
ML-DTD: Machine Learning-Based Drug Target Discovery for the Potential Treatment of COVID-19

 ,
,  , and
, and
Abstract
:1. Introduction
2. Material & Methods
2.1. Dataset
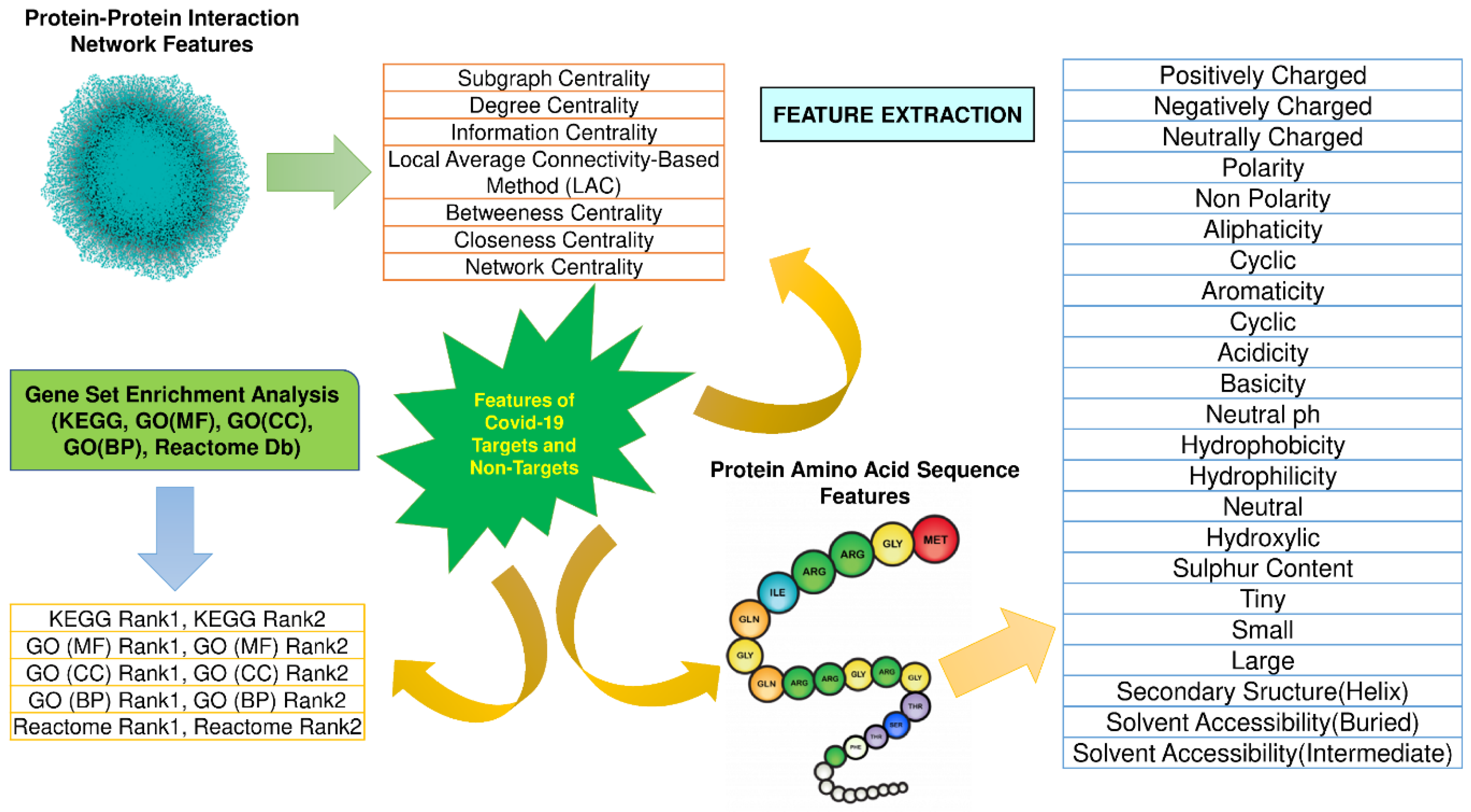
2.2. Workflow of ML-DTD
- (a)
- Feature Extraction
- (b)
- Feature Selection
- (c)
- Data Cleaning and Preprocessing
- (d)
- Classification Algorithms
- (e)
- Prediction of novel COVID-19 human drug targets
- (f)
- Drug-Repurposing Study of novel COVID-19 human drug targets
- (g)
- Docking study of the repurposed drugs detected through novel COVID-19 human drug targets
3. Results & Discussion
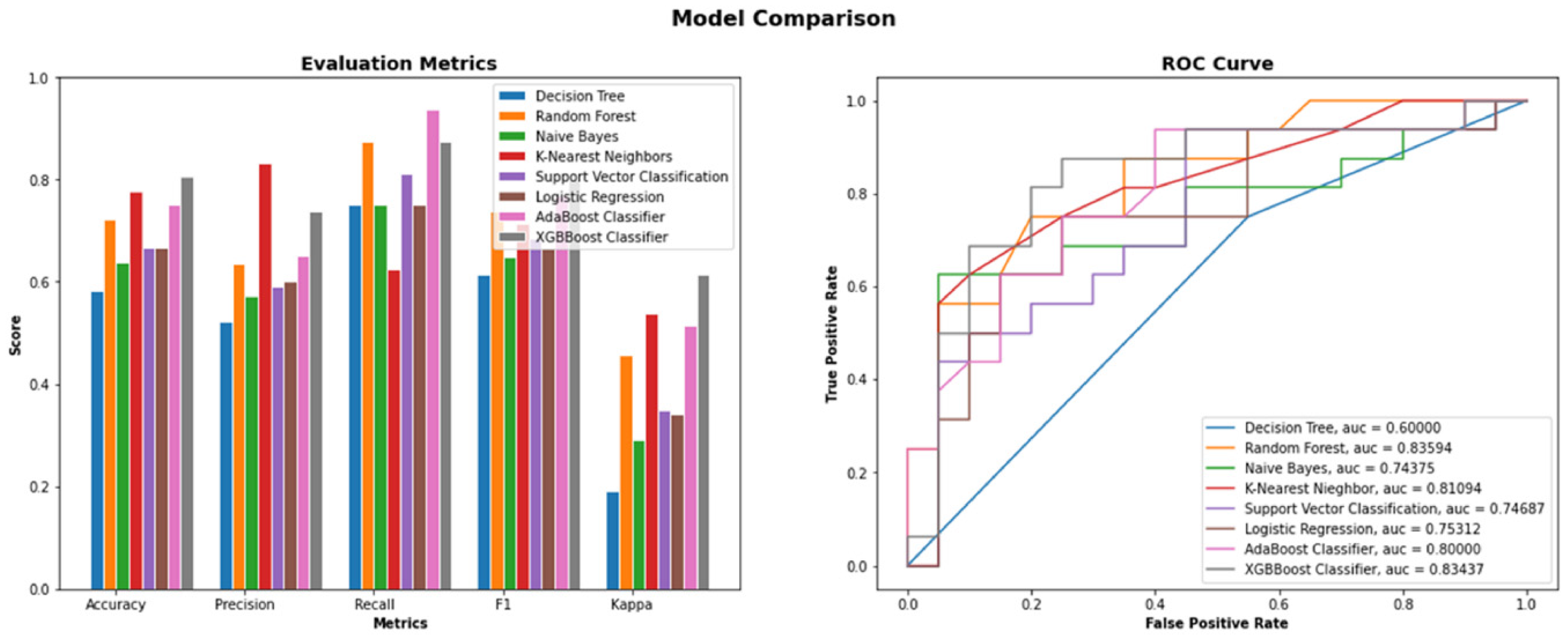
3.1. Performance Analysis of the Machine Learning Models Used in ML-DTD
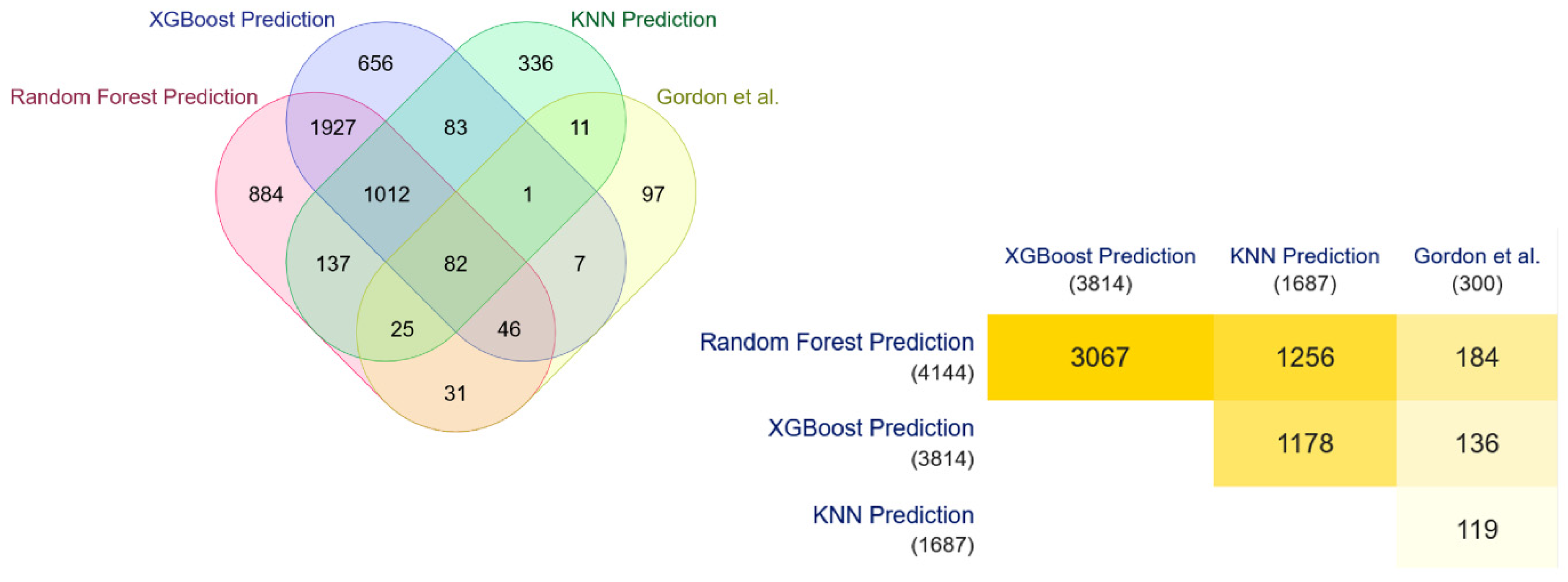
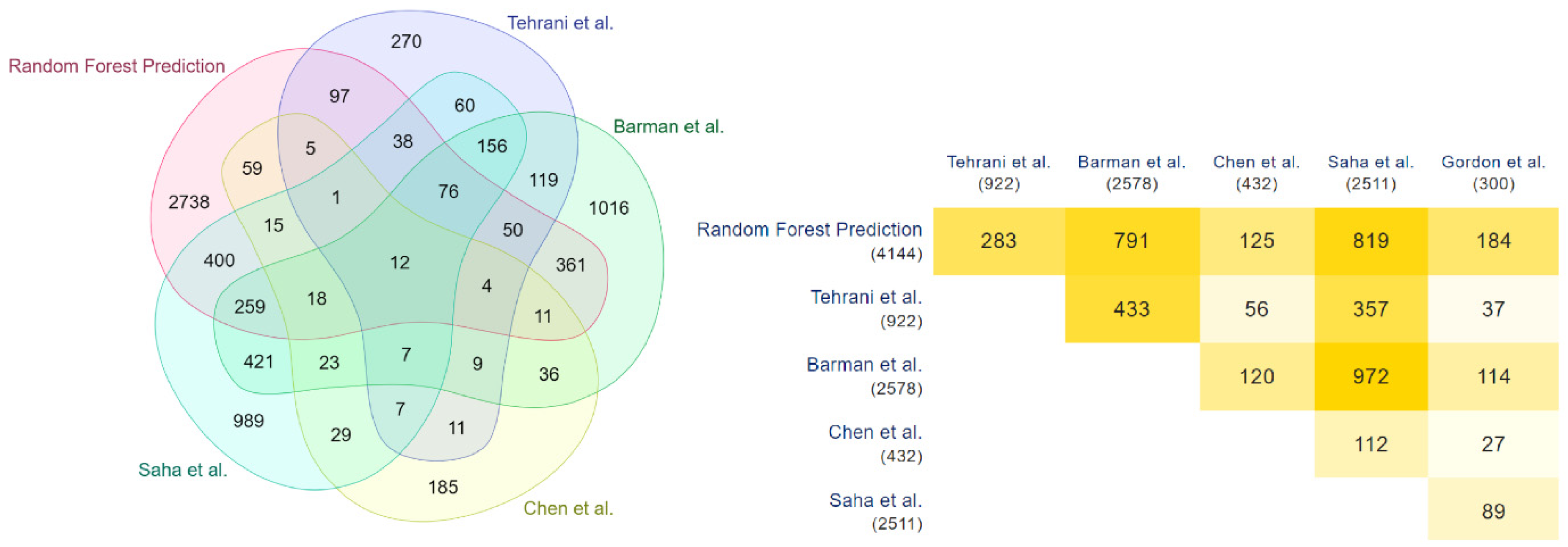
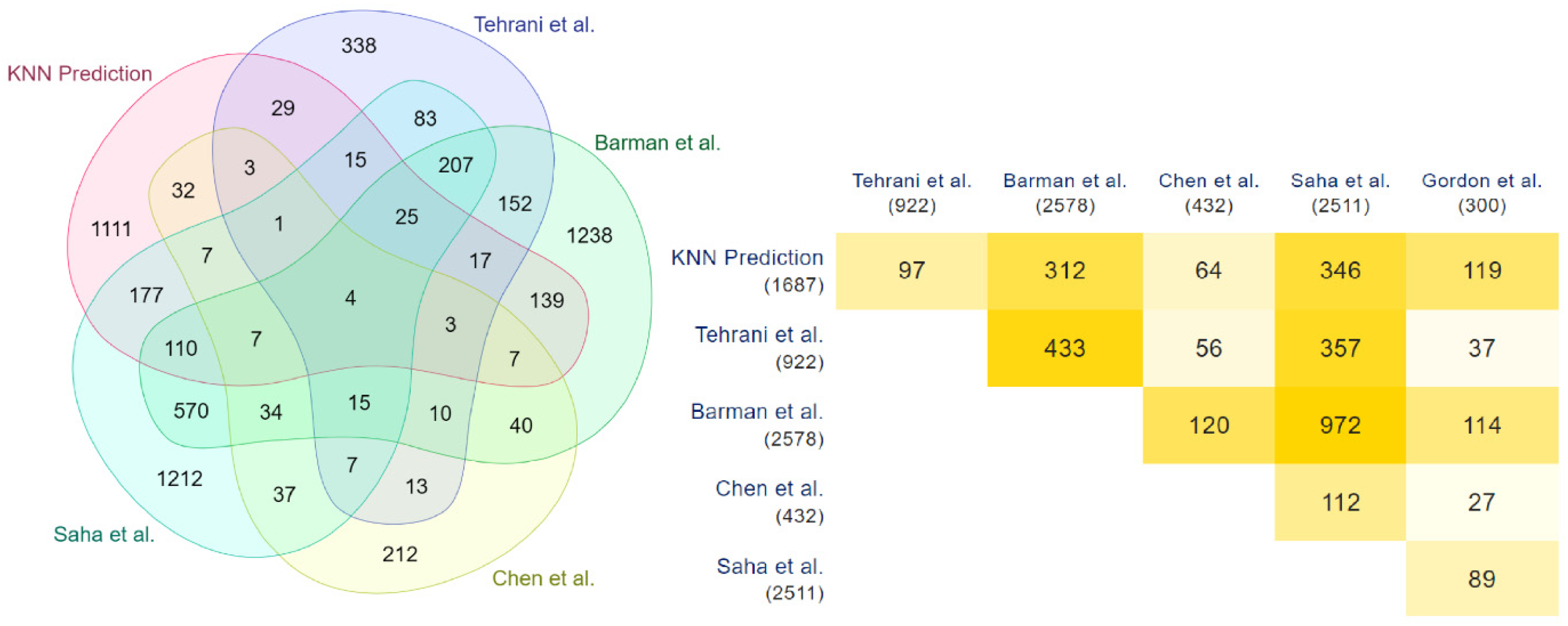
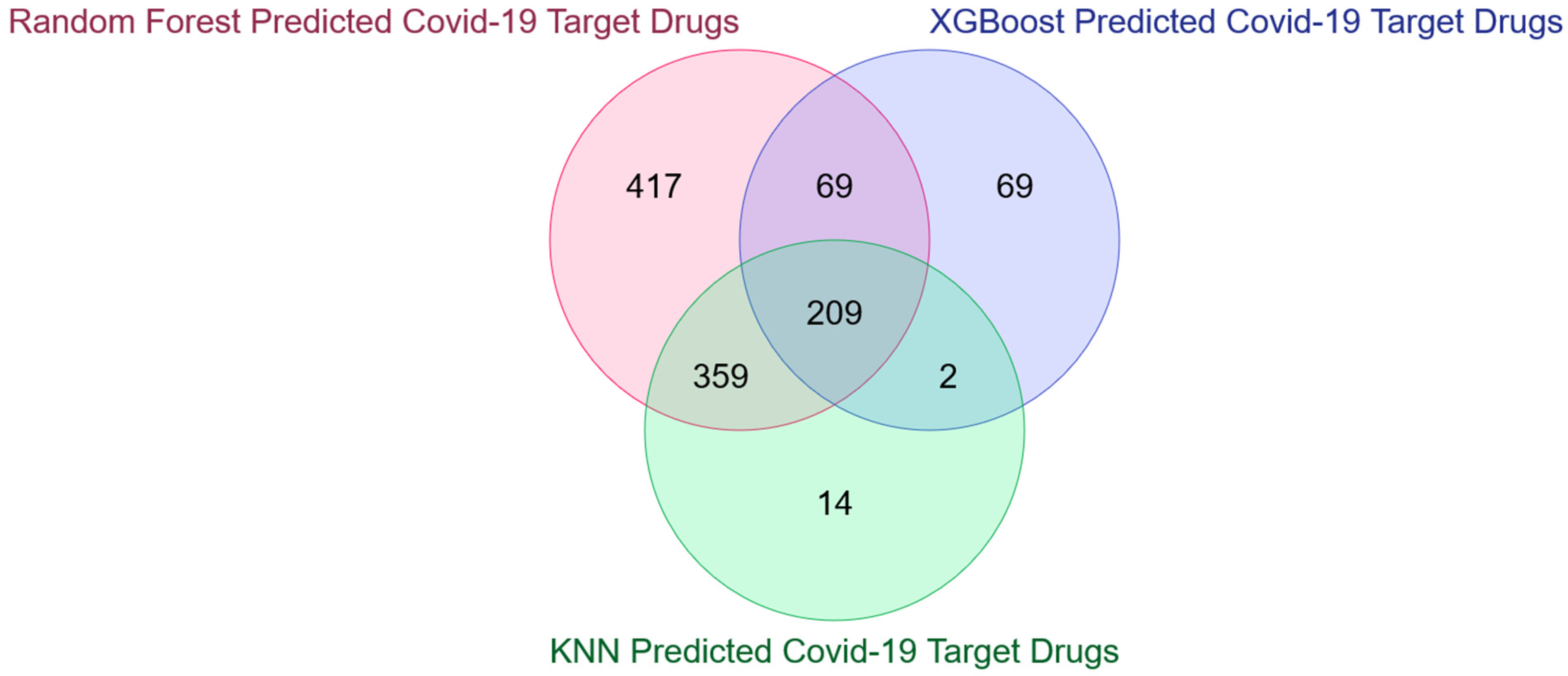
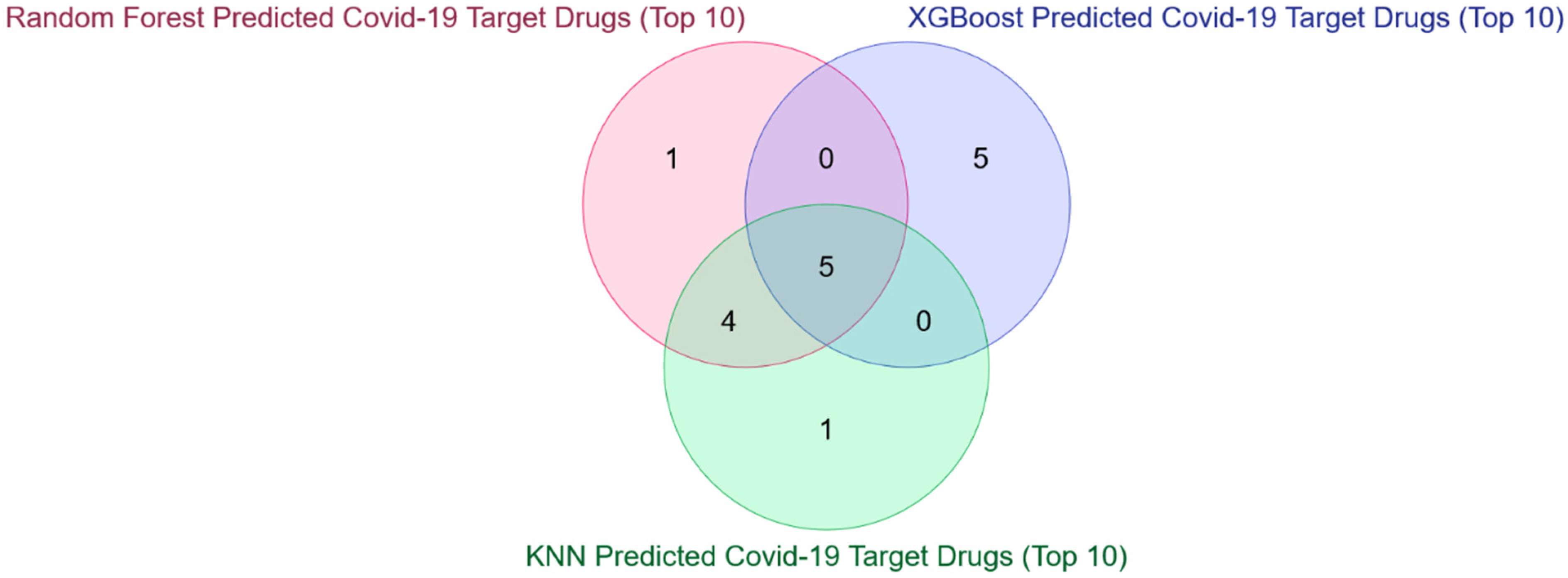
3.2. Detection and Validation of Novel COVID-19 Human Drug Targets
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barman, R.K.; Mukhopadhyay, A.; Maulik, U.; Das, S. A network biology approach to identify crucial host targets for COVID-19. Methods 2022, 203, 108–115. [Google Scholar] [CrossRef]
- McFarland, J.M.; Ho, Z.V.; Kugener, G.; Dempster, J.M.; Montgomery, P.G.; Bryan, J.G.; Krill-Burger, J.M.; Green, T.M.; Vazquez, F.; Boehm, J.S.; et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun. 2018, 9, 4610. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Saha, S.; Halder, A.K.; Bandyopadhyay, S.S.; Chatterjee, P.; Nasipuri, M.; Bose, D.; Basu, S. Drug repurposing for COVID-19 using computational screening: Is Fostamatinib/R406 a potential candidate? Methods 2022, 203, 564–574. [Google Scholar] [CrossRef]
