

Immunoinformatics Studies and Design of a Potential Multi-Epitope Peptide Vaccine to Combat the Fatal Visceral Leishmaniasis

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Methodology
2.1. Retrieval of L. donovani Disulfide Isomerase Protein Sequence for Vaccine Prediction and Antigenicity Testing
2.2. Prediction of Cytotoxic t Lymphocyte and Helper t Lymphocyte Epitopes
2.3. B-Cell Epitope Prediction for L. donovani Proteins
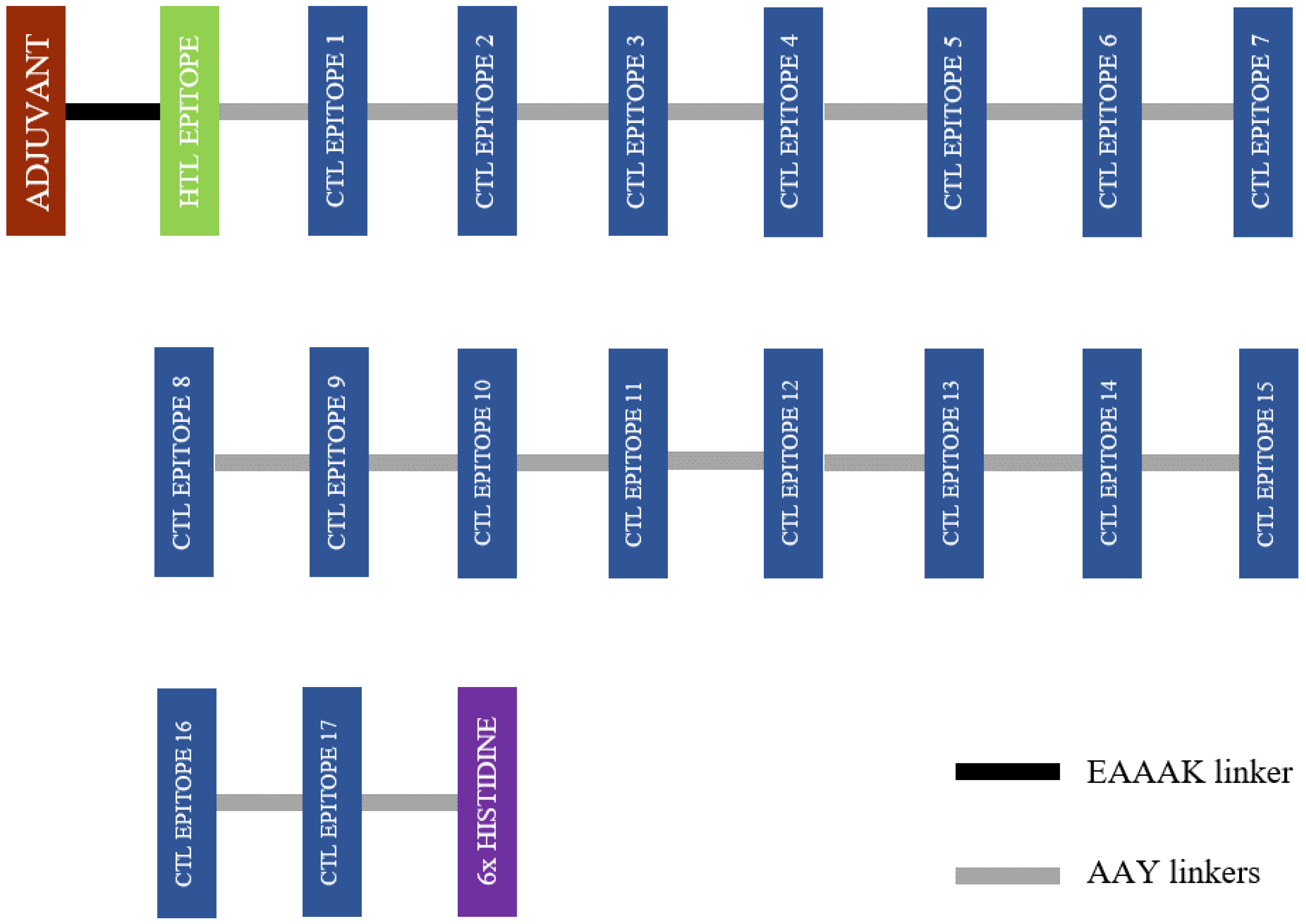
