
New Insights into SARS-CoV-2 and Cancer Cross-Talk: Does a Novel Oncogenesis Driver Emerge?

 , , and
, , and 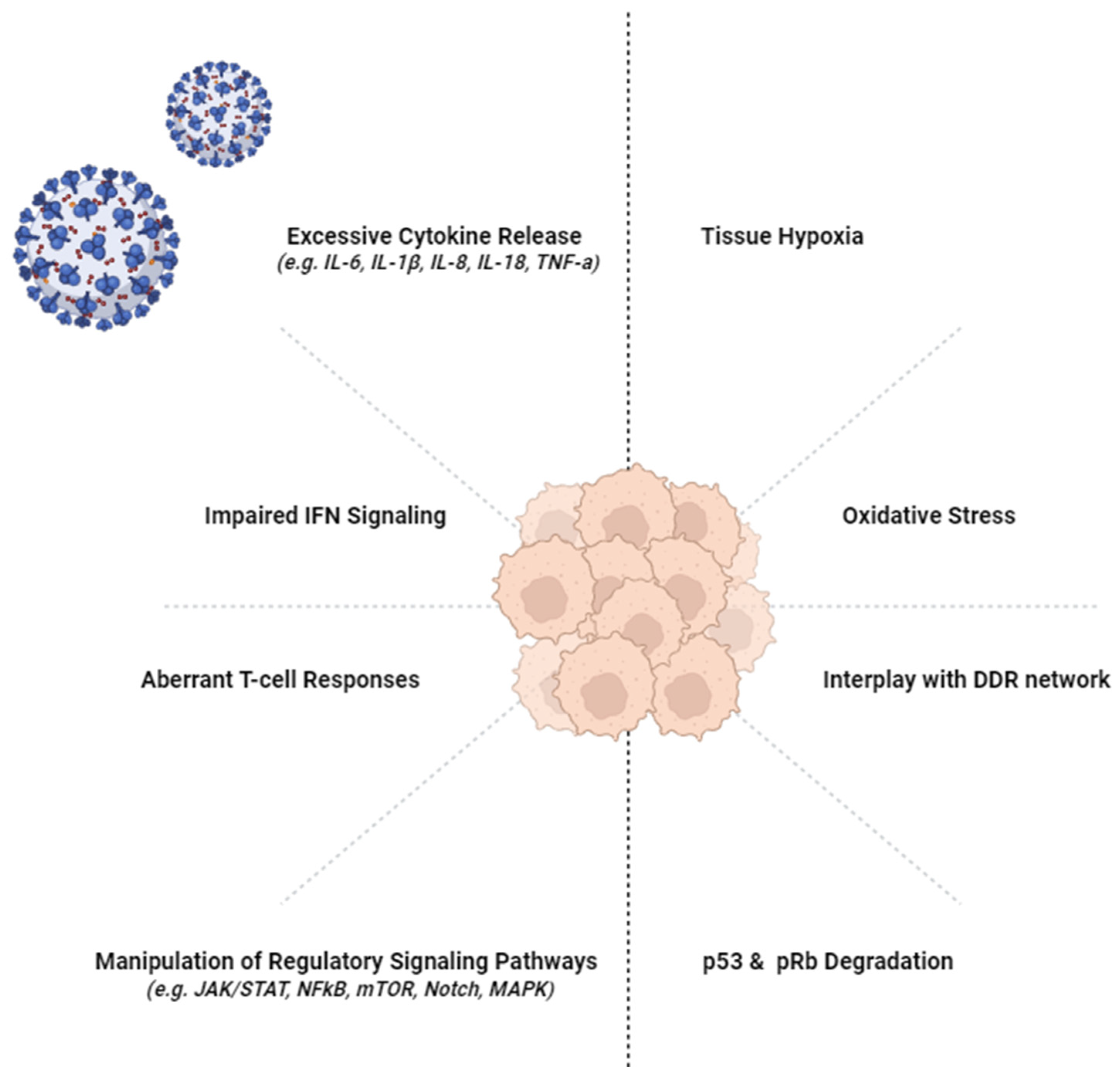{kind=link}
Abstract
1. Introduction
2. Viral Infections and Cancer
3. Chronic Inflammation and Cancer
Cytokine Release
4. Immune Escape and Cancer
5. Gut Microbiota and Cancer
6. Cytokine Release and Long-COVID
7. T-Cell Response and Long-COVID
8. Tissue Damage and Long-COVID
9. Gut Microbiota and Long-COVID
9.1. Compositional Changes of the Gut Bacterial Microbiome
9.2. The Gut Mycobiome in COVID-19
9.3. The Gut Virome in COVID-19
10. Oncogenic Pathways and SARS-CoV-2
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guarner, J. Three Emerging Coronaviruses in Two Decades. Am. J. Clin. Pathol. 2020, 153, 420–421. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Perelman, C.; Sepulveda, R.; Rebolledo, P.A.; Cuapio, A.; Villapol, S. More than 50 long-term effects of COVID-19: A systematic review and meta-analysis. Sci. Rep. 2021, 11, 16144. [Google Scholar] [CrossRef] [PubMed]
- Yelin, D.; Wirtheim, E.; Vetter, P.; Kalil, A.C.; Bruchfeld, J.; Runold, M.; Guaraldi, G.; Mussini, C.; Gudiol, C.; Pujol, M.; et al. Long-term consequences of COVID-19: Research needs. Lancet Infect. Dis. 2020, 20, 1115–1117. [Google Scholar] [CrossRef]
- Stingi, A.; Cirillo, L. SARS-CoV-2 infection and cancer: Evidence for and against a role of SARS-CoV-2 in cancer onset. BioEssays 2021, 43, e2000289. [Google Scholar] [CrossRef]
- Alpalhão, M.; Ferreira, J.A.; Filipe, P. Persistent SARS-CoV-2 infection and the risk for cancer. Med. Hypotheses 2020, 143, 109882. [Google Scholar] [CrossRef]
- Bora, V.R.; Patel, B.M. The Deadly Duo of COVID-19 and Cancer! Front. Mol. Biosci. 2021, 8, 643004. [Google Scholar] [CrossRef]
- Zong, Z.; Wei, Y.; Ren, J.; Zhang, L.; Zhou, F. The intersection of COVID-19 and cancer: Signaling pathways and treatment implications. Mol. Cancer 2021, 20, 76. [Google Scholar] [CrossRef]
- Al-Quteimat, O.M.; Amer, A.M. The Impact of the COVID-19 Pandemic on Cancer Patients. Am. J. Clin. Oncol. 2020, 43, 452–455. [Google Scholar] [CrossRef]
- Robilotti, E.V.; Babady, N.E.; Mead, P.A.; Rolling, T.; Perez-Johnston, R.; Bernardes, M.; Bogler, Y.; Caldararo, M.; Figueroa, C.J.; Glickman, M.S.; et al. Determinants of COVID-19 disease severity in patients with cancer. Nat. Med. 2020, 26, 1218–1223. [Google Scholar] [CrossRef]
- Rugge, M.; Zorzi, M.; Guzzinati, S. SARS-CoV-2 infection in the Italian Veneto region: Adverse outcomes in patients with cancer. Nat. Cancer 2020, 1, 784–788. [Google Scholar] [CrossRef]
- Russell, B.; Moss, C.L.; Shah, V.; Ko, T.K.; Palmer, K.; Sylva, R.; George, G.; Monroy-Iglesias, M.J.; Patten, P.; Ceesay, M.M.; et al. Risk of COVID-19 death in cancer patients: An analysis from Guy’s Cancer Centre and King’s College Hospital in London. Br. J. Cancer 2021, 125, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, F.; Cattaneo, C.; Arcaini, L.; Bruna, R.; Cavo, M.; Merli, F.; Angelucci, E.; Krampera, M.; Cairoli, R.; Della Porta, M.G.; et al. Clinical characteristics and risk factors associated with COVID-19 severity in patients with haematological malignancies in Italy: A retrospective, multicentre, cohort study. Lancet Haematol. 2020, 7, e737–e745. [Google Scholar] [CrossRef]
- Luo, J.; Rizvi, H.; Preeshagul, I.R.; Egger, J.V.; Hoyos, D.; Bandlamudi, C.; McCarthy, C.G.; Falcon, C.J.; Schoenfeld, A.J.; Arbour, K.C.; et al. COVID-19 in patients with lung cancer. Ann. Oncol. 2020, 31, 1386–1396. [Google Scholar] [CrossRef] [PubMed]
- Derosa, L.; Melenotte, C.; Griscelli, F.; Gachot, B.; Marabelle, A.; Kroemer, G.; Zitvogel, L. The immuno-oncological challenge of COVID-19. Nat. Cancer 2020, 1, 946–964. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Krump, N.A.; You, J. Molecular mechanisms of viral oncogenesis in humans. Nat. Rev. Microbiol. 2018, 16, 684–698. [Google Scholar] [PubMed]
- Kellogg, C.; Kouznetsova, V.L.; Tsigelny, I.F. Implications of viral infection in cancer development. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188622. [Google Scholar] [CrossRef]
- Tempera, I.; Lieberman, P.M. Oncogenic Viruses as Entropic Drivers of Cancer Evolution. Front. Virol. 2021, 1, 753366. [Google Scholar] [CrossRef]
- Pietropaolo, V.; Prezioso, C.; Moens, U. Role of Virus-Induced Host Cell Epigenetic Changes in Cancer. Int. J. Mol. Sci. 2021, 22, 8346. [Google Scholar] [CrossRef]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef]
- Akram, N.; Imran, M.; Noreen, M.; Ahmed, F.; Atif, M.; Fatima, Z.; Bilal Waqar, A. Oncogenic Role of Tumor Viruses in Humans. Viral Immunol. 2017, 30, 20–27. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Munger, K. Viruses associated with human cancer. Biochim. Biophys. Acta 2008, 1782, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Morales-Sánchez, A.; Fuentes-Pananá, E.M. Human viruses and cancer. Viruses 2014, 6, 4047–4079. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef]
- Gruhne, B.; Sompallae, R.; Marescotti, D.; Kamranvar, S.A.; Gastaldello, S.; Masucci, M.G. The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2009, 106, 2313–2318. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Weitzman, J.B. What’s the damage? The impact of pathogens on pathways that maintain host genome integrity. Cell Host Microbe 2014, 15, 283–294. [Google Scholar] [CrossRef]
- Desfarges, S.; Ciuffi, A. Viral Integration and Consequences on Host Gene Expression. In Viruses: Essential Agents of Life; Witzany, G., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 147–175. [Google Scholar]
