
IntegralVac: A Machine Learning-Based Comprehensive Multivalent Epitope Vaccine Design Method


Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection and Selection
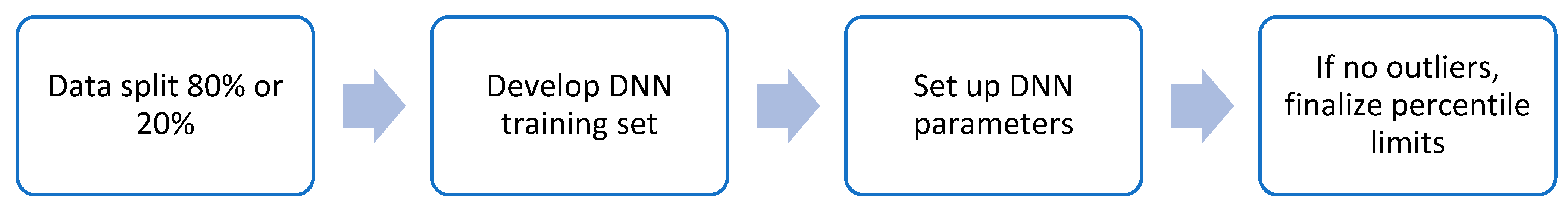
2.2. Machine Learning Model Selection for IntegralVac
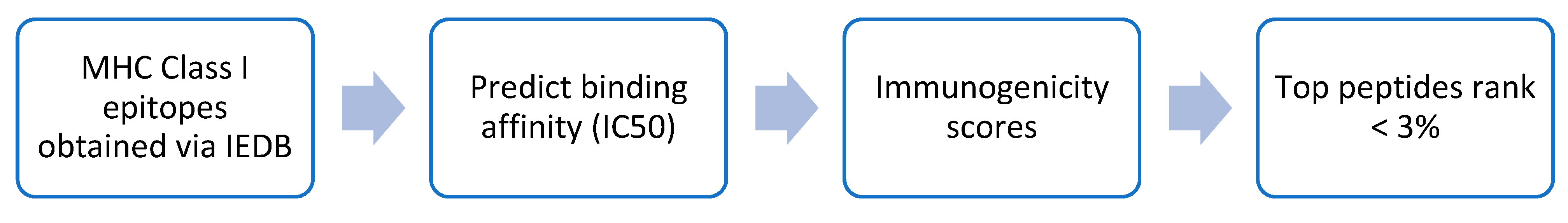
2.3. Comparison of IntegralVac MHC 1 Peptide Prediction with the NetMHCPan
2.4. Increasing Coverage of IntegralVac Using CTL Epitopes from the DeepVacPred Model
- Learning rate: [0.0001, 0.001, 0.002];
- Optimizers: [SGD, RMSProp, Adam];
- Epochs: [2000, 4000, 6000, 8000, 10,000];
- Batch size: [512, 1024, 2048, 4096]. [7]
2.5. Training IntegralVac for Multiple Known MHC Sequence Datasets with MHCSeqNet
2.6. Determining Peptide Characteristics in IntegralVac Using HemoPI Peptide Vaccine Methods
2.7. Antigenicity and Allergenicity Prediction Tool Selection
3. Results
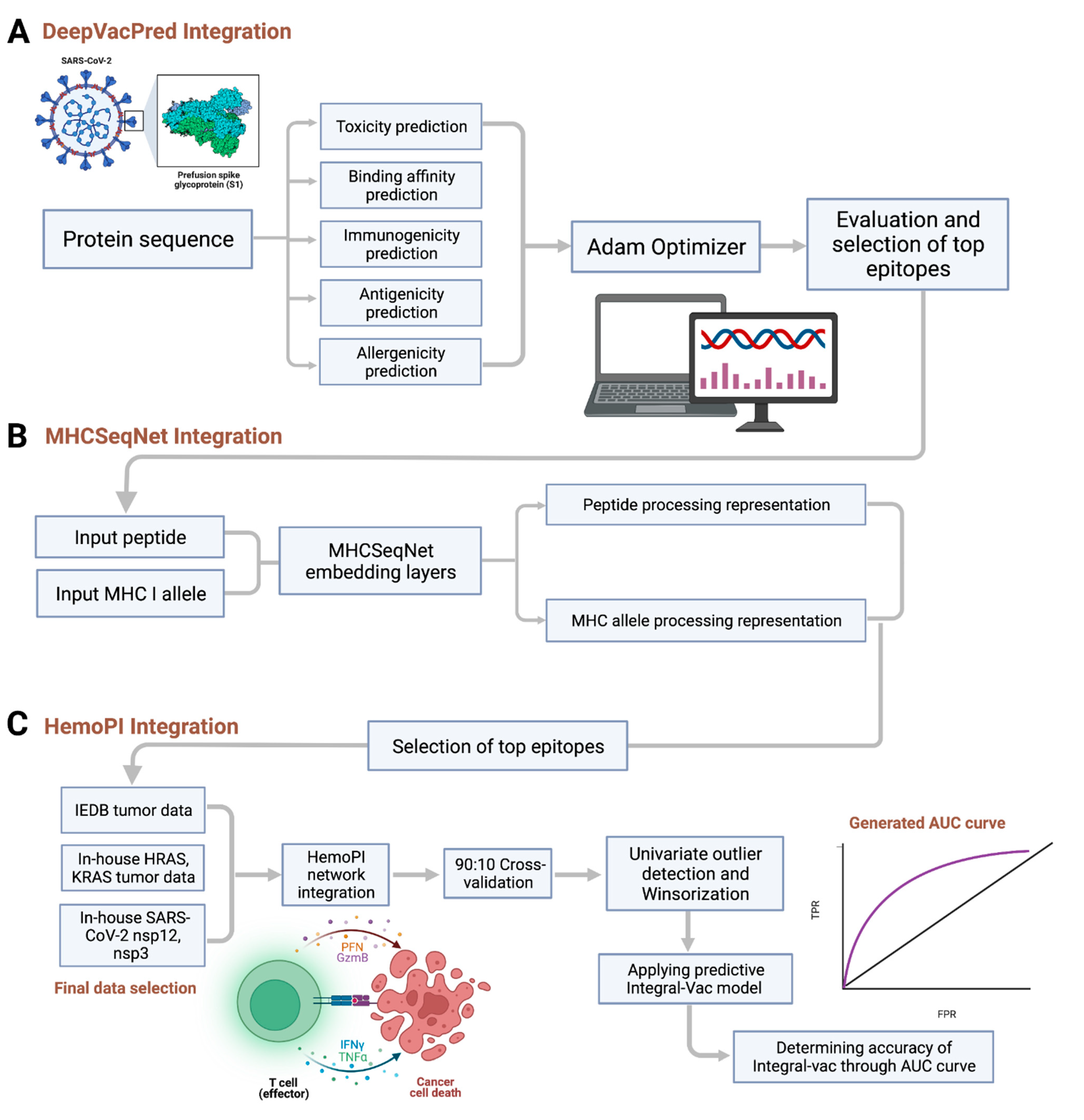
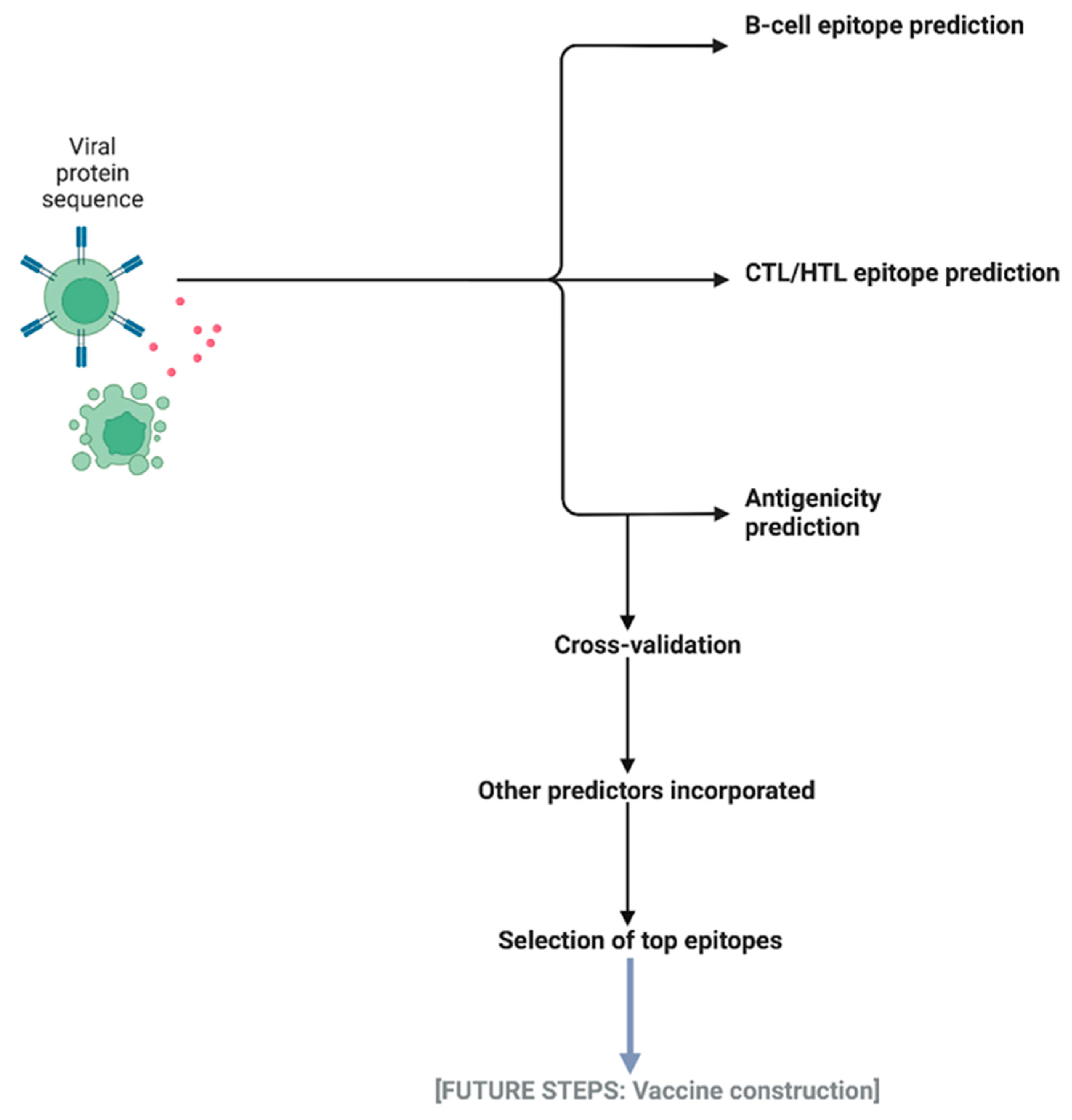
3.1. The IntegralVac Method
3.1.1. DeepVacPred Integration
3.1.2. MHCSeqNet Integration
3.1.3. HemoPI Integration
3.2. Integration of DeepVacPred into IntegralVac: Immunogenicity Predictions with Clinical Checkpoint Filters
