73 | 11 | 1011TP | TpMuguga_03g00152:TpMuguga_04g00321 | 2:01201 | KTAPVTGGVKK | 0.16 | negative | 0.00 | 0.32 | 0.00 | 0.48 |
| GQIQDVFKRL | 33.14 | 10 | 641TP | TpMuguga_02g00123 | 6:01301 | GQIQDVFKRL | 0.25 | positive | 0.00 | 0.00 | 0.00 | 19.10 |
| KTAPVTGGVK | 20.38 | 10 | 1011TP | TpMuguga_03g00152:TpMuguga_04g00321 | 2:01201 | KTAPVTGGVK | 0.46 | negative | 0.00 | 0.32 | 0.00 | 0.00 |
| SGWEGRTLI | 30.79 | 9 | 1011TP | TpMuguga_03g00428 | 3:00201 | SGWEGRTLI | 0.67 | positive | 0.00 | 0.00 | 0.30 | 0.00 |
| RQMQVKLNLP | 23.76 | 10 | 641TP | TpMuguga_01g00980 | 6:01301 | RQMQVKLNL | 0.79 | positive | 0.00 | 0.00 | 0.00 | 10.25 |
| RMDDKSGGLL | 42.72 | 10 | 641TP | TpMuguga_01g00736 | 6:01301 | RMDDKSGGLL | 1.20 | positive | 0.00 | 0.00 | 0.00 | 11.75 |
| NSFVTDTFEKL | 43.45 | 11 | 1011TP | TpMuguga_04g00404 | 3:00201 | NSFVTDTFEKL | 1.68 | positive | 0.00 | 0.00 | 0.00 | 0.00 |
| RLFNFATKRI | 37.63 | 10 | 641TP | TpMuguga_04g00503 | 6:01301 | RLFNFATKRI | 2.32 | positive | 0.00 | 0.00 | 0.00 | 6.76 |
| SLKSALIDTLI | 25.47 | 11 | 641TP | TpMuguga_02g00333 | 6:01301 | SLKSALIDTLI | 2.47 | positive | 0.00 | 0.00 | 0.00 | 7.89 |
| YGDYGEFDRKTK | 47.74 | 12 | 1011TP | TpMuguga_02g00758 | 2:01201 | YGDYGEFDRKTK | 13.40 | positive | 0.00 | 0.10 | 0.00 | 0.00 |
| EIKERLAALAL | 35.54 | 11 | 1011TP | TpMuguga_02g00758 | 3:00201 | EIKERLAALAL | 14.51 | positive | 0.00 | 0.00 | 0.00 | 0.00 |
| YGDYGEFDRKT | 52.75 | 11 | 1011TP | TpMuguga_02g00758 | 3:00201 | YGDYGEFDRK | 16.16 | positive | 0.00 | 0.00 | 0.00 | 0.00 |
| SLKSALIDT | 21.52 | 9 | 641TP | TpMuguga_02g00333 | 6:01301 | SLKSALIDT | 22.80 | positive | 0.00 | 0.00 | 0.00 | 0.00 |
| Negative control (no peptide) | 0.00 | 0.00 | 0.00 | 0.00 | ||||||||
| Positive control | 3.3 | 2.00 | 4.50 | 7.30 |
| Accession | Protein Description | Comments | Peptide | Length | Sample | BoLA_MHCI Allele | %Rank |
|---|---|---|---|---|---|---|---|
| TpMuguga_01g00075 | hypothetical protein | NQPKNVVEF | 9 | 5350TP_rpt | 3:02701 | 0.04 | |
| TpMuguga_01g00100 | Protein arginine N-methyltransferase 5 | SEIDVKDVL | 9 | 2824TP_rpt | 6:01401 | 0.01 | |
| TpMuguga_01g00151 | Insulinase (Peptidase family M16) family protein | SEVAVSAMGPL | 11 | 2824TP_rpt | 6:01401 | 1.87 | |
| TpMuguga_01g00176 | putative integral membrane protein | VEDEAAYHVQL | 11 | 2824_TP | 2:01601 | 0.03 | |
| TpMuguga_01g00188 | Tp6—Prohibitin-2 | IGSAIKDNPAFITL | 14 | 2229TP | 6:04001 | 1.66 | |
| TpMuguga_01g00235 | Eukaryotic translation initiation factor 3 subunit A | AEKEIVELV | 9 | 2123TP_rpt | 1:01901 | 0.01 | |
| TpMuguga_01g00386 | DNA polymerase family A family protein | QAYQQKVDL | 9 | 2229TP | 2:02501 | 0.45 | |
| TpMuguga_01g00421 | RWD domain protein | Identifications in A10 and A18 samples | SSISSSLLSVK | 11 | 1011TP | 2:01201 | 0.05 |
| TpMuguga_01g00421 | RWD domain protein | Identifications in A10 and A18 samples | KLIWRFIRHL | 10 | 641TP_rpt | 6:01301 | 0.29 |