- Soliman, S.H.A.; Orlacchio, A.; Verginelli, F. Viral Manipulation of the Host Epigenome as a Driver of Virus-Induced Oncogenesis. Microorganisms 2021, 9, 1179. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.W.; Housseau, F. The ‘kiss of death’ by dendritic cells to cancer cells. Cell Death Differ. 2008, 15, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- DeVito, N.C.; Plebanek, M.P.; Theivanthiran, B.; Hanks, B.A. Role of Tumor-Mediated Dendritic Cell Tolerization in Immune Evasion. Front. Immunol. 2019, 10, 2876. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Piotrowski, I.; Kulcenty, K.; Suchorska, W. Interplay between inflammation and cancer. Rep. Pract. Oncol. Radiother. 2020, 25, 422–427. [Google Scholar] [CrossRef]
- Prud’homme, G.J. Pathobiology of transforming growth factor beta in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab. Investig. 2007, 87, 1077–1091. [Google Scholar] [CrossRef]
- Yang, L.; Huang, J.; Ren, X.; Gorska, A.E.; Chytil, A.; Aakre, M.; Carbone, D.P.; Matrisian, L.M.; Richmond, A.; Lin, P.C.; et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008, 13, 23–35. [Google Scholar] [CrossRef]
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H. The role of TNF in cancer. Results Probl. Cell Differ. 2009, 49, 1–15. [Google Scholar] [PubMed]
- Grivennikov, S.I.; Karin, M. Inflammatory cytokines in cancer: Tumour necrosis factor and interleukin 6 take the stage. Ann. Rheum. Dis. 2011, 70, i104–i108. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Briukhovetska, D.; Dörr, J.; Endres, S.; Libby, P.; Dinarello, C.A.; Kobold, S. Interleukins in cancer: From biology to therapy. Nat. Rev. Cancer 2021, 21, 481–499. [Google Scholar] [CrossRef]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef]
- Qian, S.; Golubnitschaja, O.; Zhan, X. Chronic inflammation: Key player and biomarker-set to predict and prevent cancer development and progression based on individualized patient profiles. EPMA J. 2019, 10, 365–381. [Google Scholar] [CrossRef]
- Chen, X.; Cai, G.; Liu, C.; Zhao, J.; Gu, C.; Wu, L.; Hamilton, T.A.; Zhang, C.J.; Ko, J.; Zhu, L.; et al. IL-17R-EGFR axis links wound healing to tumorigenesis in Lrig1+ stem cells. J. Exp. Med. 2019, 216, 195–214. [Google Scholar] [CrossRef]
- Choi, M.R.; Sosman, J.A.; Zhang, B. The Janus Face of IL-33 Signaling in Tumor Development and Immune Escape. Cancers 2021, 13, 3281. [Google Scholar] [CrossRef]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 2015, 43, 435–449. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Lin, C.F.; Lin, C.M.; Lee, K.Y.; Wu, S.Y.; Feng, P.H.; Chen, K.Y.; Chuang, H.C.; Chen, C.L.; Wang, Y.C.; Tseng, P.C.; et al. Escape from IFN-γ-dependent immunosurveillance in tumorigenesis. J. Biomed. Sci. 2017, 24, 10. [Google Scholar] [CrossRef]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef]
- Kim, S.K.; Cho, S.W. The Evasion Mechanisms of Cancer Immunity and Drug Intervention in the Tumor Microenvironment. Front. Pharmacol. 2022, 13, 868695. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- So, T.; Takenoyama, M.; Mizukami, M.; Ichiki, Y.; Sugaya, M.; Hanagiri, T.; Sugio, K.; Yasumoto, K. Haplotype loss of HLA class I antigen as an escape mechanism from immune attack in lung cancer. Cancer Res. 2005, 65, 5945–5952. [Google Scholar] [CrossRef] [PubMed]
- Atkins, D.; Breuckmann, A.; Schmahl, G.E.; Binner, P.; Ferrone, S.; Krummenauer, F.; Störkel, S.; Seliger, B. MHC class I antigen processing pathway defects, ras mutations and disease stage in colorectal carcinoma. Int. J. Cancer 2004, 109, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Ochsenbein, A.F. Immunological ignorance of solid tumors. Semin. Immunopathol. 2005, 27, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Medema, J.P.; de Jong, J.; Peltenburg, L.T.; Verdegaal, E.M.; Gorter, A.; Bres, S.A.; Franken, K.L.; Hahne, M.; Albar, J.P.; Melief, C.J.; et al. Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 11515–11520. [Google Scholar] [CrossRef]
- Bronte, V.; Zanovello, P. Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 2005, 5, 641–654. [Google Scholar] [CrossRef]
- Wu, A.A.; Drake, V.; Huang, H.S.; Chiu, S.; Zheng, L. Reprogramming the tumor microenvironment: Tumor-induced immunosuppressive factors paralyze T cells. Oncoimmunology 2015, 4, e1016700. [Google Scholar] [CrossRef]
- Yu, C.R.; Mahdi, R.M.; Ebong, S.; Vistica, B.P.; Gery, I.; Egwuagu, C.E. Suppressor of cytokine signaling 3 regulates proliferation and activation of T-helper cells. J. Biol. Chem. 2003, 278, 29752–29759. [Google Scholar] [CrossRef]
- Egwuagu, C.E.; Yu, C.R.; Zhang, M.; Mahdi, R.M.; Kim, S.J.; Gery, I. Suppressors of cytokine signaling proteins are differentially expressed in Th1 and Th2 cells: Implications for Th cell lineage commitment and maintenance. J. Immunol. 2002, 168, 3181–3187. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef]
- Terness, P.; Bauer, T.M.; Röse, L.; Dufter, C.; Watzlik, A.; Simon, H.; Opelz, G. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: Mediation of suppression by tryptophan metabolites. J. Exp. Med. 2002, 196, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The inflammatory micro-environment in tumor progression: The role of tumor-associated macrophages. Crit. Rev. Oncol. Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Khaled, Y.S.; Ammori, B.J.; Elkord, E. Myeloid-derived suppressor cells in cancer: Recent progress and prospects. Immunol. Cell Biol. 2013, 91, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Huang, Y.; Obholzer, N.; Fayad, R.; Qiao, L. Turning on/off tumor-specific CTL response during progressive tumor growth. J. Immunol. 2005, 175, 3110–3116. [Google Scholar] [CrossRef]
- Zhou, G.; Lu, Z.; McCadden, J.D.; Levitsky, H.I.; Marson, A.L. Reciprocal changes in tumor antigenicity and antigen-specific T cell function during tumor progression. J. Exp. Med. 2004, 200, 1581–1592. [Google Scholar] [CrossRef]
