Genetic Polymorphisms Complicate COVID-19 Therapy: Pivotal Role of HO-1 in Cytokine Storm

 ,
, 
Abstract
1. Introduction
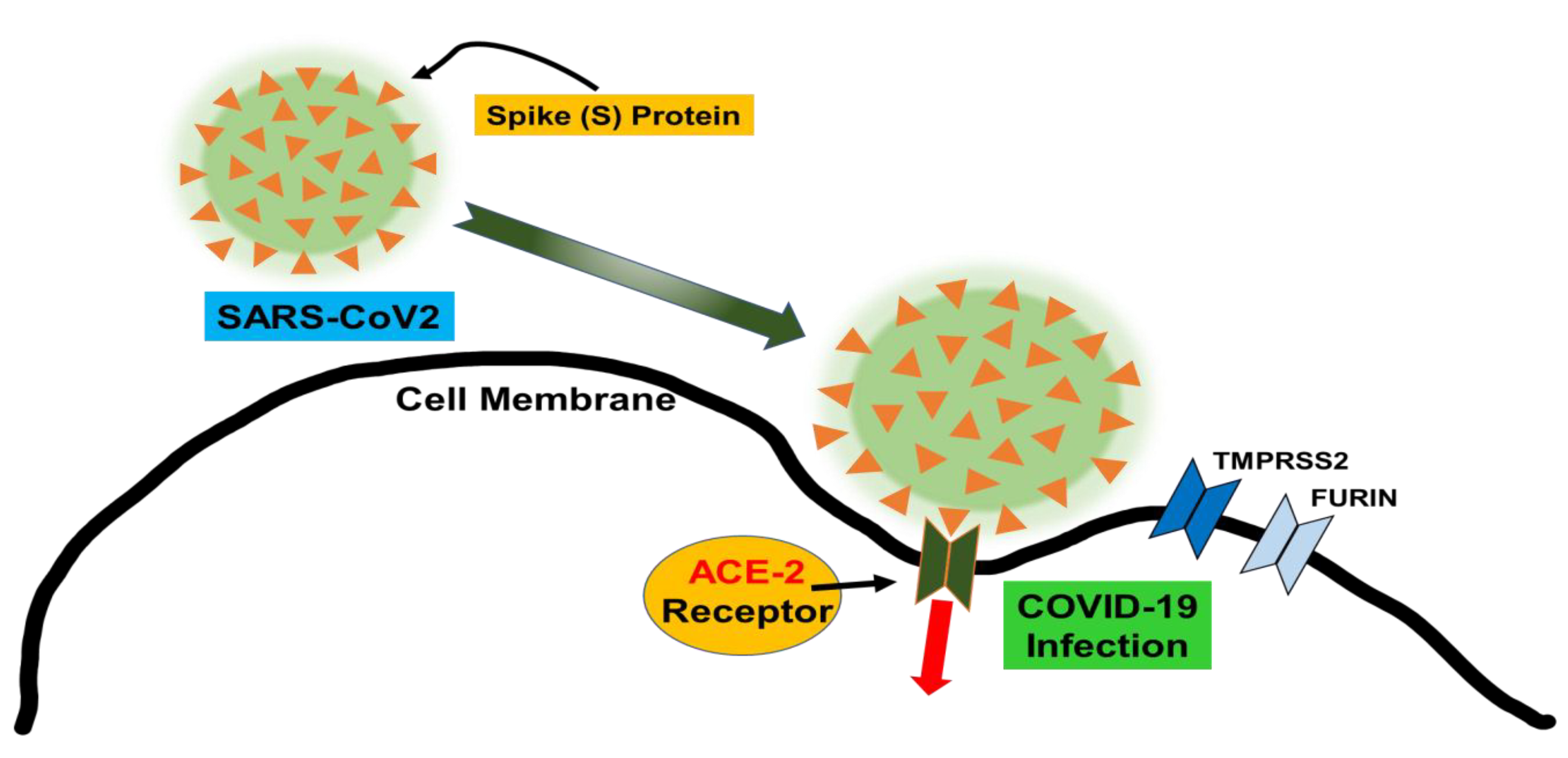
2. COVID-19 Increases Free Heme and Decreases Functional Hemoprotein
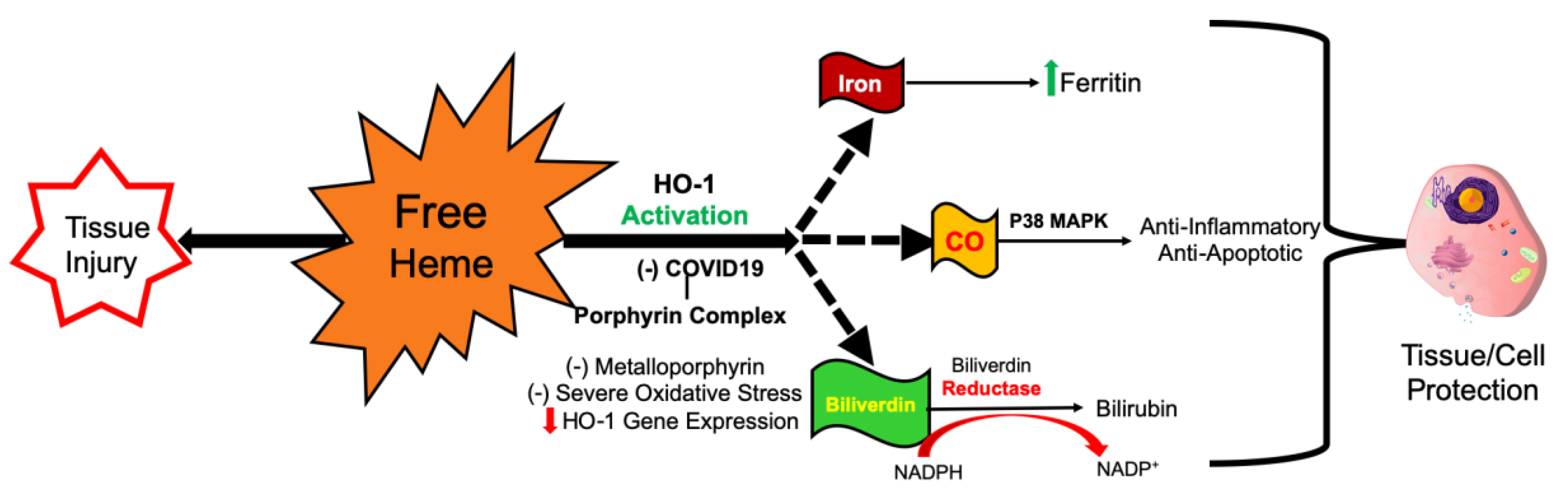
3. How COVID-19-Cytokine Storm Inhibits HO-1
4. COVID-19 Induction of ALAS-1/2
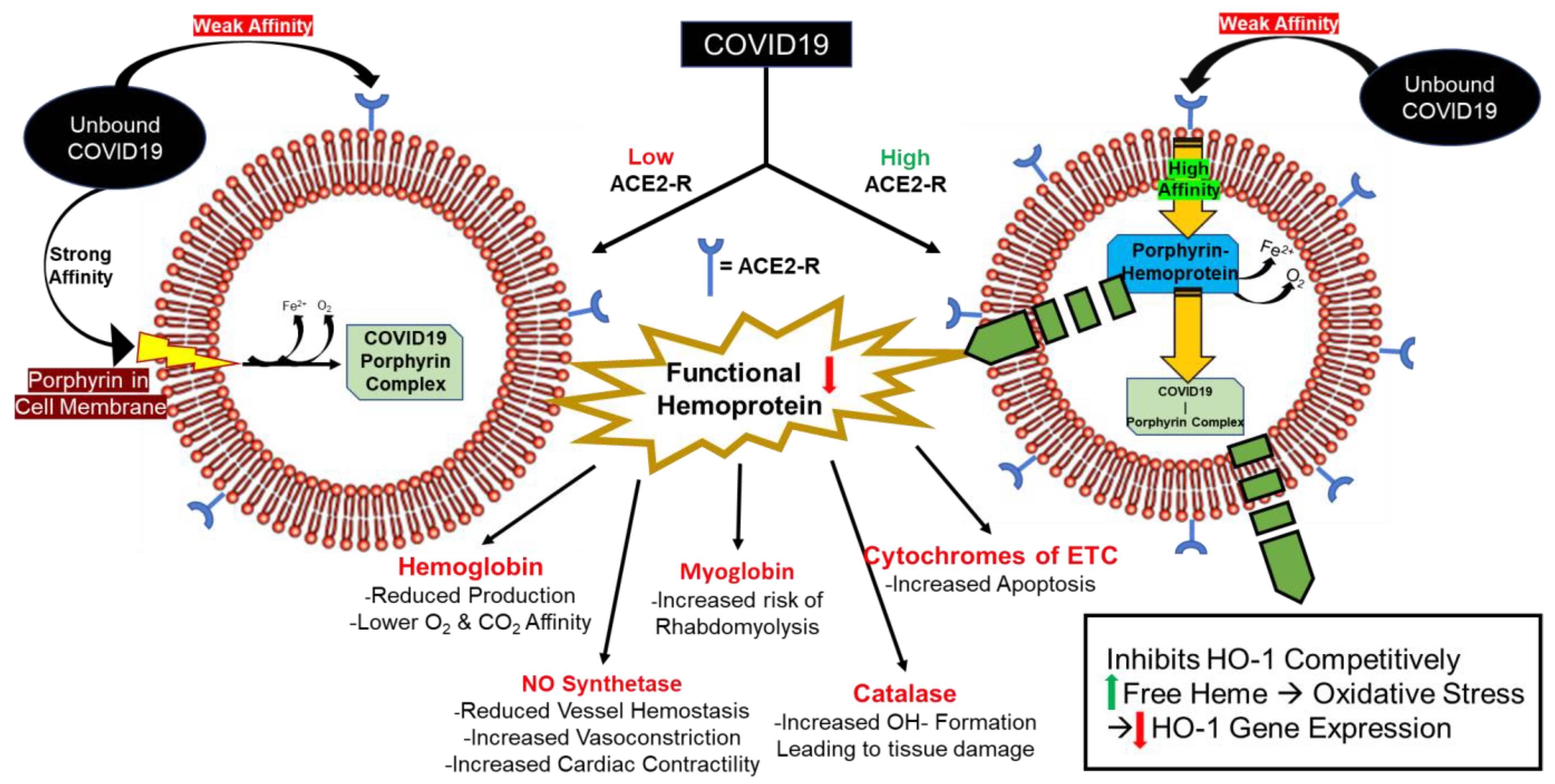
5. Consequences of Hemoprotein Malfunction/Deficiency
6. HO-1 Genetic Polymorphisms and COVID-19′s Cytokine Storm
7. Clinical Presentation
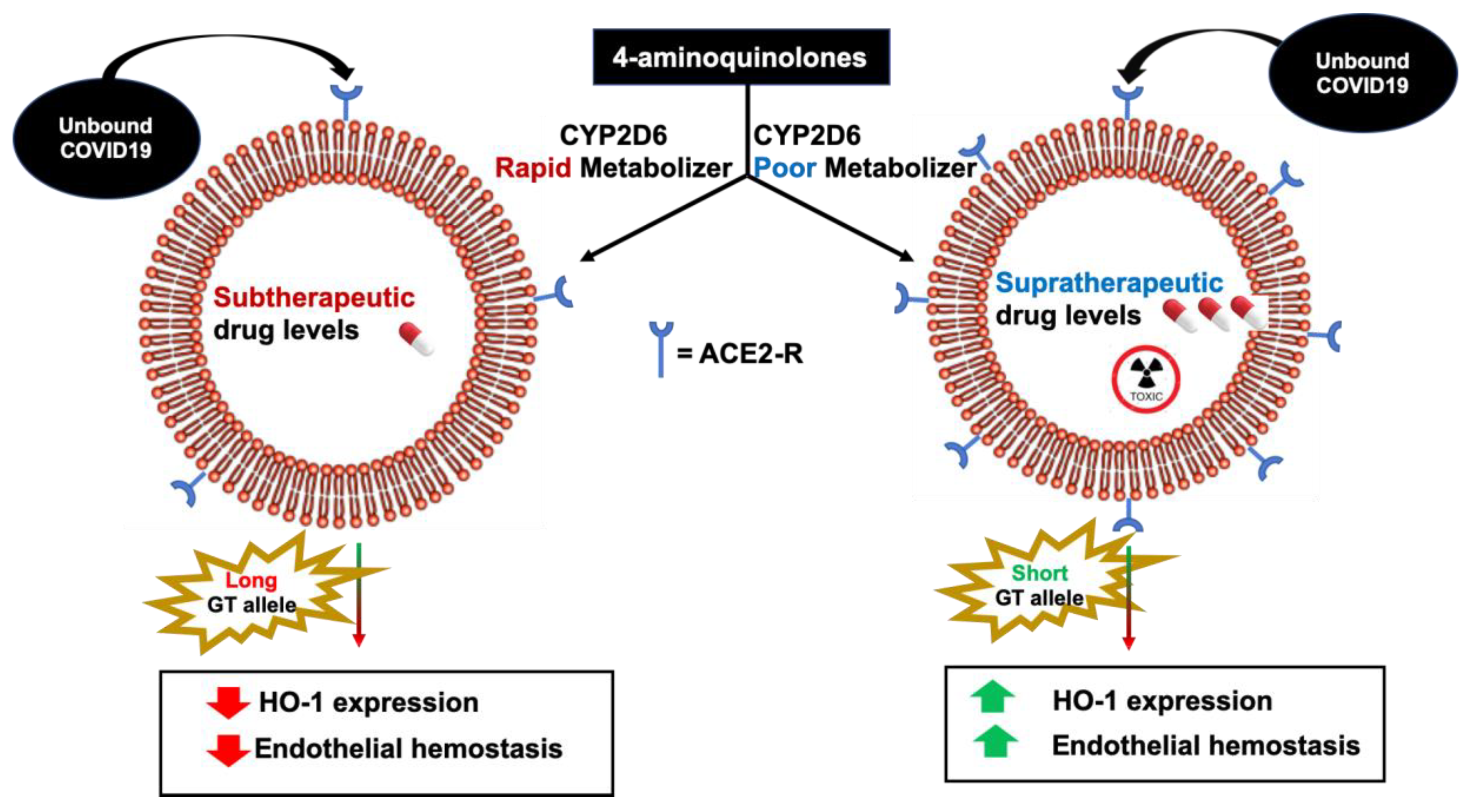
8. Cytochrome 2D6 Genetic Polymorphisms COVID-19
9. Therapeutic Strategies-COVID-19
9.1. Antivirals
9.2. Cytokine Inhibitors
9.3. Antiretrovirals, Monoclonal Antibodies, Convalescent Plasma, and Corticosteroids
10. Heme Arginate-HO-1 Levels—Inflammation
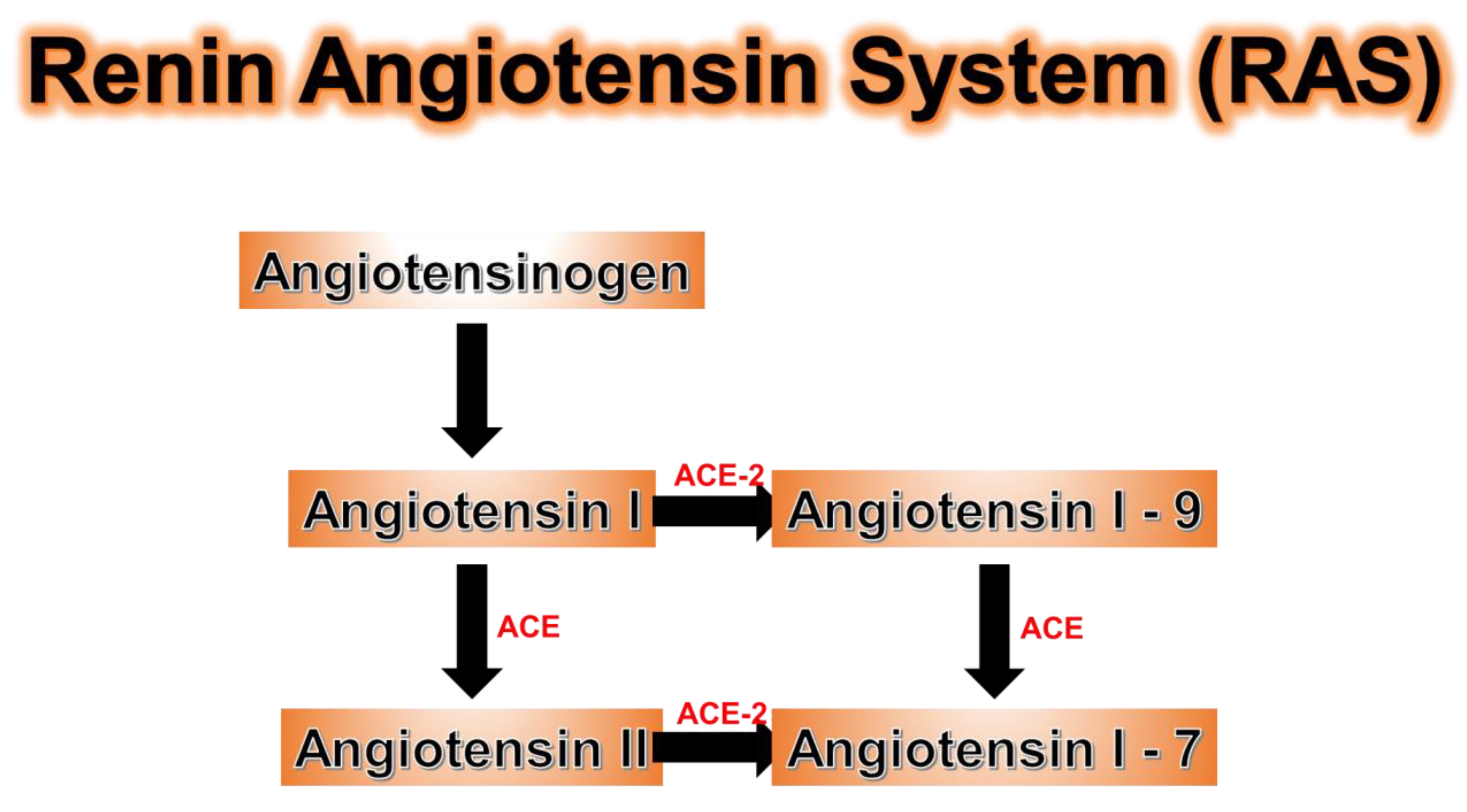
11. ACE Genetic Polymorphisms
12. Conclusions
Funding
Conflicts of Interest
Abbreviations
| ACE | angiotensin converting enzyme |
| HO | heme oxygenase |
| HIV | human immunodeficiency virus |
