The Role of Bilirubin and the Other “Yellow Players” in Neurodegenerative Diseases




Abstract
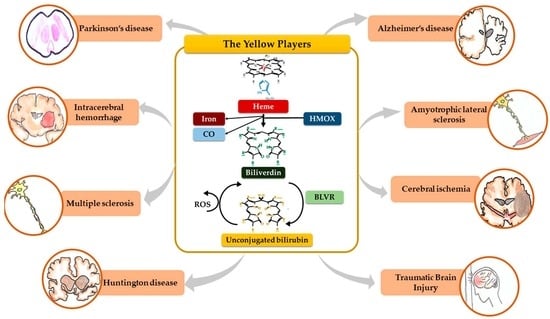
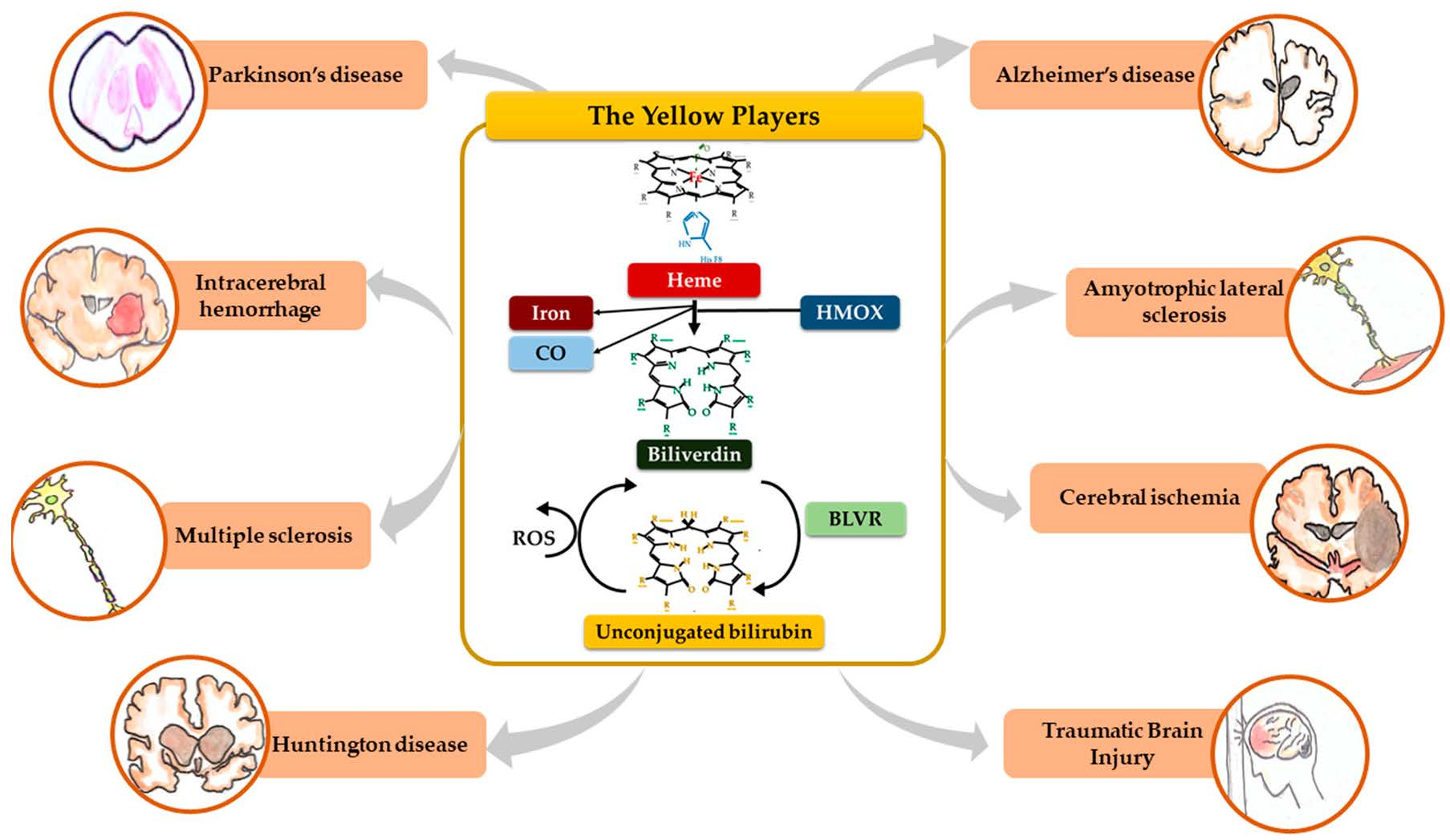
:
1. Introduction
2. The Yellow Players (YP)
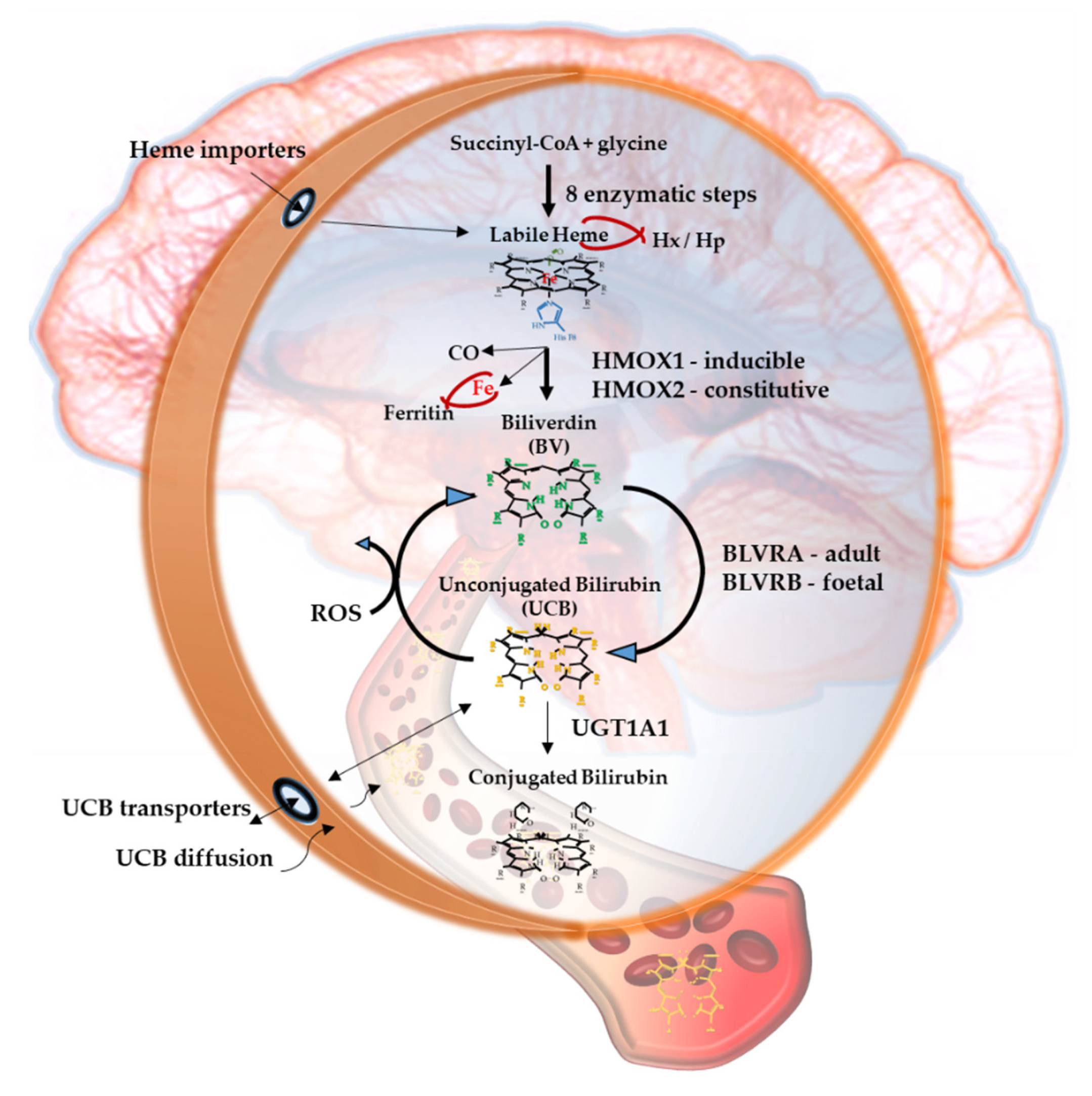
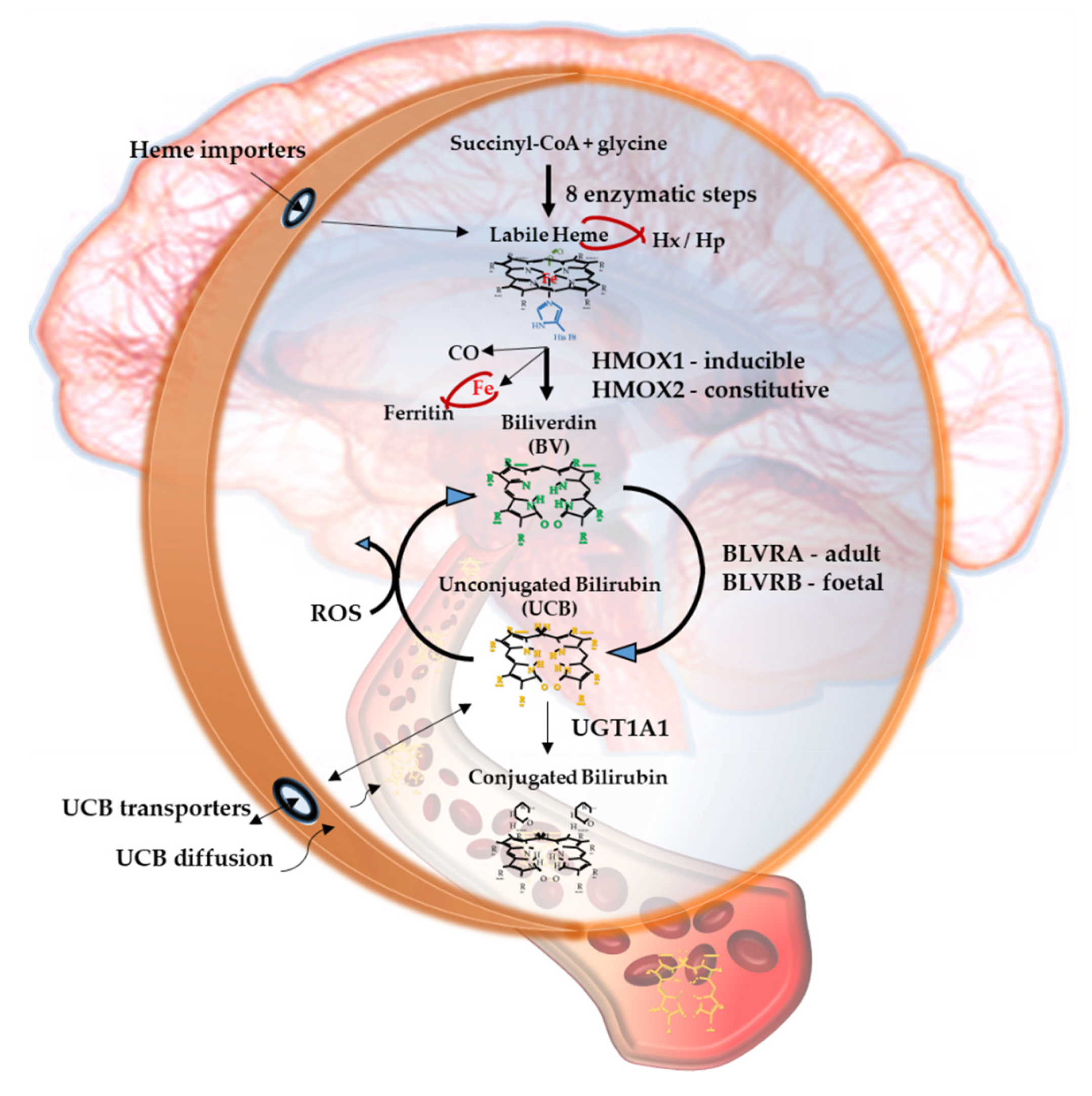
2.1. Heme
2.2. Heme Oxygenase (HMOX), Carbon Monoxide (CO) and Iron
2.3. Biliverdin
2.4. Biliverdin Reductase (BLVR)
2.5. Unconjugated Bilirubin (UCB)
2.6. UCB Degradation Products
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gazzin, S.; Vitek, L.; Watchko, J.; Shapiro, S.M.; Tiribelli, C. A Novel Perspective on the Biology of Bilirubin in Health and Disease. Trends Mol. Med. 2016, 22, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Gazzin, S.; Masutti, F.; Vítek, L.; Tiribelli, C. The molecular basis of jaundice: An old symptom revisited. Liver Int. 2016, 37, 1094–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vítek, L.; Ostrow, J.D. Bilirubin Chemistry and Metabolism; Harmful and Protective Aspects. Available online: https://www.eurekaselect.com/69920/article (accessed on 27 July 2020).