- Sakaguchi, S. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 2004, 22, 531–562. [Google Scholar] [CrossRef]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Durack, J.; Lynch, S.V. The gut microbiome: Relationships with disease and opportunities for therapy. J. Exp. Med. 2019, 216, 20–40. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef]
- Fulbright, L.E.; Ellermann, M.; Arthur, J.C. The microbiome and the hallmarks of cancer. PLoS Pathog. 2017, 13, e1006480. [Google Scholar] [CrossRef]
- Gagnaire, A.; Nadel, B.; Raoult, D.; Neefjes, J.; Gorvel, J.P. Collateral damage: Insights into bacterial mechanisms that predispose host cells to cancer. Nat. Rev. Microbiol. 2017, 15, 109–128. [Google Scholar] [CrossRef]
- Vivarelli, S.; Salemi, R.; Candido, S.; Falzone, L.; Santagati, M.; Stefani, S.; Torino, F.; Banna, G.L.; Tonini, G.; Libra, M. Gut Microbiota and Cancer: From Pathogenesis to Therapy. Cancers 2019, 11, 38. [Google Scholar] [CrossRef]
- Zitvogel, L.; Daillère, R.; Roberti, M.P.; Routy, B.; Kroemer, G. Anticancer effects of the microbiome and its products. Nat. Rev. Microbiol. 2017, 15, 465–478. [Google Scholar] [CrossRef]
- Ge, Y.; Wang, X.; Guo, Y.; Yan, J.; Abuduwaili, A.; Aximujiang, K.; Yan, J.; Wu, M. Gut microbiota influence tumor development and Alter interactions with the human immune system. J. Exp. Clin. Cancer Res. 2021, 40, 42. [Google Scholar] [CrossRef]
- Bhatt, A.P.; Redinbo, M.R.; Bultman, S.J. The role of the microbiome in cancer development and therapy. CA Cancer J. Clin. 2017, 67, 326–344. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, C.; Carlini, F.; Germinario, E.A.P.; Maroccia, Z.; Travaglione, S.; Fabbri, A. Gut Microbiota and Colon Cancer: A Role for Bacterial Protein Toxins? Int. J. Mol. Sci. 2020, 21, 6201. [Google Scholar] [CrossRef] [PubMed]
- Frisan, T. Bacterial genotoxins: The long journey to the nucleus of mammalian cells. Biochim. Biophys. Acta 2016, 1858, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Lara-Tejero, M.; Galán, J.E. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 2000, 290, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Dai, W. Genomic Instability and Cancer. J. Carcinog. Mutagen. 2014, 5, 1000165. [Google Scholar] [PubMed]
- Wada, Y.; Takemura, K.; Tummala, P.; Uchida, K.; Kitagaki, K.; Furukawa, A.; Ishige, Y.; Ito, T.; Hara, Y.; Suzuki, T.; et al. Helicobacter pylori induces somatic mutations in TP53 via overexpression of CHAC1 in infected gastric epithelial cells. FEBS Open Bio 2018, 8, 671–679. [Google Scholar] [CrossRef]
- Bergounioux, J.; Elisee, R.; Prunier, A.L.; Donnadieu, F.; Sperandio, B.; Sansonetti, P.; Arbibe, L. Calpain activation by the Shigella flexneri effector VirA regulates key steps in the formation and life of the bacterium’s epithelial niche. Cell Host Microbe 2012, 11, 240–252. [Google Scholar] [CrossRef]
- Buti, L.; Spooner, E.; Van der Veen, A.G.; Rappuoli, R.; Covacci, A.; Ploegh, H.L. Helicobacter pylori cytotoxin-associated gene A (CagA) subverts the apoptosis-stimulating protein of p53 (ASPP2) tumor suppressor pathway of the host. Proc. Natl. Acad. Sci. USA 2011, 108, 9238–9243. [Google Scholar] [CrossRef]
- Chaturvedi, R.; Asim, M.; Romero-Gallo, J.; Barry, D.P.; Hoge, S.; de Sablet, T.; Delgado, A.G.; Wroblewski, L.E.; Piazuelo, M.B.; Yan, F.; et al. Spermine oxidase mediates the gastric cancer risk associated with Helicobacter pylori CagA. Gastroenterology 2011, 141, 1696–1708. [Google Scholar] [CrossRef]
- Goodwin, A.C.; Destefano Shields, C.E.; Wu, S.; Huso, D.L.; Wu, X.; Murray-Stewart, T.R.; Hacker-Prietz, A.; Rabizadeh, S.; Woster, P.M.; Sears, C.L.; et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15354–15359. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Elinav, E.; Huber, S.; Strowig, T.; Hao, L.; Hafemann, A.; Jin, C.; Wunderlich, C.; Wunderlich, T.; Eisenbarth, S.C.; et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 9862–9867. [Google Scholar] [CrossRef] [PubMed]
- Caesar, R.; Tremaroli, V.; Kovatcheva-Datchary, P.; Cani, P.D.; Backhed, F. Crosstalk between gut microbiota and dietary lipids aggravates WAT inflammation through TLR signaling. Cell Metab. 2015, 22, 658–668. [Google Scholar] [CrossRef]
- Gur, C.; Ibrahim, Y.; Isaacson, B.; Yamin, R.; Abed, J.; Gamliel, M.; Enk, J.; Bar-On, Y.; Stanietsky-Kaynan, N.; Coppenhagen-Glazer, S.; et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 2015, 42, 344–355. [Google Scholar] [CrossRef]
- Chang, S.H.; Minn, D.; Kim, S.W.; Kim, Y.K. Inflammatory Markers and Cytokines in Moderate and Critical Cases of COVID-19. Clin. Lab. 2021, 67. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Cao, J.; Yu, Y.; Ding, J.; Eshak, E.S.; Liu, K.; Mubarik, S.; Shi, F.; Wen, H.; Zeng, Z.; et al. Epidemiological characteristics of patients with severe COVID-19 infection in Wuhan, China: Evidence from a retrospective observational study. Int. J. Epidemiol. 2021, 49, 1940–1950. [Google Scholar] [CrossRef]
- da Silva Torres, M.K.; Bichara, C.D.A.; de Almeida, M.; Vallinoto, M.C.; Queiroz, M.A.F.; Vallinoto, I.; Dos Santos, E.J.M.; de Carvalho, C.A.M.; Vallinoto, A.C.R. The Complexity of SARS-CoV-2 Infection and the COVID-19 Pandemic. Front. Microbiol. 2022, 13, 789882. [Google Scholar] [CrossRef]
- Kaur, S.; Bansal, R.; Kollimuttathuillam, S.; Gowda, A.M.; Singh, B.; Mehta, D.; Maroules, M. The looming storm: Blood and cytokines in COVID-19. Blood Rev. 2021, 46, 100743. [Google Scholar] [CrossRef]
- Chen, Y.; Klein, S.L.; Garibaldi, B.T.; Li, H.; Wu, C.; Osevala, N.M.; Li, T.; Margolick, J.B.; Pawelec, G.; Leng, S.X. Aging in COVID-19: Vulnerability, immunity and intervention. Ageing Res. Rev. 2021, 65, 101205. [Google Scholar] [CrossRef]