| TpMuguga_01g00461 | Cytokine-induced anti-apoptosis inhibitor 1, Fe-S biogenesis family protein | GQVTKASFFSSL | 12 | 641TP_rpt | 6:01301 | 0.23 | |
| TpMuguga_01g00471 | Bifunctional thioredoxin reductase/thioredoxin | Identification in three different A15 samples | FEYEFPINH | 9 | 2123TP_rpt/2408TP/2408_TP_rpt | 1:00901 | 0.03 |
| TpMuguga_01g00566 | Brix domain protein | NKKRPISIGF | 10 | 5350TP_rpt | 2:02601 | 0.13 | |
| TpMuguga_01g00736 | hypothetical protein | Identification in duplicate A18 samples | RMDDKSGGLL | 10 | 641TP | 6:01301 | 1.20 |
| TpMuguga_01g00757 | Ras family protein | Identification in duplicate A18 samples | GEFEKKYIPTL | 11 | 641TP | 6:01301 | 0.15 |
| TpMuguga_01g00808 | Ubiquitin elongating factor core family protein | SKKDLFIQF | 9 | 5350TP_rpt | 3:02701 | 0.01 | |
| TpMuguga_01g00926 | Cofilin/tropomyosin-type actin-binding family protein | VEDHDEVRGALA | 12 | 2824TP_rpt | 2:01601 | 1.95 | |
| TpMuguga_01g00934 | Heat shock cognate 90 kDa protein | SQFVKYPIQL | 10 | 641TP_rpt | 6:01301 | 0.00 | |
| TpMuguga_01g01188 | Translation initiation factor IF-2 | NNPIGRVGF | 9 | 5350TP_rpt | 3:02701 | 0.10 | |
| TpMuguga_01g01198 | hypothetical protein | RALFDDLHRY | 10 | 2229TP | 4:02401 | 0.28 | |
| TpMuguga_01g01207 | Pre-mRNA-splicing factor srp2 | FGPINRIDF | 9 | 5350TP_rpt | 3:02701 | 0.90 | |
| TpMuguga_01g01210 | Mitochondrial carrier family protein | TQYERIKERL | 10 | 641TP | 6:01301 | 0.01 | |
| TpMuguga_01g02005 | Rab-GTPase-TBC domain protein | KLNEQKILSL | 10 | 641TP_rpt | 6:01301 | 0.08 | |
| TpMuguga_01g02030 | hypothetical protein | Identifications in A12 and A15 samples | EEIAHVLHY | 9 | 2123TP_rpt | 1:01901/1:00901 | 0.39/1.00 |
| TpMuguga_01g02175 | Sas10 C-terminal domain protein | YLHEFHNFI | 9 | 2123TP_rpt | 2:02501 | 1.69 | |
| TpMuguga_02g00113 | DNA replication licensing factor MCM6 | SSLLKLTNK | 9 | A10TP_rpt | 2:01201 | 0.01 | |
| TpMuguga_02g00123 | DEAD/DEAH box helicase | GQIQDVFKRL | 10 | 641TP | 6:01301 | 0.25 | |
| TpMuguga_02g00142 | Polyubiquitin | SKDEHKKLY | 9 | 2229TP | 1:02301 | 0.07 | |
| TpMuguga_02g00248 | Nucleolar GTP-binding protein 1 | HMFSGKRTL | 9 | 641TP_rpt | 6:01301 | 0.05 | |
| TpMuguga_02g00488 | ATP synthase subunit D | AEDFKSLVI | 9 | 2824TP_rpt | 2:01601 | 0.01 | |
| TpMuguga_02g00543 | putative integral membrane protein | KLANSKNVSL | 10 | 641TP_rpt | 6:01301 | 0.11 | |
| TpMuguga_02g00551 | 23 kDa piroplasm membrane protein | SKATDRLVV | 9 | 5350TP_rpt | 3:02701 | 0.22 | |
| TpMuguga_02g00613 | DEAD/DEAH box helicase | AKKITELGF | 9 | 5350TP_rpt | 2:02601 | 0.06 | |
| TpMuguga_02g00703 | putative integral membrane protein | three different identifications in one A20 sample | VKKLKESLL | 9 | 5350TP_rpt | 2:02601 | 0.02 |
