Microbiota-Mitochondria Inter-Talk: A Potential Therapeutic Strategy in Obesity and Type 2 Diabetes

 ,
,  and
and 
Abstract
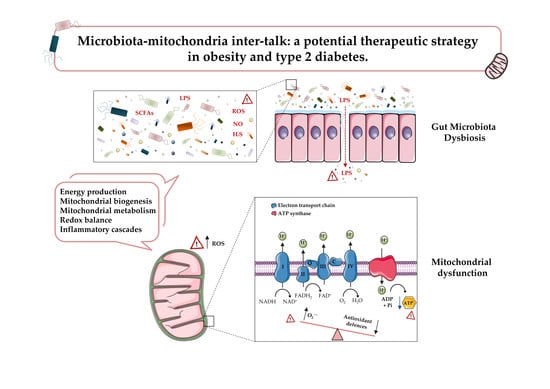
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Mitochondrial Dysfunction in Obesity and T2D
2.1. Mitochondrial ROS
2.2. Oxidative/Nitrosative Stress Signaling
2.3. Mitochondrial Dysfunction in Obesity and T2D
3. Gut Microbiota: A Novel Key Player in Obesity and T2D
4. Microbiota and Mitochondria in Obesity and T2D: Friends or Foes?
5. SCFAs, Secondary Bile Acids and Mitochondria
6. Microbiota, Mitochondria and the Immune System
7. Microbiota and Mitochondria: Potential Therapeutic Strategies in Obesity and T2D
8. Concluding Remarks and Future Perspectives
Survey Methodology
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 22 June 2020).
- Abarca-Gómez, L.; Abdeen, Z.A.; Hamid, Z.A.; Abu-Rmeileh, N.M.; Acosta-Cazares, B.; Acuin, C.; Adams, R.J.; Aekplakorn, W.; Afsana, K.; Aguilar-Salinas, C.A. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: A pooled analysis of 2416 population-based measurement studies in 1289 million children, adolescents, and adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef]
- Collaborators, G.O. Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Dyson, P.A. The therapeutics of lifestyle management on obesity. Diabetes Obes. Metab. 2010, 12, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Taylor, V.H.; Forhan, M.; Vigod, S.N.; McIntyre, R.S.; Morrison, K.M. The impact of obesity on quality of life. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 139–146. [Google Scholar] [CrossRef]
- Fruhbeck, G.; Toplak, H.; Woodward, E.; Yumuk, V.; Maislos, M.; Oppert, J.M. Executive Committee of the European Association for the Study of, O. Obesity: The gateway to ill health—An EASO position statement on a rising public health, clinical and scientific challenge in Europe. Obes. Facts 2013, 6, 117–120. [Google Scholar] [CrossRef]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef]
- Das, U.N. Obesity: Genes, brain, gut, and environment. Nutrition 2010, 26, 459–473. [Google Scholar] [CrossRef]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef]
- Rabe, K.; Lehrke, M.; Parhofer, K.G.; Broedl, U.C. Adipokines and insulin resistance. Mol. Med. 2008, 14, 741–751. [Google Scholar] [CrossRef]
- Fernandez-Sanchez, A.; Madrigal-Santillan, E.; Bautista, M.; Esquivel-Soto, J.; Morales-Gonzalez, A.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sanchez-Rivera, G.; Valadez-Vega, C.; Morales-Gonzalez, J.A. Inflammation, oxidative stress, and obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef] [PubMed]
- Osborn, O.; Olefsky, J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012, 18, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Zmora, N.; Bashiardes, S.; Levy, M.; Elinav, E. The Role of the Immune System in Metabolic Health and Disease. Cell Metab. 2017, 25, 506–521. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef]
- Marseglia, L.; Manti, S.; D’Angelo, G.; Nicotera, A.; Parisi, E.; Di Rosa, G.; Gitto, E.; Arrigo, T. Oxidative stress in obesity: A critical component in human diseases. Int. J. Mol. Sci. 2014, 16, 378–400. [Google Scholar] [CrossRef]
- Gao, C.L.; Zhu, C.; Zhao, Y.P.; Chen, X.H.; Ji, C.B.; Zhang, C.M.; Zhu, J.G.; Xia, Z.K.; Tong, M.L.; Guo, X.R. Mitochondrial dysfunction is induced by high levels of glucose and free fatty acids in 3T3-L1 adipocytes. Mol. Cell. Endocrinol. 2010, 320, 25–33. [Google Scholar] [CrossRef]
