
Antitumor Peptide-Based Vaccine in the Limelight

 ,
, 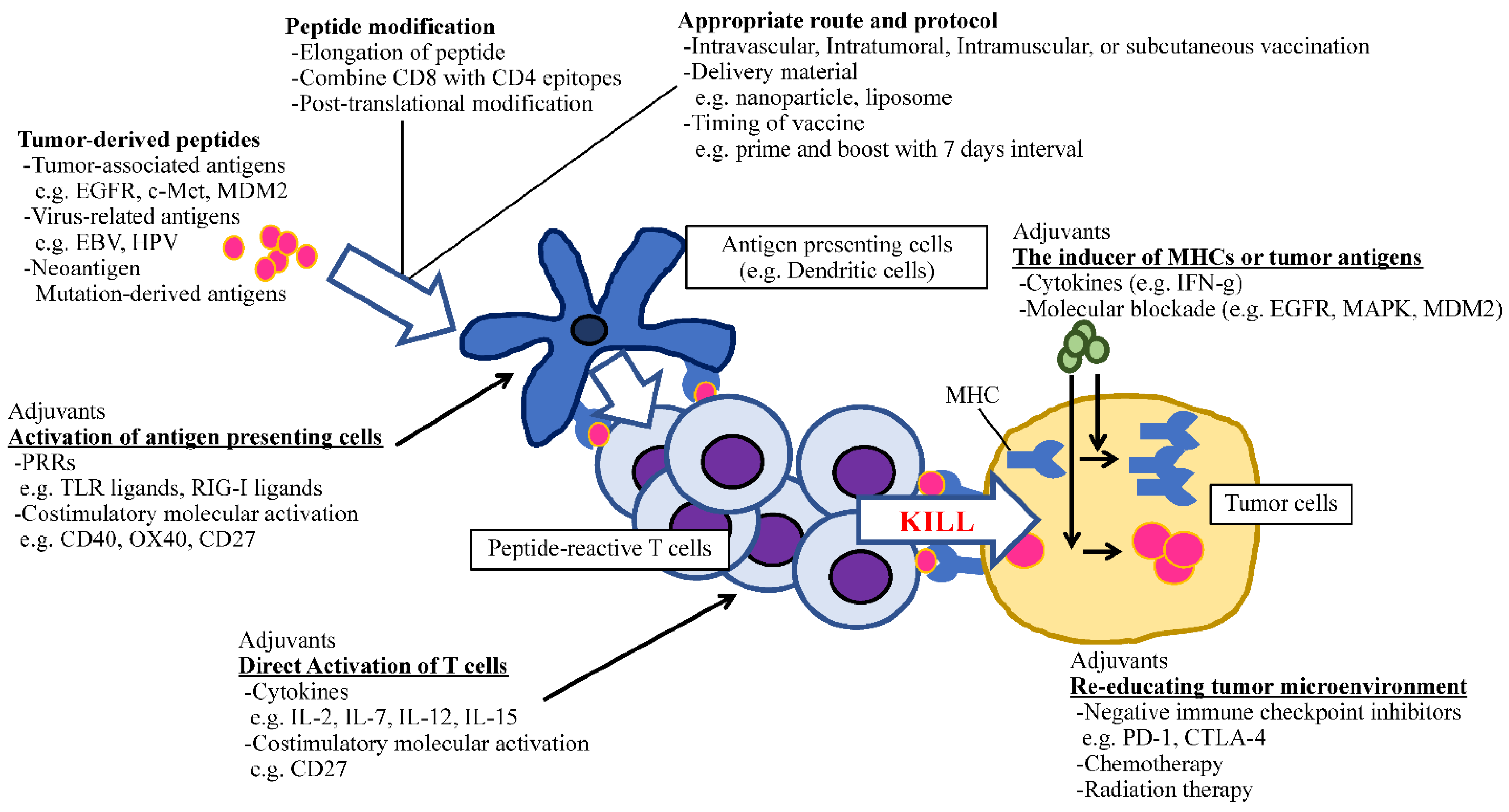{kind=link}
Abstract
1. Introduction
2. Selecting Optimal Peptides for Cancer Peptide Vaccines
2.1. Selecting Tumor Antigens as a Source of Vaccines
2.2. The Factors That Modulate Peptide Immunogenicity
3. Adjuvants and Delivery Timing of Vaccines to Induce Optimal Immune Responses
3.1. Conventional Adjuvants
3.2. Modern Adjuvants
3.3. The Timing of Vaccine
3.4. The Delivery of Vaccine
4. Clinical Evidence of Cancer Peptide Vaccines
5. Future Perspective
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef]
- Sultan, H.; Kumai, T.; Fesenkova, V.I.; Fan, A.E.; Wu, J.; Cho, H.-I.; Kobayashi, H.; Harabuchi, Y.; Celis, E. Sustained Persistence of IL2 Signaling Enhances the Antitumor Effect of Peptide Vaccines through T-cell Expansion and Preventing PD-1 Inhibition. Cancer Immunol. Res. 2018, 6, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Ademuyiwa, F.O.; Chen, I.; Luo, J.; Rimawi, M.F.; Hagemann, I.S.; Fisk, B.; Jeffers, G.; Skidmore, Z.L.; Basu, A.; Richters, M.; et al. Immunogenomic profiling and pathological response results from a clinical trial of docetaxel and carboplatin in triple-negative breast cancer. Breast Cancer Res. Treat. 2021, 189, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, A.L.; Kyritsis, C.; Tampé, R.; Cresswell, P. Early phagosomes in dendritic cells form a cellular compartment sufficient for cross presentation of exogenous antigens. Proc. Natl. Acad. Sci. USA 2003, 100, 12889–12894. [Google Scholar] [CrossRef] [PubMed]
- Fleri, W.; Paul, S.; Dhanda, S.; Mahajan, S.; Xu, X.; Peters, B.; Sette, A. The Immune Epitope Database and Analysis Resource in Epitope Discovery and Synthetic Vaccine Design. Front. Immunol. 2017, 8, 278. [Google Scholar] [CrossRef]
- Ishibashi, K.; Kumai, T.; Ohkuri, T.; Kosaka, A.; Nagato, T.; Hirata, Y.; Ohara, K.; Oikawa, K.; Aoki, N.; Akiyama, N.; et al. Epigenetic modification augments the immunogenicity of human leukocyte antigen G serving as a tumor antigen for T cell-based immunotherapy. OncoImmunology 2016, 5, e1169356. [Google Scholar] [CrossRef]
- Rammensee, H.-G.; Bachmann, J.; Emmerich, N.P.N.; Bachor, O.A.; Stevanović, S. SYFPEITHI: Database for MHC ligands and peptide motifs. Immunogenetics 1999, 50, 213–219. [Google Scholar] [CrossRef]
- Peters, B.; Sette, A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinform. 2005, 6, 132. [Google Scholar] [CrossRef]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef]
- Chaves, F.A.; Lee, A.H.; Nayak, J.; Richards, K.A.; Sant, A.J. The Utility and Limitations of Current Web-Available Algorithms To Predict Peptides Recognized by CD4 T Cells in Response to Pathogen Infection. J. Immunol. 2012, 188, 4235–4248. [Google Scholar] [CrossRef]
- Pritchard, A.L.; Burel, J.G.; Neller, M.; Hayward, N.; Lopez, J.A.; Fatho, M.; Lennerz, V.; Wölfel, T.; Schmidt, C. Exome Sequencing to Predict Neoantigens in Melanoma. Cancer Immunol. Res. 2015, 3, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Chikamatsu, K.; Albers, A.E.; Stanson, J.; Kwok, W.W.; Appella, E.; Whiteside, T.L.; DeLeo, A.B. P53(110-124)-specific human CD4+ T-helper cells enhance in vitro generation and antitumor function of tumor-reactive CD8+ T cells. Cancer Res. 2003, 63, 3675–3681. [Google Scholar]
- Kumai, T.; Matsuda, Y.; Ohkuri, T.; Oikawa, K.; Ishibashi, K.; Aoki, N.; Kimura, S.; Harabuchi, Y.; Celis, E.; Kobayashi, H. c-Met is a novel tumor associated antigen for T-cell based immunotherapy against NK/T cell lymphoma. OncoImmunology 2015, 4, e976077. [Google Scholar] [CrossRef]
- Kumai, T.; Matsuda, Y.; Oikawa, K.; Aoki, N.; Kimura, S.; Harabuchi, Y.; Celis, E.; Kobayashi, H. EGFR inhibitors augment antitumour helper T-cell responses of HER family-specific immunotherapy. Br. J. Cancer 2013, 109, 2155–2166. [Google Scholar] [CrossRef]
- Aruga, A.; Takeshita, N.; Kotera, Y.; Okuyama, R.; Matsushita, N.; Ohta, T.; Takeda, K.; Yamamoto, M. Long-term Vaccination with Multiple Peptides Derived from Cancer-Testis Antigens Can Maintain a Specific T-cell Response and Achieve Disease Stability in Advanced Biliary Tract Cancer. Clin. Cancer Res. 2013, 19, 2224–2231. [Google Scholar] [CrossRef]
- Nishizawa, S.; Hirohashi, Y.; Torigoe, T.; Takahashi, A.; Tamura, Y.; Mori, T.; Kanaseki, T.; Kamiguchi, K.; Asanuma, H.; Morita, R.; et al. HSP DNAJB8 Controls Tumor-Initiating Ability in Renal Cancer Stem–like Cells. Cancer Res. 2012, 72, 2844–2854. [Google Scholar] [CrossRef]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Löwer, M.; Van De Roemer, N.; De Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the Mutanome for Tumor Vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef]
- Rusch, V.; Baselga, J.; Cordon-Cardo, C.; Orazem, J.; Zaman, M.; Hoda, S.; McIntosh, J.; Kurie, J.; Dmitrovsky, E. Differential expression of the epidermal growth factor receptor and its ligands in primary non-small cell lung cancers and adjacent benign lung. Cancer Res. 1993, 53, 2379–2385. [Google Scholar]