- Java, A.; Apicelli, A.J.; Liszewski, M.K.; Coler-Reilly, A.; Atkinson, J.P.; Kim, A.H.; Kulkarni, H.S. The complement system in COVID-19: Friend and foe? JCI Insight 2020, 5, e140711. [Google Scholar] [CrossRef]
- Teijaro, J.R. Cytokine storms in infectious diseases. Semin. Immunopathol. 2017, 39, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.C.; Quach, T.T.T. COVID-19 cytokine storm syndrome: A threshold concept. Lancet Microbe 2021, 2, e49–e50. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Kunnumakkara, A.B.; Rana, V.; Parama, D.; Banik, K.; Girisa, S.; Henamayee, S.; Thakur, K.K.; Dutta, U.; Garodia, P.; Gupta, S.C.; et al. COVID-19, cytokines, inflammation, and spices: How are they related? Life Sci. 2021, 284, 119201. [Google Scholar] [CrossRef] [PubMed]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000.e3. [Google Scholar] [CrossRef]
- Logue, J.K.; Franko, N.M.; McCulloch, D.J.; McDonald, D.; Magedson, A.; Wolf, C.R.; Chu, H.Y. Sequelae in Adults at 6 Months After COVID-19 Infection. JAMA Netw. Open 2021, 4, e210830. [Google Scholar] [CrossRef]
- Al-Aly, Z.; Xie, Y.; Bowe, B. High-dimensional characterization of post-acute sequelae of COVID-19. Nature 2021, 594, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Huang, L.; Wang, Y.; Li, X.; Ren, L.; Gu, X.; Kang, L.; Guo, L.; Liu, M.; Zhou, X.; et al. 6-month consequences of COVID-19 in patients discharged from hospital: A cohort study. Lancet 2021, 397, 220–232. [Google Scholar] [CrossRef]
- Montefusco, L.; Ben Nasr, M.; D’Addio, F.; Loretelli, C.; Rossi, A.; Pastore, I.; Daniele, G.; Abdelsalam, A.; Maestroni, A.; Dell’Acqua, M.; et al. Acute and long-term disruption of glycometabolic control after SARS-CoV-2 infection. Nat. Metab. 2021, 3, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, P.; Salzman, S.H. Six-Minute Walk Test: Clinical Role, Technique, Coding, and Reimbursement. Chest 2020, 157, 603–611. [Google Scholar] [CrossRef]
- Blomberg, B.; Mohn, K.G.; Brokstad, K.A.; Zhou, F.; Linchausen, D.W.; Hansen, B.A.; Lartey, S.; Onyango, T.B.; Kuwelker, K.; Saevik, M.; et al. Long COVID in a prospective cohort of home-isolated patients. Nat. Med. 2021, 27, 1607–1613. [Google Scholar] [CrossRef] [PubMed]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-acute COVID-19 syndrome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Parums, D.V. Editorial: Long COVID, or Post-COVID Syndrome, and the Global Impact on Health Care. Med. Sci. Monit. 2021, 27, e933446. [Google Scholar] [CrossRef] [PubMed]
- Sykes, D.L.; Holdsworth, L.; Jawad, N.; Gunasekera, P.; Morice, A.H.; Crooks, M.G. Post-COVID-19 Symptom Burden: What is Long-COVID and How Should We Manage It? Lung 2021, 199, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Naeije, R.; Caravita, S. Phenotyping long COVID. Eur. Respir. J. 2021, 58, 2101763. [Google Scholar] [CrossRef]
- Proal, A.D.; VanElzakker, M.B. Long COVID or Post-acute Sequelae of COVID-19 (PASC): An Overview of Biological Factors That May Contribute to Persistent Symptoms. Front. Microbiol. 2021, 12, 698169. [Google Scholar] [CrossRef]
- Queiroz, M.A.F.; Neves, P.; Lima, S.S.; Lopes, J.D.C.; Torres, M.; Vallinoto, I.; Bichara, C.D.A.; Dos Santos, E.F.; de Brito, M.; da Silva, A.L.S.; et al. Cytokine Profiles Associated With Acute COVID-19 and Long COVID-19 Syndrome. Front. Cell Infect. Microbiol. 2022, 12, 922422. [Google Scholar] [CrossRef]
- Moreno-Perez, O.; Merino, E.; Leon-Ramirez, J.M.; Andres, M.; Ramos, J.M.; Arenas-Jimenez, J.; Asensio, S.; Sanchez, R.; Ruiz-Torregrosa, P.; Galan, I.; et al. Post-acute COVID-19 syndrome. Incidence and risk factors: A Mediterranean cohort study. J. Infect. 2021, 82, 378–383. [Google Scholar] [CrossRef]
- Sapir, T.; Averch, Z.; Lerman, B.; Bodzin, A.; Fishman, Y.; Maitra, R. COVID-19 and the Immune Response: A Multi-Phasic Approach to the Treatment of COVID-19. Int. J. Mol. Sci. 2022, 23, 8606. [Google Scholar] [CrossRef]
- Lechner-Scott, J.; Levy, M.; Hawkes, C.; Yeh, A.; Giovannoni, G. Long COVID or post COVID-19 syndrome. Mult. Scler. Relat. Dis. 2021, 55, 103268. [Google Scholar] [CrossRef]
- Schultheiß, C.; Willscher, E.; Paschold, L.; Gottschick, C.; Klee, B.; Henkes, S.S.; Bosurgi, L.; Dutzmann, J.; Sedding, D.; Frese, T.; et al. The IL-1β, IL-6, and TNF cytokine triad is associated with post-acute sequelae of COVID-19. Cell Rep. Med. 2022, 3, 100663. [Google Scholar] [CrossRef]
- Peluso, M.J.; Lu, S.; Tang, A.F.; Durstenfeld, M.S.; Ho, H.E.; Goldberg, S.A.; Forman, C.A.; Munter, S.E.; Hoh, R.; Tai, V.; et al. Markers of Immune Activation and Inflammation in Individuals With Postacute Sequelae of Severe Acute Respiratory Syndrome Coronavirus 2 Infection. J. Infect. Dis. 2021, 224, 1839–1848. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Ampudia, Y.; Monsalve, D.M.; Rojas, M.; Rodríguez, Y.; Zapata, E.; Ramírez-Santana, C.; Anaya, J.M. Persistent Autoimmune Activation and Proinflammatory State in Post-Coronavirus Disease 2019 Syndrome. J. Infect. Dis. 2022, 225, 2155–2162. [Google Scholar] [CrossRef] [PubMed]
- Phetsouphanh, C.; Darley, D.R.; Wilson, D.B.; Howe, A.; Munier, C.M.L.; Patel, S.K.; Juno, J.A.; Burrell, L.M.; Kent, S.J.; Dore, G.J.; et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat. Immunol. 2022, 23, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Sette, A.; Crotty, S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 2021, 184, 861–880. [Google Scholar] [CrossRef]
- Arunachalam, P.S.; Wimmers, F.; Mok, C.K.P.; Perera, R.; Scott, M.; Hagan, T.; Sigal, N.; Feng, Y.; Bristow, L.; Tak-Yin Tsang, O.; et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 2020, 369, 1210–1220. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Beziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Laing, A.G.; Lorenc, A.; Del Molino Del Barrio, I.; Das, A.; Fish, M.; Monin, L.; Munoz-Ruiz, M.; McKenzie, D.R.; Hayday, T.S.; Francos-Quijorna, I.; et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020, 26, 1623–1635. [Google Scholar] [CrossRef]