2.4. Construction of Multi-Epitope Subunit Vaccine
2.5. Prediction of Antigenicity, Allergenicity and Physiochemical Properties of Vaccine Protein
2.6. Tertiary Structure Prediction
2.7. Tertiary Structure Refinement and Validation
2.8. Protein-Protein Docking
2.9. Molecular Dynamics of Vaccine-TLRs Complex
3. Results
3.1. Identification of Disulfide Isomerase as a Potential Vaccine Candidate for L. donovani Infections
3.2. Immunogenic CTL and HTL Epitopes of the Disulfide Isomerase as a Potential Vaccine Candidate for L. donovani Infections
3.3. Combining CTL and HTL Epitopes and Adjuvants—The Vaccine Construct
3.4. B Cell Epitope Evaluation of the Vaccine Construct
3.5. Antigenicity and Allergenicity of the Vaccine Construct Sequence
3.6. Physiochemical Parameters of the Vaccine Construct
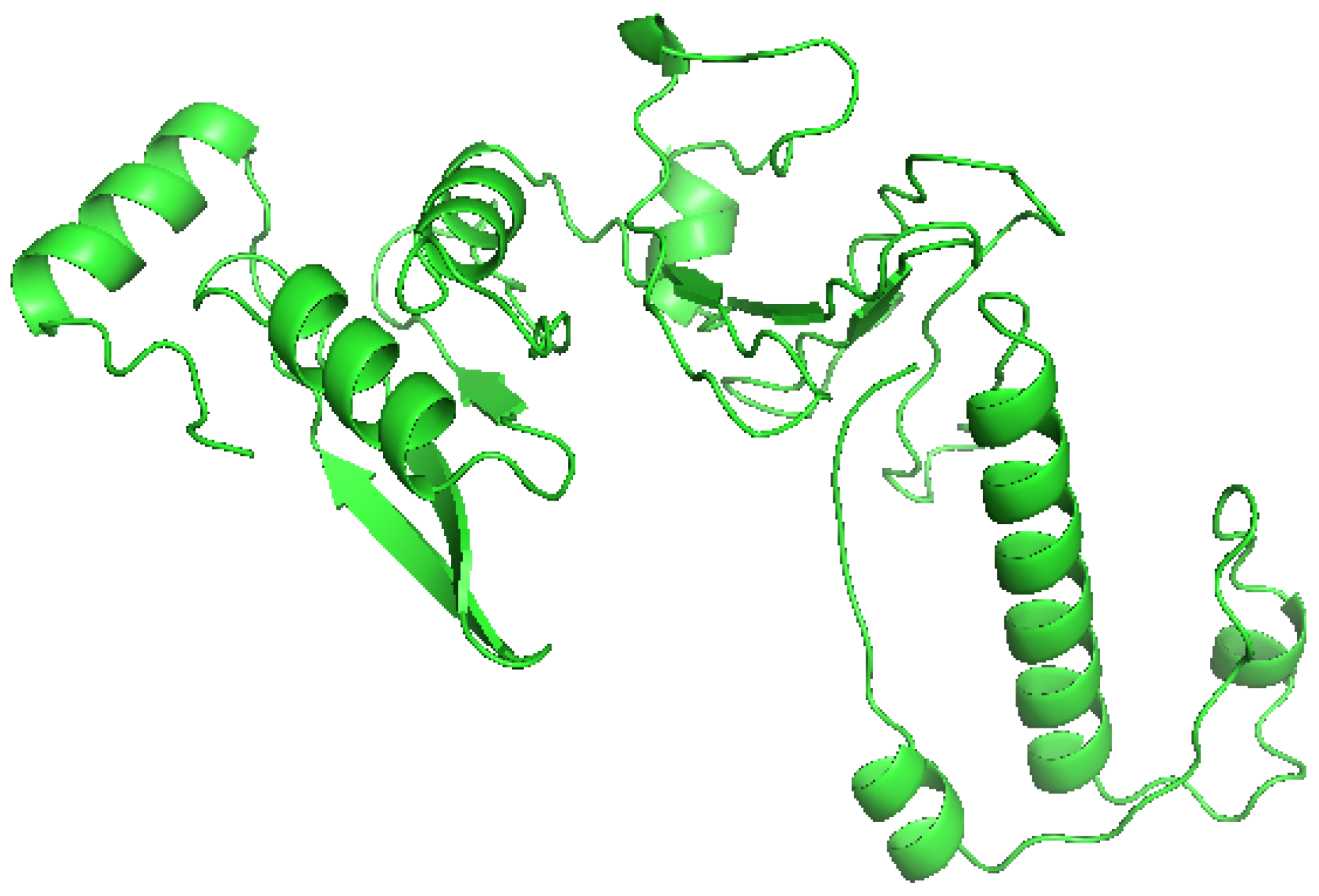
3.7. Tertiary Structure Prediction and Refinement
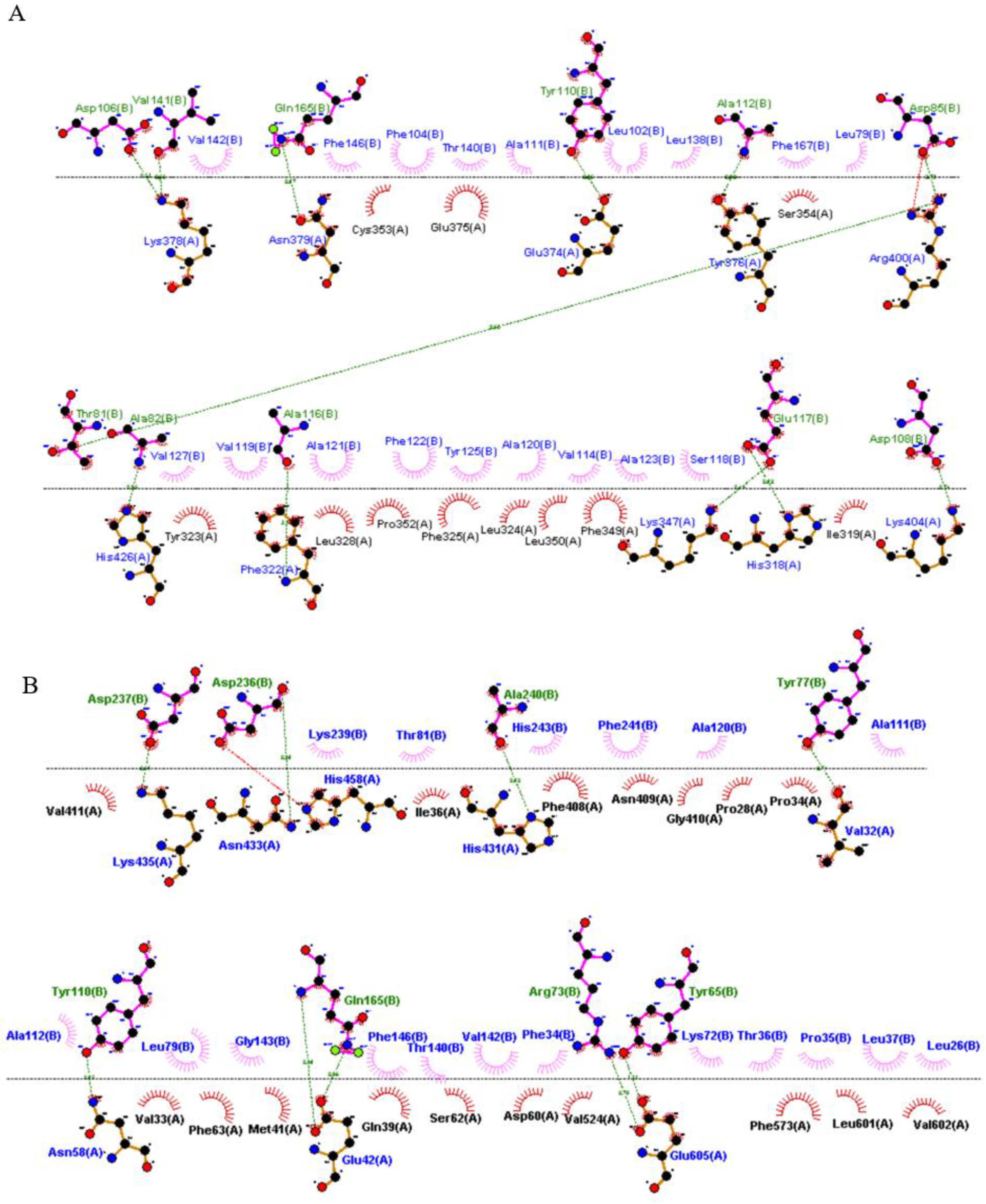