- Kurts, C.; Sutherland, R.M.; Davey, G.; Li, M.; Lew, A.; Blanas, E.; Carbone, F.R.; Miller, J.F.A.P.; Heath, W.R. CD8 T cell ignorance or tolerance to islet antigens depends on antigen dose. Proc. Natl. Acad. Sci. USA 1999, 96, 12703–12707. [Google Scholar] [CrossRef]
- Schuler, P.J.; Boeckers, P.; Engers, R.; Boelke, E.; Bas, M.; Greve, J.; Dumitru, C.A.; Lehnerdt, G.F.; Ferris, R.L.; Andrade Filho, P.A.; et al. EGFR-specific T cell frequencies correlate with EGFR expression in head and neck squamous cell carcinoma. J. Transl. Med. 2011, 9, 168. [Google Scholar] [CrossRef][Green Version]
- Simpson, A.J.G.; Caballero, O.L.; Jungbluth, A.; Chen, Y.-T.; Old, L.J. Cancer/testis antigens, gametogenesis and cancer. Nat. Cancer 2005, 5, 615–625. [Google Scholar] [CrossRef]
- Valmori, D.; Dutoit, V.; Liénard, D.; Rimoldi, D.; Pittet, M.J.; Champagne, P.; Ellefsen, K.; Sahin, U.; Speiser, D.; Lejeune, F.; et al. Naturally occurring human lymphocyte antigen-A2 restricted CD8+ T-cell response to the cancer testis antigen NY-ESO-1 in melanoma patients. Cancer Res. 2000, 60, 4499–4506. [Google Scholar] [PubMed]
- Magee, J.A.; Piskounova, E.; Morrison, S.J. Cancer Stem Cells: Impact, Heterogeneity, and Uncertainty. Cancer Cell 2012, 21, 283–296. [Google Scholar] [CrossRef]
- Jaiswal, B.S.; Kljavin, N.M.; Stawiski, E.W.; Chan, E.; Parikh, C.; Durinck, S.; Chaudhuri, S.; Pujara, K.; Guillory, J.; Edgar, K.A.; et al. Oncogenic ERBB3 Mutations in Human Cancers. Cancer Cell 2013, 23, 603–617. [Google Scholar] [CrossRef]
- Suzuki, K.; Matsubara, H. Recent Advances in p53 Research and Cancer Treatment. J. Biomed. Biotechnol. 2011, 2011, 978312. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Han, S.Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function–structure and function–mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429. [Google Scholar] [CrossRef]
- Wu, W.; Chen, Y.; Huang, L.; Li, W.; Tao, C.; Shen, H. Point mutation screening of tumor neoantigens and peptide-induced specific cytotoxic T lymphocytes using The Cancer Genome Atlas database. Oncol. Lett. 2020, 20, 1. [Google Scholar] [CrossRef]
- Jensen, S.M.; Twitty, C.G.; Maston, L.D.; Antony, P.A.; Lim, M.; Hu, H.-M.; Petrausch, U.; Restifo, N.P.; Fox, B.A. Increased Frequency of Suppressive Regulatory T Cells and T Cell-Mediated Antigen Loss Results in Murine Melanoma Recurrence. J. Immunol. 2012, 189, 767–776. [Google Scholar] [CrossRef]
- Kono, M.; Kumai, T.; Hayashi, R.; Yamaki, H.; Komatsuda, H.; Wakisaka, R.; Nagato, T.; Ohkuri, T.; Kosaka, A.; Ohara, K.; et al. Interruption of MDM2 signaling augments MDM2-targeted T cell-based antitumor immunotherapy through antigen-presenting machinery. Cancer Immunol. Immunother. 2021, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, R.; Nagato, T.; Kumai, T.; Ohara, K.; Ohara, M.; Ohkuri, T.; Hirata-Nozaki, Y.; Harabuchi, S.; Kosaka, A.; Nagata, M.; et al. Expression of placenta-specific 1 and its potential for eliciting anti-tumor helper T-cell responses in head and neck squamous cell carcinoma. OncoImmunology 2021, 10, 1856545. [Google Scholar] [CrossRef]
- Hayashi, S.; Kumai, T.; Matsuda, Y.; Aoki, N.; Sato, K.; Kimura, S.; Kitada, M.; Tateno, M.; Celis, E.; Kobayashi, H. Six-transmembrane epithelial antigen of the prostate and enhancer of zeste homolog 2 as immunotherapeutic targets for lung cancer. J. Transl. Med. 2011, 9, 191. [Google Scholar] [CrossRef]
- Spitzer, M.H.; Carmi, Y.; Reticker-Flynn, N.; Kwek, S.S.; Madhireddy, D.; Martins, M.M.; Gherardini, P.F.; Prestwood, T.R.; Chabon, J.; Bendall, S.C.; et al. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell 2017, 168, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Xu, N.; Saito, S.; Zhou, L.; Ozgenc, I.; Webb, J.; Fu, C.; Zolkind, P.; Egloff, A.M.; Uppaluri, R. Integrating CD4+ T cell help for therapeutic cancer vaccination in a preclinical head and neck cancer model. OncoImmunology 2021, 10, 1958589. [Google Scholar] [CrossRef]
- Giuntoli, R.L.; Lu, J.; Kobayashi, H.; Kennedy, R.; Celis, E. Direct costimulation of tumor-reactive CTL by helper T cells potentiate their proliferation, survival, and effector function. Clin. Cancer Res. 2002, 8, 922–931. [Google Scholar]
- Perez-Diez, A.; Joncker, N.T.; Choi, K.; Chan, W.F.N.; Anderson, C.C.; Lantz, O.; Matzinger, P. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood 2007, 109, 5346–5354. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Cuss, S.M.; Singh, V.; Gurusamy, D.; Shoe, J.L.; Leighty, R.; Bronte, V.; Hurwitz, A.A. CD4+ T Cell Help Selectively Enhances High-Avidity Tumor Antigen-Specific CD8+ T Cells. J. Immunol. 2015, 195, 3482–3489. [Google Scholar] [CrossRef]
- Masuko, K.; Wakita, D.; Togashi, Y.; Kita, T.; Kitamura, H.; Nishimura, T. Artificially synthesized helper/killer-hybrid epitope long peptide (H/K-HELP): Preparation and immunological analysis of vaccine efficacy. Immunol. Lett. 2015, 163, 102–112. [Google Scholar] [CrossRef]
- de Goer de Herve, M.G.; Cariou, A.; Simonetta, F.; Taoufik, Y. Heterospecific CD4 help to rescue CD8 T cell killers. J. Immunol. 2008, 181, 5974–5980. [Google Scholar] [CrossRef]