3.3. Human Cancer and COVID Epitope Data Predictions from In-House Data Sets
3.4. Generating Amino Acid Predictor-Based Immunogenicity Rankings
3.5. Immunogenicity Predictions for Coronavirus Data
3.6. Validation of Integral-Vac with KRAS/HRAS Tumor Epitopes
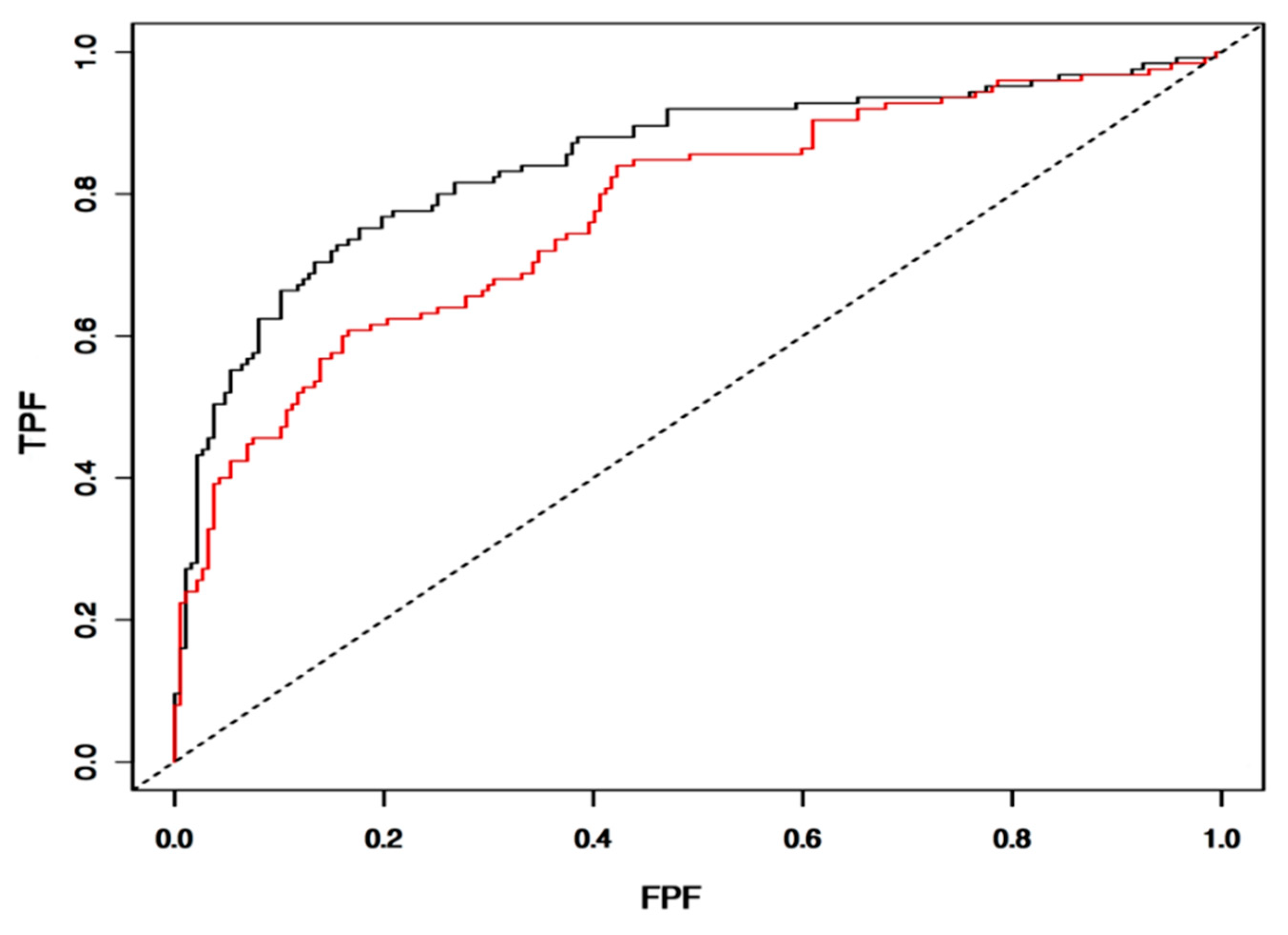
3.7. Validation of IntegralVac Predictions Using ROC Curve
4. Discussion
5. Conclusions
6. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Topuzoğullari, M.; Acar, T.; Arayici, P.P.; Uçar, B.; Uğurel, E.; Abamor, E.; Arasoğlu, T.; Turgut-Balik, D.; Derman, S. An insight into the epitope-based peptide vaccine design strategy and studies against COVID-19. Turk. J. Biol. 2020, 44, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cai, H.; Hu, J.; Lian, J.; Gu, J.; Zhang, S.; Ye, C.; Lu, Y.; Jin, C.; Yu, G.; et al. Epidemiological, clinical characteristics of cases of SARS-CoV-2 infection with abnormal imaging findings. Int. J. Infect. Dis. 2020, 94, 81–87. [Google Scholar] [CrossRef]
- Acharya, C.; Coop, A.; Polli, J.E.; MacKerell, A.D. Recent advances in ligand-based drug design: Relevance and utility of the conformationally sampled pharmacophore approach. Curr. Comput. Aided-Drug Des. 2010, 7, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Scipy.Stats.Mstats.Winsorize—SciPy v1.7.1 Manual. SciPy Documentation. 