- Rutwick Surya, U.; Praveen, N. A molecular docking study of SARS-CoV-2 main protease against phytochemicals of Boerhavia diffusa Linn. for novel COVID-19 drug discovery. Virusdisease 2021, 32, 46–54. [Google Scholar] [CrossRef]
- Hosseini, M.; Chen, W.; Xiao, D.; Wang, C. Computational molecular docking and virtual screening revealed promising SARS-CoV-2 drugs. Precis. Clin. Med. 2021, 4, 1–16. [Google Scholar] [CrossRef]
- El-Behery, H.; Attia, A.-F.; El-Fishawy, N.; Torkey, H. Efficient machine learning model for predicting drug-target interactions with case study for Covid-19. Comput. Biol. Chem. 2021, 93, 107536. [Google Scholar] [CrossRef]
- Wang, W.; Lv, H.; Zhao, Y.; Liu, D.; Wang, Y.; Zhang, Y. DLS: A Link Prediction Method Based on Network Local Structure for Predicting Drug-Protein Interactions. Front. Bioeng. Biotechnol. 2020, 8, 330. [Google Scholar] [CrossRef]
- Dezső, Z.; Ceccarelli, M. Machine learning prediction of oncology drug targets based on protein and network properties. BMC Bioinform. 2020, 21, 104. [Google Scholar] [CrossRef]
- Li, F.; Zhang, Z.; Guan, J.; Zhou, S. Effective drug–target interaction prediction with mutual interaction neural network. Bioinformatics 2022, 38, 3582–3589. [Google Scholar] [CrossRef]