- Kumai, T.; Kobayashi, H.; Harabuchi, Y.; Celis, E. Peptide vaccines in cancer—Old concept revisited. Curr. Opin. Immunol. 2017, 45, 1–7. [Google Scholar] [CrossRef]
- Wells, D.K.; van Buuren, M.M.; Dang, K.K.; Hubbard-Lucey, V.M.; Sheehan, K.C.; Campbell, K.M.; Lamb, A.; Ward, J.P.; Sidney, J.; Blazquez, A.B.; et al. Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell 2020, 183, 818–834. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.-H.; Sherman, L.A. N-terminal trimer extension of nominal CD8 T cell epitopes is sufficient to promote cross-presentation to cognate CD8 T cells in vivo. J. Immunol. 2007, 179, 8280–8286. [Google Scholar] [CrossRef] [PubMed]
- Bijker, M.S.; Eeden, S.J.F.V.D.; Franken, K.L.; Melief, C.J.M.; van der Burg, S.H.; Offringa, R. Superior induction of anti-tumor CTL immunity by extended peptide vaccines involves prolonged, DC-focused antigen presentation. Eur. J. Immunol. 2008, 38, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Quakkelaar, E.D.; Fransen, M.F.; van Maren, W.W.; Vaneman, J.; Loof, N.M.; van Heiningen, S.H.; Verbeek, J.S.; Ossendorp, F.; Melief, C.J. IgG-Mediated Anaphylaxis to a Synthetic Long Peptide Vaccine Containing a B Cell Epitope Can Be Avoided by Slow-Release Formulation. J. Immunol. 2014, 192, 5813–5820. [Google Scholar] [CrossRef]
- León, B.; Ballesteros-Tato, A.; Randall, T.D.; Lund, F.E. Prolonged antigen presentation by immune complex–binding dendritic cells programs the proliferative capacity of memory CD8 T cells. J. Exp. Med. 2014, 211, 1637–1655. [Google Scholar] [CrossRef]
- Rad-Malekshahi, M.; Fransen, M.F.; Krawczyk, M.; Mansourian, M.; Bourajjaj, M.; Chen, J.; Ossendorp, F.; Hennink, W.E.; Mastrobattista, E.; Amidi, M. Self-Assembling Peptide Epitopes as Novel Platform for Anticancer Vaccination. Mol. Pharm. 2017, 14, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Sultan, H.; Kumai, T.; Nagato, T.; Wu, J.; Salazar, A.M.; Celis, E. The route of administration dictates the immunogenicity of peptide-based cancer vaccines in mice. Cancer Immunol. Immunother. 2019, 68, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Park, J.; Jiang, Y.; Woodrow, K.A. Rational design of charged peptides that self-assemble into robust nanofibers as immune-functional scaffolds. Acta Biomater. 2017, 55, 183–193. [Google Scholar] [CrossRef]
- Lynn, G.M.; Sedlik, C.; Baharom, F.; Zhu, Y.; Ramirez-Valdez, R.A.; Coble, V.L.; Tobin, K.; Nichols, S.R.; Itzkowitz, Y.; Zaidi, N.; et al. Peptide-TLR-7/8a conjugate vaccines chemically programmed for nanoparticle self-assembly enhance CD8 T-cell immunity to tumor antigens. Nat. Biotechnol. 2020, 38, 320–332. [Google Scholar] [CrossRef]
- Gupta, S.; Singh, I.; Sharma, A.K.; Kumar, P. Ultrashort Peptide Self-Assembly: Front-Runners to Transport Drug and Gene Cargos. Front. Bioeng. Biotechnol. 2020, 8, 504. [Google Scholar] [CrossRef]
- Sultan, H.; Wu, J.; Kumai, T.; Salazar, A.M.; Celis, E. Role of MDA5 and interferon-I in dendritic cells for T cell expansion by anti-tumor peptide vaccines in mice. Cancer. Immunol. Immunother. 2018, 67, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, T.; Valadan, R.; Rafiei, A.; Abbasi, A.; Haghshenas, M.R. A novel recombinant protein vaccine containing the different E7 proteins of the HPV16, 18, 6, 11 E7 linked to the HIV-1 Tat (47–57) improve cytotoxic immune responses. Biotechnol. Lett. 2021, 43, 1933–1944. [Google Scholar] [CrossRef]
- Piantavigna, S.; McCubbin, G.A.; Boehnke, S.; Graham, B.; Spiccia, L.; Martin, L.L. A mechanistic investigation of cell-penetrating Tat peptides with supported lipid membranes. Biochim. Biophys. Acta 2011, 1808, 1811–1817. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Kovalchin, J.T.; Muhlenkamp, P.; Chandawarkar, R.Y. Exogenous heat shock protein 70 binds macrophage lipid raft microdomain and stimulates phagocytosis, processing, and MHC-II presentation of antigens. Blood 2006, 107, 1636–1642. [Google Scholar] [CrossRef]
- Ohara, K.; Ohkuri, T.; Kumai, T.; Nagato, T.; Nozaki, Y.; Ishibashi, K.; Kosaka, A.; Nagata, M.; Harabuchi, S.; Ohara, M.; et al. Targeting phosphorylated p53 to elicit tumor-reactive T helper responses against head and neck squamous cell carcinoma. Oncoimmunology 2018, 7, e1466771. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Papadimitriou, J.; Burchell, J.M.; Graham, R.; Beatson, R. Latest developments in MUC1 immunotherapy. Biochem. Soc. Trans. 2018, 46, 659–668. [Google Scholar] [CrossRef]
- Kumai, T.; Ishibashi, K.; Oikawa, K.; Matsuda, Y.; Aoki, N.; Kimura, S.; Hayashi, S.; Kitada, M.; Harabuchi, Y.; Celis, E.; et al. Induction of tumor-reactive T helper responses by a posttranslational modified epitope from tumor protein p53. Cancer Immunol. Immunother. 2014, 63, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Raposo, B.; Merky, P.; Lundqvist, C.; Yamada, H.; Urbonaviciute, V.; Niaudet, C.; Viljanen, J.; Kihlberg, J.; Kyewski, B.; Ekwall, O.; et al. T cells specific for post-translational modifications escape intrathymic tolerance induction. Nat. Commun. 2018, 9, 353. [Google Scholar] [CrossRef]