2021. Available online: docs.scipy.org/doc/scipy/reference/generated/scipy.stats.mstats.winsorize.html (accessed on 1 July 2021).
- Saha, S.; Raghava, G.P.S. BcePred: Prediction of Continuous B-Cell Epitopes in Antigenic Sequences Using Physico-chemical Properties; ICARIS 2004, LNCS 3239; Nicosia, G., Cutello, V., Bentley, P.J., Timis, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 197–204. [Google Scholar]
- Yang, Z.; Bogdan, P.; Nazarian, S. An in silico deep learning approach to multi-epitope vaccine design: A SARS-CoV-2 case study. Sci. Rep. 2021, 11, 3238. [Google Scholar] [CrossRef] [PubMed]
- Phloyphisut, P.; Pornputtapong, N.; Sriswasdi, S.; Chuangsuwanich, E. MHCSeqNet: A deep neural network model for universal MHC binding prediction. BMC Bioinform. 2019, 20, 270. [Google Scholar] [CrossRef] [PubMed]
- Plisson, F.; Ramírez-Sánchez, O.; Martínez-Hernández, C. Machine learning-guided discovery and design of non-hemolytic peptides. Sci. Rep. 2020, 10, 16581. [Google Scholar] [CrossRef]
- Savsani, K.; Jabbour, G.; Dakshanamurthy, S. A New Epitope Selection Method: Application to Design a Multi-Valent Epitope Vaccine Targeting HRAS Oncogene in Squamous Cell Carcinoma. Vaccines. 2022, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Parn, S.; Jabbour, G.; Nguyenkhoa, V.; Dakshanamurthy, S. Design of Peptide Vaccine for COVID19: CD8+ and CD4+ T cell epitopes from SARS-CoV-2 open-reading-frame protein variants. BioRxiv 2021, 1–38. [Google Scholar] [CrossRef]
- Zeng, S.; Pöttler, M.; Lan, B.; Grützmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Leung, K.; Leung, G. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: A modelling study. Lancet 2020, 395, 689–697. [Google Scholar] [CrossRef]
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A.; et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015, 43, 405–412. [Google Scholar] [CrossRef]
- Ray, S. Improve Your Model Performance Using Cross Validation (in Python and R). Improve Your Model Performance Using Cross Validation, Analysis Vidhya. 30 April 2018. Available online: www.analyticsvidhya.com/blog/2018/05/improve-model-performance-cross-validation-in-python-r (accessed on 10 June 2021).