- Adhami, M.; Sadeghi, B.; Rezapour, A.; Haghdoost, A.A.; MotieGhader, H. Repurposing novel therapeutic candidate drugs for coronavirus disease-19 based on protein-protein interaction network analysis. BMC Biotechnol. 2021, 21, 22. [Google Scholar] [CrossRef]
- Chen, X.; Ji, Z.L.; Chen, Y.Z. TTD: Therapeutic Target Database. Nucleic Acids Res. 2002, 30, 412–415. [Google Scholar] [CrossRef]
- Drews, J. Drug Discovery: A Historical Perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Wuchty, S.; Stadler, P.F. Centers of complex networks. J. Theor. Biol. 2003, 223, 45–53. [Google Scholar] [CrossRef]
- Joy, M.P.; Brock, A.; Ingber, D.E.; Huang, S. High-Betweenness Proteins in the Yeast Protein Interaction Network. J. Biomed. Biotechnol. 2005, 2005, 594674. [Google Scholar] [CrossRef]
- Tang, Y.; Li, M.; Wang, J.; Pan, Y.; Wu, F.-X. CytoNCA: A cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems 2015, 127, 67–72. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Pande, A.; Patiyal, S.; Lathwal, A.; Arora, C.; Kaur, D.; Dhall, A.; Mishra, G.; Kaur, H.; Sharma, N.; Jain, S.; et al. Computing wide range of protein/peptide features from their sequence and Structure. BioRxiv 2019. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Consortium, T.G.O. The Gene Ontology project in 2008. Nucleic Acids Res. 2007, 36, D440–D444. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob, H. Reactome pathway analysis: A high-performance in-memory approach. BMC Bioinform. 2017, 18, 142. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Schapire, R.E. A brief introduction to boosting. In Proceedings of the 16th International Joint Conference on Artificial Intelligence, Stockholm, Sweden, 31 July–6 August 1999; Volume 2, pp. 1401–1406. [Google Scholar]