| TpMuguga_02g00703 | putative integral membrane protein | three different identifications in one A20 sample | NKLGDPLTL | 9 | 5350TP_rpt | 3:02701 | 0.12 |
| TpMuguga_02g00703 | putative integral membrane protein | three different identifications in one A20 sample | YKPEGMEYPF | 10 | 5350TP_rpt | 3:02701 | 1.59 |
| TpMuguga_02g00706 | hypothetical protein | YQKNSNNPFM | 10 | 5350TP_rpt | 2:02601 | 1.11 | |
| TpMuguga_02g00718 | hypothetical protein | GKNSVLLQV | 9 | 5350TP_rpt | 2:02601 | 0.15 | |
| TpMuguga_02g00723 | hypothetical protein | RTFNDVSKRKH | 11 | 2408_TP | 1:00901 | 0.71 | |
| TpMuguga_02g00753 | chaperone protein DnaK | TQVGIKVY | 8 | 2408_TP | 1:00901 | 0.15 | |
| TpMuguga_02g00895 | Tp9—Hypothetical protein | two overlapping identifications in one A10 sample | (RDDLG)SSFTSGYTK | 9/14 | 1011TP | 2:01201 | 0.01/0.58 |
| TpMuguga_02g00896 | hypothetical protein | AQGDPVFL | 8 | 2408_TP | 2:02501 | 0.16 | |
| TpMuguga_03g00253 | hypothetical protein | Identifications in A10, A15 and A20 samples | LQSEVFPNY | 9 | 2408_TP | 1:00901 | 0.11 |
| TpMuguga_03g00253 | hypothetical protein | Identifications in A10, A15, and A20 samples | FNFSESKLTF | 10 | 5350_TP | 3:02701 | 1.38 |
| TpMuguga_03g00253 | hypothetical protein | Identifications in A10, A15, and A20 samples | LNTSIGGSL | 9 | A10_TP | 3:00201 | 1.00 |
| TpMuguga_03g00257 | hypothetical protein | SQNNRSEMSNL | 11 | 641TP_rpt | 6:01301 | 0.07 | |
| TpMuguga_03g00330 | hypothetical protein | SSMRDALNPPPTH | 13 | 2408_TP | 1:00901 | 1.16 | |
| TpMuguga_03g00388 | hypothetical protein | SQKRKNKPL | 9 | 641TP_rpt | 6:01301 | 0.01 | |
| TpMuguga_03g00469 | High mobility group protein homolog NHP1 | AKKDPNAPKRAL | 12 | 5350TP_rpt | 2:02601 | 0.13 | |
| TpMuguga_03g00478 | DEAD/DEAH box helicase family protein | YVGKAPTLW | 9 | 5350TP_rpt | 3:02701 | 1.32 | |
| TpMuguga_03g00507 | CCA tRNA nucleotidyltransferase mitochondrial | SKVDRVSL | 8 | 2229TP | 1:02301 | 0.09 | |
| TpMuguga_03g00544 | putative integral membrane protein | NVFPLILGK | 9 | A10_TP | 2:01201 | 0.08 | |
| TpMuguga_03g00577 | Cwf15/Cwc15 cell cycle control family protein | SQQPPSFLNDAVRTDFH | 17 | 2408_TP | 1:00901 | 0.52 | |
| TpMuguga_03g00655 | N4—Hypothetical protein | GVDVDQLLH | 9 | 2123TP_rpt | 1:00901 | 0.36 | |
| TpMuguga_03g00747 | DEAD/DEAH box helicase | SMKGKHELTL | 10 | 641TP | 6:01301 | 0.01 | |
| TpMuguga_03g00852 | Protein transport protein Sec61 subunit alpha | two different identifications in one A10 sample | SSMVMQLLAGSK | 12 | A10TP_rpt | 2:01201 | 1.27 |
| TpMuguga_03g00852 | Protein transport protein Sec61 subunit alpha | two different identifications in one A10 sample | KGTEFEGALISL | 12 | A10TP_rpt | 3:00201 | 1.43 |
| TpMuguga_03g00858 | Multiprotein-bridging factor 1c | Identifications in duplicate A10 samples | AGVELDTQKKFL | 12 | A10_TP | 3:00201 | 1.36 |
| TpMuguga_03g00861 | p150—Hypothetical protein | LGPILIYEDL | 10 | A10TP_rpt | 3:00201 | 0.10 | |