- Sutherland, L.N.; Capozzi, L.C.; Turchinsky, N.J.; Bell, R.C.; Wright, D.C. Time course of high-fat diet-induced reductions in adipose tissue mitochondrial proteins: Potential mechanisms and the relationship to glucose intolerance. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1076–E1083. [Google Scholar] [CrossRef]
- Koh, E.H.; Park, J.Y.; Park, H.S.; Jeon, M.J.; Ryu, J.W.; Kim, M.; Kim, S.Y.; Kim, M.S.; Kim, S.W.; Park, I.S.; et al. Essential role of mitochondrial function in adiponectin synthesis in adipocytes. Diabetes 2007, 56, 2973–2981. [Google Scholar] [CrossRef]
- Feldmann, H.M.; Golozoubova, V.; Cannon, B.; Nedergaard, J. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 2009, 9, 203–209. [Google Scholar] [CrossRef]
- Rains, J.L.; Jain, S.K. Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med. 2011, 50, 567–575. [Google Scholar] [CrossRef]
- Bloch-Damti, A.; Bashan, N. Proposed mechanisms for the induction of insulin resistance by oxidative stress. Antioxid. Redox Signal. 2005, 7, 1553–1567. [Google Scholar] [CrossRef]
- Festi, D.; Schiumerini, R.; Eusebi, L.H.; Marasco, G.; Taddia, M.; Colecchia, A. Gut microbiota and metabolic syndrome. World J. Gastroenterol. 2014, 20, 16079–16094. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.T.; Nieuwdorp, M.; Backhed, F. Microbial modulation of insulin sensitivity. Cell Metab. 2014, 20, 753–760. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M.; et al. The gut microbiota and host health: A new clinical frontier. Gut 2016, 65, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Saint-Georges-Chaumet, Y.; Edeas, M. Microbiota-mitochondria inter-talk: Consequence for microbiota-host interaction. Pathog. Dis. 2016, 74, ftv096. [Google Scholar] [CrossRef] [PubMed]
- Franco-Obregon, A.; Gilbert, J.A. The Microbiome-Mitochondrion Connection: Common Ancestries, Common Mechanisms, Common Goals. mSystems 2017, 2, e00017–e00018. [Google Scholar] [CrossRef]
- Lucattini, R.; Likic, V.A.; Lithgow, T. Bacterial proteins predisposed for targeting to mitochondria. Mol. Biol. Evol. 2004, 21, 652–658. [Google Scholar] [CrossRef]
- Lobet, E.; Letesson, J.J.; Arnould, T. Mitochondria: A target for bacteria. Biochem. Pharmacol. 2015, 94, 173–185. [Google Scholar] [CrossRef]
- Neish, A.S.; Rheinallt, M.J. Redox signaling mediates symbiosis between the gut microbiota and the intestine. Gut Microbes 2014, 5, 250–253. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxidative Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef] [PubMed]
- Tauffenberger, A.; Fiumelli, H.; Almustafa, S.; Magistretti, P.J. Lactate and pyruvate promote oxidative stress resistance through hormetic ROS signaling. Cell Death Dis. 2019, 10, 653. [Google Scholar] [CrossRef] [PubMed]
- Cerdan, S.; Rodrigues, T.B.; Sierra, A.; Benito, M.; Fonseca, L.L.; Fonseca, C.P.; Garcia-Martin, M.L. The redox switch/redox coupling hypothesis. Neurochem. Int. 2006, 48, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Gutierrez, C.; Bonora, N.; Bobo-Jimenez, V.; Jimenez-Blasco, D.; Lopez-Fabuel, I.; Fernandez, E.; Josephine, C.; Bonvento, G.; Enriquez, J.A.; Almeida, A. Astrocytic mitochondrial ROS modulate brain metabolism and mouse behaviour. Nat. Metab. 2019, 1, 201–211. [Google Scholar] [CrossRef]
- Tormos, K.V.; Anso, E.; Hamanaka, R.B.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.; Chandel, N.S. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011, 14, 537–544. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxidative Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef]