- Le Pichon, J.-B.; Riordan, S.M.; Watchko, J.; Shapiro, S.M. The Neurological Sequelae of Neonatal Hyperbilirubinemia: Definitions, Diagnosis and Treatment of the Kernicterus Spectrum Disorders (KSDs). Curr. Pediatr. Rev. 2017, 13, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.A.; Robinson, D.L.; Vreman, H.J.; Puffenberger, E.G.; Hart, G.; Morton, D.H. Management of hyperbilirubinemia and prevention of kernicterus in 20 patients with Crigler-Najjar disease. Eur. J. Pediatr. 2006, 165, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Watchko, J.F.; Tiribelli, C. Bilirubin-Induced Neurologic Damage—Mechanisms and Management Approaches. N. Engl. J. Med. 2013, 369, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- Diamond, I.D.; Schmid, R.S. Experimental bilirubin encephalopathy. The mode of entry of bilirubin-14C into the central nervous system. J. Clin. Investig. 1966, 45, 678–689. [Google Scholar] [CrossRef] [Green Version]
- Wennberg, R.P.; Ahlfors, C.E.; Bhutani, V.K.; Johnson, L.H.; Shapiro, S.M. Toward Understanding Kernicterus: A Challenge to Improve the Management of Jaundiced Newborns. Pediatrics 2006, 117, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef]
- Baranano, D.E.; Rao, M.; Ferris, C.D.; Snyder, S.H. Biliverdin reductase: A major physiologic cytoprotectant. Proc. Natl. Acad. Sci. USA 2002, 99, 16093–16098. [Google Scholar] [CrossRef] [Green Version]
- Abraham, N.G.; Kappas, A. Pharmacological and Clinical Aspects of Heme Oxygenase. Pharmacol. Rev. 2008, 60, 79–127. [Google Scholar] [CrossRef] [Green Version]
- Gozzelino, R. The Pathophysiology of Heme in the Brain. Available online: https://www.eurekaselect.com/135089/article (accessed on 27 July 2020).
- Maines, M.D. New Insights into Biliverdin Reductase Functions: Linking Heme Metabolism to Cell Signaling. Physiology 2005, 20, 382–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitti, M.; Piras, S.; Brondolo, L.; Marinari, U.M.; Pronzato, M.A.; Furfaro, A.L. Heme Oxygenase 1 in the Nervous System: Does It Favor Neuronal Cell Survival or Induce Neurodegeneration? Int. J. Mol. Sci. 2018, 19, 2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryter, S.W.; Alam, J.; Choi, A.M.K. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Song, W.; Tavitian, A.; Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Prog. Neurobiol. 2019, 172, 40–70. [Google Scholar] [CrossRef]
- Wagner, K.-H.; Wallner, M.; Mölzer, C.; Gazzin, S.; Bulmer, A.C.; Tiribelli, C.; Vitek, L. Looking to the horizon: The role of bilirubin in the development and prevention of age-related chronic diseases. Clin. Sci. 2015, 129, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tu, Y.; Moon, C.; Nagata, E.; Ronnett, G.V. Heme oxygenase-1 and heme oxygenase-2 have distinct roles in the proliferation and survival of olfactory receptor neurons mediated by cGMP and bilirubin, respectively. J. Neurochem. 2003, 85, 1247–1261. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Nam, E.; Lee, H.-K.; Lim, M.H.; Rhee, H.-W. In Cellulo Mapping of Subcellular Localized Bilirubin. ACS Chem. Biol. 2016, 11, 2177–2185. [Google Scholar] [CrossRef]
- Takeda, T.; Mu, A.; Tai, T.T.; Kitajima, S.; Taketani, S. Continuous de novo biosynthesis of haem and its rapid turnover to bilirubin are necessary for cytoprotection against cell damage. Sci. Rep. 2015, 5, 10488. [Google Scholar] [CrossRef] [Green Version]
- Funahashi, A.; Komatsu, M.; Furukawa, T.; Yoshizono, Y.; Yoshizono, H.; Orikawa, Y.; Takumi, S.; Shiozaki, K.; Hayashi, S.; Kaminishi, Y.; et al. Eel green fluorescent protein is associated with resistance to oxidative stress. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2016, 181–182, 35–39. [Google Scholar] [CrossRef]
- Kumagai, A.; Ando, R.; Miyatake, H.; Greimel, P.; Kobayashi, T.; Hirabayashi, Y.; Shimogori, T.; Miyawaki, A. A Bilirubin-Inducible Fluorescent Protein from Eel Muscle. Cell 2013, 153, 1602–1611. [Google Scholar] [CrossRef] [Green Version]
- Vítek, L.; Schwertner, H.A. The heme catabolic pathway and its protective effects on oxidative stress-mediated diseases. Adv. Clin. Chem. 2007, 43, 1–57. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Fiorito, V.; Petrillo, S.; Tolosano, E. Unraveling the Role of Heme in Neurodegeneration. Front. Neurosci. 2018, 12, 712. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Shan, Y.; Tang, Z.; Wu, X.; Bi, C.; Zhang, Y.; Gao, Y.; Liu, H. The Neuroprotective Effect of Hemin and the Related Mechanism in Sevoflurane Exposed Neonatal Rats. Front. Neurosci. 2019, 13, 537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Zhang, Y.; Tang, Z.; Shan, Y.; Wu, X.; Liu, H. Hemin treatment protects neonatal rats from sevoflurane-induced neurotoxicity via the phosphoinositide 3-kinase/Akt pathway. Life Sci. 2020, 242, 117151. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Li, X.; Liu, Y.; Chang, W.; Liu, W.; Yuan, J.; Chen, J. Hemin provides protection against lead neurotoxicity through heme oxygenase 1/carbon monoxide activation. J. Appl. Toxicol. 2018, 38, 1353–1364. [Google Scholar] [CrossRef]
- Dang, T.N.; Robinson, S.R.; Dringen, R.; Bishop, G.M. Uptake, metabolism and toxicity of hemin in cultured neurons. Neurochem. Int. 2011, 58, 804–811. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Mancuso, C.; Butterfield, D.A. The Janus face of the heme oxygenase/biliverdin reductase system in Alzheimer disease: It’s time for reconciliation. Neurobiol. Dis. 2014, 62, 144–159. [Google Scholar] [CrossRef] [Green Version]
- Bulters, D.; Gaastra, B.; Zolnourian, A.; Alexander, S.; Ren, D.; Blackburn, S.L.; Borsody, M.; Doré, S.; Galea, J.; Iihara, K.; et al. Haemoglobin scavenging in intracranial bleeding: Biology and clinical implications. Nat. Rev. Neurol. 2018, 14, 416–432. [Google Scholar] [CrossRef]
- Van Acker, Z.P.; Luyckx, E.; Dewilde, S. Neuroglobin Expression in the Brain: A Story of Tissue Homeostasis Preservation. Mol. Neurobiol. 2019, 56, 2101–2122. [Google Scholar] [CrossRef]
- Khan, A.; Jamwal, S.; Bijjem, K.R.V.; Prakash, A.; Kumar, P. Neuroprotective effect of hemeoxygenase-1/glycogen synthase kinase-3β modulators in 3-nitropropionic acid-induced neurotoxicity in rats. Neuroscience 2015, 287, 66–77. [Google Scholar] [CrossRef]
- Chen, J. Heme oxygenase in neuroprotection: From mechanisms to therapeutic implications. Rev. Neurosci. 2014, 25, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Jazwa, J.A.; Cuadrado, C.A. Targeting Heme Oxygenase-1 for Neuroprotection and Neuroinflammation in Neurodegenerative Diseases. Curr. Drug Targets 2010, 11, 1517–1531. [Google Scholar] [CrossRef]
- Ahmad, A.S.; Zhuang, H.; Doré, S. Heme oxygenase-1 protects brain from acute excitotoxicity. Neuroscience 2006, 141, 1703–1708. [Google Scholar] [CrossRef] [PubMed]
- Colín-González, A.L.; Orozco-Ibarra, M.; Chánez-Cárdenas, M.E.; Rangel-López, E.; Santamaría, A.; Pedraza-Chaverri, J.; Barrera-Oviedo, D.; Maldonado, P.D. Heme oxygenase-1 (HO-1) upregulation delays morphological and oxidative damage induced in an excitotoxic/pro-oxidant model in the rat striatum. Neuroscience 2013, 231, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Ku, B.M.; Joo, Y.; Mun, J.; Roh, G.S.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Kim, H.J. Heme oxygenase protects hippocampal neurons from ethanol-induced neurotoxicity. Neurosci. Lett. 2006, 405, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Orozco-Ibarra, M.; Estrada-Sánchez, A.M.; Massieu, L.; Pedraza-Chaverrí, J. Heme oxygenase-1 induction prevents neuronal damage triggered during mitochondrial inhibition: Role of CO and bilirubin. Int. J. Biochem. Cell Biol. 2009, 41, 1304–1314. [Google Scholar] [CrossRef]
- Sferrazzo, G.; Di Rosa, M.; Barone, E.; Li Volti, G.; Musso, N.; Tibullo, D.; Barbagallo, I. Heme Oxygenase-1 in Central Nervous System Malignancies. J. Clin. Med. 2020, 9, 1562. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Sultana, R.; Coccia, R.; Mancuso, C.; Perluigi, M.; Butterfield, D.A. Heme oxygenase-1 posttranslational modifications in the brain of subjects with Alzheimer disease and mild cognitive impairment. Free Radic. Biol. Med. 2012, 52, 2292–2301. [Google Scholar] [CrossRef] [Green Version]
- Chang, E.F.; Wong, R.J.; Vreman, H.J.; Igarashi, T.; Galo, E.; Sharp, F.R.; Stevenson, D.K.; Noble-Haeusslein, L.J. Heme Oxygenase-2 Protects against Lipid Peroxidation-Mediated Cell Loss and Impaired Motor Recovery after Traumatic Brain Injury. J. Neurosci. 2003, 23, 3689–3696. [Google Scholar] [CrossRef]
- Doré, S.; Snyder, S.H. Neuroprotective action of bilirubin against oxidative stress in primary hippocampal cultures. Ann. N. Y. Acad. Sci. 1999, 890, 167–172. [Google Scholar] [CrossRef]
- Doré, S.; Goto, S.; Sampei, K.; Blackshaw, S.; Hester, L.D.; Ingi, T.; Sawa, A.; Traystman, R.J.; Koehler, R.C.; Snyder, S.H. Heme oxygenase-2 acts to prevent neuronal death in brain cultures and following transient cerebral ischemia. Neuroscience 2000, 99, 587–592. [Google Scholar] [CrossRef]
- Doré, S.; Takahashi, M.; Ferris, C.D.; Hester, L.D.; Guastella, D.; Snyder, S.H. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. USA 1999, 96, 2445–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade, V.M.; Aschner, M.; Marreilha dos Santos, A.P. Neurotoxicity of Metal Mixtures. In Neurotoxicity of Metals; Aschner, M., Costa, L.G., Eds.; Advances in Neurobiology; Springer International Publishing: Cham, Switzerland, 2017; pp. 227–265. ISBN 978-3-319-60189-2. [Google Scholar]
- Schipper, H.M. Brain iron deposition and the free radical-mitochondrial theory of ageing. Ageing Res. Rev. 2004, 3, 265–301. [Google Scholar] [CrossRef]
- Zhang, J.; Piantadosi, C.A. Mitochondrial oxidative stress after carbon monoxide hypoxia in the rat brain. J. Clin. Investig. 1992, 90, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Stockard-Sullivan, J.E.; Korsak, R.A.; Webber, D.S.; Edmond, J. Mild carbon monoxide exposure and auditory function in the developing rat. J. Neurosci. Res. 2003, 74, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Webber, D.S.; Korsak, R.A.; Sininger, L.K.; Sampogna, S.L.; Edmond, J. Mild carbon monoxide exposure impairs the developing auditory system of the rat. J. Neurosci. Res. 2003, 74, 655–665. [Google Scholar] [CrossRef]
- Deguchi, K.; Hayashi, T.; Nagotani, S.; Sehara, Y.; Zhang, H.; Tsuchiya, A.; Ohta, Y.; Tomiyama, K.; Morimoto, N.; Miyazaki, M.; et al. Reduction of cerebral infarction in rats by biliverdin associated with amelioration of oxidative stress. Brain Res. 2008, 1188, 1–8. [Google Scholar] [CrossRef]