- Overwijk, W.W.; Tsung, A.; Irvine, K.R.; Parkhurst, M.R.; Goletz, T.J.; Tsung, K.; Carroll, M.W.; Liu, C.; Moss, B.; Rosenberg, S.A.; et al. gp100/pmel 17 is a murine tumor rejection antigen: Induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J. Exp. Med. 1998, 188, 277–286. [Google Scholar] [CrossRef]
- Powell, J.D. The induction and maintenance of T cell anergy. Clin. Immunol. 2006, 120, 239–246. [Google Scholar] [CrossRef]
- Ballesteros-Tato, A.; Leon, B.; Lee, B.O.; Lund, F.E.; Randall, T.D. Epitope-specific regulation of memory programming by differential duration of antigen presentation to influenza-specific CD8+ T cells. Immunity 2014, 41, 127–140. [Google Scholar] [CrossRef]
- Curtsinger, J.M.; Schmidt, C.S.; Mondino, A.; Lins, D.C.; Kedl, R.M.; Jenkins, M.; Mescher, M.F. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J. Immunol. 1999, 162, 3256–3262. [Google Scholar]
- van der Burg, S.H.; Arens, R.; Ossendorp, F.; van Hall, T.; Melief, C.J. Vaccines for established cancer: Overcoming the challenges posed by immune evasion. Nat. Cancer 2016, 16, 219–233. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Restifo, N.P. Cancer immunotherapy: Moving beyond current vaccines. Nat. Med. 2004, 10, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Hailemichael, Y.; Dai, Z.; Jaffarzad, N.; Ye, Y.; Medina, M.A.; Huang, X.F.; Dorta-Estremera, S.M.; Greeley, N.R.; Nitti, G.; Peng, W.; et al. Persistent antigen at vaccination sites induces tumor-specific CD8+ T cell sequestration, dysfunction and deletion. Nat. Med. 2013, 19, 465–472. [Google Scholar] [CrossRef]
- Pashine, A.; Valiante, N.M.; Ulmer, J.B. Targeting the innate immune response with improved vaccine adjuvants. Nat. Med. 2005, 11, S63–S68. [Google Scholar] [CrossRef] [PubMed]
- Sultan, H.; Fesenkova, V.I.; Addis, D.; Fan, A.E.; Kumai, T.; Wu, J.; Salazar, A.M.; Celis, E. Designing therapeutic cancer vaccines by mimicking viral infections. Cancer Immunol. Immunother. 2017, 66, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Nair-Gupta, P.; Baccarini, A.; Tung, N.; Seyffer, F.; Florey, O.; Huang, Y.; Banerjee, M.; Overholtzer, M.; Roche, P.; Tampé, R.; et al. TLR Signals Induce Phagosomal MHC-I Delivery from the Endosomal Recycling Compartment to Allow Cross-Presentation. Cell 2014, 158, 506–521. [Google Scholar] [CrossRef] [PubMed]
- Pahlavanneshan, S.; Sayadmanesh, A.; Ebrahimiyan, H.; Basiri, M. Toll-Like Receptor-Based Strategies for Cancer Immunotherapy. J. Immunol. Res. 2021, 2021, 9912188. [Google Scholar] [CrossRef]
- Kumai, T.; Lee, S.; Cho, H.-I.; Sultan, H.; Kobayashi, H.; Harabuchi, Y.; Celis, E. Optimization of Peptide Vaccines to Induce Robust Antitumor CD4 T-cell Responses. Cancer Immunol. Res. 2016, 5, 72–83. [Google Scholar] [CrossRef]
- Patel, S.P.; Petroni, G.R.; Roszik, J.; Olson, W.C.; Wages, N.A.; Chianese-Bullock, K.A.; Smolkin, M.; Varhegyi, N.; Gaughan, E.; Smith, K.T.; et al. Phase I/II trial of a long peptide vaccine (LPV7) plus toll-like receptor (TLR) agonists with or without incomplete Freund’s adjuvant (IFA) for resected high-risk melanoma. J. Immunother. Cancer 2021, 9, e003220. [Google Scholar] [CrossRef] [PubMed]
- Garçon, N.; Chomez, P.; Van Mechelen, M. GlaxoSmithKline Adjuvant Systems in vaccines: Concepts, achievements and perspectives. Expert Rev. Vaccines 2007, 6, 723–739. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.S.; Abrams, J.; Thompson, J.A.; Kashani-Sabet, M.; DeConti, R.C.; Hwu, W.J.; Atkins, M.B.; Whitman, E.; Ernstoff, M.S.; Haluska, F.G.; et al. Randomized trial of an allogeneic melanoma lysate vaccine with low-dose interferon Alfa-2b compared with high-dose interferon Alfa-2b for Resected stage III cutaneous melanoma. J. Clin. Oncol. 2007, 25, 2078–2085. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, C.; Rocha, B.; Tanchot, C. A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science 2002, 297, 2060–2063. [Google Scholar] [CrossRef] [PubMed]
- Stevens, A.D.; Bullock, T.N.J. Therapeutic vaccination targeting CD40 and TLR3 controls melanoma growth through existing intratumoral CD8 T cells without new T cell infiltration. Cancer Immunol. Immunother. 2021, 70, 2139–2150. [Google Scholar] [CrossRef]
- Pulko, V.; Liu, X.; Krco, C.J.; Harris, K.J.; Frigola, X.; Kwon, E.D.; Dong, H. TLR3-Stimulated Dendritic Cells Up-regulate B7-H1 Expression and Influence the Magnitude of CD8 T Cell Responses to Tumor Vaccination. J. Immunol. 2009, 183, 3634–3641. [Google Scholar] [CrossRef]
- Varthaman, A.; Moreau, H.D.; Maurin, M.; Benaroch, P. TLR3-Induced Maturation of Murine Dendritic Cells Regulates CTL Responses by Modulating PD-L1 Trafficking. PLoS ONE 2016, 11, e0167057. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu-Lieskovan, S.; Chmielowski, B.; Govindan, R.; Naing, A.; Bhardwaj, N.; Margolin, K.; Awad, M.M.; Hellmann, M.D.; Lin, J.J.; et al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-small Cell Lung Cancer, or Bladder Cancer. Cell 2020, 183, 347–362. [Google Scholar] [CrossRef]