- Li, S.; Jiang, L.; Li, X.; Lin, F.; Wang, Y.; Li, B.; Jiang, T.; An, W.; Liu, S.; Liu, H.; et al. Clinical and pathological investigation of patients with severe COVID-19. JCI Insight 2020, 5, e138070. [Google Scholar] [CrossRef]
- Schurink, B.; Roos, E.; Radonic, T.; Barbe, E.; Bouman, C.S.C.; de Boer, H.H.; de Bree, G.J.; Bulle, E.B.; Aronica, E.M.; Florquin, S.; et al. Viral presence and immunopathology in patients with lethal COVID-19: A prospective autopsy cohort study. Lancet Microbe 2020, 1, e290–e299. [Google Scholar] [CrossRef]
- Magleby, R.; Westblade, L.F.; Trzebucki, A.; Simon, M.S.; Rajan, M.; Park, J.; Goyal, P.; Safford, M.M.; Satlin, M.J. Impact of Severe Acute Respiratory Syndrome Coronavirus 2 Viral Load on Risk of Intubation and Mortality Among Hospitalized Patients With Coronavirus Disease 2019. Clin. Infect. Dis. 2021, 73, e4197–e4205. [Google Scholar] [CrossRef] [PubMed]
- Kuri-Cervantes, L.; Pampena, M.B.; Meng, W.; Rosenfeld, A.M.; Ittner, C.A.G.; Weisman, A.R.; Agyekum, R.S.; Mathew, D.; Baxter, A.E.; Vella, L.A.; et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020, 5, eabd7114. [Google Scholar] [CrossRef] [PubMed]
- Galani, I.E.; Rovina, N.; Lampropoulou, V.; Triantafyllia, V.; Manioudaki, M.; Pavlos, E.; Koukaki, E.; Fragkou, P.C.; Panou, V.; Rapti, V.; et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 2021, 22, 32–40. [Google Scholar] [CrossRef]
- Files, J.K.; Sarkar, S.; Fram, T.R.; Boppana, S.; Sterrett, S.; Qin, K.; Bansal, A.; Long, D.M.; Sabbaj, S.; Kobie, J.J.; et al. Duration of post-COVID-19 symptoms is associated with sustained SARS-CoV-2-specific immune responses. JCI Insight 2021, 6, e151544. [Google Scholar] [CrossRef]
- Loretelli, C.; Abdelsalam, A.; D’Addio, F.; Ben Nasr, M.; Assi, E.; Usuelli, V.; Maestroni, A.; Seelam, A.J.; Ippolito, E.; Di Maggio, S.; et al. PD-1 blockade counteracts post-COVID-19 immune abnormalities and stimulates the anti-SARS-CoV-2 immune response. JCI Insight 2021, 6, e146701. [Google Scholar] [CrossRef]
- Glynne, P.; Tahmasebi, N.; Gant, V.; Gupta, R. Long COVID following mild SARS-CoV-2 infection: Characteristic T cell alterations and response to antihistamines. J. Investig. Med. 2022, 70, 61–67. [Google Scholar] [CrossRef]
- Galán, M.; Vigón, L.; Fuertes, D.; Murciano-Antón, M.A.; Casado-Fernández, G.; Domínguez-Mateos, S.; Mateos, E.; Ramos-Martín, F.; Planelles, V.; Torres, M.; et al. Persistent Overactive Cytotoxic Immune Response in a Spanish Cohort of Individuals With Long-COVID: Identification of Diagnostic Biomarkers. Front. Immunol. 2022, 13, 848886. [Google Scholar] [CrossRef]
- Peluso, M.J.; Deitchman, A.N.; Torres, L.; Iyer, N.S.; Munter, S.E.; Nixon, C.C.; Donatelli, J.; Thanh, C.; Takahashi, S.; Hakim, J.; et al. Long-term SARS-CoV-2-specific immune and inflammatory responses in individuals recovering from COVID-19 with and without post-acute symptoms. Cell Rep. 2021, 36, 109518. [Google Scholar] [CrossRef]
- Wiech, M.; Chroscicki, P.; Swatler, J.; Stepnik, D.; De Biasi, S.; Hampel, M.; Brewinska-Olchowik, M.; Maliszewska, A.; Sklinda, K.; Durlik, M.; et al. Remodeling of T Cell Dynamics During Long COVID Is Dependent on Severity of SARS-CoV-2 Infection. Front. Immunol. 2022, 13, 886431. [Google Scholar] [CrossRef]
- Jarjour, N.N.; Masopust, D.; Jameson, S.C. T cell memory: Understanding COVID-19. Immunity 2021, 54, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; Ural, B.B.; Farber, D.L. Tissue-specific immunity for a changing world. Cell 2021, 184, 1517–1529. [Google Scholar] [CrossRef] [PubMed]
- Sasson, S.C.; Gordon, C.L.; Christo, S.N.; Klenerman, P.; Mackay, L.K. Local heroes or villains: Tissue-resident memory T cells in human health and disease. Cell. Mol. Immunol. 2020, 17, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, S.; Goplen, N.P.; Li, C.; Cheon, I.S.; Dai, Q.; Huang, S.; Shan, J.; Ma, C.; Ye, Z.; et al. PD-1hi CD8+ resident memory T cells balance immunity and fibrotic sequelae. Sci. Immunol. 2019, 4, eaaw1217. [Google Scholar] [CrossRef]
- Goplen, N.P.; Wu, Y.; Son, Y.M.; Li, C.; Wang, Z.; Cheon, I.S.; Jiang, L.; Zhu, B.; Ayasoufi, K.; Chini, E.N.; et al. Tissue-resident CD8(+) T cells drive age-associated chronic lung sequelae after viral pneumonia. Sci. Immunol. 2020, 5, eabc4557. [Google Scholar] [CrossRef]
- Cheon, I.S.; Li, C.; Son, Y.M.; Goplen, N.P.; Wu, Y.; Cassmann, T.; Wang, Z.; Wei, X.; Tang, J.; Li, Y.; et al. Immune signatures underlying post-acute COVID-19 lung sequelae. Sci. Immunol. 2021, 6, eabk1741. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Burnham, E.L.; Janssen, W.J.; Riches, D.W.; Moss, M.; Downey, G.P. The fibroproliferative response in acute respiratory distress syndrome: Mechanisms and clinical significance. Eur. Respir. J. 2014, 43, 276–285. [Google Scholar] [CrossRef]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef]
- Budinger, G.R.S.; Misharin, A.V.; Ridge, K.M.; Singer, B.D.; Wunderink, R.G. Distinctive features of severe SARS-CoV-2 pneumonia. J. Clin. Investig. 2021, 131, e149412. [Google Scholar] [CrossRef]
- Østergaard, L. SARS-CoV-2 related microvascular damage and symptoms during and after COVID-19: Consequences of capillary transit-time changes, tissue hypoxia and inflammation. Physiol. Rep. 2021, 9, e14726. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yu, C.; Jing, H.; Wu, X.; Novakovic, V.A.; Xie, R.; Shi, J. Long COVID: The Nature of Thrombotic Sequelae Determines the Necessity of Early Anticoagulation. Front. Cell Infect. Microbiol. 2022, 12, 861703. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B.; Laubscher, G.J.; Pretorius, E. A central role for amyloid fibrin microclots in long COVID/PASC: Origins and therapeutic implications. Biochem. J. 2022, 479, 537–559. [Google Scholar] [CrossRef]