- Zou, Z.-Y.; Liu, J.; Chang, C.; Li, J.-J.; Luo, J.; Jin, Y.; Ma, Z.; Wang, T.-H.; Shao, J.-L. Biliverdin administration regulates the microRNA-mRNA expressional network associated with neuroprotection in cerebral ischemia reperfusion injury in rats. Int. J. Mol. Med. 2019, 43, 1356–1372. [Google Scholar] [CrossRef] [Green Version]
- Rice, A.C.; Shapiro, S.M. Biliverdin-induced brainstem auditory evoked potential abnormalities in the jaundiced Gunn rat. Brain Res. 2006, 1107, 215–221. [Google Scholar] [CrossRef]
- Cunningham, O.; Gore, M.G.; Mantle, T.J. Initial-rate kinetics of the flavin reductase reaction catalysed by human biliverdin-IXbeta reductase (BVR-B). Biochem. J. 2000, 345, 393–399. [Google Scholar] [CrossRef]
- Shalloe, F.; Elliott, G.; Ennis, O.; Mantle, T.J. Evidence that biliverdin-IXβ reductase and flavin reductase are identical. Biochem. J. 1996, 316, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Atukeren, P.; Oner, S.; Baran, O.; Kemerdere, R.; Eren, B.; Cakatay, U.; Tanriverdi, T. Oxidant and anti-oxidant status in common brain tumors: Correlation to TP53 and human biliverdin reductase. Clin. Neurol. Neurosurg. 2017, 158, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Tetzlaff, W.; Paty, D.W.; Cynader, M.S. Biliverdin reductase, a major physiologic cytoprotectant, suppresses experimental autoimmune encephalomyelitis. Free Radic. Biol. Med. 2006, 40, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Cini, C.; Preziosi, P.; Perluigi, M.; Mancuso, C.; Butterfield, D.A. Biliverdin reductase--a protein levels and activity in the brains of subjects with Alzheimer disease and mild cognitive impairment. Biochim. Biophys. Acta 2011, 1812, 480–487. [Google Scholar] [CrossRef]
- Di Domenico, F.; Barone, E.; Mancuso, C.; Perluigi, M.; Cocciolo, A.; Mecocci, P.; Butterfield, D.A.; Coccia, R. HO-1/BVR-a system analysis in plasma from probable Alzheimer’s disease and mild cognitive impairment subjects: A potential biochemical marker for the prediction of the disease. J. Alzheimers Dis. 2012, 32, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ding, Y.; Lu, T.; Zhang, Y.; Xu, N.; Yu, L.; McBride, D.W.; Flores, J.J.; Tang, J.; Zhang, J.H. Bliverdin reductase-A improves neurological function in a germinal matrix hemorrhage rat model. Neurobiol. Dis. 2018, 110, 122–132. [Google Scholar] [CrossRef]
- Mueller, C.; Zhou, W.; VanMeter, A.; Heiby, M.; Magaki, S.; Ross, M.M.; Espina, V.; Schrag, M.; Dickson, C.; Liotta, L.A.; et al. The Heme Degradation Pathway is a Promising Serum Biomarker Source for the Early Detection of Alzheimer’s Disease. J. Alzheimer’s Dis. 2010, 19, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Matic, L.P.; Jesus Iglesias, M.; Vesterlund, M.; Lengquist, M.; Hong, M.-G.; Saieed, S.; Sanchez-Rivera, L.; Berg, M.; Razuvaev, A.; Kronqvist, M.; et al. Novel Multiomics Profiling of Human Carotid Atherosclerotic Plaques and Plasma Reveals Biliverdin Reductase B as a Marker of Intraplaque Hemorrhage. JACC Basic Transl. Sci. 2018, 3, 464–480. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, B.; Wang, X.; Luo, L.; Li, P.; Paty, D.W.; Cynader, M.S. Bilirubin as a potent antioxidant suppresses experimental autoimmune encephalomyelitis: Implications for the role of oxidative stress in the development of multiple sclerosis. J. Neuroimmunol. 2003, 139, 27–35. [Google Scholar] [CrossRef]
- Yu, M.; Su, D.; Yang, Y.; Qin, L.; Hu, C.; Liu, R.; Zhou, Y.; Yang, C.; Yang, X.; Wang, G.; et al. D-T7 Peptide-Modified PEGylated Bilirubin Nanoparticles Loaded with Cediranib and Paclitaxel for Antiangiogenesis and Chemotherapy of Glioma. ACS Appl. Mater. Interfaces 2019, 11, 176–186. [Google Scholar] [CrossRef]
- Oda, E.; Kawai, R. A possible cross-sectional association of serum total bilirubin with coronary heart disease and stroke in a Japanese health screening population. Heart Vessels 2012, 27, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, M.; Edelenbos, J.; Doré, S. Bilirubin and Ischemic Stroke: Rendering the Current Paradigm to Better Understand the Protective Effects of Bilirubin. Mol. Neurobiol. 2019, 56, 5483–5496. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.-Y.; Liou, H.-C.; Kang, K.-H.; Wu, R.-M.; Wen, C.-C.; Fu, W.-M. Over-expression of Heme oxygenase-1 protects dopaminergic neurons against 1-methyl-4-phenylpyridinium-induced neurotoxicity. Mol. Pharmacol. 2008, 74, 1564–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Choi, Y.K. Regenerative Effects of Heme Oxygenase Metabolites on Neuroinflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 78. [Google Scholar] [CrossRef] [Green Version]
- Zhong, K.; Wang, X.; Ma, X.; Ji, X.; Sang, S.; Shao, S.; Zhao, Y.; Xiang, Y.; Li, J.; Wang, G.; et al. Association between serum bilirubin and asymptomatic intracranial atherosclerosis: Results from a population-based study. Neurol. Sci. 2020, 41, 1531–1538. [Google Scholar] [CrossRef]
- Yang, F.-C.; Riordan, S.M.; Winter, M.; Gan, L.; Smith, P.G.; Vivian, J.L.; Shapiro, S.M.; Stanford, J.A. Fate of Neural Progenitor Cells Transplanted into Jaundiced and Nonjaundiced Rat Brains. Cell Transpl. 2017, 26, 605–611. [Google Scholar] [CrossRef]
- Loftspring, M.C.; Johnson, H.L.; Feng, R.; Johnson, A.J.; Clark, J.F. Unconjugated Bilirubin Contributes to Early Inflammation and Edema after Intracerebral Hemorrhage. J. Cereb. Blood Flow Metab. 2010, 31, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Marques, J.G.; Pedro, I.; Ouakinin, S. Unconjugated bilirubin and acute psychosis: A five years retrospective observational and controlled study in patients with schizophrenia, schizoaffective and bipolar disorders. Int. J. Psychiatry Clin. Pract. 2019, 23, 281–285. [Google Scholar] [CrossRef]
- Bin-Nun, A.; Mimouni, F.B.; Kasirer, Y.; Schors, I.; Schimmel, M.S.; Kaplan, M.; Hammerman, C. Might Bilirubin Serve as a Natural Antioxidant in Response to Neonatal Encephalopathy? Am. J. Perinatol. 2018, 35, 1107–1112. [Google Scholar] [CrossRef]
- Dani, C.; Poggi, C.; Fancelli, C.; Pratesi, S. Changes in bilirubin in infants with hypoxic–ischemic encephalopathy. Eur. J. Pediatr. 2018, 177, 1795–1801. [Google Scholar] [CrossRef]
- Fereshtehnejad, S.M.; Poorsattar Bejeh Mir, K.; Poorsattar Bejeh Mir, A.; Mohagheghi, P. Evaluation of the possible antioxidative role of bilirubin protecting from free radical related illnesses in neonates. Acta Med. Iran 2012, 50, 153–163. [Google Scholar] [PubMed]
- Fujiwara, R.; Haag, M.; Schaeffeler, E.; Nies, A.T.; Zanger, U.M.; Schwab, M. Systemic regulation of bilirubin homeostasis: Potential benefits of hyperbilirubinemia. Hepatology 2018, 67, 1609–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brites, D. The evolving landscape of neurotoxicity by unconjugated bilirubin: Role of glial cells and inflammation. Front. Pharmacol. 2012, 3, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jašprová, J.; Dal Ben, M.; Hurný, D.; Hwang, S.; Žížalová, K.; Kotek, J.; Wong, R.J.; Stevenson, D.K.; Gazzin, S.; Tiribelli, C.; et al. Neuro-inflammatory effects of photodegradative products of bilirubin. Sci. Rep. 2018, 8, 7444. [Google Scholar] [CrossRef]
- Luan, H.; Liu, L.-F.; Tang, Z.; Mok, V.C.T.; Li, M.; Cai, Z. Elevated excretion of biopyrrin as a new marker for idiopathic Parkinson’s disease. Parkinsonism Relat. Disord. 2015, 21, 1371–1372. [Google Scholar] [CrossRef]
- Alexander, J.; Marcel, R.; Niklas, L.; Andreas, S.R.; Diana, F.; Karl-Heinz, H.; Anna, S.; Marvin, R.; Milena, G.; Charline, S.; et al. Propentdyopents as Heme Degradation Intermediates Constrict Mouse Cerebral Arterioles and Are Present in the Cerebrospinal Fluid of Patients With Subarachnoid Hemorrhage. Circ. Res. 2019, 124, e101–e114. [Google Scholar] [CrossRef]
- Clark, J.F.; Loftspring, M.; Wurster, W.L.; Pyne-Geithman, G.J. Chemical and biochemical oxidations in spinal fluid after subarachnoid hemorrhage. Front. Biosci. 2008, 13, 1806–1812. [Google Scholar] [CrossRef] [Green Version]
- Righy, C.; Bozza, M.T.; Oliveira, M.F.; Bozza, F.A. Molecular, Cellular and Clinical Aspects of Intracerebral Hemorrhage: Are the Enemies Within? Curr. Neuropharmacol. 2016, 14, 392–402. [Google Scholar] [CrossRef] [Green Version]
- Vaya, J.; Song, W.; Khatib, S.; Geng, G.; Schipper, H.M. Effects of heme oxygenase-1 expression on sterol homeostasis in rat astroglia. Free Radic. Biol. Med. 2007, 42, 864–871. [Google Scholar] [CrossRef]
- Lin, W.-P.; Xiong, G.-P.; Lin, Q.; Chen, X.-W.; Zhang, L.-Q.; Shi, J.-X.; Ke, Q.-F.; Lin, J.-H. Heme oxygenase-1 promotes neuron survival through down-regulation of neuronal NLRP1 expression after spinal cord injury. J. Neuroinflamm. 2016, 13, 52. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Doré, S.; Ferris, C.D.; Tomita, T.; Sawa, A.; Wolosker, H.; Borchelt, D.R.; Iwatsubo, T.; Kim, S.-H.; Thinakaran, G.; et al. Amyloid Precursor Proteins Inhibit Heme Oxygenase Activity and Augment Neurotoxicity in Alzheimer’s Disease. Neuron 2000, 28, 461–473. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Tu, Y.; Connolly, E.C.; Ronnett, G.V. Heme oxygenase-2 protects against glutathione depletion-induced neuronal apoptosis mediated by bilirubin and cyclic GMP. Curr. Neurovasc. Res. 2005, 2, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A.I. Heme oxygenase-1 as a therapeutic target in neurodegenerative diseases and brain infections. Curr. Pharm. Des. 2008, 14, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Hettiarachchi, N.; Dallas, M.; Al-Owais, M.; Griffiths, H.; Hooper, N.; Scragg, J.; Boyle, J.; Peers, C. Heme oxygenase-1 protects against Alzheimer’s amyloid-β(1-42)-induced toxicity via carbon monoxide production. Cell Death Dis. 2014, 5, e1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hettiarachchi, N.T.; Boyle, J.P.; Dallas, M.L.; Al-Owais, M.M.; Scragg, J.L.; Peers, C. Heme oxygenase-1 derived carbon monoxide suppresses Aβ1-42 toxicity in astrocytes. Cell Death Dis. 2017, 8, e2884. [Google Scholar] [CrossRef]
- Lin, S.-H.; Song, W.; Cressatti, M.; Zukor, H.; Wang, E.; Schipper, H.M. Heme oxygenase-1 modulates microRNA expression in cultured astroglia: Implications for chronic brain disorders. Glia 2015, 63, 1270–1284. [Google Scholar] [CrossRef]
- Mancuso, C.; Barone, E.; Guido, P.; Miceli, F.; Di Domenico, F.; Perluigi, M.; Santangelo, R.; Preziosi, P. Inhibition of lipid peroxidation and protein oxidation by endogenous and exogenous antioxidants in rat brain microsomes in vitro. Neurosci. Lett. 2012, 518, 101–105. [Google Scholar] [CrossRef]