- Woo, S.R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef]
- Wang, Z.; Celis, E. STING activator c-di-GMP enhances the anti-tumor effects of peptide vaccines in melanoma-bearing mice. Cancer Immunol. Immunother. 2015, 64, 1057–1066. [Google Scholar] [CrossRef]
- Ramanjulu, J.M.; Pesiridis, G.S.; Yang, J.; Concha, N.; Singhaus, R.; Zhang, S.-Y.; Tran, J.-L.; Moore, P.; Lehmann, S.; Eberl, H.C.; et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 2018, 564, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Gardner, T.A.; Elzey, B.D.; Hahn, N.M. Sipuleucel-T (Provenge) autologous vaccine approved for treatment of men with asymptomatic or minimally symptomatic castrate-resistant metastatic prostate cancer. Hum. Vaccines Immunother. 2012, 8, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Moro-García, M.A.; Alonso-Arias, R.; Lopez-Larrea, C. When aging reaches CD4+ T-cells: Phenotypic and functional changes. Front. Immunol. 2013, 4, 107. [Google Scholar] [CrossRef] [PubMed]
- Chick, R.C.; Clifton, G.T.; Hale, D.F.; Vreeland, T.J.; Hickerson, A.T.; Bohan, P.M.K.; McCarthy, P.M.; Litton, J.K.; Alatrash, G.; Murthy, R.K.; et al. Subgroup analysis of nelipepimut-S plus GM-CSF combined with trastuzumab versus trastuzumab alone to prevent recurrences in patients with high-risk, HER2 low-expressing breast cancer. Clin. Immunol. 2021, 225, 108679. [Google Scholar] [CrossRef] [PubMed]
- Voskens, C.J.; Sewell, D.; Hertzano, R.; DeSanto, J.; Rollins, S.; Lee, M.; Taylor, R.; Wolf, J.; Suntharalingam, M.; Gastman, B.; et al. Induction of MAGE-A3 and HPV-16 immunity by Trojan vaccines in patients with head and neck carcinoma. Head Neck 2012, 34, 1734–1746. [Google Scholar] [CrossRef]
- Hoeller, C.; Michielin, O.; Ascierto, P.A.; Szabo, Z.; Blank, C.U. Systematic review of the use of granulocyte-macrophage colony-stimulating factor in patients with advanced melanoma. Cancer Immunol Immunother 2016, 65, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Slingluff, C.L.; Petroni, G.R.; Olson, W.C.; Smolkin, M.E.; Ross, M.I.; Haas, N.B.; Grosh, W.W.; Boisvert, M.E.; Kirkwood, J.M.; Chianese-Bullock, K.A. Effect of Granulocyte/Macrophage Colony-Stimulating Factor on Circulating CD8+ and CD4+ T-Cell Responses to a Multipeptide Melanoma Vaccine: Outcome of a Multicenter Randomized Trial. Clin. Cancer Res. 2009, 15, 7036–7044. [Google Scholar] [CrossRef] [PubMed]
- Parmiani, G.; Castelli, C.; Pilla, L.; Santinami, M.; Colombo, M.P.; Rivoltini, L. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann. Oncol. 2007, 18, 226–232. [Google Scholar] [CrossRef]
- Serafini, P.; Carbley, R.; Noonan, K.A.; Tan, G.; Bronte, V.; Borrello, I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004, 64, 6337–6343. [Google Scholar] [CrossRef]
- Xue, D.; Hsu, E.; Fu, Y.-X.; Peng, H. Next-generation cytokines for cancer immunotherapy. Antib. Ther. 2021, 4, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, N.; Zhou, L.; Wang, J.; Zhou, Y.; Zhang, T.; Fang, Y.; Deng, J.; Gao, Y.; Liang, X.; et al. IL-2 regulates tumor-reactive CD8+ T cell exhaustion by activating the aryl hydrocarbon receptor. Nat. Immunol. 2021, 22, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-I.; Reyes-Vargas, E.; Delgado, J.C.; Celis, E. A Potent Vaccination Strategy That Circumvents Lymphodepletion for Effective Antitumor Adoptive T-cell Therapy. Cancer Res. 2012, 72, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Hsu, E.J.; Cao, X.; Moon, B.; Bae, J.; Sun, Z.; Liu, Z.; Fu, Y.-X. A cytokine receptor-masked IL2 prodrug selectively activates tumor-infiltrating lymphocytes for potent antitumor therapy. Nat. Commun. 2021, 12, 2768. [Google Scholar] [CrossRef]
- Macdonald, G. HLA-DR expression is associated with better prognosis in sporadic Australian clinicopathological Stage C colorectal cancers. Int. J. Cancer 2009, 125, 1231–1237. [Google Scholar]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Hotz, C.; Bourquin, C. Systemic cancer immunotherapy with Toll-like receptor 7 agonists: Timing is everything. Oncoimmunology 2012, 1, 227–228. [Google Scholar] [CrossRef]
- Koga-Yamakawa, E.; Murata, M.; Dovedi, S.J.; Wilkinson, R.W.; Ota, Y.; Umehara, H.; Sugaru, E.; Hirose, Y.; Harada, H.; Jewsbury, P.J.; et al. TLR7 tolerance is independent of the type I IFN pathway and leads to loss of anti-tumor efficacy in mice. Cancer Immunol. Immunother. 2015, 64, 1229–1239. [Google Scholar] [CrossRef]
- Bourquin, C.; Hotz, C.; Noerenberg, D.; Voelkl, A.; Heidegger, S.; Roetzer, L.C.; Storch, B.; Sandholzer, N.; Wurzenberger, C.; Anz, D.; et al. Systemic Cancer Therapy with a Small Molecule Agonist of Toll-like Receptor 7 Can Be Improved by Circumventing TLR Tolerance. Cancer Res. 2011, 71, 5123–5133. [Google Scholar] [CrossRef]
- Prlic, M.; Bevan, M.J. Exploring regulatory mechanisms of CD8+ T cell contraction. Proc. Natl. Acad. Sci. USA 2008, 105, 16689–16694. [Google Scholar] [CrossRef] [PubMed]