- Libby, P.; Luscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef] [PubMed]
- Yong, S.J. Long COVID or post-COVID-19 syndrome: Putative pathophysiology, risk factors, and treatments. Infect. Dis. 2021, 53, 737–754. [Google Scholar] [CrossRef]
- Yeoh, Y.K.; Zuo, T.; Lui, G.C.; Zhang, F.; Liu, Q.; Li, A.Y.; Chung, A.C.; Cheung, C.P.; Tso, E.Y.; Fung, K.S.; et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 2021, 70, 698–706. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Liang, W.; Feng, Z.; Rao, S.; Xiao, C.; Xue, X.; Lin, Z.; Zhang, Q.; Qi, W. Diarrhoea may be underestimated: A missing link in 2019 novel coronavirus. Gut 2020, 69, 1141–1143. [Google Scholar] [CrossRef]
- Wolfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Muller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef]
- Xu, Y.; Li, X.; Zhu, B.; Liang, H.; Fang, C.; Gong, Y.; Guo, Q.; Sun, X.; Zhao, D.; Shen, J.; et al. Characteristics of pediatric SARS-CoV-2 infection and potential evidence for persistent fecal viral shedding. Nat. Med. 2020, 26, 502–505. [Google Scholar] [CrossRef]
- Ng, S.C.; Tilg, H. COVID-19 and the gastrointestinal tract: More than meets the eye. Gut 2020, 69, 973–974. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Kumar, M.; Baker, M.T.; Singh, V.; Vijay-Kumar, M. Mammalian gut immunity. Biomed. J. 2014, 37, 246–258. [Google Scholar] [PubMed]
- Zuo, T.; Kamm, M.A.; Colombel, J.F.; Ng, S.C. Urbanization and the gut microbiota in health and inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 440–452. [Google Scholar] [CrossRef]
- Zuo, T.; Liu, Q.; Zhang, F.; Yeoh, Y.K.; Wan, Y.; Zhan, H.; Lui, G.C.Y.; Chen, Z.; Li, A.Y.L.; Cheung, C.P.; et al. Temporal landscape of human gut RNA and DNA virome in SARS-CoV-2 infection and severity. Microbiome 2021, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Zhan, H.; Zhang, F.; Liu, Q.; Tso, E.Y.K.; Lui, G.C.Y.; Chen, N.; Li, A.; Lu, W.; Chan, F.K.L.; et al. Alterations in Fecal Fungal Microbiome of Patients With COVID-19 During Time of Hospitalization until Discharge. Gastroenterology 2020, 159, 1302–1310.e5. [Google Scholar] [CrossRef]
- Zuo, T.; Zhang, F.; Lui, G.C.Y.; Yeoh, Y.K.; Li, A.Y.L.; Zhan, H.; Wan, Y.; Chung, A.C.K.; Cheung, C.P.; Chen, N.; et al. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology 2020, 159, 944–955.e8. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, L.; Wang, Y.; Dai, T.; Qin, Z.; Zhou, F.; Zhang, L. Alterations in microbiota of patients with covid-19: Potential mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2022, 7, 143. [Google Scholar] [CrossRef]
- Zuo, T.; Wu, X.; Wen, W.; Lan, P. Gut Microbiome Alterations in COVID-19. Genom. Proteom. Bioinform. 2021, 19, 679–688. [Google Scholar] [CrossRef]
- Tang, L.; Gu, S.; Gong, Y.; Li, B.; Lu, H.; Li, Q.; Zhang, R.; Gao, X.; Wu, Z.; Zhang, J.; et al. Clinical Significance of the Correlation between Changes in the Major Intestinal Bacteria Species and COVID-19 Severity. Engineering 2020, 6, 1178–1184. [Google Scholar] [CrossRef]
- Gu, S.; Chen, Y.; Wu, Z.; Chen, Y.; Gao, H.; Lv, L.; Guo, F.; Zhang, X.; Luo, R.; Huang, C.; et al. Alterations of the Gut Microbiota in Patients With Coronavirus Disease 2019 or H1N1 Influenza. Clin. Infect. Dis. 2020, 71, 2669–2678. [Google Scholar] [CrossRef]
- Weng, J.; Li, Y.; Li, J.; Shen, L.; Zhu, L.; Liang, Y.; Lin, X.; Jiao, N.; Cheng, S.; Huang, Y.; et al. Gastrointestinal sequelae 90 days after discharge for COVID-19. Lancet Gastroenterol. Hepatol. 2021, 6, 344–346. [Google Scholar] [CrossRef]
- Gaebler, C.; Wang, Z.; Lorenzi, J.C.C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y.; et al. Evolution of antibody immunity to SARS-CoV-2. Nature 2021, 591, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Sang, L.; Ye, F.; Ruan, S.; Zhong, B.; Song, T.; Alshukairi, A.N.; Chen, R.; Zhang, Z.; et al. Kinetics of viral load and antibody response in relation to COVID-19 severity. J. Clin. Investig. 2020, 130, 5235–5244. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gu, S.; Chen, Y.; Lu, H.; Shi, D.; Guo, J.; Wu, W.R.; Yang, Y.; Li, Y.; Xu, K.J.; et al. Six-month follow-up of gut microbiota richness in patients with COVID-19. Gut 2022, 71, 222–225. [Google Scholar] [CrossRef]
- Sokol, H.; Contreras, V.; Maisonnasse, P.; Desmons, A.; Delache, B.; Sencio, V.; Machelart, A.; Brisebarre, A.; Humbert, L.; Deryuter, L.; et al. SARS-CoV-2 infection in nonhuman primates alters the composition and functional activity of the gut microbiota. Gut Microbes 2021, 13, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Mak, J.W.Y.; Su, Q.; Yeoh, Y.K.; Lui, G.C.; Ng, S.S.S.; Zhang, F.; Li, A.Y.L.; Lu, W.; Hui, D.S.; et al. Gut microbiota dynamics in a prospective cohort of patients with post-acute COVID-19 syndrome. Gut 2022, 71, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Lau, R.I.; Liu, Q.; Chan, F.K.L.; Ng, S.C. Post-acute COVID-19 syndrome and gut dysbiosis linger beyond 1 year after SARS-CoV-2 clearance. Gut, 2022; Online ahead of print. [Google Scholar] [CrossRef]
- Guo, Y.; Xie, J.P.; Deng, K.; Li, X.; Yuan, Y.; Xuan, Q.; Xie, J.; He, X.M.; Wang, Q.; Li, J.J.; et al. Prophylactic Effects of Bifidobacterium adolescentis on Anxiety and Depression-Like Phenotypes After Chronic Stress: A Role of the Gut Microbiota-Inflammation Axis. Front. Behav. Neurosci. 2019, 13, 126. [Google Scholar] [CrossRef]