3.8. Protein-Protein Docking Analysis
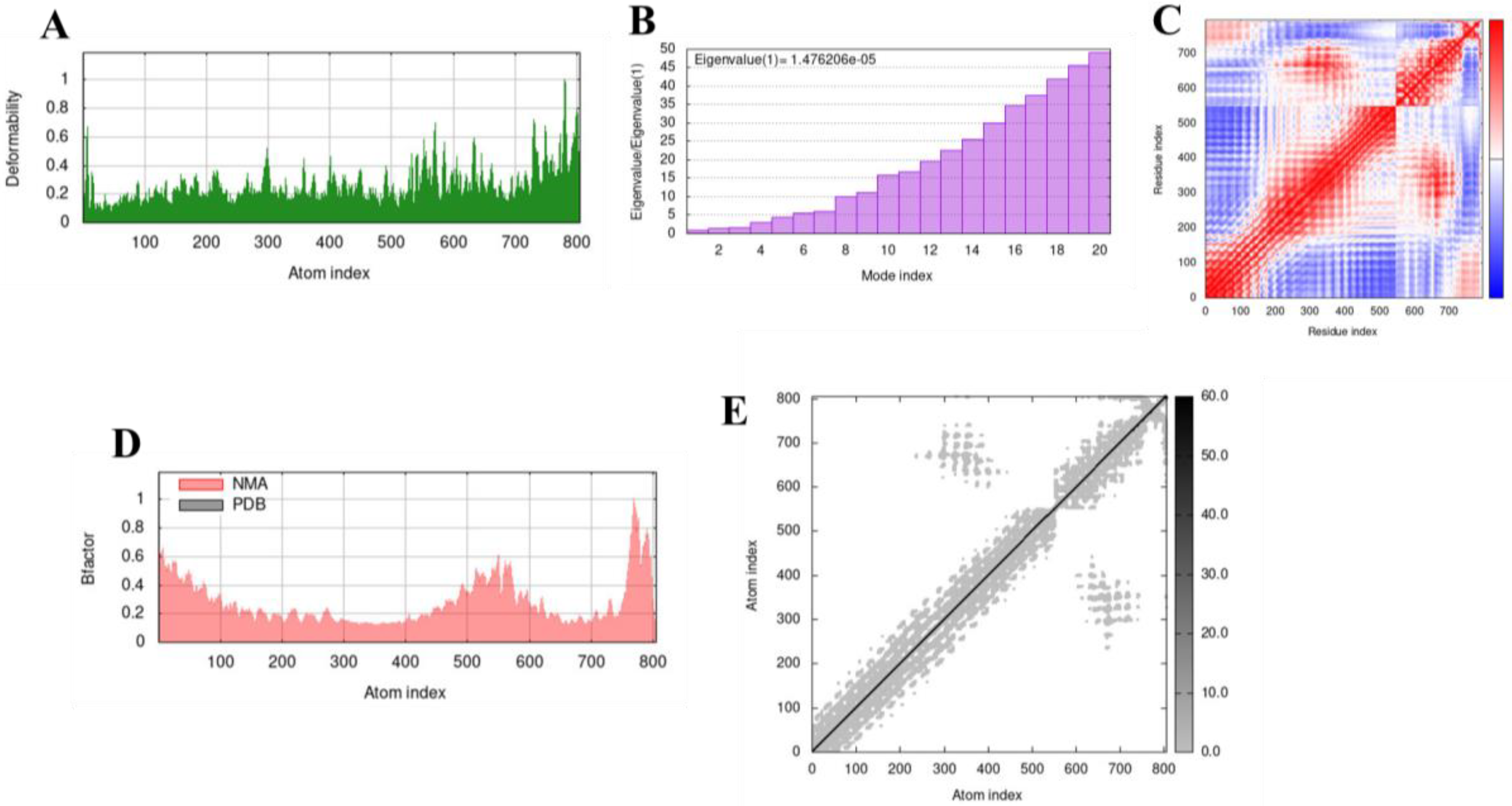
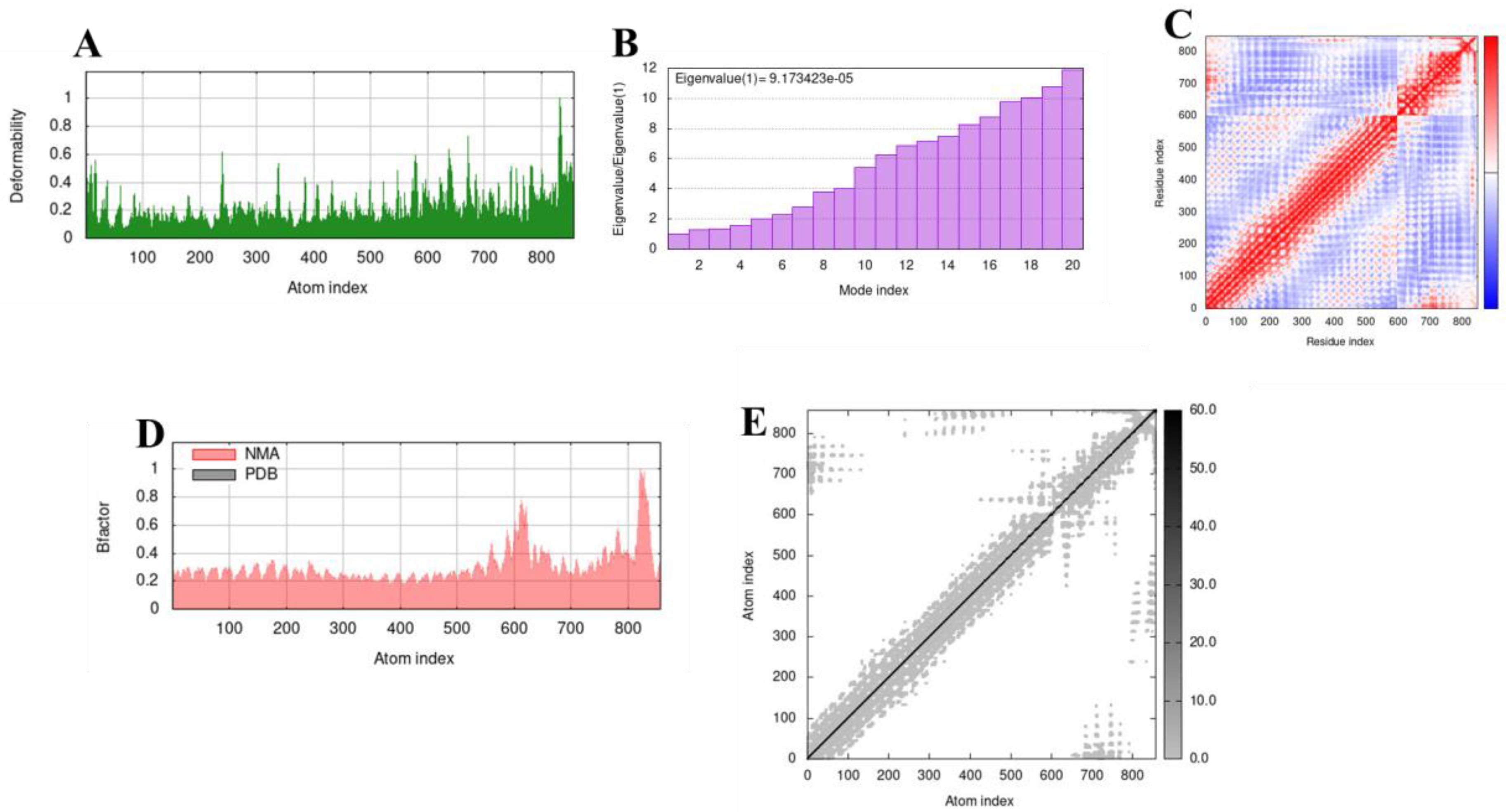
3.9. Molecular Dynamics Simulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aruleba, R.T.; Carter, K.; Brombacher, F.; Hurdayal, R. Can we harness immune responses to improve drug treatment in leishmaniasis? Microorganisms 2020, 8, 1069. [Google Scholar] [CrossRef] [PubMed]
- WHO. Leishmaniasis. 2022. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 11 May 2022).