- Brand, M.D.; Orr, A.L.; Perevoshchikova, I.V.; Quinlan, C.L. The role of mitochondrial function and cellular bioenergetics in ageing and disease. Br. J. Dermatol. 2013, 169 (Suppl. 2), 1–8. [Google Scholar] [CrossRef]
- Valera-Alberni, M.; Canto, C. Mitochondrial stress management: A dynamic journey. Cell Stress 2018, 2, 253–274. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Chandhok, G.; Lazarou, M.; Neumann, B. Structure, function, and regulation of mitofusin-2 in health and disease. Biol. Rev. Camb. Philos. Soc. 2018, 93, 933–949. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.; Naon, D.; Pich, S.; Soriano, F.X.; Vega, N.; Rieusset, J.; Laville, M.; Guillet, C.; Boirie, Y.; Wallberg-Henriksson, H.; et al. Expression of Mfn2, the Charcot-Marie-Tooth neuropathy type 2A gene, in human skeletal muscle: Effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin-6. Diabetes 2005, 54, 2685–2693. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, D.; Hernandez-Alvarez, M.I.; Segales, J.; Sorianello, E.; Munoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 5523–5528. [Google Scholar] [CrossRef]
- Han, W.; Fessel, J.P.; Sherrill, T.; Kocurek, E.G.; Yull, F.E.; Blackwell, T.S. Enhanced Expression of Catalase in Mitochondria Modulates NF-κB–Dependent Lung Inflammation through Alteration of Metabolic Activity in Macrophages. J. Immunol. 2020, 205, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Sun, Y.; Long, J.; Sui, X.; Yang, J.; Wang, Q.; Wang, S.; He, H.; Luo, Y.; Qiu, Z. Involvement of the Nrf2-Keap1 signaling pathway in protection against thallium-induced oxidative stress and mitochondrial dysfunction in primary hippocampal neurons. Toxicol. Lett. 2020, 319, 66–73. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Kaludercic, N.; Giorgio, V. The Dual Function of Reactive Oxygen/Nitrogen Species in Bioenergetics and Cell Death: The Role of ATP Synthase. Oxidative Med. Cell. Longev. 2016, 2016, 3869610. [Google Scholar] [CrossRef]
- Riobo, N.A.; Clementi, E.; Melani, M.; Boveris, A.; Cadenas, E.; Moncada, S.; Poderoso, J.J. Nitric oxide inhibits mitochondrial NADH:ubiquinone reductase activity through peroxynitrite formation. Biochem. J. 2001, 359, 139–145. [Google Scholar] [CrossRef]
- Poderoso, J.J.; Helfenberger, K.; Poderoso, C. The effect of nitric oxide on mitochondrial respiration. Nitric Oxide Biol. Chem. 2019, 88, 61–72. [Google Scholar] [CrossRef]
- Andrades, M.E.; Morina, A.; Spasic, S.; Spasojevic, I. Bench-to-bedside review: Sepsis—from the redox point of view. Crit. Care 2011, 15, 230. [Google Scholar] [CrossRef]
- Mantzarlis, K.; Tsolaki, V.; Zakynthinos, E. Role of Oxidative Stress and Mitochondrial Dysfunction in Sepsis and Potential Therapies. Oxidative Med. Cell. Longev. 2017, 2017, 5985209. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.W.; Dixit, V.D. Mechanisms of disease: Inflammasome activation and the development of type 2 diabetes. Front. Immunol. 2013, 4, 50. [Google Scholar] [CrossRef]
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef]
- Pallavi, M.; Suchitra, M.M.; Alok Sachan, L.A.; Srinivasa Rao, P.V.L.N. Role of adipokines, oxidative stress, and endotoxins in the pathogenesis of non-alcoholic fatty liver disease in patients with type 2 diabetes mellitus. Int. J. Res. Med Sci. 2019, 7, 1644. [Google Scholar] [CrossRef]
- Han, C.Y. Roles of Reactive Oxygen Species on Insulin Resistance in Adipose Tissue. Diabetes Metab. J. 2016, 40, 272–279. [Google Scholar] [CrossRef]