- Bacaër, N. Verhulst and the logistic equation (1838). In A Short History of Mathematical Population Dynamics; Bacaër, N., Ed.; Springer London: London, UK, 2011; pp. 35–39. [Google Scholar]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Kamiński, B.; Jakubczyk, M.; Szufel, P. A framework for sensitivity analysis of decision trees. Cent. Eur. J. Oper. Res. 2018, 26, 135–159. [Google Scholar] [CrossRef]
- Tin Kam, H. Random decision forests. In Proceedings of the 3rd International Conference on Document Analysis and Recognition, Montreal, QC, Canada, 14–16 August 1995; Volume 271, pp. 278–282. [Google Scholar]
- Hand, D.J.; Yu, K. Idiot’s Bayes: Not So Stupid after All? Int. Stat. Rev. Rev. Int. De Stat. 2001, 69, 385–398. [Google Scholar] [CrossRef]
- Fix, E.; Hodges, J.L. Discriminatory Analysis. Nonparametric Discrimination: Consistency Properties. Int. Stat. Rev. Rev. Int. De Stat. 1989, 57, 238–247. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, C.; Feng, X.; Nie, L.; Tang, M.; Zhang, H.; Xiong, Y.; Swisher, S.K.; Srivastava, M.; Chen, J. Interactomes of SARS-CoV-2 and human coronaviruses reveal host factors potentially affecting pathogenesis. EMBO J. 2021, 40, e107776. [Google Scholar] [CrossRef]