- Gibbs, P.E.M.; Maines, M.D. Biliverdin inhibits activation of NF-κB: Reversal of inhibition by human biliverdin reductase. Int. J. Cancer 2007, 121, 2567–2574. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bisht, K.; Wegiel, B.; Tampe, J.; Neubauer, O.; Wagner, K.-H.; Otterbein, L.E.; Bulmer, A.C. Biliverdin modulates the expression of C5aR in response to endotoxin in part via mTOR signaling. Biochem. Biophys. Res. Commun. 2014, 449, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Nakao, A.; Murase, N.; Ho, C.; Toyokawa, H.; Billiar, T.R.; Kanno, S. Biliverdin Administration Prevents the Formation of Intimal Hyperplasia Induced by Vascular Injury. Circulation 2005, 112, 587–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegiel, B.; Baty, C.J.; Gallo, D.; Csizmadia, E.; Scott, J.R.; Akhavan, A.; Chin, B.Y.; Kaczmarek, E.; Alam, J.; Bach, F.H.; et al. Cell Surface Biliverdin Reductase Mediates Biliverdin-induced Anti-inflammatory Effects via Phosphatidylinositol 3-Kinase and Akt. J. Biol. Chem. 2009, 284, 21369–21378. [Google Scholar] [CrossRef] [Green Version]
- Wegiel, B.; Gallo, D.; Csizmadia, E.; Roger, T.; Kaczmarek, E.; Harris, C.; Zuckerbraun, B.S.; Otterbein, L.E. Biliverdin inhibits Toll-like receptor-4 (TLR4) expression through nitric oxide-dependent nuclear translocation of biliverdin reductase. Proc. Natl. Acad. Sci. USA 2011, 108, 18849–18854. [Google Scholar] [CrossRef] [Green Version]
- Gurba, P.E.; Zand, R. Bilirubin binding to myelin basic protein, histones and its inhibition in vitro of cerebellar protein synthesis. Biochem. Biophys. Res. Commun. 1974, 58, 1142–1147. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.J.; Braak, E. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J. Neuropathol. Exp. Neurol. 1999, 58, 1010–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, M.; Garrido, J.J.; Wandosell, F.G. Modulation of GSK-3 as a Therapeutic Strategy on Tau Pathologies. Front. Mol. Neurosci. 2011, 4, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miralem, T.; Lerner-Marmarosh, N.; Gibbs, P.E.M.; Jenkins, J.L.; Heimiller, C.; Maines, M.D. Interaction of human biliverdin reductase with Akt/protein kinase B and phosphatidylinositol-dependent kinase 1 regulates glycogen synthase kinase 3 activity: A novel mechanism of Akt activation. FASEB J. 2016, 30, 2926–2944. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Tramutola, A.; Lanzillotta, C.; Arena, A.; Blarzino, C.; Cassano, T.; Butterfield, D.A.; Di Domenico, F.; Perluigi, M.; Barone, E. Loss of biliverdin reductase-A favors Tau hyper-phosphorylation in Alzheimer’s disease. Neurobiol. Dis. 2019, 125, 176–189. [Google Scholar] [CrossRef]
- Kim, S.J.; Shin, M.J.; Kim, D.W.; Yeo, H.J.; Yeo, E.J.; Choi, Y.J.; Sohn, E.J.; Han, K.H.; Park, J.; Lee, K.W.; et al. Tat-Biliverdin Reductase A Exerts a Protective Role in Oxidative Stress-Induced Hippocampal Neuronal Cell Damage by Regulating the Apoptosis and MAPK Signaling. Int. J. Mol. Sci. 2020, 21, 2672. [Google Scholar] [CrossRef]
- Triani, F.; Tramutola, A.; Di Domenico, F.; Sharma, N.; Butterfield, D.A.; Head, E.; Perluigi, M.; Barone, E. Biliverdin reductase-A impairment links brain insulin resistance with increased Aβ production in an animal model of aging: Implications for Alzheimer disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 3181–3194. [Google Scholar] [CrossRef]
- Mancuso, C. Bilirubin and brain: A pharmacological approach. Neuropharmacology 2017, 118, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di Domenico, F.; Cassano, T.; Arena, A.; Tramutola, A.; Lavecchia, M.A.; Coccia, R.; Butterfield, D.A.; Perluigi, M. Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: A new paradigm. Free Radic. Biol. Med. 2016, 91, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Puri, B.K.; Walker, A.J.; Berk, M.; Walder, K.; Bortolasci, C.C.; Marx, W.; Carvalho, A.F.; Maes, M. The compensatory antioxidant response system with a focus on neuroprogressive disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 95, 109708. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, P.; Lu, J.; Xiong, W.; Oger, J.; Tetzlaff, W.; Cynader, M. Bilirubin Possesses Powerful Immunomodulatory Activity and Suppresses Experimental Autoimmune Encephalomyelitis. J. Immunol. 2008, 181, 1887–1897. [Google Scholar] [CrossRef] [Green Version]
- Vianello, E.; Zampieri, S.; Marcuzzo, T.; Tordini, F.; Bottin, C.; Dardis, A.; Zanconati, F.; Tiribelli, C.; Gazzin, S. Histone acetylation as a new mechanism for bilirubin-induced encephalopathy in the Gunn rat. Sci. Rep. 2018, 8, 13690. [Google Scholar] [CrossRef]
- Qaisiya, M.; Brischetto, C.; Jašprová, J.; Vitek, L.; Tiribelli, C.; Bellarosa, C. Bilirubin-induced ER stress contributes to the inflammatory response and apoptosis in neuronal cells. Arch. Toxicol. 2017, 91, 1847–1858. [Google Scholar] [CrossRef]
- Qaisiya, M.; Coda Zabetta, C.D.; Bellarosa, C.; Tiribelli, C. Bilirubin mediated oxidative stress involves antioxidant response activation via Nrf2 pathway. Cell. Signal. 2014, 26, 512–520. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Hanieh, H.; Nakahama, T.; Kishimoto, T. The roles of aryl hydrocarbon receptor in immune responses. Int. Immunol. 2013, 25, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Phelan, D.; Winter, G.M.; Rogers, W.J.; Lam, J.C.; Denison, M.S. Activation of the Ah Receptor Signal Transduction Pathway by Bilirubin and Biliverdin. Arch. Biochem. Biophys. 1998, 357, 155–163. [Google Scholar] [CrossRef]
- Vítek, V.L. Bilirubin as a signaling molecule. Med. Res. Rev. 2020, 40, 1335–1351. [Google Scholar] [CrossRef]
- Datla Srinivasa, R.; Dusting Gregory, J.; Mori Trevor, A.; Taylor Caroline, J.; Croft Kevin, D. Jiang Fan Induction of Heme Oxygenase-1 In Vivo Suppresses NADPH Oxidase–Derived Oxidative Stress. Hypertension 2007, 50, 636–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, F.; Deng, X.; Yu, Y.; Chen, X.; Shen, L.; Zhong, X.; Qiu, W.; Jiang, Y.; Zhang, J.; Hu, X. Serum bilirubin concentrations and multiple sclerosis. J. Clin. Neurosci. 2011, 18, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Gennuso, F.; Fernetti, C.; Tirolo, C.; Testa, N.; L’Episcopo, F.; Caniglia, S.; Morale, M.C.; Ostrow, J.D.; Pascolo, L.; Tiribelli, C.; et al. Bilirubin protects astrocytes from its own toxicity by inducing up-regulation and translocation of multidrug resistance-associated protein 1 (Mrp1). Proc. Natl. Acad. Sci. USA 2004, 101, 2470–2475. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.M.P.; Solá, S.; Brito, M.A.; Brites, D.; Moura, J.J.G. Bilirubin directly disrupts membrane lipid polarity and fluidity, protein order, and redox status in rat mitochondria. J. Hepatol. 2002, 36, 335–341. [Google Scholar] [CrossRef]
- Rodrigues, C.M.P.; Solá, S.; Brites, D. Bilirubin induces apoptosis via the mitochondrial pathway in developing rat brain neurons. Hepatology 2002, 35, 1186–1195. [Google Scholar] [CrossRef]
- Fernandes, A.; Falcão, A.S.; Silva, R.F.M.; Gordo, A.C.; Gama, M.J.; Brito, M.A.; Brites, D. Inflammatory signalling pathways involved in astroglial activation by unconjugated bilirubin. J. Neurochem. 2006, 96, 1667–1679. [Google Scholar] [CrossRef]
- Fernandes, A.; Falcão, A.S.; Silva, R.F.M.; Brito, M.A.; Brites, D. MAPKs are key players in mediating cytokine release and cell death induced by unconjugated bilirubin in cultured rat cortical astrocytes. Eur. J. Neurosci. 2007, 25, 1058–1068. [Google Scholar] [CrossRef]
- Chang, F.-Y.; Lee, C.-C.; Huang, C.-C.; Hsu, K.-S. Unconjugated Bilirubin Exposure Impairs Hippocampal Long-Term Synaptic Plasticity. PLoS ONE 2009, 4, e5876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grojean, S.; Koziel, V.; Vert, P.; Daval, J.L. Bilirubin induces apoptosis via activation of NMDA receptors in developing rat brain neurons. Exp. Neurol. 2000, 166, 334–341. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, W.; Tanswell, A.K.; Luo, X. The Effects of Bilirubin on Evoked Potentials and Long-Term Potentiation in Rat Hippocampus In Vivo. Pediatric Res. 2003, 53, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, C.; Capone, C.; Ranieri, S.C.; Fusco, S.; Calabrese, V.; Eboli, M.L.; Preziosi, P.; Galeotti, T.; Pani, G. Bilirubin as an endogenous modulator of neurotrophin redox signaling. J. Neurosci. Res. 2008, 86, 2235–2249. [Google Scholar] [CrossRef] [PubMed]
- Gazzin, S.; Berengeno, A.L.; Strazielle, N.; Fazzari, F.; Raseni, A.; Ostrow, J.D.; Wennberg, R.; Ghersi-Egea, J.-F.; Tiribelli, C. Modulation of Mrp1 (ABCc1) and Pgp (ABCb1) by Bilirubin at the Blood-CSF and Blood-Brain Barriers in the Gunn Rat. PLoS ONE 2011, 6, e16165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawat, V.; Bortolussi, G.; Gazzin, S.; Tiribelli, C.; Muro, A.F. Bilirubin-Induced Oxidative Stress Leads to DNA Damage in the Cerebellum of Hyperbilirubinemic Neonatal Mice and Activates DNA Double-Strand Break Repair Pathways in Human Cells. Oxid. Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Robert, M.C.; Furlan, G.; Rosso, N.; Gambaro, S.E.; Apitsionak, F.; Vianello, E.; Tiribelli, C.; Gazzin, S. Alterations in the Cell Cycle in the Cerebellum of Hyperbilirubinemic Gunn Rat: A Possible Link with Apoptosis? PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Neis, V.B.; Rosa, P.B.; Moretti, M.; Rodrigues, A.L.S. Involvement of Heme Oxygenase-1 in Neuropsychiatric and Neurodegenerative Diseases. Curr. Pharm. Des. 2018, 24, 2283–2302. [Google Scholar] [CrossRef]
- Schipper, H.M. Heme oxygenase expression in human central nervous system disorders. Free Radic. Biol. Med. 2004, 37, 1995–2011. [Google Scholar] [CrossRef]
- González-Reyes, S.; Orozco-Ibarra, M.; Guzmán-Beltrán, S.; Molina-Jijón, E.; Massieu, L.; Pedraza-Chaverri, J. Neuroprotective role of heme-oxygenase 1 against iodoacetate-induced toxicity in rat cerebellar granule neurons: Role of bilirubin. Free Radic. Res. 2009, 43, 214–223. [Google Scholar] [CrossRef]
- Schipper, H.M. Heme oxygenase-1: Role in brain aging and neurodegeneration. Exp. Gerontol. 2000, 35, 821–830. [Google Scholar] [CrossRef]
- Doré, S.; Sampei, K.; Goto, S.; Alkayed, N.J.; Guastella, D.; Blackshaw, S.; Gallagher, M.; Traystman, R.J.; Hurn, P.D.; Koehler, R.C.; et al. Heme oxygenase-2 is neuroprotective in cerebral ischemia. Mol. Med. 1999, 5, 656–663. [Google Scholar]
- Nam, J.; Lee, Y.; Yang, Y.; Jeong, S.; Kim, W.; Yoo, J.-W.; Moon, J.-O.; Lee, C.; Chung, H.Y.; Kim, M.-S.; et al. Is it worth expending energy to convert biliverdin into bilirubin? Free Radic. Biol. Med. 2018, 124, 232–240. [Google Scholar] [CrossRef]
- Mancuso, C.; Barone, E. The Heme Oxygenase/Biliverdin Reductase Pathway in Drug Research and Development. Available online: https://www.eurekaselect.com/70167/article (accessed on 27 July 2020).