- Manna, P.; Jain, S.K. Obesity, Oxidative Stress, Adipose Tissue Dysfunction, and the Associated Health Risks: Causes and Therapeutic Strategies. Metab. Syndr. Relat. Disord. 2015, 13, 423–444. [Google Scholar] [CrossRef]
- Lefranc, C.; Friederich-Persson, M.; Palacios-Ramirez, R.; Nguyen Dinh Cat, A. Mitochondrial oxidative stress in obesity: Role of the mineralocorticoid receptor. J. Endocrinol. 2018, 238, R143–R159. [Google Scholar] [CrossRef]
- Wang, C.H.; Wang, C.C.; Huang, H.C.; Wei, Y.H. Mitochondrial dysfunction leads to impairment of insulin sensitivity and adiponectin secretion in adipocytes. FEBS J. 2013, 280, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Fujimoto, S.; Sato, Y.; Nishi, Y.; Mukai, E.; Yamano, G.; Sato, H.; Tahara, Y.; Ogura, K.; Nagashima, K.; et al. Reduction of reactive oxygen species ameliorates metabolism-secretion coupling in islets of diabetic GK rats by suppressing lactate overproduction. Diabetes 2013, 62, 1996–2003. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.; Apostolova, N.; Diaz-Rua, R.; Muntane, J.; Victor, V.M. Mitochondria and T2D: Role of Autophagy, ER Stress, and Inflammasome. Trends Endocrinol. Metab. 2020. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.W.; Lee, M.S. Mitochondria and the NLRP3 inflammasome: Physiological and pathological relevance. Arch. Pharmacal Res. 2016, 39, 1503–1518. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.; Persechini, P.M.; Ojcius, D.M. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 2007, 282, 2871–2879. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- David, J.A.; Rifkin, W.J.; Rabbani, P.S.; Ceradini, D.J. The Nrf2/Keap1/ARE Pathway and Oxidative Stress as a Therapeutic Target in Type II Diabetes Mellitus. J. Diabetes Res. 2017, 2017, 4826724. [Google Scholar] [CrossRef]
- Bitar, M.S.; Al-Mulla, F. A defect in Nrf2 signaling constitutes a mechanism for cellular stress hypersensitivity in a genetic rat model of type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E1119–E1129. [Google Scholar] [CrossRef]
- Rabbani, P.S.; Soares, M.A.; Hameedi, S.G.; Kadle, R.L.; Mubasher, A.; Kowzun, M.; Ceradini, D.J. Dysregulation of Nrf2/Keap1 Redox Pathway in Diabetes Affects Multipotency of Stromal Cells. Diabetes 2019, 68, 141–155. [Google Scholar] [CrossRef]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Satokari, R.; Grönroos, T.; Laitinen, K.; Salminen, S.; Isolauri, E. Bifidobacterium and Lactobacillus DNA in the human placenta. Lett. Appl. Microbiol. 2009, 48, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The placenta harbors a unique microbiome. Sci. Transl. Med. 2014, 6, 237ra65. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.M.; Murphy, K.; Stanton, C.; Ross, R.P.; Kober, O.I.; Juge, N.; Avershina, E.; Rudi, K.; Narbad, A.; Jenmalm, M.C. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Health Dis. 2015, 26, 26050. [Google Scholar] [CrossRef]
- Matamoros, S.; Gras-Leguen, C.; Le Vacon, F.; Potel, G.; de La Cochetiere, M.-F. Development of intestinal microbiota in infants and its impact on health. Trends Microbiol. 2013, 21, 167–173. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef]
- Dudek-Wicher, R.K.; Junka, A.; Bartoszewicz, M. The influence of antibiotics and dietary components on gut microbiota. Prz. Gastroenterol. 2018, 13, 85–92. [Google Scholar] [CrossRef]
- Li, Y.; Hao, Y.; Fan, F.; Zhang, B. The role of microbiome in insomnia, circadian disturbance and depression. Front. Psychiatry 2018, 9, 669. [Google Scholar] [CrossRef]