- Bilgic-Temel, A.; Murrell, D.F.; Uzun, S. Cutaneous leishmaniasis: A neglected disfiguring disease for women. Int. J. Women’s Dermatol. 2019, 5, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Singh, A. Chemotherapeutics of visceral leishmaniasis: Present and future developments. Parasitology 2018, 145, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Kaye, P.; Aebischer, T. Visceral leishmaniasis: Immunology and prospects for a vaccine. Clin. Microbiol. Infect. 2011, 17, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Singh, O.P.; Hasker, E.; Boelaert, M.; Sundar, S. Elimination of visceral leishmaniasis on the Indian subcontinent. Lancet Infect. Dis. 2016, 16, e304–e309. [Google Scholar] [CrossRef]
- Ehreth, J. The global value of vaccination. Vaccine 2003, 21, 596–600. [Google Scholar] [CrossRef]
- Normile, D. Driven to extinction. Am. Assoc. Adv. Sci. 2008, 319, 1606–1609. [Google Scholar] [CrossRef]
- Siegrist, C.-A. Vaccine immunology. Vaccines 2008, 5, 17–36. [Google Scholar]
- Onile, O.S.; Ojo, G.J.; Oyeyemi, B.F.; Agbowuro, G.O.; Fadahunsi, A.I. Development of multiepitope subunit protein vaccines against toxoplasma gondii using an immunoinformatics approach. NAR Genom. Bioinform. 2020, 2, lqaa048. [Google Scholar] [CrossRef]
- Adekiya, T.A.; Aruleba, R.T.; Khanyile, S.; Masamba, P.; Oyinloye, B.E.; Kappo, A.P. Structural analysis and epitope prediction of MHC class-1-chain related protein-a for cancer vaccine development. Vaccines 2017, 6, 1. [Google Scholar] [CrossRef]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 8285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezediuno, L.O.; Onile, O.S.; Oladipo, E.K.; Majolagbe, O.N.; Jimah, E.M.; Senbadejo, T.Y. Designing multi-epitope subunit vaccine for ocular trachoma infection using chlamydia trachomatis polymorphic membrane proteins G. Inform. Med. Unlocked 2021, 26, 100764. [Google Scholar] [CrossRef]
- Denkers, E.Y. Toll-like receptor initiated host defense against toxoplasma gondii. J. Biomed. Biotechnol. 2009, 2010, 737125. [Google Scholar] [PubMed]
- Siegrist, C.-A. Vaccine Immunology; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Pandey, R.K.; Bhatt, T.K.; Prajapati, V.K. Novel immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating anopheles salivary protein. Sci. Rep. 2018, 8, 1125. [Google Scholar] [CrossRef] [PubMed]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- Sanchez-Trincado, J.L.; Gomez-Perosanz, M.; Reche, P.A. Fundamentals and methods for T-and B-cell epitope prediction. J. Immunol. Res. 2017, 2017, 2680160. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef]
- Shey, R.A.; Ghogomu, S.M.; Esoh, K.K.; Nebangwa, N.D.; Shintouo, C.M.; Nongley, N.F.; Asa, B.F.; Ngale, F.N.; Vanhamme, L.; Souopgui, J. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci. Rep. 2019, 9, 4409. [Google Scholar] [CrossRef]
- Vaure, C.; Liu, Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front. Immunol. 2014, 5, 316. [Google Scholar] [CrossRef]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity prediction by descriptor fingerprints. Bioinformatics 2014, 30, 846–851. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. Proteom. Protoc. Handb. 2005, 571–607. [Google Scholar]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Li, W.; Liu, S.; Xu, J. RaptorX-property: A web server for protein structure property prediction. Nucleic Acids Res. 2016, 44, W430–W435. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Park, H.; Heo, L.; Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef] [PubMed]