- Saha, S.; Chatterjee, P.; Nasipuri, M.; Basu, S. Detection of spreader nodes in human-SARS-CoV protein-protein interaction network. PeerJ 2021, 9, e12117. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Samavarchi-Tehrani, P.; Abdouni, H.; Knight, J.; Astori, A.; Samson, R.; Lin, Z.-Y.; Kim, D.-K.; Knapp, J.; St-Germain, J.; Go, C.; et al. A SARS-CoV-2—host proximity interactome. BioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, Y.; Min, Z.; Mo, J.; Ju, Z.; Guan, W.; Zeng, B.; Liu, Y.; Chen, J.; Zhang, Q.; et al. COVID19db: A comprehensive database platform to discover potential drugs and targets of COVID-19 at whole transcriptomic scale. Nucleic Acids Res. 2022, 50, D747–D757. [Google Scholar] [CrossRef]
- Avram, S.; Bologa, C.G.; Holmes, J.; Bocci, G.; Wilson, T.B.; Nguyen, D.-T.; Curpan, R.; Halip, L.; Bora, A.; Yang, J.J.; et al. DrugCentral 2021 supports drug discovery and repositioning. Nucleic Acids Res. 2021, 49, D1160–D1169. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef]
- Chen, T.-F.; Chang, Y.-C.; Hsiao, Y.; Lee, K.-H.; Hsiao, Y.-C.; Lin, Y.-H.; Tu, Y.-C.E.; Huang, H.-C.; Chen, C.-Y.; Juan, H.-F. DockCoV2: A drug database against SARS-CoV-2. Nucleic Acids Res. 2021, 49, D1152–D1159. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; El Oualid, F.; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti–COVID-19 drug design. Sci. Adv. 2020, 6, eabd4596. [Google Scholar] [CrossRef]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.-D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Yang, M.; Hong, Z.; Zhang, L.; Huang, Z.; Chen, X.; He, S.; Zhou, Z.; Zhou, Z.; Chen, Q.; et al. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm. Sin. B 2020, 10, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-Ray Structures Reveal a Large Hinge-bending Motion Important for Inhibitor Binding and Catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef] [PubMed]