| SARS | acute respiratory distress syndrome |
| MERS | middle East respiratory syndrome |
| ROS | reactive oxygen species |
| NO | nitric oxide |
| cAMP | cyclic adenosine monophosphate |
| TMPRSS-2 | transmembrane serine protease 2 |
| ETC | electron transport chain |
| OH− | hydroxide |
| p38 MAPK | p38 mitogen-activated protein kinases |
| PBGD | porphobilinogen deaminase |
| ALA | delta-aminolevulinic acid |
| HMB | hydroxymethylbilane |
| ETC | electron transport chain |
| AKI | acute kidney injury |
| ARF | acute renal failure |
| CHF | congestive heart failure |
| AOCI | anemia of chronic inflammation |
| Ox-HDL | oxidized high-density lipoprotein |
| VLDL | very low-density lipoprotein |
| IL | interleukins |
| TNF | tumor necrosis factor |
| NADH | nicotinamide adenine dinucleotide |
| FADH2 | flavin adenine dinucleotide |
| ANTs | adenine nucleotide translocases |
| UCP | uncoupling proteins |
| INOS | inducible nitric oxide synthetase |
| ALT | aspartate transaminase |
| AST | alanine aminotransferase |
| CRP | c-reactive protein |
| LPV/RTV | Lopinavir/ritonavir |
| RTC | randomized clinical trial |
| AZT | azidothymidine |
References
- Sohrabi, C.; Alsafi, Z.; O’Neill, N.; Khan, M.; Kerwan, A.; Al-Jabir, A.; Iosifidis, C.; Agha, R. World Health Organization declares global emergency: A review of the 2019 novel coronavirus (COVID-19). Int. J. Surg. 2020, 76, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Paules, C.I.; Marston, H.D.; Fauci, A.S. Coronavirus Infections-More than Just the Common Cold. JAMA 2020. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.E.L.; Leo, Y.S.; Tan, C.C. COVID-19 in Singapore-Current Experience: Critical Global Issues That Require Attention and Action. JAMA 2020. [Google Scholar] [CrossRef]
- Arutyunov, G.P.; Koziolova, N.A.; Tarlovskaya, E.I.; Arutyunov, A.G.; Grigorjeva, N.Y.; Dzhunusbekova, G.A.; Malchikova, S.V.; Mitkovskaya, N.P.; Orlova, Y.A.; Petrova, M.M.; et al. The Agreed Experts’ Position of the Eurasian Association of Therapists on Some new Mechanisms of COVID-19 Pathways: Focus on Hemostasis, Hemotransfusion