- Cuzzubbo, S.; Mangsbo, S.; Nagarajan, D.; Habra, K.; Pockley, A.G.; McArdle, S.E.B. Cancer Vaccines: Adjuvant Potency, Importance of Age, Lifestyle, and Treatments. Front. Immunol. 2021, 11, 615240. [Google Scholar] [CrossRef]
- Wu, X.; Feng, Q.-M.; Wang, Y.; Shi, J.; Ge, H.-L.; Di, W. The immunologic aspects in advanced ovarian cancer patients treated with paclitaxel and carboplatin chemotherapy. Cancer Immunol. Immunother. 2009, 59, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Welters, M.J.; van der Sluis, T.C.; van Meir, H.; Loof, N.M.; van Ham, V.J.; van Duikeren, S.; Santegoets, S.J.; Arens, R.; de Kam, M.L.; Cohen, A.F.; et al. Vaccination during myeloid cell depletion by cancer chemotherapy fosters robust T cell responses. Sci. Transl. Med. 2016, 8, 334ra352. [Google Scholar] [CrossRef] [PubMed]
- Berinstein, N.L.; Karkada, M.; Oza, A.; Odunsi, K.; Villella, J.A.; Nemunaitis, J.J.; Morse, M.A.; Pejovic, T.; Bentley, J.; Buyse, M.; et al. Survivin-targeted immunotherapy drives robust polyfunctional T cell generation and differentiation in advanced ovarian cancer patients. OncoImmunology 2015, 4, e1026529. [Google Scholar] [CrossRef] [PubMed]
- Tagliamonte, M.; Petrizzo, A.; Napolitano, M.; Luciano, A.; Arra, C.; Maiolino, P.; Izzo, F.; Tornesello, M.L.; Aurisicchio, L.; Ciliberto, G.; et al. Novel metronomic chemotherapy and cancer vaccine combinatorial strategy for hepatocellular carcinoma in a mouse model. Cancer Immunol. Immunother. 2015, 64, 1305–1314. [Google Scholar] [CrossRef]
- Matsumura, S.; Wang, B.; Kawashima, N.; Braunstein, S.; Badura, M.; Cameron, T.O.; Babb, J.; Schneider, R.; Formenti, S.C.; Dustin, M.; et al. Radiation-Induced CXCL16 Release by Breast Cancer Cells Attracts Effector T Cells. J. Immunol. 2008, 181, 3099–3107. [Google Scholar] [CrossRef]
- Aliru, M.L.; Schoenhals, J.E.; Venkatesulu, B.P.; Anderson, C.C.; Barsoumian, H.B.; Younes, A.I.; KMahadevan, L.S.; Soeung, M.; Aziz, K.E.; Welsh, J.W.; et al. Radiation therapy and immunotherapy: What is the optimal timing or sequencing? Immunotherapy 2018, 10, 299–316. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hu, X.; Xiang, D. Nanoparticle drug delivery systems: An excellent carrier for tumor peptide vaccines. Drug Deliv. 2018, 25, 1319–1327. [Google Scholar] [CrossRef]
- Li, H.; Li, Y.; Jiao, J.; Hu, H.-M. Alpha-alumina nanoparticles induce efficient autophagy-dependent cross-presentation and potent antitumour response. Nat. Nanotechnol. 2011, 6, 645–650. [Google Scholar] [CrossRef]
- Sun, B.; Xia, T. Nanomaterial-based vaccine adjuvants. J. Mater. Chem. B 2016, 4, 5496–5509. [Google Scholar] [CrossRef]
- Niikura, K.; Matsunaga, T.; Suzuki, T.; Kobayashi, S.; Yamaguchi, H.; Orba, Y.; Kawaguchi, A.; Hasegawa, H.; Kajino, K.; Ninomiya, T.; et al. Gold Nanoparticles as a Vaccine Platform: Influence of Size and Shape on Immunological Responses in Vitro and in Vivo. ACS Nano 2013, 7, 3926–3938. [Google Scholar] [CrossRef] [PubMed]
- van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Ardavanis, A.; Litton, J.K.; Shumway, N.M.; Hale, D.F.; Murray, J.L.; Perez, S.A.; Ponniah, S.; Baxevanis, C.N.; Papamichail, M.; et al. Primary analysis of a prospective, randomized, single-blinded phase II trial evaluating the HER2 peptide GP2 vaccine in breast cancer patients to prevent recurrence. Oncotarget 2016, 7, 66192–66201. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Ardavanis, A.; Symanowski, J.; Murray, J.L.; Shumway, N.M.; Litton, J.K.; Hale, D.F.; Perez, S.A.; Anastasopoulou, E.A.; Pistamaltzian, N.F.; et al. Primary analysis of a prospective, randomized, single-blinded phase II trial evaluating the HER2 peptide AE37 vaccine in breast cancer patients to prevent recurrence. Ann. Oncol. 2016, 27, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Clifton, G.T.; Holmes, J.P.; Schneble, E.; van Echo, D.; Ponniah, S.; Peoples, G.E. Final report of the phase I/II clinical trial of the E75 (nelipepimut-S) vaccine with booster inoculations to prevent disease recurrence in high-risk breast cancer patients. Ann. Oncol. 2014, 25, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Lu, B.; Melisko, M.; Hiller, J.P.; Bondarenko, I.; Brunt, A.M.; Sergii, G.; Petrakova, K.; Peoples, G.E. Efficacy and Safety Analysis of Nelipepimut-S Vaccine to Prevent Breast Cancer Recurrence: A Randomized, Multicenter, Phase III Clinical Trial. Clin. Cancer Res. 2019, 25, 4248–4254. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Schwartzentruber, D.J.; Hwu, P.; Marincola, F.M.; Topalian, S.L.; Restifo, N.P.; Dudley, M.E.; Schwarz, S.L.; Spiess, P.J.; et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat. Med. 1998, 4, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, J.M.; Lee, S.; Moschos, S.J.; Albertini, M.R.; Michalak, J.C.; Sander, C.; Whiteside, T.; Butterfield, L.H.; Weiner, L. Immunogenicity and antitumor effects of vaccination with peptide vaccine+/− granulocyte-monocyte colony-stimulating factor and/or IFN-α2b in advanced metastatic melanoma: Eastern Cooperative Oncology Group Phase II Trial E1696. Clin. Cancer Res. 2009, 15, 1443–1451. [Google Scholar] [CrossRef]