- McHugh, M.L. Interrater reliability: The kappa statistic. Biochem. Med. 2012, 22, 276–282. [Google Scholar] [CrossRef]
- Kumar, R.; Indrayan, A. Receiver operating characteristic (ROC) curve for medical researchers. Indian Pediatrics 2011, 48, 277–287. [Google Scholar] [CrossRef]
- Available online: https://molbiotools.com/ (accessed on 1 May 2022).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl. No. | Specifications/Features | SelectKBest Score |
|---|---|---|
| 1 | GO (BP) Rank2 | 7.62 |
| 2 | GO (MF) Rank1 | 6.73 |
| 3 | GO (BP) Rank1 | 5.25 |
| 4 | GO (MF) Rank2 | 3.94 |
| 5 | GO (CC) Rank1 | 3.34 |
| 6 | Reactome Rank2 | 2.89 |
| 7 | GO (CC) Rank2 | 2.82 |
| 8 | Reactome Rank1 | 1.61 |
| 9 | KEGG Rank1 | 1.59 |
| 10 | KEGG Rank2 | 1.27 |
| 11 | Betweenness | 1.14 |
| 12 | Tiny (PCP_TN) | 0.72 |
| 13 | Degree | 0.6 |
| 14 | Network | 0.56 |
| 15 | Sulphur Content (PCP_SC) | 0.35 |
| 16 | Solvent Accessibility (Intermediate) (PCP_SA_IN) | 0.32 |
| 17 | Cyclic (PCP_CY) | 0.25 |
| 18 | Small (PCP_SM) | 0.21 |
| 19 | Subgraph | 0.212 |
| 20 | Aromaticity (PCP_AR) | 0.18 |
| Sl. No. | Models | Accuracy | Precision | Recall | F1-Score | Cohens Kappa Score | Area under Curve |
|---|---|---|---|---|---|---|---|
| 1 | XGBBoost | 0.80 | 0.73 | 0.87 | 0.79 | 0.61 | 0.83 |
| 2 | AdaBoost | 0.75 | 0.65 | 0.93 | 0.76 | 0.51 | 0.79 |
| 3 | LR | 0.66 | 0.60 | 0.75 | 0.66 | 0.34 | 0.75 |