Issues and Blood gas Exchange. Kardiologiia 2020, 60, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Read, R. Flawed methods in COVID-19: Attacks the 1-Beta Chain of Hemoglobin and Captures the Porphyrin to Inhibit Human Heme Metabolism. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Abraham, N.G.; Kappas, A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol. Rev. 2008, 60, 79–127. [Google Scholar] [CrossRef]
- Liu, W.; Li, H. COVID-19: Attacks the 1-Beta Chain of Hemoglobin and Captures the Porphyrinto Inhibit Human Heme Metabolism. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Gross, S.; Jahn, C.; Cushman, S.; Bar, C.; Thum, T. SARS-CoV-2 receptor ACE2-dependent implications on the cardiovascular system: From basic science to clinical implications. J. Mol. Cell. Cardiol. 2020, 144, 47–53. [Google Scholar] [CrossRef]
- Martinez, J.C.; Garcia, H.O.; Otheguy, L.E.; Drummond, G.S.; Kappas, A. Control of severe hyperbilirubinemia in full-term newborns with the inhibitor bilirubin production Sn-mesoporphrin. Pediatrics 1999, 103, 1–5. [Google Scholar] [CrossRef]
- Berglund, L.; Angelin, B.; Blomstrand, R.; Drummond, G.; Kappas, A. Sn-protoporphyrin lowers serum bilirubin levels, decreases biliary bilirubin output, enhances biliary heme excretion and potently inhibits hepatic heme oxygenase activity in normal human subjects. Hepatology 1988, 8, 625–631. [Google Scholar] [CrossRef]
- Drummond, G.S.; Galbraith, R.A.; Sardana, M.K.; Kappas, A. Reduction of the C2 and C4 vinyl groups of Sn-protoporphyrin to form Sn- mesoporphyrin markedly enhances the ability of the metalloporphyrin to inhibit in vivo heme catabolism. Arch. Biochem. Biophys. 1987, 255, 64–74. [Google Scholar] [CrossRef]
- Drummond, G.S.; Kappas, A. Sn-protoporphyrin inhibition of fetal and neonatal brain heme oxygenase. Transplacental passage of the metalloporphyrin and prenatal suppression of hyperbilirubinemia in the newborn animal. J. Clin. Investig. 