- Maines, M.D. The Heme Oxygenase System: A Regulator of Second Messenger Gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, A.F.; Palma, L.A.; Schmid, R. Reduction of biliverdin and placental transfer of bilirubin and biliverdin in the pregnant guinea pig. Biochem. J. 1981, 194, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, S.; Kondo, M.; Imai, T.; Kusaka, T.; Isobe, K.; Onishi, S. Relationships between serum (ZZ)-bilirubin, its subfractions and biliverdin concentrations in infants at 1-month check-ups. Ann. Clin. Biochem. 2001, 38, 323–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei-Wei, C.; Zhang, X.; Wen-Juan, H. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [Green Version]
- Azam, S.; Jakaria, M.; Kim, I.-S.; Kim, J.; Haque, M.E.; Choi, D.-K. Regulation of Toll-Like Receptor (TLR) Signaling Pathway by Polyphenols in the Treatment of Age-Linked Neurodegenerative Diseases: Focus on TLR4 Signaling. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Cao, C.-X.; Yang, Q.-W.; Lv, F.-L.; Cui, J.; Fu, H.-B.; Wang, J.-Z. Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochem. Biophys. Res. Commun. 2007, 353, 509–514. [Google Scholar] [CrossRef]
- Lotz, M.; Ebert, S.; Esselmann, H.; Iliev, A.I.; Prinz, M.; Wiazewicz, N.; Wiltfang, J.; Gerber, J.; Nau, R. Amyloid beta peptide 1–40 enhances the action of Toll-like receptor-2 and -4 agonists but antagonizes Toll-like receptor-9-induced inflammation in primary mouse microglial cell cultures. J. Neurochem. 2005, 94, 289–298. [Google Scholar] [CrossRef]
- Mellanby, R.J.; Cambrook, H.; Turner, D.G.; O’Connor, R.A.; Leech, M.D.; Kurschus, F.C.; MacDonald, A.S.; Arnold, B.; Anderton, S.M. TLR-4 ligation of dendritic cells is sufficient to drive pathogenic T cell function in experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2012, 9, 248. [Google Scholar] [CrossRef] [Green Version]
- Minoretti, P.; Gazzaruso, C.; Vito, C.D.; Emanuele, E.; Bianchi, M.; Coen, E.; Reino, M.; Geroldi, D. Effect of the functional toll-like receptor 4 Asp299Gly polymorphism on susceptibility to late-onset Alzheimer’s disease. Neurosci. Lett. 2006, 391, 147–149. [Google Scholar] [CrossRef]
- Noelker, C.; Morel, L.; Lescot, T.; Osterloh, A.; Alvarez-Fischer, D.; Breloer, M.; Henze, C.; Depboylu, C.; Skrzydelski, D.; Michel, P.P.; et al. Toll like receptor 4 mediates cell death in a mouse MPTP model of Parkinson disease. Sci. Rep. 2013, 3, 1393. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.; Letiembre, M.; Liu, Y.; Heine, H.; Penke, B.; Hao, W.; Bode, B.; Manietta, N.; Walter, J.; Schulz-Schüffer, W.; et al. Role of the Toll-Like Receptor 4 in Neuroinflammation in Alzheimer’s Disease. CPB 2007, 20, 947–956. [Google Scholar] [CrossRef]
- Ager, R.R.; Fonseca, M.I.; Chu, S.-H.; Sanderson, S.D.; Taylor, S.M.; Woodruff, T.M.; Tenner, A.J. Microglial C5aR (CD88) expression correlates with amyloid-β deposition in murine models of Alzheimer’s disease. J. Neurochem. 2010, 113, 389–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, X.; Xi, W.; Gu, C.; Huang, X. Complement protein C5a enhances the β-amyloid-induced neuro-inflammatory response in microglia in Alzheimer’s disease. Med. Sci. (Paris) 2018, 34, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Nizami, S.; Hall-Roberts, H.; Warrier, S.; Cowley, S.A.; Daniel, E.D. Microglial inflammation and phagocytosis in Alzheimer’s disease: Potential therapeutic targets. Br. J. Pharmacol. 2019, 176, 3515–3532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, H.; Hughes, M.N.; Green, C.J.; Naughton, P.; Foresti, R.; Motterlini, R. Interaction of bilirubin and biliverdin with reactive nitrogen species. FEBS Lett. 2003, 543, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Sanchez, E.; Perez, M.J.; Nytofte, N.S.; Briz, O.; Monte, M.J.; Lozano, E.; Serrano, M.A.; Marin, J.J.G. Protective role of biliverdin against bile acid-induced oxidative stress in liver cells. Free Radic. Biol. Med. 2016, 97, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, S.G.; Stucker, T.; Rasmussen, R.D.; Ikeda, R.M.; Ruebner, B.H.; Bergstrom, D.E.; Hanson, F.W. Changes in bilirubins in human prenatal development. Biochem. J. 1980, 186, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Komuro, A.; Tobe, T.; Nakano, Y.; Yamaguchi, T.; Tomita, M. Cloning and characterization of the cDNA encoding human biliverdin-IX alpha reductase. Biochim. Biophys. Acta 1996, 1309, 89–99. [Google Scholar] [CrossRef]
- Kapitulnik, J.; Maines, M.D. Pleiotropic functions of biliverdin reductase: Cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Trends Pharmacol. Sci. 2009, 30, 129–137. [Google Scholar] [CrossRef]
- O’Brien, L.; Hosick, P.A.; John, K.; Stec, D.E.; Hinds, T.D. Biliverdin reductase isozymes in metabolism. Trends Endocrinol. Metab. 2015, 26, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Tudor, C.; Lerner-Marmarosh, N.; Engelborghs, Y.; Gibbs, P.E.M.; Maines, M.D. Biliverdin reductase is a transporter of haem into the nucleus and is essential for regulation of HO-1 gene expression by haematin. Biochem. J. 2008, 413, 405–416. [Google Scholar] [CrossRef] [Green Version]
- Derada Troletti, C.; de Goede, P.; Kamermans, A.; de Vries, H.E. Molecular alterations of the blood–brain barrier under inflammatory conditions: The role of endothelial to mesenchymal transition. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Galaris, D.; Pantopoulos, K. Oxidative Stress and Iron Homeostasis: Mechanistic and Health Aspects. Crit. Rev. Clin. Lab. Sci. 2008, 45, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Jazwa, A.; Grochot-Przeczek, A.; Rutkowski, A.J.; Cisowski, J.; Agarwal, A.; Jozkowicz, A.; Dulak, J. Heme Oxygenase-1 and the Vascular Bed: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal. 2008, 10, 1767–1812. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [Green Version]
- Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Coccia, R.; Preziosi, P.; Perluigi, M.; Mancuso, C.; Butterfield, D.A. Oxidative and Nitrosative Modifications of Biliverdin Reductase-A in the Brain of Subjects with Alzheimer’s Disease and Amnestic Mild Cognitive Impairment. J. Alzheimer’s Dis. 2011, 25, 623–633. [Google Scholar] [CrossRef] [Green Version]
- Lerner-Marmarosh, N.; Shen, J.; Torno, M.D.; Kravets, A.; Hu, Z.; Maines, M.D. Human biliverdin reductase: A member of the insulin receptor substrate family with serine/threonine/tyrosine kinase activity. Proc. Natl. Acad. Sci. USA 2005, 102, 7109–7114. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, P.E.M.; Lerner-Marmarosh, N.; Poulin, A.; Farah, E.; Maines, M.D. Human biliverdin reductase-based peptides activate and inhibit glucose uptake through direct interaction with the kinase domain of insulin receptor. FASEB J. 2014, 28, 2478–2491. [Google Scholar] [CrossRef] [Green Version]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; de la Monte, S.M. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J. Alzheimers Dis. 2005, 8, 247–268. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [Green Version]
- Talbot, K.; Wang, H.-Y.; Kazi, H.; Han, L.-Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [Green Version]
- Barone, E.; Tramutola, A.; Triani, F.; Calcagnini, S.; Di Domenico, F.; Ripoli, C.; Gaetani, S.; Grassi, C.; Butterfield, D.A.; Cassano, T.; et al. Biliverdin Reductase-A Mediates the Beneficial Effects of Intranasal Insulin in Alzheimer Disease. Mol. Neurobiol. 2019, 56, 2922–2943. [Google Scholar] [CrossRef]
- Stocker, R. Antioxidant Activities of Bile Pigments. Antioxid. Redox Signal. 2004, 6, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Sedlak, T.W.; Saleh, M.; Higginson, D.S.; Paul, B.D.; Juluri, K.R.; Snyder, S.H. Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc. Natl. Acad. Sci. USA 2009, 106, 5171–5176. [Google Scholar] [CrossRef] [Green Version]
- Vasavda, C.; Kothari, R.; Malla, A.P.; Tokhunts, R.; Lin, A.; Ji, M.; Ricco, C.; Xu, R.; Saavedra, H.G.; Sbodio, J.I.; et al. Bilirubin Links Heme Metabolism to Neuroprotection by Scavenging Superoxide. Cell Chem. Biol. 2019, 26, 1450–1460.e7. [Google Scholar] [CrossRef] [PubMed]
- Jayanti, S.; Moretti, R.; Tiribelli, C.; Gazzin, S. Bilirubin and inflammation in neurodegenerative and other neurological diseases. Neuroimmunol. Neuroinflamm. 2020, 7, 92–108. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, S.; Lee, D.Y.; Yu, B.; Miao, W.; Jon, S. Multistimuli-Responsive Bilirubin Nanoparticles for Anticancer Therapy. Angew. Chem. Int. Ed. 2016, 55, 10676–10680. [Google Scholar] [CrossRef]
- Aycicek, A.; Erel, O. Total oxidant/antioxidant status in jaundiced newborns before and after phototherapy. J. Pediatr. 2007, 83, 319–322. [Google Scholar] [CrossRef]