| 4 | SVM | 0.66 | 0.59 | 0.81 | 0.68 | 0.34 | 0.74 |
| 5 | Decision Tree | 0.58 | 0.52 | 0.75 | 0.61 | 0.19 | 0.60 |
| 6 | Random Forest | 0.72 | 0.63 | 0.87 | 0.73 | 0.45 | 0.83 |
| 7 | Naive Bayes | 0.63 | 0.57 | 0.75 | 0.64 | 0.29 | 0.74 |
| 8 | KNN | 0.77 | 0.83 | 0.62 | 0.71 | 0.53 | 0.81 |
| Drug Name | The Number of Target-Pathway Interactions | The Number of Targets | The Number of Pathways | Evidence for COVID-19 |
|---|---|---|---|---|
| Sunitinib | 91 | 24 | 44 | Link to PubMed |
| Bosutinib | 66 | 20 | 43 | Link to PubMed |
| Crizotinib | 35 | 15 | 25 | Link to PubMed |
| Midostaurin | 47 | 6 | 39 | Link to PubMed |
| Nintedanib | 24 | 6 | 22 | Link to PubMed |
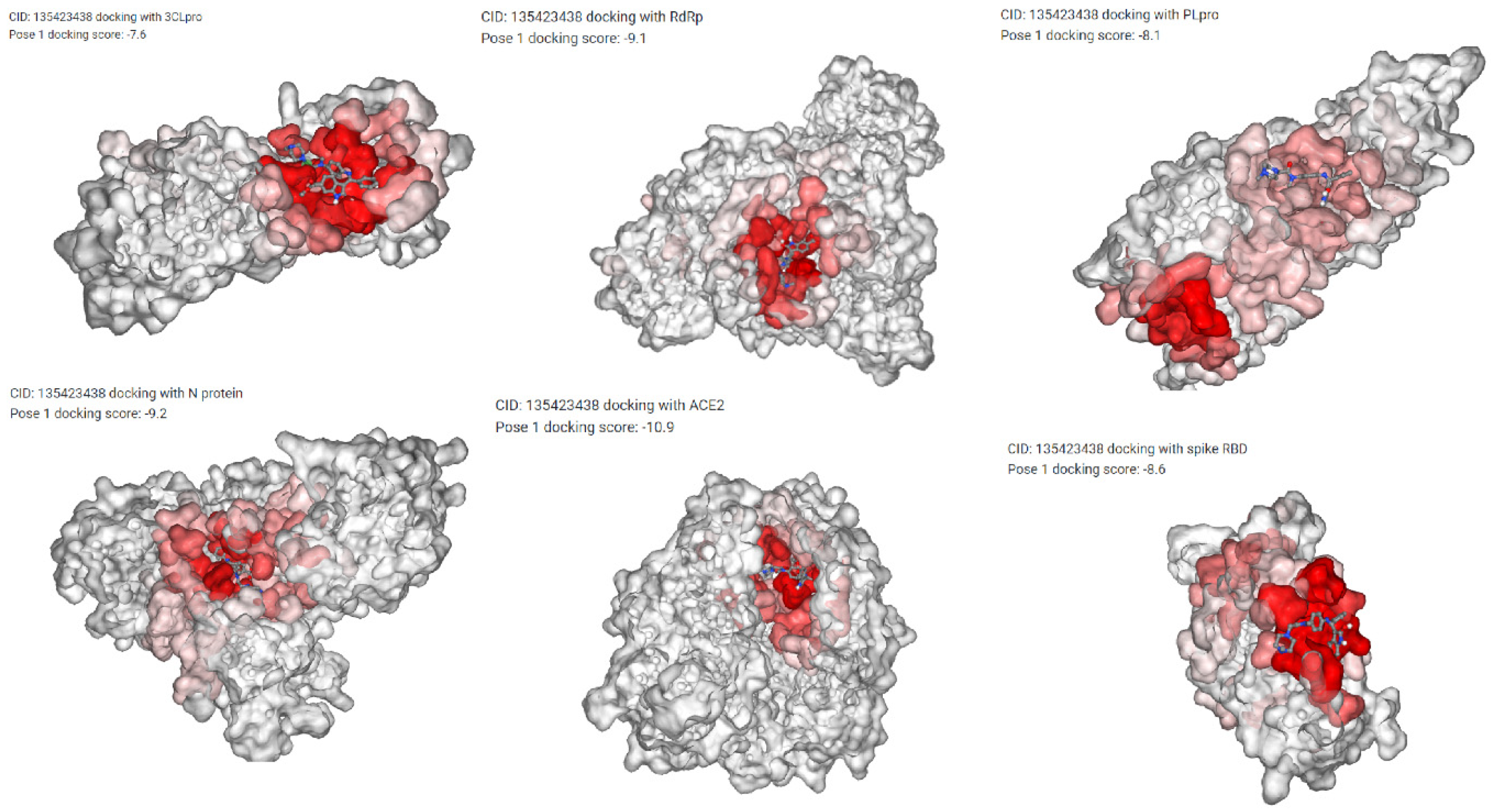
| COVID-19 Structure Details | Predicted Drugs Docking Score by DockCoV2 | |||||
|---|---|---|---|---|---|---|
| Structure Name | Structure ID | Bosutinib | Crizotinib | Midostaurin | Nintedanib | Sunitinib |
| 3C-like protease | 3CLpro | −7 | −7.4 | −8.5 | −7.6 | −7.1 |
| RNA-dependent RNA polymerase | RdRp | −8.4 | −7.9 | −8.9 | −9.1 | −7.6 |
| Papain-like protease | PLpro | −7.7 | −7.3 | −8.8 | −8.1 | −6.9 |
| nucleocapsid protein | N | −9 | −9.5 | −10.1 | −9.2 | −8.8 |
| ACE2 Receptor Protein | ACE2 | −8.8 | −9.2 | −14.5 | −10.9 | −8 |
| Spike Protein | Spike RBD | −7 | −7 | −8.8 | −8.6 | −8.3 |
| Predicted Drugs Best Pose by DockCoV2 | |||||
|---|---|---|---|---|---|
| Structure ID | Bosutinib | Crizotinib | Midostaurin | Nintedanib | Sunitinib |
| 3CLpro | Best Pose Link | Best Pose Link | Best Pose Link | Best Pose Link | Best Pose Link |