- Schwartzentruber, D.J.; Lawson, D.H.; Richards, J.M.; Conry, R.M.; Miller, D.M.; Treisman, J.; Gailani, F.; Riley, L.; Conlon, K.; Pockaj, B.; et al. gp100 Peptide Vaccine and Interleukin-2 in Patients with Advanced Melanoma. N. Engl. J. Med. 2011, 364, 2119–2127. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.-C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient with Epithelial Cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; Van Der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Mehrotra, S.; Britten, C.D.; Chin, S.; Garrett-Mayer, E.; Cloud, C.A.; Li, M.; Scurti, G.; Salem, M.; Nelson, M.H.; Thomas, M.B.; et al. Vaccination with poly(IC:LC) and peptide-pulsed autologous dendritic cells in patients with pancreatic cancer. J. Hematol. Oncol. 2017, 10, 82. [Google Scholar] [CrossRef]
- Migliorini, D.; Dutoit, V.; Allard, M.; Grandjean Hallez, N.; Marinari, E.; Widmer, V.; Philippin, G.; Corlazzoli, F.; Gustave, R.; Kreutzfeldt, M.; et al. Phase I/II trial testing safety and immunogenicity of the multipeptide IMA950/poly-ICLC vaccine in newly diagnosed adult malignant astrocytoma patients. Neuro-oncology 2019, 21, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.; Weinschenk, T.; Stenzl, A.; Zdrojowy, R.; Pluzanska, A.; Szczylik, C.; Staehler, M.; Brugger, W.; Dietrich, P.Y.; Mendrzyk, R.; et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat. Med. 2012, 18, 1254–1261. [Google Scholar] [CrossRef]
- Rini, B.I.; Stenzl, A.; Zdrojowy, R.; Kogan, M.; Shkolnik, M.; Oudard, S.; Weikert, S.; Bracarda, S.; Crabb, S.; Bedke, J.; et al. IMA901, a multipeptide cancer vaccine, plus sunitinib versus sunitinib alone, as first-line therapy for advanced or metastatic renal cell carcinoma (IMPRINT): A multicentre, open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2016, 17, 1599–1611. [Google Scholar] [CrossRef]
- Miyazawa, M.; Ohsawa, R.; Tsunoda, T.; Hirono, S.; Kawai, M.; Tani, M.; Nakamura, Y.; Yamaue, H. Phase I clinical trial using peptide vaccine for human vascular endothelial growth factor receptor 2 in combination with gemcitabine for patients with advanced pancreatic cancer. Cancer Sci. 2010, 101, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Yamaue, H.; Tsunoda, T.; Tani, M.; Miyazawa, M.; Yamao, K.; Mizuno, N.; Okusaka, T.; Ueno, H.; Boku, N.; Fukutomi, A.; et al. Randomized phase II / III clinical trial of elpamotide for patients with advanced pancreatic cancer: PEGASUS-PC Study. Cancer Sci. 2015, 106, 883–890. [Google Scholar] [CrossRef]
- Middleton, G.; Silcocks, P.; Cox, T.; Valle, J.; Wadsley, J.; Propper, D.; Coxon, F.; Ross, P.; Madhusudan, S.; Roques, T.; et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): An open-label, randomised, phase 3 trial. Lancet Oncol. 2014, 15, 829–840. [Google Scholar] [CrossRef]
- Hasegawa, K.; Ikeda, Y.; Kunugi, Y.; Kurosaki, A.; Imai, Y.; Kohyama, S.; Nagao, S.; Kozawa, E.; Yoshida, K.; Tsunoda, T.; et al. Phase I Study of Multiple Epitope Peptide Vaccination in Patients With Recurrent or Persistent Cervical Cancer. J. Immunother. 2018, 41, 201–207. [Google Scholar] [CrossRef]
- Kjeldsen, J.W.; Iversen, T.Z.; Engell-Noerregaard, L.; Mellemgaard, A.; Andersen, M.H.; Svane, I.M. Durable Clinical Responses and Long-Term Follow-Up of Stage III–IV Non-Small-Cell Lung Cancer (NSCLC) Patients Treated With IDO Peptide Vaccine in a Phase I Study—A Brief Research Report. Front. Immunol. 2018, 9, 2145. [Google Scholar] [CrossRef]
- Hacohen, N.; Fritsch, E.F.; Carter, T.A.; Lander, E.S.; Wu, C.J. Getting Personal with Neoantigen-Based Therapeutic Cancer Vaccines. Cancer Immunol. Res. 2013, 1, 11–15. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Fang, Y.; Mo, F.; Shou, J.; Wang, H.; Luo, K.; Zhang, S.; Han, N.; Li, H.; Ye, S.; Zhou, Z.; et al. A pan-cancer clinical study of personalized neoantigen vaccine monotherapy in treating patients with various types of advanced solid tumors. Clin. Cancer Res. 2020, 26, 4511–4520. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. Erratum: Corrigendum: An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2018, 555, 402. [Google Scholar] [CrossRef]
- Correale, P.; Botta, C.; Martino, E.C.; Ulivieri, C.; Battaglia, G.; Carfagno, T.; Rossetti, M.G.; Fioravanti, A.; Guidelli, G.M.; Cheleschi, S.; et al. Phase Ib study of poly-epitope peptide vaccination to thymidylate synthase (TSPP) and GOLFIG chemo-immunotherapy for treatment of metastatic colorectal cancer patients. OncoImmunology 2015, 5, e1101205. [Google Scholar] [CrossRef] [PubMed]
- Murahashi, M.; Hijikata, Y.; Yamada, K.; Tanaka, Y.; Kishimoto, J.; Inoue, H.; Marumoto, T.; Takahashi, A.; Okazaki, T.; Takeda, K.; et al. Phase I clinical trial of a five-peptide cancer vaccine combined with cyclophosphamide in advanced solid tumors. Clin. Immunol. 2016, 166, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, J.; Sun, Z.; Pagliano, O.; Chauvin, J.-M.; Sander, C.; Janjic, B.; Tarhini, A.A.; Tawbi, H.A.; Kirkwood, J.M.; Moschos, S.; et al. PD-1 and Tim-3 Regulate the Expansion of Tumor Antigen–Specific CD8+ T Cells Induced by Melanoma Vaccines. Cancer Res. 2014, 74, 1045–1055. [Google Scholar] [CrossRef]