- Nugent, T.; Cozzetto, D.; Jones, D.T. Evaluation of predictions in the CASP10 model refinement category. Proteins Struct. Funct. Bioinform. 2014, 82, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Runthala, A. Protein structure prediction: Challenging targets for CASP10. J. Biomol. Struct. Dyn. 2012, 30, 607–615. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like receptors activation, signaling, and targeting: An overview. Bull. Natl. Res. Cent. 2019, 43, 187. [Google Scholar] [CrossRef]
- Tuon, F.F.; Amato, V.S.; Bacha, H.A.; AlMusawi, T.; Duarte, M.I.; Neto, V.A. Toll-like receptors and leishmaniasis. Infect. Immun. 2008, 76, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast interaction refinement in molecular docking. Proteins Struct. Funct. Bioinform. 2007, 69, 139–159. [Google Scholar] [CrossRef] [PubMed]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef]
- Gregory, R.L.; Filler, S. Protective secretory immunoglobulin A antibodies in humans following oral immunization with Streptococcus mutans. Infect. Immun. 1987, 55, 2409–2415. [Google Scholar] [CrossRef]
- Meek, B.; Back, J.W.; Klaren, V.N.; Speijer, D.; Peek, R. Protein disulfide isomerase of toxoplasma gondii is targeted by mucosal IgA antibodies in humans. FEBS Lett. 2002, 522, 104–108. [Google Scholar] [CrossRef]
- Gupta, S.K.; Sisodia, B.S.; Sinha, S.; Hajela, K.; Naik, S.; Shasany, A.K.; Dube, A. Proteomic approach for identification and characterization of novel immunostimulatory proteins from soluble antigens of leishmania donovani promastigotes. Proteomics 2007, 7, 816–823. [Google Scholar] [CrossRef]
- Cruse, J.M.; Lewis, R.E. Atlas of Immunology; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Geginat, J.; Paroni, M.; Maglie, S.; Alfen, J.S.; Kastirr, I.; Gruarin, P.; De Simone, M.; Pagani, M.; Abrignani, S. Plasticity of human CD4 T cell subsets. Front. Immunol. 2014, 5, 630. [Google Scholar] [CrossRef]
- Toussi, D.N.; Massari, P. Immune adjuvant effect of molecularly-defined toll-like receptor ligands. Vaccines 2014, 2, 323–353. [Google Scholar] [CrossRef]
- Shamriz, S.; Ofoghi, H.; Moazami, N. Effect of linker length and residues on the structure and stability of a fusion protein with malaria vaccine application. Comput. Biol. Med. 2016, 76, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zaro, J.L.; Shen, W.-C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, R.; Chu, Z.; Yu, F.; Zha, Y. Contriving multi-epitope subunit of vaccine for COVID-19: Immunoinformatics approaches. Front. Immunol. 2020, 11, 1784. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Stenzel, D.; Liu, A.; Liu, D.; Brown, D.; Ambrogelly, A. Quantification of a recombinant antigen in an immuno-stimulatory whole yeast cell-based therapeutic vaccine. Anal. Biochem. 2018, 545, 65–71. [Google Scholar] [CrossRef]
- Zobayer, N.; Hossain, A.A.; Rahman, M.A. A combined view of B-cell epitope features in antigens. Bioinformation 2019, 15, 530. [Google Scholar] [CrossRef]
- Onile, O.S.; Fadahunsi, A.I.; Adekunle, A.A.; Oyeyemi, B.F.; Anumudu, C.I. An immunoinformatics approach for the design of a multi-epitope subunit vaccine for urogenital schistosomiasis. PeerJ 2020, 8, e8795. [Google Scholar] [CrossRef]
- Aruleba, R.T.; Adekiya, T.A.; Molefe, P.F.; Ikwegbue, P.C.; Oyinloye, B.E.; Kappo, A.P. Insights into functional amino acids of ULBP2 as potential immunogens against cancer. Sci. Afr. 2020, 10, e00581. [Google Scholar] [CrossRef]
- Aruleba, R.T.; Adekiya, T.A.; Oyinloye, B.E.; Kappo, A.P. Structural studies of predicted ligand binding sites and molecular docking analysis of Slc2a4 as a therapeutic target for the treatment of cancer. Int. J. Mol. Sci. 2018, 19, 386. [Google Scholar] [CrossRef]
- Osero, B.O.; Aruleba, R.T.; Brombacher, F.; Hurdayal, R. Unravelling the unsolved paradoxes of cytokine families in host resistance and susceptibility to leishmania infection. Cytokine X 2020, 2, 100043. [Google Scholar] [CrossRef]