| RdRp | Best Pose Link | Best Pose Link | Best Pose Link | Best Pose Link | Best Pose Link |
| PLpro | Best Pose Link | Best Pose Link | Best Pose Link | Best Pose Link | Best Pose Link |
| N | Best Pose Link | Best Pose Link | Best Pose Link | Best Pose Link | Best Pose Link |
| ACE2 | Best Pose Link | Best Pose Link | Best Pose Link | Best Pose Link | Best Pose Link |
| Spike RBD | Best Pose Link | Best Pose Link | Best Pose Link | Best Pose Link | Best Pose Link |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saha, S.; Chatterjee, P.; Halder, A.K.; Nasipuri, M.; Basu, S.; Plewczynski, D. ML-DTD: Machine Learning-Based Drug Target Discovery for the Potential Treatment of COVID-19. Vaccines 2022, 10, 1643. https://doi.org/10.3390/vaccines10101643
Saha S, Chatterjee P, Halder AK, Nasipuri M, Basu S, Plewczynski D. ML-DTD: Machine Learning-Based Drug Target Discovery for the Potential Treatment of COVID-19. Vaccines. 2022; 10(10):1643. https://doi.org/10.3390/vaccines10101643
Chicago/Turabian StyleSaha, Sovan, Piyali Chatterjee, Anup Kumar Halder, Mita Nasipuri, Subhadip Basu, and Dariusz Plewczynski. 2022. "ML-DTD: Machine Learning-Based Drug Target Discovery for the Potential Treatment of COVID-19" Vaccines 10, no. 10: 1643. https://doi.org/10.3390/vaccines10101643
APA StyleSaha, S., Chatterjee, P., Halder, A. K., Nasipuri, M., Basu, S., & Plewczynski, D. (2022). ML-DTD: Machine Learning-Based Drug Target Discovery for the Potential Treatment of COVID-19. Vaccines, 10(10), 1643. https://doi.org/10.3390/vaccines10101643