- Belizário, J.E.; Faintuch, J.; Garay-Malpartida, M. Gut microbiome dysbiosis and immunometabolism: New frontiers for treatment of metabolic diseases. Mediat. Inflamm. 2018, 2018, 2037838. [Google Scholar] [CrossRef]
- Sun, L.; Ma, L.; Ma, Y.; Zhang, F.; Zhao, C.; Nie, Y. Insights into the role of gut microbiota in obesity: Pathogenesis, mechanisms, and therapeutic perspectives. Protein Cell 2018, 9, 397–403. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Yang, W.; Chen, G.; Shafiq, M.; Javed, S.; Ali Zaidi, S.S.; Shahid, R.; Liu, C.; Bokhari, H. Analysis of gut microbiota of obese individuals with type 2 diabetes and healthy individuals. PLoS ONE 2019, 14, e0226372. [Google Scholar] [CrossRef] [PubMed]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Blandino, G.; Inturri, R.; Lazzara, F.; Di Rosa, M.; Malaguarnera, L. Impact of gut microbiota on diabetes mellitus. Diabetes Metab. 2016, 42, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef]
- Geerlings, S.Y.; Kostopoulos, I.; de Vos, W.M.; Belzer, C. Akkermansia muciniphila in the Human Gastrointestinal Tract: When, Where, and How? Microorganisms 2018, 6, 75. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Degli Esposti, M.; Chouaia, B.; Comandatore, F.; Crotti, E.; Sassera, D.; Lievens, P.M.-J.; Daffonchio, D.; Bandi, C. Evolution of mitochondria reconstructed from the energy metabolism of living bacteria. PLoS ONE 2014, 9, e96566. [Google Scholar] [CrossRef]
- Kalghatgi, S.; Spina, C.S.; Costello, J.C.; Liesa, M.; Morones-Ramirez, J.R.; Slomovic, S.; Molina, A.; Shirihai, O.S.; Collins, J.J. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in mammalian cells. Sci. Transl. Med. 2013, 5, 192ra85. [Google Scholar] [CrossRef]
- Mottawea, W.; Chiang, C.K.; Muhlbauer, M.; Starr, A.E.; Butcher, J.; Abujamel, T.; Deeke, S.A.; Brandel, A.; Zhou, H.; Shokralla, S.; et al. Altered intestinal microbiota-host mitochondria crosstalk in new onset Crohn’s disease. Nat. Commun. 2016, 7, 13419. [Google Scholar] [CrossRef] [PubMed]
- Saint-Georges-Chaumet, Y.; Attaf, D.; Pelletier, E.; Edeas, M. Targeting microbiota-mitochondria inter-talk: Microbiota control mitochondria metabolism. Cell. Mol. Biol. 2015, 61, 121–124. [Google Scholar]
- Cook, S.; Sellin, J. Short chain fatty acids in health and disease. Aliment. Pharmacol. Ther. 1998, 12, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.S.; Preston, T.; Frost, G.; Morrison, D.J. Role of Gut Microbiota-Generated Short-Chain Fatty Acids in Metabolic and Cardiovascular Health. Curr. Nutr. Rep. 2018, 7, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.V.; Frassetto, A.; Kowalik, E.J., Jr.; Nawrocki, A.R.; Lu, M.M.; Kosinski, J.R.; Hubert, J.A.; Szeto, D.; Yao, X.; Forrest, G.; et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS ONE 2012, 7, e35240. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef]
- Lumeng, L.; Davis, E.J. The oxidation of acetate by liver mitochondria. FEBS Lett. 1973, 29, 124–126. [Google Scholar] [CrossRef]
- Galmozzi, A.; Mitro, N.; Ferrari, A.; Gers, E.; Gilardi, F.; Godio, C.; Cermenati, G.; Gualerzi, A.; Donetti, E.; Rotili, D.; et al. Inhibition of class I histone deacetylases unveils a mitochondrial signature and enhances oxidative metabolism in skeletal muscle and adipose tissue. Diabetes 2013, 62, 732–742. [Google Scholar] [CrossRef]
- Mollica, M.P.; Mattace Raso, G.; Cavaliere, G.; Trinchese, G.; De Filippo, C.; Aceto, S.; Prisco, M.; Pirozzi, C.; Di Guida, F.; Lama, A.; et al. Butyrate Regulates Liver Mitochondrial Function, Efficiency, and Dynamics in Insulin-Resistant Obese Mice. Diabetes 2017, 66, 1405–1418. [Google Scholar] [CrossRef]