- Araújo, A.R.; Rosso, N.; Bedogni, G.; Tiribelli, C.; Bellentani, S. Global epidemiology of non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: What we need in the future. Liver Int. 2018, 38 (Suppl. 1), 47–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, H.; Golabi, P.; Younossi, Z.M. Pediatric Non-Alcoholic Fatty Liver Disease. Children (Basel) 2017, 4, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, R.; Caruso, P.; Gazzin, S. Non-alcoholic fatty liver disease and neurological defects. Ann. Hepatol. 2019, 18, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; Fargion, S.; Fracanzani, A.L. Brain involvement in non-alcoholic fatty liver disease (NAFLD): A systematic review. Dig. Liver Dis. 2019, 51, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.; Brace, C.S.; Ben-Josef, G.; West, T.; Wozniak, D.F.; Holtzman, D.M.; Herzog, E.D.; Imai, S. SIRT1 Promotes the Central Adaptive Response to Diet Restriction through Activation of the Dorsomedial and Lateral Nuclei of the Hypothalamus. J. Neurosci. 2010, 30, 10220–10232. [Google Scholar] [CrossRef]
- Moraes, D.S.; Moreira, D.C.; Andrade, J.M.O.; Santos, S.H.S. Sirtuins, brain and cognition: A review of resveratrol effects. IBRO Rep. 2020, 9, 46–51. [Google Scholar] [CrossRef]
- Radak, Z.; Suzuki, K.; Posa, A.; Petrovszky, Z.; Koltai, E.; Boldogh, I. The systemic role of SIRT1 in exercise mediated adaptation. Redox Biol. 2020, 35, 101467. [Google Scholar] [CrossRef]
- Jang, B.K. Elevated serum bilirubin levels are inversely associated with nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2012, 18, 357–359. [Google Scholar] [CrossRef]
- Kumar, R.; Rastogi, A.; Maras, J.S.; Sarin, S.K. Unconjugated hyperbilirubinemia in patients with non-alcoholic fatty liver disease: A favorable endogenous response. Clin. Biochem. 2012, 45, 272–274. [Google Scholar] [CrossRef]
- Lin, L.-Y.; Kuo, H.-K.; Hwang, J.-J.; Lai, L.-P.; Chiang, F.-T.; Tseng, C.-D.; Lin, J.-L. Serum bilirubin is inversely associated with insulin resistance and metabolic syndrome among children and adolescents. Atherosclerosis 2009, 203, 563–568. [Google Scholar] [CrossRef]
- Puri, K.; Nobili, V.; Melville, K.; Corte, C.D.; Sartorelli, M.R.; Lopez, R.; Feldstein, A.E.; Alkhouri, N. Serum bilirubin level is inversely associated with nonalcoholic steatohepatitis in children. J. Pediatr. Gastroenterol. Nutr. 2013, 57, 114–118. [Google Scholar] [CrossRef]
- Herskovits, A.Z.; Guarente, L. SIRT1 in neurodevelopment and brain senescence. Neuron 2014, 81, 471–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekaran, K.; Salimian, M.; Konduru, S.R.; Choi, J.; Kumar, P.; Long, A.; Klimova, N.; Ho, C.-Y.; Kristian, T.; Russell, J.W. Overexpression of Sirtuin 1 protein in neurons prevents and reverses experimental diabetic neuropathy. Brain 2019, 142, 3737–3752. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Ibdah, J.A. Sirtuins and nonalcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 10084–10092. [Google Scholar] [CrossRef] [PubMed]
- Vítek, L.; Majer, F.; Muchová, L.; Zelenka, J.; Jirásková, A.; Branný, P.; Malina, J.; Ubik, K. Identification of bilirubin reduction products formed by Clostridium perfringens isolated from human neonatal fecal flora. J. Chromatogr. B 2006, 833, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Sedlak, T.; Snyder, S. Bilirubin Benefits: Cellular Protection by a Biliverdin Reductase Antioxidant Cycle. Pediatrics 2004, 113, 1776–1782. [Google Scholar] [CrossRef] [PubMed]
- Jasprova, J.; Dal Ben, M.; Vianello, E.; Goncharova, I.; Urbanova, M.; Vyroubalova, K.; Gazzin, S.; Tiribelli, C.; Sticha, M.; Cerna, M.; et al. The Biological Effects of Bilirubin Photoisomers. PLoS ONE 2016, 11, e0148126. [Google Scholar] [CrossRef]
- Garcia, E.; Aguilar-Cevallos, J.; Silva-Garcia, R.; Ibarra, A. Cytokine and Growth Factor Activation In Vivo and In Vitro after Spinal Cord Injury. Available online: https://www.hindawi.com/journals/mi/2016/9476020/ (accessed on 27 July 2020).
- Kempuraj, D.; Thangavel, R.; Natteru, P.; Selvakumar, G.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar] [PubMed]
- Dietzschold, B.; Richt, J.A. (Eds.) Protective and Pathological Immune Responses in the CNS; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2002; ISBN 978-3-540-42668-4. [Google Scholar]
- Hirota, H. Accelerated Nerve Regeneration in Mice by upregulated expression of interleukin (IL) 6 and IL-6 receptor after trauma. J. Exp. Med. 1996, 183, 2627–2634. [Google Scholar] [CrossRef] [Green Version]
- Hagman, S.; Mäkinen, A.; Ylä-Outinen, L.; Huhtala, H.; Elovaara, I.; Narkilahti, S. Effects of inflammatory cytokines IFN-γ, TNF-α and IL-6 on the viability and functionality of human pluripotent stem cell-derived neural cells. J. Neuroimmunol. 2019, 331, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Vodret, S.; Bortolussi, G.; Jašprová, J.; Vitek, L.; Muro, A.F. Inflammatory signature of cerebellar neurodegeneration during neonatal hyperbilirubinemia in Ugt1 (-/-) mouse model. J. Neuroinflamm. 2017, 14, 64. [Google Scholar] [CrossRef] [Green Version]
- Vodret, S.; Bortolussi, G.; Iaconcig, A.; Martinelli, E.; Tiribelli, C.; Muro, A.F. Attenuation of neuro-inflammation improves survival and neurodegeneration in a mouse model of severe neonatal hyperbilirubinemia. Brain Behav. Immun. 2018, 70, 166–178. [Google Scholar] [CrossRef]
- Shimoharada, K.; Inoue, S.; Nakahara, M.; Kanzaki, N.; Shimizu, S.; Kang, D.; Hamasaki, N.; Kinoshita, S. Urine Concentration of Biopyrrins: A New Marker for Oxidative Stress in Vivo. Clin. Chem. 1998, 44, 2554–2555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vítek, L.; Kráslová, I.; Muchová, L.; Novotný, L.; Yamaguchi, T. Urinary excretion of oxidative metabolites of bilirubin in subjects with Gilbert syndrome. J. Gastroenterol. Hepatol. 2007, 22, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Jašprová, J.; Dvořák, A.; Vecka, M.; Leníček, M.; Lacina, O.; Valášková, P.; Zapadlo, M.; Plavka, R.; Klán, P.; Vítek, L. A novel accurate LC-MS/MS method for quantitative determination of Z-lumirubin. Sci. Rep. 2020, 10, 4411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ndayisaba, A.; Kaindlstorfer, C.; Wenning, G.K. Iron in Neurodegeneration—Cause or Consequence? Front. Neurosci. 2019, 13, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-J.; Zou, Z.-Y.; Liu, J.; Xiong, L.-L.; Jiang, H.-Y.; Wang, T.-H.; Shao, J.-L. Biliverdin administration ameliorates cerebral ischemia reperfusion injury in rats and is associated with proinflammatory factor downregulation. Exp. Ther. Med. 2017, 14, 671–679. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Gu, R.; Hu, W.; Sun, Z.; Wang, G.; Wang, L.; Xu, Y. Upregulation of heme oxygenase-1 protected against brain damage induced by transient cerebral ischemia-reperfusion injury in rats. Exp. Ther. Med. 2018, 15, 4629–4636. [Google Scholar] [CrossRef]
- Pehar, M.; Vargas, M.R.; Cassina, P.; Barbeito, A.G.; Beckman, J.S.; Barbeito, L. Complexity of Astrocyte-Motor Neuron Interactions in Amyotrophic Lateral Sclerosis. NDD 2005, 2, 139–146. [Google Scholar] [CrossRef]
- van Horssen, J.; Schreibelt, G.; Drexhage, J.; Hazes, T.; Dijkstra, C.D.; van der Valk, P.; de Vries, H.E. Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radic. Biol. Med. 2008, 45, 1729–1737. [Google Scholar] [CrossRef]
- Parfenova, H.; Leffler, C.W. Cerebroprotective functions of HO-2. Curr. Pharm. Des. 2008, 14, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Gandini, N.A.; Fermento, M.E.; Salomón, D.G.; Obiol, D.J.; Andrés, N.C.; Zenklusen, J.C.; Arevalo, J.; Blasco, J.; López Romero, A.; Facchinetti, M.M.; et al. Heme oxygenase-1 expression in human gliomas and its correlation with poor prognosis in patients with astrocytoma. Tumor Biol. 2014, 35, 2803–2815. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
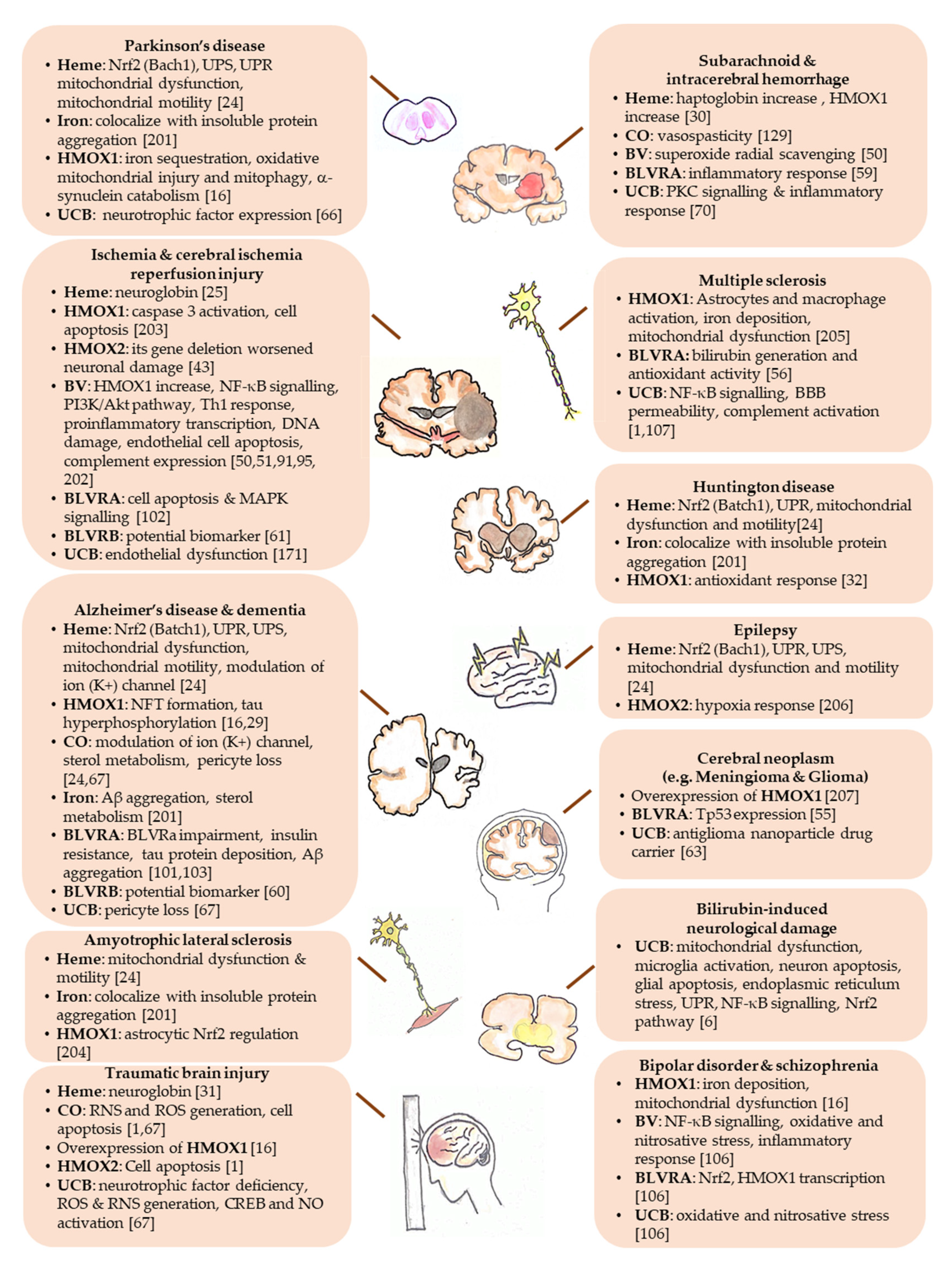
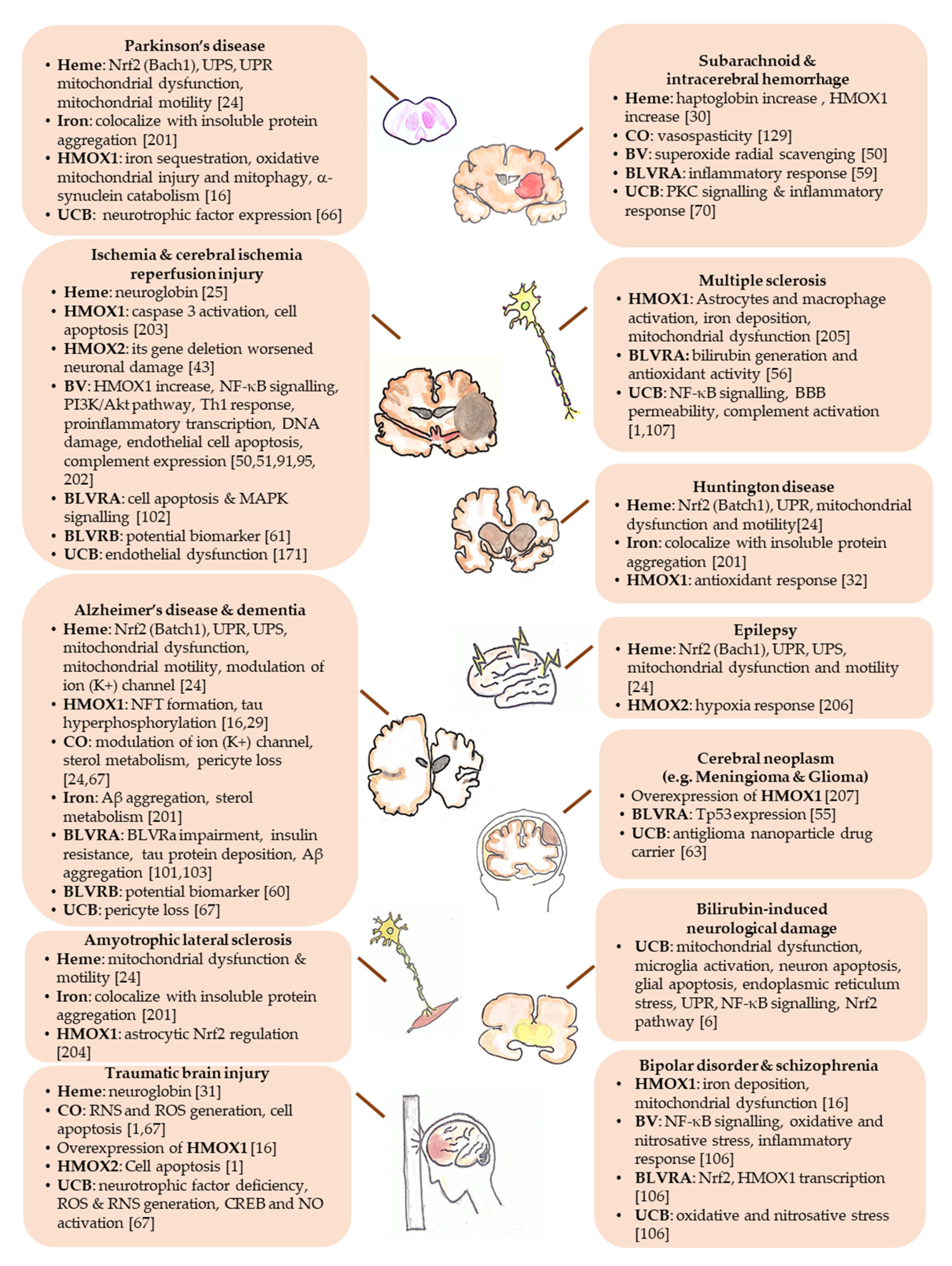
| Yellow Players | Pathological Condition | Ref. |
|---|---|---|
| Heme | Essential for oxygen storage, neurogenesis, cell survival, differentiation, circadian rhythms regulation, cellular energy production, gene and micro RNA (miRNA) processing. | [12,24] |
| Accumulating in the brain in course of hemorrhage, traumatic brain injury, stroke, ischemia, and diseases with increased BBB permeability (such as Parkinson’s, Alzheimer’s, and Huntington’s disease). | [12] | |
| Neuroprotective against xenobiotic toxicity. | [25,26,27] | |
| Neuroprotective against ischemic, traumatic and neurodegenerative insults, by inducing neuroglobin. | [31] | |
| Protective in a pharmacological model of Huntington’s disease. | [32] | |
| Contributing to the progression of brain diseases (such as intracerebral/subarachnoid hemorrhage; neuropathic porphyria; Friedreich ataxia; posterior column ataxia, retinitis pigmentosa, hereditary sensory and autonomic neuropathy). | [12,24] | |