- Oladipo, E.K.; Ajayi, A.F.; Ariyo, O.E.; Onile, S.O.; Jimah, E.M.; Ezediuno, L.O.; Adebayo, O.I.; Adebayo, E.T.; Odeyemi, A.N.; Oyeleke, M.O.; et al. Exploration of surface glycoprotein to design multi-epitope vaccine for the prevention of COVID-19. Inform. Med. Unlocked 2020, 21, 100438. [Google Scholar] [CrossRef]
- Fadaka, A.O.; Aruleba, R.T.; Sibuyi, N.R.S.; Klein, A.; Madiehe, A.M.; Meyer, M. Inhibitory potential of repurposed drugs against the SARS-CoV-2 main protease: A computational-aided approach. J. Biomol. Struct. Dyn. 2022, 40, 3416–3427. [Google Scholar] [CrossRef] [PubMed]
- Brito, R.C.F.D.; Ruiz, J.C.; Cardoso, J.M.D.O.; Ostolin, T.L.V.D.P.; Reis, L.E.S.; Mathias, F.A.S.; Aguiar-Soares, R.D.D.O.; Roatt, B.M.; Corrêa-Oliveira, R.; Resende, D.D.M.; et al. Chimeric vaccines designed by immunoinformatics-activated polyfunctional and memory T cells that trigger protection against experimental visceral leishmaniasis. Vaccines 2020, 8, 252. [Google Scholar] [CrossRef] [PubMed]
- Vakili, B.; Eslami, M.; Hatam, G.R.; Zare, B.; Erfani, N.; Nezafat, N.; Ghasemi, Y. Immunoinformatics-aided design of a potential multi-epitope peptide vaccine against Leishmania infantum. Int. J. Biol. Macromol. 2018, 120, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Foroutan, M.; Barati, M.; Ghaffarifar, F. Enhancing immune responses by a novel multi-epitope ROP8 DNA vaccine plus interleukin-12 plasmid as a genetic adjuvant against acute toxoplasma gondii infection in BALB/c mice. Microb. Pathog. 2020, 147, 104435. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, W.; Negre, N.N.; White, K.P.; Li, C.; Shah, P.K. Determinants of antigenicity and specificity in immune response for protein sequences. BMC Bioinform. 2011, 12, 251. [Google Scholar] [CrossRef]
- Weber, C.A.; Mehta, P.J.; Ardito, M.; Moise, L.; Martin, B.; De Groot, A.S. T cell epitope: Friend or foe? Immunogenicity of biologics in context. Adv. Drug Deliv. Rev. 2009, 61, 965–976. [Google Scholar] [CrossRef]
- Kima, P.E.; Soong, L. Interferon gamma in leishmaniasis. Front. Immunol. 2013, 4, 156. [Google Scholar] [CrossRef]
- Dayakar, A.; Chandrasekaran, S.; Kuchipudi, S.V.; Kalangi, S.K. Cytokines: Key determinants of resistance or disease progression in visceral leishmaniasis: Opportunities for novel diagnostics and immunotherapy. Front. Immunol. 2019, 10, 670. [Google Scholar] [CrossRef]
- Nezafat, N.; Ghasemi, Y.; Javadi, G.; Khoshnoud, M.J.; Omidinia, E. A novel multi-epitope peptide vaccine against cancer: An in silico approach. J. Theor. Biol. 2014, 349, 121–134. [Google Scholar] [CrossRef]
- Coffman, R.L.; Sher, A.; Seder, R.A. Vaccine adjuvants: Putting innate immunity to work. Immunity 2010, 33, 492–503. [Google Scholar] [CrossRef]
- Hashimoto, C.; Hudson, K.L.; Anderson, K.V. The toll gene of drosophila, required for dorsal-ventral embryonic polarity, appears to encode a transmembrane protein. Cell 1988, 52, 269–279. [Google Scholar] [CrossRef]
- Kim, H.M.; Park, B.S.; Kim, J.-I.; Kim, S.E.; Lee, J.; Oh, S.C.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.J.; et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist eritoran. Cell 2007, 130, 906–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Aruleba, R.T.; Tincho, M.B.; Pretorius, A.; Kappo, A.P. In silico prediction of new antimicrobial peptides and proteins as druggable targets towards alternative anti-schistosomal therapy. Sci. Afr. 2021, 12, e00804. [Google Scholar] [CrossRef]
- Nosrati, M.C.; Ghasemi, E.; Shams, M.; Shamsinia, S.; Yousefi, A.; Nourmohammadi, H.; Javanmardi, E.; Kordi, B.; Majidiani, H.; Ghaffari, A.D.; et al. Toxoplasma gondii ROP38 protein: Bioinformatics analysis for vaccine design improvement against toxoplasmosis. Microb. Pathog. 2020, 149, 104488. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serial No. | Accession Id | Protein Name | Antigenicity Score | Selected/Non-Selected |