- Hu, S.; Kuwabara, R.; de Haan, B.J.; Smink, A.M.; de Vos, P. Acetate and Butyrate Improve β-cell Metabolism and Mitochondrial Respiration under Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 1542. [Google Scholar] [CrossRef]
- Canfora, E.E.; Jocken, J.W.; Blaak, E.E. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 2015, 11, 577–591. [Google Scholar] [CrossRef] [PubMed]
- den Besten, G.; Gerding, A.; van Dijk, T.H.; Ciapaite, J.; Bleeker, A.; van Eunen, K.; Havinga, R.; Groen, A.K.; Reijngoud, D.J.; Bakker, B.M. Protection against the Metabolic Syndrome by Guar Gum-Derived Short-Chain Fatty Acids Depends on Peroxisome Proliferator-Activated Receptor gamma and Glucagon-Like Peptide-1. PLoS ONE 2015, 10, e0136364. [Google Scholar] [CrossRef] [PubMed]
- Lantier, L.; Fentz, J.; Mounier, R.; Leclerc, J.; Treebak, J.T.; Pehmoller, C.; Sanz, N.; Sakakibara, I.; Saint-Amand, E.; Rimbaud, S.; et al. AMPK controls exercise endurance, mitochondrial oxidative capacity, and skeletal muscle integrity. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 3211–3224. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.A.; Uguccioni, G.; Vainshtein, A.; D’Souza, D. Mechanisms of exercise-induced mitochondrial biogenesis in skeletal muscle: Implications for health and disease. Compr. Physiol. 2011, 1, 1119–1134. [Google Scholar] [CrossRef]
- Richter, E.A.; Ruderman, N.B. AMPK and the biochemistry of exercise: Implications for human health and disease. Biochem. J. 2009, 418, 261–275. [Google Scholar] [CrossRef]
- Gerard, P. Metabolism of cholesterol and bile acids by the gut microbiota. Pathogens 2013, 3, 14–24. [Google Scholar] [CrossRef]
- Nie, Y.F.; Hu, J.; Yan, X.H. Cross-talk between bile acids and intestinal microbiota in host metabolism and health. J. Zhejiang Univ. Sci. B 2015, 16, 436–446. [Google Scholar] [CrossRef]
- Velazquez-Villegas, L.A.; Perino, A.; Lemos, V.; Zietak, M.; Nomura, M.; Pols, T.W.H.; Schoonjans, K. TGR5 signalling promotes mitochondrial fission and beige remodelling of white adipose tissue. Nat. Commun. 2018, 9, 245. [Google Scholar] [CrossRef]
- Kuipers, F.; Bloks, V.W.; Groen, A.K. Beyond intestinal soap—Bile acids in metabolic control. Nat. Rev. Endocrinol. 2014, 10, 488–498. [Google Scholar] [CrossRef]
- Joyce, S.A.; Gahan, C.G. Bile Acid Modifications at the Microbe-Host Interface: Potential for Nutraceutical and Pharmaceutical Interventions in Host Health. Annu. Rev. Food Sci. Technol. 2016, 7, 313–333. [Google Scholar] [CrossRef]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Korecka, A.; de Wouters, T.; Cultrone, A.; Lapaque, N.; Pettersson, S.; Dore, J.; Blottiere, H.M.; Arulampalam, V. ANGPTL4 expression induced by butyrate and rosiglitazone in human intestinal epithelial cells utilizes independent pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G1025–G1037. [Google Scholar] [CrossRef] [PubMed]
- Alex, S.; Lichtenstein, L.; Dijk, W.; Mensink, R.P.; Tan, N.S.; Kersten, S. ANGPTL4 is produced by entero-endocrine cells in the human intestinal tract. Histochem. Cell Biol. 2014, 141, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Caron, A.Z.; He, X.; Mottawea, W.; Seifert, E.L.; Jardine, K.; Dewar-Darch, D.; Cron, G.O.; Harper, M.E.; Stintzi, A.; McBurney, M.W. The SIRT1 deacetylase protects mice against the symptoms of metabolic syndrome. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Kazgan, N.; Metukuri, M.R.; Purushotham, A.; Lu, J.; Rao, A.; Lee, S.; Pratt-Hyatt, M.; Lickteig, A.; Csanaky, I.L.; Zhao, Y.; et al. Intestine-specific deletion of SIRT1 in mice impairs DCoH2-HNF-1alpha-FXR signaling and alters systemic bile acid homeostasis. Gastroenterology 2014, 146, 1006–1016. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.; Wang, H.; Liang, F. Mechanisms Linking Inflammation to Insulin Resistance. Int. J. Endocrinol. 2015, 2015, 508409. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Zeng, M.Y.; Inohara, N.; Nunez, G. Mechanisms of inflammation-driven bacterial dysbiosis in the gut. Mucosal Immunol. 2017, 10, 18–26. [Google Scholar] [CrossRef]