| Neurotoxic in brain hemorrhage. | [30] | |
| HMOX1/2 | ||
| HMOX1 | Protective in neurodegenerative and other neurological diseases (such as Alzheimer’s and Parkinson’s disease, ischemic brain injury). | [33,34] |
| Protective against glutamatergic/aspartatergic excitotoxicity. | [35,36] | |
| Protective against ethanol-induced neurotoxicity. | [37] | |
| Protective against mitochondrial toxicity. | [32,38] | |
| Protective in a pharmacological model of Huntington’s disease. | [32] | |
| Reduces the progression of neuropsychiatric syndrome. | [12] | |
| Protective against ROS via the production of bilirubin from heme. | [3] | |
| Depending on specific conditions, inducing apoptosis and cell cycle arrest (thus being protective), but also capable of increasing chemoresistance and worsening the diagnosis in CNS malignancies. | [39] | |
| Involved in neurodegeneration (such as cerebral infarction; Alzheimer’s and Parkinson’s disease, Down syndrome, schizophrenia; stroke and CNS trauma) and brain aging when excessively expressed. | [16,40] | |
| HMOX2 | Protective from cerebral ischemia-reperfusion injury; and traumatic brain injury. | [12,41,42,43] |
| Protective from oxidative stress-mediated brain injury, such as epileptic seizures | [44] | |
| Deletion of HMOX2 increases redox stress damage in epileptic seizures (protective). | [12] | |
| Iron | Neurotoxic and involved in neurodegenerative diseases when accumulating in the brain (almost all neurological conditions). | [12,45,46] |
| CO | Neuroprotective (in low concentrations) against vasospastic reaction accompanying subarachnoid bleeding. | [46] |
| Neuroprotective (in low concentrations) against toxic noxious substances. | [38] | |
| Neurotoxic in high concentrations. | [47] | |
| May impair auditory functions. | [48,49] | |
| May impair cognitive and olfactory functions as well as the neuroendocrine system. | [12] | |
| Biliverdin | Protective against cerebral infarction; cerebral ischemia-reperfusion. | [50,51] |
| Inducing brainstem auditory evoked potential abnormalities | [52]. | |
| Induces fetal toxicity. | [53,54] | |
| BLVRA/B | ||
| BLVRA | Protective in meningioma and glioma. | [55] |
| Ameliorating autoimmune encephalomyelitis (a model for multiple sclerosis). | [56] | |
| Involved in the pathogenesis of Alzheimer’s disease. | [29,57,58] | |
| Improving neurological function in germinal matrix hemorrhage (a disease of premature infants which could bring complications like developmental delay, mental retardation, hydrocephalus and cerebral palsy). | [59] | |
| BLVRB | Potentially protective during fetal life. | [53,54] |
| Biomarker for Alzheimer’s disease and ischemic stroke. | [60,61] | |
| Bilirubin | Neuroprotective in cellular and animal models of experimental autoimmune encephalomyelitis. | [44,62,63] |
| Protective in stroke and ischemia. | [64,65] | |
| Protective against ethanol-induced neurotoxicity. | [37] | |
| Protective against mitochondrial toxicity. | [38] | |
| Reducing tumor size and improving survival in glioma. | [63] | |
| Protective against neurotoxicity in Parkinson’s disease model. | [66] | |
| Protective in traumatic brain injury. | [67] | |
| Protective in asymptomatic intracranial atherosclerosis. | [68] | |
| Improving survival of grafted neural stem cells. | [69] | |
| Contributing to inflammation in intracerebral hemorrhage. | [70] | |
| Correlating negatively with the neuropsychiatric/neurodegenerative disorders (bipolar disorder, schizophrenia, schizoaffective disorder, Alzheimer’s disease, dementia, multiple sclerosis, cerebral infarction in adults.) | [1,2,71,72] | |
| Correlating with intraventricular hemorrhage, retinopathy and greater vision loss; hypoxic-ischemic encephalopathy; and neonatal encephalopathy due to hepatic injury in infants. | [72,73,74,75] | |
| Responsible for brain damage in severe neonatal hyperbilirubinemia (kernicterus spectrum disorder: KSD) and Crigler-Najjar type I syndrome. | [4,5,6,76] | |
| Bilirubin degradation products | ||
| Bilirubin photoisomers | Pro-inflammatory activities. | [77] |
| Biopyrrins | Increased urinary excretion in Parkinson’s disease. | [78] |
| Propentdyopents | Increased in cerebrospinal fluid in subarachnoid bleeding. | [79] |
| Z-BOX A/B | Increased in cerebrospinal fluid in subarachnoid bleeding. | [80] |
| Yellow Players | Target | Effect | Ref |
|---|---|---|---|
| Heme | Generation of ROS/RNS | Vascular hypertension and vasoconstriction. In turn, the pro-oxidant milieu increases the oxidation of hemoglobin, enhancing heme release, protein carbonylation, lipids oxidation, MMP9 release, and tissue damage. | [12,30,81] |
| Activation of TLR4 | Proinflammatory activity: neutrophil migration, secretion of IL8, TNFα (activating NF-κB); increased vascular permeability; edema. | [30,81] | |
| Nrf2/Bach1/Keap1 | Inhibiting the antioxidant response. | [24] | |
| Activation of PI3K/Akt | Reducing apoptosis, increasing the expression of antioxidant enzymes (SOD, and HMOX1). | [26] | |
| Binding to hemopexin (Hx) | Chelating heme. | [12,30] | |
| Binding to haptoglobin (Hp) | Chelating heme via binding of hemoglobin. If not sufficient: increased CNS hemorrhage, oxidative stress, impaired brain performance, and reduced neurological activity. Marker of BBB disruption. | [12,30] | |
| Modulation of proteasome activity. | Impairing the activity of the ubiquitin-proteasome system; impairing the unfolded protein response (PERK/ATF6/IRE1a); and leading to the accumulation of unfolded proteins. | [24] | |
| Cofactor for cytochrome c and the mitochondrial electron transport chain (complexes II, III, IV) | Mitochondrial dysfunction, impairing ATP translocation into the cytoplasm; mitophagy and apoptosis. Impairing mitochondrial trafficking (especially relevant for neurons). | [24] | |
| Binding to Slo1 BK ion channel | Inhibiting the cellular excitability. | [12] | |
| Induction of neuroglobin expression | Reducing the apoptosis, cytochrome c, and mitochondrial dysfunction. | [25] | |
| Induction of ferritin | Chelating Fe. | [28] | |
| Inducing HMOX1 | Increasing cell survival and reducing redox stress. | [27] | |
| Decreasing lipid peroxidation, increasing the expression of the anti-apoptotic Bcl2, decreasing damage. | [46] | ||
| Possibly increasing Fe influx in mitochondria worsening the damage, increasing redox stress and inflammation. | [46] | ||
| HMOX1/2 | |||
| HMOX1 | Production of CO, BV and Fe. | Promoting proliferation trough synthesis of cGMP (maybe acting on CREB). | [18] |
| Increasing VEGF in astrocytes, leading to angiogenesis. Activating of the BDNF–TrkB–PI3K/Akt signaling with increased neuronal survival, and reduced inflammation. | [67] | ||
| When overexpressed, increasing cholesterol synthesis and cellular efflux, with an increased presence of oxysterols (products of cholesterol oxidation). The same result is obtained by the addition of CO or iron to the culture, suggesting one or both the HMOX1 products as the real effectors. | [16,82] | ||
| Decreasing oxidative and nitrosative stress; increasing (restored) GSH and catalase activity; reducing the release of TNFα and IL1β; reducing (restored) the GSK3 activity. | [32] | ||
| Increasing Fe production and deposition into astroglial mitochondria, with cellular bioenergetics failure. Increasing DNA damage (8-OHdG), protein oxidation (carbonyls), and lipid peroxidation. Altering the mitochondria morphology and cellular distribution, with mitophagy and autophagy. Enhancing the conversion of catecholamines and catechol-estrogens to neurotoxic radicals, making neurons more sensitive to H2O2 and dopamine insult. | [16] | ||
| Acutely induced after stimuli, mainly in glial cells (astrocytes, microglia). Acute up-regulation might be protective, while a chronic up-regulation may cause toxicity. Inducing Fe cell export. | [33] | ||
| Decreasing the expression of NLRP1, possibly through the inhibition of ATF4, inhibiting the inflammasome, reducing the neuronal death by apoptosis, and improving functional recovery. | [83] | ||
| Increasing miRNA expression (miR16, 17 and 140) | Downregulating the mitochondrial functions (including ATP production; mitochondrial antioxidant enzymes level; intrinsic apoptotic pathway; enhancement of TNFα synthesis; up-regulation of MAPK signaling to compromise he oxidative phosphorylation). | [16] | |
| Migrating into nuclei | HMOX1 can migrate into nuclei and act as a transcription factor of the genes involved in the cellular antioxidant response, immunity and inflammation, autophagy, hypoxia, tumor resistance, etc. However, this mechanism has not been studied in CNS diseases so far. | [1] | |
| HMOX1/2 | Amyloid protein precursor binding to HMOX1/2 | Reduced HMOX1/2 activity, reduced UCB production, and increased cellular sensitivity to H2O2 and hemin toxicity. | [84] |