- Lugade, A.A.; Moran, J.P.; Gerber, S.A.; Rose, R.C.; Frelinger, J.G.; Lord, E.M. Local Radiation Therapy of B16 Melanoma Tumors Increases the Generation of Tumor Antigen-Specific Effector Cells That Traffic to the Tumor. J. Immunol. 2005, 174, 7516–7523. [Google Scholar] [CrossRef]
- Karyampudi, L.; Lamichhane, P.; Scheid, A.D.; Kalli, K.R.; Shreeder, B.; Krempski, J.W.; Behrens, M.D.; Knutson, K.L. Accumulation of Memory Precursor CD8 T Cells in Regressing Tumors following Combination Therapy with Vaccine and Anti-PD-1 Antibody. Cancer Res. 2014, 74, 2974–2985. [Google Scholar] [CrossRef]
- Nakata, J.; Nakae, Y.; Kawakami, M.; Morimoto, S.; Motooka, D.; Hosen, N.; Fujiki, F.; Nakajima, H.; Hasegawa, K.; Nishida, S.; et al. Wilms tumour 1 peptide vaccine as a cure-oriented post-chemotherapy strategy for patients with acute myeloid leukaemia at high risk of relapse. Br. J. Haematol. 2018, 182, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.A.; Mittendorf, E.A.; Hale, D.F.; Myers, J.W.; Peace, K.M.; Jackson, D.O.; Greene, J.M.; Vreeland, T.J.; Clifton, G.T.; Ardavanis, A.; et al. Prospective, randomized, single-blinded, multi-center phase II trial of two HER2 peptide vaccines, GP2 and AE37, in breast cancer patients to prevent recurrence. Breast Cancer Res. Treat. 2020, 181, 391–401. [Google Scholar] [CrossRef]
- Shen, J.; Wang, L.-F.; Zou, Z.-Y.; Kong, W.-W.; Yan, J.; Meng, F.-Y.; Chen, F.-J.; Du, J.; Shao, J.; Xu, Q.-P.; et al. Phase I clinical study of personalized peptide vaccination combined with radiotherapy for advanced hepatocellular carcinoma. World J. Gastroenterol. 2017, 23, 5395–5404. [Google Scholar] [CrossRef] [PubMed]
- Ciccolini, J.; Barbolosi, D.; André, N.; Benzekry, S.; Barlesi, F. Combinatorial immunotherapy strategies: Most gods throw dice, but fate plays chess. Ann. Oncol. 2019, 30, 1690–1691. [Google Scholar] [CrossRef] [PubMed]
- Schlom, J.; Gulley, J.L. Vaccines as an Integral Component of Cancer Immunotherapy. JAMA 2018, 320, 2195–2196. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Gatti-Mays, M.E.; Redman, J.M.; Collins, J.M.; Bilusic, M. Cancer vaccines: Enhanced immunogenic modulation through therapeutic combinations. Hum. Vaccines Immunother. 2017, 13, 2561–2574. [Google Scholar] [CrossRef]
- Shemesh, C.S.; Hsu, J.C.; Hosseini, I.; Shen, B.-Q.; Rotte, A.; Twomey, P.; Girish, S.; Wu, B. Personalized Cancer Vaccines: Clinical Landscape, Challenges, and Opportunities. Mol. Ther. 2021, 29, 555–570. [Google Scholar] [CrossRef]
- Shi, J.; Tricot, G.J.; Garg, T.K.; Malaviarachchi, P.A.; Szmania, S.M.; Kellum, R.E.; Storrie, B.; Mulder, A.; Shaughnessy, J.J.D.; Barlogie, B.; et al. Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell–mediated lysis of myeloma. Blood 2008, 111, 1309–1317. [Google Scholar] [CrossRef]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef]
- Yang, F.; Shi, K.; Jia, Y.-P.; Hao, Y.; Peng, J.; Qian, Z.-Y. Advanced biomaterials for cancer immunotherapy. Acta Pharmacol. Sin. 2020, 41, 911–927. [Google Scholar] [CrossRef]
- Butts, C.; Socinski, M.A.; Mitchell, P.L.; Thatcher, N.; Havel, L.; Krzakowski, M.; Nawrocki, S.; Ciuleanu, T.E.; Bosquée, L.; Trigo, J.M.; et al. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer (START): A randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 59–68. [Google Scholar] [CrossRef]
- Singer, C.F.; Pfeiler, G.; Hubalek, M.; Bartsch, R.; Stöger, H.; Pichler, A.; Petru, E.; Bjelic-Radisic, V.; Greil, R.; Rudas, M.; et al. Efficacy and safety of the therapeutic cancer vaccine tecemotide (L-BLP25) in early breast cancer: Results from a prospective, randomised, neoadjuvant phase II study (ABCSG 34). Eur. J. Cancer 2020, 132, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Mazières, J.; Planchard, D.; Stinchcombe, T.E.; Dy, G.K.; Antonia, S.J.; Horn, L.; Lena, H.; Minenza, E.; Mennecier, B.; et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): A phase 2, single-arm trial. Lancet Oncol. 2015, 16, 257–265. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumai, T.; Yamaki, H.; Kono, M.; Hayashi, R.; Wakisaka, R.; Komatsuda, H. Antitumor Peptide-Based Vaccine in the Limelight. Vaccines 2022, 10, 70. https://doi.org/10.3390/vaccines10010070
Kumai T, Yamaki H, Kono M, Hayashi R, Wakisaka R, Komatsuda H. Antitumor Peptide-Based Vaccine in the Limelight. Vaccines. 2022; 10(1):70. https://doi.org/10.3390/vaccines10010070
Chicago/Turabian StyleKumai, Takumi, Hidekiyo Yamaki, Michihisa Kono, Ryusuke Hayashi, Risa Wakisaka, and Hiroki Komatsuda. 2022. "Antitumor Peptide-Based Vaccine in the Limelight" Vaccines 10, no. 1: 70. https://doi.org/10.3390/vaccines10010070
APA StyleKumai, T., Yamaki, H., Kono, M., Hayashi, R., Wakisaka, R., & Komatsuda, H. (2022). Antitumor Peptide-Based Vaccine in the Limelight. Vaccines, 10(1), 70. https://doi.org/10.3390/vaccines10010070