- Gao, Q.; Bao, L.; Mao, H.; Wang, L.; Xu, K. Rapid development of an inactivated vaccine for SARS-CoV-2. BioRxiv 2020. [Google Scholar] [CrossRef]
- Jabbour, G.; Rego, S.; Nguyenkhoa, V.; Dakshanamurthy, S. Design of T-cell epitope-based vaccine candidate for SARS-CoV-2 targeting nucleocapsid and spike protein escape variants. BioRxiv 2021, 1–31. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Sequence | DeepVacPred Subunit | Vaxijen | AntigenPro | AllergenFP | ToxinPred |
|---|---|---|---|---|---|
| FVFKNIDGYFKIYSKHTPINLVRDLPQGFS | 5 | 0.476 | 0.477 | - | - |
| LGQSKRVDFCGKGYHLMSFPQSAPHGVVFL | 23 | 0.671 | 0.736 | - | - |
| LGVYYHKNNKSWMESEFRVYSSANNCTFEY | 4 | 0.390 | 0.736 | - | - |
| ILDITPCSFGGVSVITPGTNTSNQVAVLYQ | 13 | 0.832 | 0.403 | - | - |
| LPDPSKPSKRSFIEDLLFNKVTLADAGFIK | 19 | 0.361 | 0.499 | - | - |
| Validation | AUC | Threshold | Accuracy | Sensitivity | Specificity |
|---|---|---|---|---|---|
| Train set | 0.9608 | 0.32 | 0.960 | 0.95 | 0.95 |
| Test set | 0.9046 | 0.5 | 0.90 | 0.90 | 0.90 |
| Top 27 Population Alleles from which Epitopes Are Extracted | |
|---|---|
| HLA-A | HLA-B |
| HLA-A*01:01 | HLA-B*07:02 |
| HLA-A*02:01 | HLA-B*08:01 |
| HLA-A*02:03 | HLA-B*15:01 |
| HLA-A*02:06 | HLA-B*35:01 |
| HLA-A*03:01 | HLA-B*40:01 |
| HLA-A*11:01 | HLA-B*44:02 |
| HLA-A*23:01 | HLA-B*44:03 |
| HLA-A*24:02 | HLA-B*51:01 |
| HLA-A*26:01 | HLA-B*53:01 |
| HLA-A*30:01 | HLA-B*57:01 |
| HLA-A*30:02 | HLA-B*58:01 |
| HLA-A*31:01 | |
| HLA-A*32:01 | |
| HLA-A*33:01 | |
| HLA-A*68:01 | |
| HLA-A*68:02 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suri, S.; Dakshanamurthy, S. IntegralVac: A Machine Learning-Based Comprehensive Multivalent Epitope Vaccine Design Method. Vaccines 2022, 10, 1678. https://doi.org/10.3390/vaccines10101678
Suri S, Dakshanamurthy S. IntegralVac: A Machine Learning-Based Comprehensive Multivalent Epitope Vaccine Design Method. Vaccines. 2022; 10(10):1678. https://doi.org/10.3390/vaccines10101678
Chicago/Turabian StyleSuri, Sadhana, and Sivanesan Dakshanamurthy. 2022. "IntegralVac: A Machine Learning-Based Comprehensive Multivalent Epitope Vaccine Design Method" Vaccines 10, no. 10: 1678. https://doi.org/10.3390/vaccines10101678
APA StyleSuri, S., & Dakshanamurthy, S. (2022). IntegralVac: A Machine Learning-Based Comprehensive Multivalent Epitope Vaccine Design Method. Vaccines, 10(10), 1678. https://doi.org/10.3390/vaccines10101678