| TpMuguga_03g02030 | N-terminal region of Chorein, a TM vesicle-mediated sorter family protein | SSIDVNVKL | 9 | 1011TP | 3:00201 | 0.09 | |
| TpMuguga_03g02350 | RF-1 domain protein | KQVVRDAMVEQDML | 14 | 641TP | 6:01301 | 1.58 | |
| TpMuguga_03g02680 | putative integral membrane protein | FGIPLVTK | 8 | A10TP_rpt | 2:01201 | 0.38 | |
| TpMuguga_04g00144 | hypothetical protein | NSNELKDIK | 9 | A10TP_rpt | 2:01201 | 1.60 | |
| TpMuguga_04g00206 | hypothetical protein | IEDEVCKVI | 9 | 2824TP_rpt | 2:01601 | 0.09 | |
| TpMuguga_04g00229 | Pescadillo N-terminus family protein | SLMPKKHKRLL | 11 | 641TP_rpt | 6:01301 | 0.08 | |
| TpMuguga_04g00233 | Protein IWS1 homolog | SQMSRNIESKH | 11 | 2123TP_rpt | 1:00901 | 0.03 | |
| TpMuguga_04g00368 | 26S proteasome non-ATPase regulatory subunit 4 homolog | IGLIASAKL | 9 | A10TP_rpt | 3:00201 | 0.27 | |
| TpMuguga_04g00439 | hypothetical protein | NSQLRQKIRSM | 11 | 641TP_rpt | 6:01301 | 1.09 | |
| TpMuguga_04g00484 | OST-HTH Associated domain protein | SGINLGNVNSL | 11 | A10_TP | 3:00201 | 0.09 | |
| TpMuguga_04g00662 | putative integral membrane protein | DKKALTVAL | 9 | 5350TP_rpt | 3:02701 | 0.01 | |
| TpMuguga_04g00754 | hypothetical protein | SQFPRNPVDSLL | 12 | 641TP_rpt | 6:01301 | 0.01 | |
| TpMuguga_04g00785 | Helicase C-terminal domain | NNFNHSLL | 8 | 5350TP_rpt | 3:02701 | 0.28 | |
| TpMuguga_04g00790 | AP2-coincident C-terminal family protein | SQSEEIEKYLH | 11 | 2408_TP | 1:00901 | 0.07 | |
| TpMuguga_04g02060 | hypothetical protein | HQQDSQYYLTQH | 12 | 2408_TP | 1:00901 | 0.10 | |
| TpMuguga_04g02625 | Importin subunit alpha-6 | KQLRNENEI | 9 | 641TP_rpt | 6:01301 | 0.41 |
| Accession | Protein Description | Comments | Peptide | Length | Sample | BoLA_DR Allele | %RPS |
|---|---|---|---|---|---|---|---|
| TpMuguga_01g00016 | Vacuolar protein sorting/targeting protein 10 | LDEKSYTILDTSEGAVI | 17 | 641TP_DR | DRB3_2002 | 0.23 | |
| TpMuguga_01g00324 | Tubulin/FtsZ family, GTPase domain protein | four nested peptides in one sample | (EFQ)TNLVPYPRIHFML | 13–16 | 5072TP_DR | DRB3_1001 | 0.1/0.10/0.15/0.34 |
| TpMuguga_01g00552 | hypothetical protein | RAKEYNFISKIVYRS | 15 | 495TP_DR | DRB3_1001 | 0.06 | |
| TpMuguga_01g00701 | Rhoptry-associated protein 1 (RAP-1) family protein | two nested peptides in one sample | (SE)VQHVVFSFLNDPYK | 14, 16 | 5350TP_DR | DRB3_1201 | 0.01/0.03 |
| TpMuguga_01g00937 | Cell division control protein 48 homolog E | two nested peptides in one sample | (RPG)RLDQLIYIPLPDLPAR | 16, 19 | 641TP_DR | DRB3_2002 | 0.00/0.01 |
| TpMuguga_01g00972 | hypothetical protein | LPVWEAVNDERVDEA | 15 | 2824TP_DR | DRB3_1601 | 0.04 | |
| TpMuguga_01g00987 | P104 | VAPKDTTLEYLKVFLNK | 17 | 641TP_DR | DRB3_2002 | 0.46 | |
| TpMuguga_01g01056 | p32—Merozoite Antigen | two nested peptides in one sample | SEVKFETYYDDVLFKGK(S) | 17, 18 | 5350TP_DR | DRB3_1201 | 0.00/0.00 |