| HMOX2 | Generation of CO, BV and Fe2+. | Fostering cell survival (via UCB action) and proliferation (via cGMP signaling). | [18] |
| Basal production of UCB and CO in neurons. | CO: inducing cyclic guanylyl cyclase, in turn producing cGMP (possibly via ERK). | [33] | |
| Production of UCB and cGMP | Increasing neuroprotection toward redox stress. | [85] | |
| CO | Voltage-gated K+ channel | Modulating the cellular excitability. | [24] |
| Activation of guanylyl cyclase | Increasing cGMP, activating of cGMP protein kinase and p38 MAPK, preventing neurons degeneration, activating noradrenergic neurons, decreasing apoptosis, reducing inflammation. | [3,33,86] | |
| AMPK | Inhibiting AMPK activation, decreasing the toxicity of the Aβ. | [87,88] | |
| HIF1α | Activating the Ca channels, CAMK2B, AMPKα, increasing mitochondrial respiration. | [67] | |
| HMOX1 | Inducing HMOX1 (through Nrf2 signaling) | [67] | |
| miRNA | Increasing miR-140, 17, 16. Decreasing miR-297, 206, 187, 181a, 138, 29c, in turn reducing the mRNA levels of Ngfr, Vglut1, MAPK3, TNFα, and Sirt1, abnormally expressed in various central nervous system disorders. | [89] | |
| Iron | Generation of hydroxyl radicals. | Inducing a high rate of protein carbonylation, reducing SOD activity, increasing DNA damage, inhibiting the DNA repair system. Activating NF-κB and AP-1 with BBB disruption and worsening of damage. Reducing mitochondrial respiratory functions. | [81] |
| Activation of NF-κB and induction of inflammation. | Inducting the glutamate excitotoxicity (release of glutamate) with increased BBB permeability, neuronal autophagy, neuronal atrophy and death. Releasing of MMP9, TNFα, IL1β, microgliosis. | [81] | |
| miRNA | Increasing miR-140, 17, 16. Decreasing miR-297, 206, 187, 181a, 138, 29c, in turn reducing the mRNA levels of Ngfr, Vglut1, MAPK3, TNFα, and Sirt1, abnormally expressed in various CNS disorders. | [89] | |
| BV | Scavenging ROS | Lowering DNA damage (8-OHdG). | [51,90] |
| miR-204-5p, Ets1 | Lowering Th1 type response. | [51] | |
| NF-κB | Lowering NF-κB-DNA binding and pro-inflammatory factors transcription/production | [91,92] | |
| TLR4 | Inducing BLVR translocation into the nucleus, binding to TLR4 promoter, repressing the expression of TLR4, and leading to the inhibition of inflammatory cytokine production. | [93] | |
| JNK | Reducing the JNK activation, affecting JNK/AP-1 pathway, suppressing the transcription of TNFα and diminishing endothelial cell apoptosis. | [94] | |
| PI3K/Akt | Inducing the interaction of BLVRA with PI3K, activating Akt signaling, and increasing anti-inflammatory cytokine (IL10) production in macrophages. | [95] | |
| ROS, NRS formation | Preventing oxidative damage in rat brain microsomes | [50,90] | |
| Complement | Inhibiting C5aR gene and protein expression that is mediated by mTOR pathway accompanied by the reduction of pro-inflammatory cytokines (TNFα and IL6) gene expression (macrophages). | [93] | |
| BLVR | Inducing BLVR translocation into the nucleus. | [96] | |
| Histones | Possible inhibition of the histone synthesis. | [97] | |
| BLVRA | Akt gene | Modulating glycogen synthase kinase, and Tau protein deposition in the brain. | [98,99,100,101] |
| NF-κB | Direct binding, arresting the cell cycle. | [91] | |
| eNOS/NO/TLR4 pathway | Binding on the gene promoter, inhibition of transcription, reducing the inflammation. | [96] | |
| Improving hematoma resolution and neurological functions. | [59] | ||
| MAPK/PI3K | Maintaining the synaptic plasticity, memory consolidation, inducing the genes required for neuronal and synapse growth, maintenance and repair processes. | [29] | |
| MAPK/Akt | Inhibiting MAPK/Akt activation, reducing apoptosis, protecting the hippocampal neuronal cell from oxidative stress. | [102] | |
| BACE-1 protein | Reducing the BLVRA activation inducing the phosphorylation of BACE-1, promoting insulin resistance and increasing Aβ levels in the brain of an animal model of aging. | [103] | |
| MEK1-ERK1/2-Elk1 signaling | Transcriptional activation of stress-induced genes, including HMOX1. | [104] | |
| Insulin receptor | Inducing an early activation of IRS1 and improving brain insulin resistance. | [105] | |
| HMOX1 | Improving cellular antioxidant defense via HMOX1 induction. | [106] | |
| UCB | NF-κB | Direct binding, arresting the cell cycle. | [91] |
| Preventing NF-κB-DNA binding, suppressing T-cell activation. | [107] | ||
| Histone acetylation | Modulating histone 3 acetylation and the transcription of genes involved in brain development. | [108] | |
| ER | Inducing ER stress, inflammation and apoptosis | [109] | |
| Nrf2 | Activating the Nrf2 pathway, thus the antioxidant response. | [110] | |
| CREB | Increasing the phosphorylation of CREB possibly leading to BDNF production, boosting the survival and the repair processes in traumatic brain injury. | [67] | |
| AKT | Enhancing blood flow and regeneration in ischemic injury. | [67] | |
| HIF1α | Stabilizing HIF1α, activating Ca channels, CAMKβ, AMPKα, and increasing mitochondrial respiration. | [67] | |
| AhR | Modulating the transcription of genes coding for detoxification enzymes (CYP1A1, UGT1A1), acting on the cell cycle, MAPK cascade, Nrf2 pathway, and immune response. | [111,112] | |
| CAR | Involved in the disposal of exogenous and endogenous substances, and inhibition of gluconeogenesis. | [113] | |
| ApoD | A non-albumin carrier of bilirubin in human plasma, contributing to protection against oxidative stress, is highly expressed in the brain. | [113] | |
| Neurotrophic factor | Increasing the expression of BDNF in neurons and GDNF in glia, leading to reduction of neuronal loss in substantia nigra in animal model of Parkinson’s disease | [66] | |
| MRGPRX4 | Mediating the cholestatic itch in the primary sensory neurons, acting in host defense and immune reaction. | [113] | |
| Angiogenic and energy-sensing genes in astrocytes | Enhancing PGC1α and HIF1α production in astrocytes which plays a role in mitochondria biogenesis, reduction of inflammatory, and angiogenesis | [67] | |
| ROS/RNS generation | Inhibiting of NMDA excitotoxicity, preventing neuronal death. | [104,114] | |
| Affecting BBB permeability and preventing inflammatory cell invasion | [62] | ||
| Macrophages and T cells | Immuno-modulatory activity by reducing the expression of Fc receptor in macrophage and inhibiting T cell response. | [115] | |
| PKC/ICAM-1 signaling, | Contributing to neutrophil infiltration, early inflammation, and edema. Decreasing nitrate/nitrite formation, reduced perihematomal microgliosis. | [70] | |
| Mrp1/ABCc1 | Upregulating and translocating its transporter (Mrp1/ABCc1) from the Golgi apparatus to the plasma membrane. | [116] | |
| Cellular and subcellular membranes | Altering membrane polarity and fluidity, the opening of the permeability transition pores, inducing cellular energy failure, activating the mitochondrial apoptotic pathway. | [117,118] | |
| P38MAPK-JNK1/2-NFkb | Inducting of inflammation, the release of TNFα, IL1β, reduction of the cellular viability | [119,120] | |
| NMDA receptors, glutamate | Inducing glutamate excitotoxicity, reducing the expression of the NMDA receptors, impairing long-term potentiation and long-term depression | [121,122,123] | |
| ERK-Akt-CREB | Scavenging ROS, decreasing neurotrophic factor availability. | [124] | |
| Mrp1/ABCc1 and Pgp/MDR1/ABCb1 | Modulating the expression of its transporters (Mrp1/ABCc1 and Pgp/MDR1/ABCb1) at the blood-brain interfaces | [125] | |
| DNA | Inducing ROS, which in turn leads to DNA damage, despite the activation of the DNA repair pathways. | [126] | |
| Cell cycle | Inducing cell cycle arrest | [127] | |
| UCB degradation products | Ca channels | Opening of the Ca channels and decreasing the conductance of the cerebral myocytes, inducing vasoconstriction. | [79] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jayanti, S.; Vítek, L.; Tiribelli, C.; Gazzin, S. The Role of Bilirubin and the Other “Yellow Players” in Neurodegenerative Diseases. Antioxidants 2020, 9, 900. https://doi.org/10.3390/antiox9090900
Jayanti S, Vítek L, Tiribelli C, Gazzin S. The Role of Bilirubin and the Other “Yellow Players” in Neurodegenerative Diseases. Antioxidants. 2020; 9(9):900. https://doi.org/10.3390/antiox9090900
Chicago/Turabian StyleJayanti, Sri, Libor Vítek, Claudio Tiribelli, and Silvia Gazzin. 2020. "The Role of Bilirubin and the Other “Yellow Players” in Neurodegenerative Diseases" Antioxidants 9, no. 9: 900. https://doi.org/10.3390/antiox9090900
APA StyleJayanti, S., Vítek, L., Tiribelli, C., & Gazzin, S. (2020). The Role of Bilirubin and the Other “Yellow Players” in Neurodegenerative Diseases. Antioxidants, 9(9), 900. https://doi.org/10.3390/antiox9090900