- Crouser, E.D.; Julian, M.W.; Blaho, D.V.; Pfeiffer, D.R. Endotoxin-induced mitochondrial damage correlates with impaired respiratory activity. Crit. Care Med. 2002, 30, 276–284. [Google Scholar] [CrossRef]
- Lee, I.; Huttemann, M. Energy crisis: The role of oxidative phosphorylation in acute inflammation and sepsis. Biochim. Et Biophys. Acta 2014, 1842, 1579–1586. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Marin-Penalver, J.J.; Martin-Timon, I.; Sevillano-Collantes, C.; Del Canizo-Gomez, F.J. Update on the treatment of type 2 diabetes mellitus. World J. Diabetes 2016, 7, 354–395. [Google Scholar] [CrossRef] [PubMed]
- Mazloom, K.; Siddiqi, I.; Covasa, M. Probiotics: How Effective Are They in the Fight against Obesity? Nutrients 2019, 11, 258. [Google Scholar] [CrossRef] [PubMed]
- Cerdo, T.; Garcia-Santos, J.A.; Bermúdez, M.; Campoy, C. The Role of Probiotics and Prebiotics in the Prevention and Treatment of Obesity. Nutrients 2019, 11, 635. [Google Scholar] [CrossRef]
- Tiderencel, K.A.; Hutcheon, D.A.; Ziegler, J. Probiotics for the treatment of type 2 diabetes: A review of randomized controlled trials. Diabetes Metab. Res. Rev. 2020, 36, e3213. [Google Scholar] [CrossRef] [PubMed]
- Saez-Lara, M.J.; Robles-Sanchez, C.; Ruiz-Ojeda, F.J.; Plaza-Diaz, J.; Gil, A. Effects of Probiotics and Synbiotics on Obesity, Insulin Resistance Syndrome, Type 2 Diabetes and Non-Alcoholic Fatty Liver Disease: A Review of Human Clinical Trials. Int. J. Mol. Sci. 2016, 17, 928. [Google Scholar] [CrossRef]
- Mohamadshahi, M.; Veissi, M.; Haidari, F.; Javid, A.Z.; Mohammadi, F.; Shirbeigi, E. Effects of probiotic yogurt consumption on lipid profile in type 2 diabetic patients: A randomized controlled clinical trial. J. Res. Med Sci. Off. J. Isfahan Univ. Med Sci. 2014, 19, 531. [Google Scholar]
- Tonucci, L.B.; Olbrich Dos Santos, K.M.; Licursi de Oliveira, L.; Rocha Ribeiro, S.M.; Duarte Martino, H.S. Clinical application of probiotics in type 2 diabetes mellitus: A randomized, double-blind, placebo-controlled study. Clin. Nutr. 2017, 36, 85–92. [Google Scholar] [CrossRef]
- Cani, P.D.; Van Hul, M. Novel opportunities for next-generation probiotics targeting metabolic syndrome. Curr. Opin. Biotechnol. 2015, 32, 21–27. [Google Scholar] [CrossRef]
- Wang, H.; Lu, Y.; Yan, Y.; Tian, S.; Zheng, D.; Leng, D.; Wang, C.; Jiao, J.; Wang, Z.; Bai, Y. Promising Treatment for Type 2 Diabetes: Fecal Microbiota Transplantation Reverses Insulin Resistance and Impaired Islets. Front. Cell. Infect. Microbiol. 2019, 9, 455. [Google Scholar] [CrossRef]
- Montan, P.D.; Sourlas, A.; Olivero, J.; Silverio, D.; Guzman, E.; Kosmas, C.E. Pharmacologic therapy of obesity: Mechanisms of action and cardiometabolic effects. Ann. Transl. Med. 2019, 7, 393. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Manneras-Holm, L.; Stahlman, M.; Olsson, L.M.; Serino, M.; Planas-Felix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.Z.; No, M.H.; Heo, J.W.; Park, D.H.; Kang, J.H.; Kim, J.H.; Seo, D.Y.; Han, J.; Jung, S.J.; Kwak, H.B. Effects of Acute Exercise on Mitochondrial Function, Dynamics, and Mitophagy in Rat Cardiac and Skeletal Muscles. Int. Neurourol. J. 2019, 23, S22–31. [Google Scholar] [CrossRef]