- Ren, Z.; Wang, H.; Cui, G.; Lu, H.; Wang, L.; Luo, H.; Chen, X.; Ren, H.; Sun, R.; Liu, W.; et al. Alterations in the human oral and gut microbiomes and lipidomics in covid-19. Gut 2021, 70, 1253–1265. [Google Scholar] [CrossRef]
- Lahti, L.; Salojarvi, J.; Salonen, A.; Scheffer, M.; de Vos, W.M. Tipping elements in the human intestinal ecosystem. Nat. Commun. 2014, 5, 4344. [Google Scholar] [CrossRef]
- Buffie, C.G.; Pamer, E.G. Microbiota-mediated colonization resistance against intestinal pathogens. Nat. Rev. Immunol. 2013, 13, 790–801. [Google Scholar] [CrossRef]
- Finlay, B.B.; Amato, K.R.; Azad, M.; Blaser, M.J.; Bosch, T.C.G.; Chu, H.; Dominguez-Bello, M.G.; Ehrlich, S.D.; Elinav, E.; Geva-Zatorsky, N.; et al. The hygiene hypothesis, the COVID pandemic, and consequences for the human microbiome. Proc. Natl. Acad. Sci. USA 2021, 118, e2010217118. [Google Scholar] [CrossRef] [PubMed]
- Vestad, B.; Ueland, T.; Lerum, T.V.; Dahl, T.B.; Holm, K.; Barratt-Due, A.; Kåsine, T.; Dyrhol-Riise, A.M.; Stiksrud, B.; Tonby, K.; et al. Respiratory dysfunction three months after severe COVID-19 is associated with gut microbiota alterations. J. Intern. Med. 2022, 291, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Nataf, S.; Pays, L. Molecular Insights into SARS-CoV2-Induced Alterations of the Gut/Brain Axis. Int. J. Mol. Sci. 2021, 22, 10440. [Google Scholar] [CrossRef] [PubMed]
- Santus, W.; Devlin, J.R.; Behnsen, J. Crossing Kingdoms: How the Mycobiota and Fungal-Bacterial Interactions Impact Host Health and Disease. Infect. Immun. 2021, 89, e00648-20. [Google Scholar] [CrossRef]
- van Tilburg Bernardes, E.; Pettersen, V.K.; Gutierrez, M.W.; Laforest-Lapointe, I.; Jendzjowsky, N.G.; Cavin, J.B.; Vicentini, F.A.; Keenan, C.M.; Ramay, H.R.; Samara, J.; et al. Intestinal fungi are causally implicated in microbiome assembly and immune development in mice. Nat. Commun. 2020, 11, 2577. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Gu, S.; Jiang, H.; Yan, R.; Chen, Y.; Chen, Y.; Luo, R.; Huang, C.; Lu, H.; Zheng, B.; et al. Gut mycobiota alterations in patients with COVID-19 and H1N1 infections and their associations with clinical features. Commun. Biol. 2021, 4, 480. [Google Scholar] [CrossRef]
- Cox, M.J.; Loman, N.; Bogaert, D.; O’Grady, J. Co-infections: Potentially lethal and unexplored in COVID-19. Lancet Microbe 2020, 1, e11. [Google Scholar] [CrossRef]
- Peman, J.; Ruiz-Gaitan, A.; Garcia-Vidal, C.; Salavert, M.; Ramirez, P.; Puchades, F.; Garcia-Hita, M.; Alastruey-Izquierdo, A.; Quindos, G. Fungal co-infection in COVID-19 patients: Should we be concerned? Rev. Iberoam. Micol. 2020, 37, 41–46. [Google Scholar] [CrossRef]
- Song, G.; Liang, G.; Liu, W. Fungal Co-infections Associated with Global COVID-19 Pandemic: A Clinical and Diagnostic Perspective from China. Mycopathologia 2020, 185, 599–606. [Google Scholar] [CrossRef]
- Erb Downward, J.R.; Falkowski, N.R.; Mason, K.L.; Muraglia, R.; Huffnagle, G.B. Modulation of post-antibiotic bacterial community reassembly and host response by Candida albicans. Sci. Rep. 2013, 3, 2191. [Google Scholar] [CrossRef]
- Zuo, T.; Wong, S.H.; Cheung, C.P.; Lam, K.; Lui, R.; Cheung, K.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Wu, J.C.Y.; et al. Gut fungal dysbiosis correlates with reduced efficacy of fecal microbiota transplantation in Clostridium difficile infection. Nat. Commun. 2018, 9, 3663. [Google Scholar] [CrossRef] [PubMed]
- Kumamoto, C.A. Inflammation and gastrointestinal Candida colonization. Curr. Opin. Microbiol. 2011, 14, 386–391. [Google Scholar] [CrossRef]
- Sonoyama, K.; Miki, A.; Sugita, R.; Goto, H.; Nakata, M.; Yamaguchi, N. Gut colonization by Candida albicans aggravates inflammation in the gut and extra-gut tissues in mice. Med. Mycol. 2011, 49, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Neil, J.A.; Cadwell, K. The Intestinal Virome and Immunity. J. Immunol. 2018, 201, 1615–1624. [Google Scholar] [CrossRef] [PubMed]
- Virgin, H.W. The virome in mammalian physiology and disease. Cell 2014, 157, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Liu, Q.; Zhang, F.; Lui, G.C.; Tso, E.Y.; Yeoh, Y.K.; Chen, Z.; Boon, S.S.; Chan, F.K.; Chan, P.K.; et al. Depicting SARS-CoV-2 faecal viral activity in association with gut microbiota composition in patients with COVID-19. Gut 2021, 70, 276–284. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef]
- Zuo, T.; Lu, X.J.; Zhang, Y.; Cheung, C.P.; Lam, S.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Zhao, R.; Chan, P.K.S.; et al. Gut mucosal virome alterations in ulcerative colitis. Gut 2019, 68, 1169–1179. [Google Scholar] [CrossRef]
- Diard, M.; Bakkeren, E.; Cornuault, J.K.; Moor, K.; Hausmann, A.; Sellin, M.E.; Loverdo, C.; Aertsen, A.; Ackermann, M.; De Paepe, M.; et al. Inflammation boosts bacteriophage transfer between Salmonella spp. Science 2017, 355, 1211–1215. [Google Scholar] [CrossRef]
- Cao, J.; Wang, C.; Zhang, Y.; Lei, G.; Xu, K.; Zhao, N.; Lu, J.; Meng, F.; Yu, L.; Yan, J.; et al. Integrated gut virome and bacteriome dynamics in COVID-19 patients. Gut Microbes 2021, 13, 1–21. [Google Scholar] [CrossRef]
- Tutuncuoglu, B.; Cakir, M.; Batra, J.; Bouhaddou, M.; Eckhardt, M.; Gordon, D.E.; Krogan, N.J. The Landscape of Human Cancer Proteins Targeted by SARS-CoV-2. Cancer Discov. 2020, 10, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Rahimmanesh, I.; Shariati, L.; Dana, N.; Esmaeili, Y.; Vaseghi, G.; Haghjooy Javanmard, S. Cancer Occurrence as the Upcoming Complications of COVID-19. Front. Mol. Biosci. 2022, 8, 813175. [Google Scholar] [CrossRef] [PubMed]