|---|---|---|---|---|
| 1 | ACE74539.1 | Disulfide isomerase | 0.90 | Selected |
| 2 | AYU84134.1 | Protein disulfide isomerase 2 | 0.89 | Non-selected |
| 3 | TPP51958.1 | Protein disulfide-isomerase domain | 0.50 | Non-selected |
| 4 | AYU76139.1 | Protein disulfide isomerase | 0.55 | Non-selected |
| Serial No. | Accession Id | Epitope | Combined Score | Length |
|---|---|---|---|---|
| 1 | ACE74539.1 | QIKGFPTLY | 0.94 | 9 |
| 2 | ACE74539.1 | RTAAGIASY | 2.21 | 9 |
| 3 | ACE74539.1 | DAMESVTVY | 0.91 | 9 |
| 4 | ACE74539.1 | MTAESVKRF | 0.84 | 9 |
| 5 | ACE74539.1 | FLATAVLDY | 2.96 | 9 |
| 6 | ACE74539.1 | SLVAVAEKY | 0.97 | 9 |
| 7 | ACE74539.1 | LTFIDGDQY | 1.60 | 9 |
| 8 | ACE74539.1 | VTAESVAAF | 0.93 | 9 |
| 9 | ACE74539.1 | SVAAFVEKY | 1.80 | 9 |
| 10 | ACE74539.1 | LTTVVGQTF | 0.90 | 9 |
| 11 | ACE74539.1 | VVGQTFAKY | 0.89 | 9 |
| 12 | ACE74539.1 | YTDGTQNVM | 1.73 | 9 |
| 13 | ACE74539.1 | GTQNVMLLF | 1.65 | 9 |
| 14 | ACE74539.1 | TQNVMLLFY | 1.65 | 9 |
| 15 | ACE74539.1 | KMDATTNDF | 1.21 | 9 |
| 16 | ACE74539.1 | EVSGFPTIY | 1.32 | 9 |
| 17 | ACE74539.1 | TADDIKAFV | 0.77 | 9 |
| Serial No. | Allele | Length | Start | End | Peptide | Method | Percentile Rank |
|---|---|---|---|---|---|---|---|
| 1 | H-2-Db | 14 | 81 | 94 | SLAEKYQIKGFPTL | NetMHCpan EL 4.0 | 1.10 |
| Rank | Sequence | Start Position | Score |
|---|---|---|---|
| 1 | AAYSLVAVAEKYAA | 87 | 0.91 |
| 2 | TFAKYAAYVVGQTF | 154 | 0.87 |
| 3 | AKSLAEKYQIKGFP | 11 | 0.85 |
| 4 | VVGQTFAAYVVGQT | 141 | 0.83 |
| 5 | AYLTTVVGQTFAAY | 136 | 0.81 |
| 6 | PTLYAAYRTAAGIA | 35 | 0.79 |
| 7 | RFAAYFLATAVLDY | 73 | 0.78 |
| 8 | TQNVMLLFAAYTQN | 187 | 0.77 |
| 9 | AGIASYAAYDAMES | 45 | 0.75 |
| 9 | MLLFYAAYKMDATT | 202 | 0.75 |
| 9 | AFAAYSVAAFVEKY | 121 | 0.75 |
| 10 | TQNVMAAYGTQNVM | 178 | 0.74 |
| 11 | AAYYTDGTQNVMAA | 171 | 0.72 |
| 12 | VVGQTFAKYAAYYT | 162 | 0.71 |
| 13 | ALSEAAAKSLAEKY | 5 | 0.69 |
| 13 | PTLAAYQIKGFPTL | 24 | 0.69 |
| 14 | TAESVKRFAAYFLA | 67 | 0.68 |
| 15 | QIKGFPTLAAYQIK | 19 | 0.66 |
| 16 | AAYDAMESVTVYAA | 51 | 0.65 |
| 16 | DQYAAYVTAESVAA | 108 | 0.65 |
| 16 | AYLTFIDGDQYAAY | 100 | 0.65 |
| 17 | VAVAEKYAAYLTFI | 92 | 0.64 |
| 18 | FLATAVLDYAAYSL | 78 | 0.62 |
| 19 | QIKGFPTLYAAYRT | 30 | 0.60 |
| Name of Vaccine | Name of the Targets | PDB IDs of the Targets | Binding Affinity, ∆G (kcal/mol) (ClusPro) | Global Energy (kcal/mol) (FireDock) |
|---|---|---|---|---|
| LdV vaccine | TLR-2 | 3a7c | −1071.30 | −30.84 |
| TLR-4 | 4g8a | −1175.40 | −6.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onile, O.S.; Musaigwa, F.; Ayawei, N.; Omoboyede, V.; Onile, T.A.; Oghenevovwero, E.; Aruleba, R.T. Immunoinformatics Studies and Design of a Potential Multi-Epitope Peptide Vaccine to Combat the Fatal Visceral Leishmaniasis. Vaccines 2022, 10, 1598. https://doi.org/10.3390/vaccines10101598
Onile OS, Musaigwa F, Ayawei N, Omoboyede V, Onile TA, Oghenevovwero E, Aruleba RT. Immunoinformatics Studies and Design of a Potential Multi-Epitope Peptide Vaccine to Combat the Fatal Visceral Leishmaniasis. Vaccines. 2022; 10(10):1598. https://doi.org/10.3390/vaccines10101598
Chicago/Turabian StyleOnile, Olugbenga Samson, Fungai Musaigwa, Nimibofa Ayawei, Victor Omoboyede, Tolulope Adelonpe Onile, Eyarefe Oghenevovwero, and Raphael Taiwo Aruleba. 2022. "Immunoinformatics Studies and Design of a Potential Multi-Epitope Peptide Vaccine to Combat the Fatal Visceral Leishmaniasis" Vaccines 10, no. 10: 1598. https://doi.org/10.3390/vaccines10101598
APA StyleOnile, O. S., Musaigwa, F., Ayawei, N., Omoboyede, V., Onile, T. A., Oghenevovwero, E., & Aruleba, R. T. (2022). Immunoinformatics Studies and Design of a Potential Multi-Epitope Peptide Vaccine to Combat the Fatal Visceral Leishmaniasis. Vaccines, 10(10), 1598. https://doi.org/10.3390/vaccines10101598