- Zafaranieh, S.; Choobineh, S.; Soori, R. The effect of 12 weeks of aerobic exercise on mitochondrial dynamics in cardiac myocytes of type 2 diabetic rats. Sport Sci. Health 2018, 14, 305–312. [Google Scholar] [CrossRef]
- Smith, R.A.; Hartley, R.C.; Cocheme, H.M.; Murphy, M.P. Mitochondrial pharmacology. Trends Pharmacol. Sci. 2012, 33, 341–352. [Google Scholar] [CrossRef]
- Nightingale, H.; Pfeffer, G.; Bargiela, D.; Horvath, R.; Chinnery, P.F. Emerging therapies for mitochondrial disorders. Brain 2016, 139, 1633–1648. [Google Scholar] [CrossRef]
- Serviddio, G.; Bellanti, F.; Sastre, J.; Vendemiale, G.; Altomare, E. Targeting mitochondria: A new promising approach for the treatment of liver diseases. Curr. Med. Chem. 2010, 17, 2325–2337. [Google Scholar] [CrossRef]
- Apostolova, N.; Victor, V.M. Molecular strategies for targeting antioxidants to mitochondria: Therapeutic implications. Antioxid. Redox Signal. 2015, 22, 686–729. [Google Scholar] [CrossRef]
- Escribano-Lopez, I.; Diaz-Morales, N.; Rovira-Llopis, S.; de Maranon, A.M.; Orden, S.; Alvarez, A.; Banuls, C.; Rocha, M.; Murphy, M.P.; Hernandez-Mijares, A.; et al. The mitochondria-targeted antioxidant MitoQ modulates oxidative stress, inflammation and leukocyte-endothelium interactions in leukocytes isolated from type 2 diabetic patients. Redox Biol. 2016, 10, 200–205. [Google Scholar] [CrossRef]
- Pung, Y.F.; Rocic, P.; Murphy, M.P.; Smith, R.A.; Hafemeister, J.; Ohanyan, V.; Guarini, G.; Yin, L.; Chilian, W.M. Resolution of mitochondrial oxidative stress rescues coronary collateral growth in Zucker obese fatty rats. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Lopez, I.; Banuls, C.; Diaz-Morales, N.; Iannantuoni, F.; Rovira-Llopis, S.; Gomis, R.; Rocha, M.; Hernandez-Mijares, A.; Murphy, M.P.; Victor, V.M. The Mitochondria-Targeted Antioxidant MitoQ Modulates Mitochondrial Function and Endoplasmic Reticulum Stress in Pancreatic beta Cells Exposed to Hyperglycaemia. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2019, 52, 186–197. [Google Scholar] [CrossRef]
- Rovira-Llopis, S.; Apostolova, N.; Banuls, C.; Muntane, J.; Rocha, M.; Victor, V.M. Mitochondria, the NLRP3 Inflammasome, and Sirtuins in Type 2 Diabetes: New Therapeutic Targets. Antioxid. Redox Signal. 2018, 29, 749–791. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vezza, T.; Abad-Jiménez, Z.; Marti-Cabrera, M.; Rocha, M.; Víctor, V.M. Microbiota-Mitochondria Inter-Talk: A Potential Therapeutic Strategy in Obesity and Type 2 Diabetes. Antioxidants 2020, 9, 848. https://doi.org/10.3390/antiox9090848
Vezza T, Abad-Jiménez Z, Marti-Cabrera M, Rocha M, Víctor VM. Microbiota-Mitochondria Inter-Talk: A Potential Therapeutic Strategy in Obesity and Type 2 Diabetes. Antioxidants. 2020; 9(9):848. https://doi.org/10.3390/antiox9090848
Chicago/Turabian StyleVezza, Teresa, Zaida Abad-Jiménez, Miguel Marti-Cabrera, Milagros Rocha, and Víctor Manuel Víctor. 2020. "Microbiota-Mitochondria Inter-Talk: A Potential Therapeutic Strategy in Obesity and Type 2 Diabetes" Antioxidants 9, no. 9: 848. https://doi.org/10.3390/antiox9090848
APA StyleVezza, T., Abad-Jiménez, Z., Marti-Cabrera, M., Rocha, M., & Víctor, V. M. (2020). Microbiota-Mitochondria Inter-Talk: A Potential Therapeutic Strategy in Obesity and Type 2 Diabetes. Antioxidants, 9(9), 848. https://doi.org/10.3390/antiox9090848