- Saini, G.; Aneja, R. Cancer as a prospective sequela of long COVID-19. BioEssays 2021, 43, e2000331. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Gatti, P.; Ilamathi, H.S.; Todkar, K.; Germain, M. Mitochondria Targeted Viral Replication and Survival Strategies-Prospective on SARS-CoV-2. Front. Pharmacol. 2020, 11, 578599. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.M.; Walsh, L.A.; Chan, T.A. Driver mutations of cancer epigenomes. Protein Cell 2014, 5, 265–296. [Google Scholar] [CrossRef]
- García-Carpizo, V.; Sarmentero, J.; Han, B.; Graña, O.; Ruiz-Llorente, S.; Pisano, D.G.; Serrano, M.; Brooks, H.B.; Campbell, R.M.; Barrero, M.J. NSD2 contributes to oncogenic RAS-driven transcription in lung cancer cells through long-range epigenetic activation. Sci. Rep. 2016, 6, 32952. [Google Scholar] [CrossRef]
- Sun, D.; Yu, M.; Li, Y.; Xing, H.; Gao, Y.; Huang, Z.; Hao, W.; Lu, K.; Kong, C.; Shimozato, O.; et al. Histone deacetylase 2 is involved in DNA damage-mediated cell death of human osteosarcoma cells through stimulation of the ATM/p53 pathway. FEBS Open Bio 2019, 9, 478–489. [Google Scholar] [CrossRef]
- Ma-Lauer, Y.; Carbajo-Lozoya, J.; Hein, M.Y.; Müller, M.A.; Deng, W.; Lei, J.; Meyer, B.; Kusov, Y.; Von Brunn, B.; Bairad, D.R.; et al. p53 down-regulates SARS coronavirus replication and is targeted by the SARS-unique domain and PLpro via E3 ubiquitin ligase RCHY1. Proc. Natl. Acad. Sci. USA 2016, 113, E5192–E5201. [Google Scholar] [CrossRef]
- Bhardwaj, K.; Liu, P.; Leibowitz, J.L.; Kao, C.C. The coronavirus endoribonuclease Nsp15 interacts with retinoblastoma tumor suppressor protein. J. Virol. 2012, 86, 4294–4304. [Google Scholar] [CrossRef]
- Singh, N.; Bharara Singh, A. S2 subunit of SARS-nCoV-2 interacts with tumor suppressor protein p53 and BRCA: An in silico study. Trans. Oncol. 2020, 13, 100814. [Google Scholar] [CrossRef] [PubMed]
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef]
- Martin-Hijano, L.; Sainz, B., Jr. The Interactions Between Cancer Stem Cells and the Innate Interferon Signaling Pathway. Front. Immunol. 2020, 11, 526. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Murti, A.; Pfeffer, S.R.; Basu, L.; Kim, J.G.; Pfeffer, L.M. IFNα/β promotes cell survival by activating NF-κB. Proc. Natl. Acad. Sci. USA 2000, 97, 13631–13636. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Reich, N.C. Stimulation of primary human endothelial cell proliferation by IFN. J. Immunol. 2003, 170, 5373–5381. [Google Scholar] [CrossRef]
- Beatty, G.L.; Paterson, Y. IFN-γ can promote tumor evasion of the immune system in vivo by down-regulating cellular levels of an endogenous tumor antigen. J. Immunol. 2000, 165, 5502–5508. [Google Scholar] [CrossRef]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef]
- Farahani, M.; Niknam, Z.; Mohammadi Amirabad, L.; Amiri-Dashatan, N.; Koushki, M.; Nemati, M.; Danesh Pouya, F.; Rezaei-Tavirani, M.; Rasmi, Y.; Tayebi, L. Molecular pathways involved in COVID-19 and potential pathway-based therapeutic targets. Biomed. Pharmacother. 2022, 145, 112420. [Google Scholar] [CrossRef]
- Ramaiah, M.J. mTOR inhibition and p53 activation, microRNAs: The possible therapy against pandemic COVID-19. Gene Rep. 2020, 20, 100765. [Google Scholar] [CrossRef]
- Breikaa, R.M.; Lilly, B. The Notch Pathway: A Link Between COVID-19 Pathophysiology and Its Cardiovascular Complications. Front. Cardiovasc. Med. 2021, 8, 681948. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch signaling pathway: Architecture, disease, and therapeutics. Signal. Transduct. Target. Ther. 2022, 7, 95. [Google Scholar] [CrossRef]
- Misiorek, J.O.; Przybyszewska-Podstawka, A.; Kałafut, J.; Paziewska, B.; Rolle, K.; Rivero-Müller, A.; Nees, M. Context Matters: NOTCH Signatures and Pathway in Cancer Progression and Metastasis. Cells 2021, 10, 94. [Google Scholar] [CrossRef] [PubMed]
- Kudaravalli, S.; den Hollander, P.; Mani, S.A. Role of p38 MAP kinase in cancer stem cells and metastasis. Oncogene 2022, 41, 3177–3185. [Google Scholar] [CrossRef] [PubMed]
- Grimes, J.M.; Grimes, K.V. p38 MAPK inhibition: A promising therapeutic approach for COVID-19. J. Mol. Cell Cardiol. 2020, 144, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Limoge, M.; Safina, A.; Truskinovsky, A.M.; Aljahdali, I.; Zonneville, J.; Gruevski, A.; Arteaga, C.L.; Bakin, A.V. Tumor p38MAPK signaling enhances breast carcinoma vascularization and growth by promoting expression and deposition of pro-tumorigenic factors. Oncotarget 2017, 8, 61969–61981. [Google Scholar] [CrossRef][Green Version]
- Serebrovska, Z.O.; Chong, E.Y.; Serebrovska, T.V.; Tumanovska, L.V.; Xi, L. Hypoxia, HIF-1α, and COVID-19: From pathogenic factors to potential therapeutic targets. Acta Pharmacol. Sin. 2020, 41, 1539–1546. [Google Scholar] [CrossRef]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-Modified Cancer Cell Metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rapti, V.; Tsaganos, T.; Vathiotis, I.A.; Syrigos, N.K.; Li, P.; Poulakou, G. New Insights into SARS-CoV-2 and Cancer Cross-Talk: Does a Novel Oncogenesis Driver Emerge? Vaccines 2022, 10, 1607. https://doi.org/10.3390/vaccines10101607
Rapti V, Tsaganos T, Vathiotis IA, Syrigos NK, Li P, Poulakou G. New Insights into SARS-CoV-2 and Cancer Cross-Talk: Does a Novel Oncogenesis Driver Emerge? Vaccines. 2022; 10(10):1607. https://doi.org/10.3390/vaccines10101607
Chicago/Turabian StyleRapti, Vasiliki, Thomas Tsaganos, Ioannis A. Vathiotis, Nikolaos K. Syrigos, Peifeng Li, and Garyfallia Poulakou. 2022. "New Insights into SARS-CoV-2 and Cancer Cross-Talk: Does a Novel Oncogenesis Driver Emerge?" Vaccines 10, no. 10: 1607. https://doi.org/10.3390/vaccines10101607
APA StyleRapti, V., Tsaganos, T., Vathiotis, I. A., Syrigos, N. K., Li, P., & Poulakou, G. (2022). New Insights into SARS-CoV-2 and Cancer Cross-Talk: Does a Novel Oncogenesis Driver Emerge? Vaccines, 10(10), 1607. https://doi.org/10.3390/vaccines10101607