| TpMuguga_01g01129 | hypothetical protein | TKTVSFSNKISFHYF | 15 | 495TP_DR | DRB3_1001 | 4.02 | |
| TpMuguga_02g00253 | ATP synthase, Delta/Epsilon chain, beta-sandwich domain protein | NKDLVFSLLSSHEALY | 16 | 641TP_DR | DRB3_2002 | 0.06 | |
| TpMuguga_02g00551 | 23 kDa piroplasm membrane protein | EDRLATYKPFTEDPSKKR | 18 | 5350TP_DR | DRB3_1201 | 0.01 | |
| TpMuguga_02g00753 | chaperone protein DnaK | DFDQRILNFLVDEFKK | 16 | 5350TP_DR | DRB3_1201 | 0.12 | |
| TpMuguga_04g00395 | 26S proteasome non-ATPase regulatory subunit 6 | KQGDLLINRIQKLSRIIDM | 19 | 2123TP_DR | DRB3_1501 | 4.22 | |
| TpMuguga_04g00719 | 2-oxoisovalerate dehydrogenase subunit alpha mitochondrial | two nested peptides in one sample | (E)NTKAYELLPGLFDDV | 15, 16 | 641TP_DR | DRB3_2002 | 0.01/0.06 |
| TpMuguga_04g02435 | ARF guanine-nucleotide exchange factor GNL1 | two nested peptides in one sample | KEKDFLADITKELDESQ(S) | 17, 18 | 2824TP_DR | DRB3_1601 | 0.11/0.84 |
| Accession | Protein Name | Antigens Recognised by T. parva-Specific | Identified in Peptide Elution From | ||
|---|---|---|---|---|---|
| CD8 | CD4 | DR | MHCI | ||
| TpMuguga_01g00188 | Tp6—Prohibitin-2 | CD8 | - | - | MHCI |
| TpMuguga_02g00123 | Tp32—DEAD/DEAH box helicase | CD8 | CD4 | - | MHCI |
| TpMuguga_02g00895 | Tp9—Hypothetical protein | CD8 | CD4 | - | MHCI |
| TpMuguga_03g00655 | Tp13—Hypothetical protein | - | CD4 | - | MHCI |
| TpMuguga_03g00861 | Tp20—Hypothetical protein | - | CD4 | - | MHCI |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Connelley, T.; Nicastri, A.; Sheldrake, T.; Vrettou, C.; Fisch, A.; Reynisson, B.; Buus, S.; Hill, A.; Morrison, I.; Nielsen, M.; et al. Immunopeptidomic Analysis of BoLA-I and BoLA-DR Presented Peptides from Theileria parva Infected Cells. Vaccines 2022, 10, 1907. https://doi.org/10.3390/vaccines10111907
Connelley T, Nicastri A, Sheldrake T, Vrettou C, Fisch A, Reynisson B, Buus S, Hill A, Morrison I, Nielsen M, et al. Immunopeptidomic Analysis of BoLA-I and BoLA-DR Presented Peptides from Theileria parva Infected Cells. Vaccines. 2022; 10(11):1907. https://doi.org/10.3390/vaccines10111907
Chicago/Turabian StyleConnelley, Timothy, Annalisa Nicastri, Tara Sheldrake, Christina Vrettou, Andressa Fisch, Birkir Reynisson, Soren Buus, Adrian Hill, Ivan Morrison, Morten Nielsen, and et al. 2022. "Immunopeptidomic Analysis of BoLA-I and BoLA-DR Presented Peptides from Theileria parva Infected Cells" Vaccines 10, no. 11: 1907. https://doi.org/10.3390/vaccines10111907
APA StyleConnelley, T., Nicastri, A., Sheldrake, T., Vrettou, C., Fisch, A., Reynisson, B., Buus, S., Hill, A., Morrison, I., Nielsen, M., & Ternette, N. (2022). Immunopeptidomic Analysis of BoLA-I and BoLA-DR Presented Peptides from Theileria parva Infected Cells. Vaccines, 10(11), 1907. https://doi.org/10.3390/vaccines10111907