1986, 77, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Sisson, T.R.; Drummond, G.S.; Samonte, D.; Calabio, R.; Kappas, A. Sn-protoporphyrin blocks the increase in serum bilirubin levels that develops postnatally in homozygous Gunn rats. J. Exp. Med. 1988, 167, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Valaes, T.; Drummond, G.S.; Kappas, A. Control of hyperbilirubinemia in glucose-6-phosphate dehydrogenase-deficient newborns using an inhibitor of bilirubin production, Sn-mesoporphyrin. Pediatrics 1998, 101, E1. [Google Scholar] [CrossRef] [PubMed]
- Simionatto, C.S.; Anderson, K.E.; Drummond, G.S.; Kappas, A. Studies on the mechanism of Sn-protoporphyrin suppression of hyperbilirubinemia. Inhibition of heme oxidation and bilirubin production. J. Clin. Investig. 1985, 75, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef]
- Hooper, P.L. COVID-19 and heme oxygenase: Novel insight into the disease and potential therapies. Cell Stress Chaperones 2020. [Google Scholar] [CrossRef]
- Fuller, S.J.; Wiley, J.S. Heme Biosynthesis and Its Disorders: Porphyrias and Sideroblastic Anemias. In Hematology Basic Principles and Practice, 7th ed.; Ronald, H., Leslie, E., Helen, E., Jeffrey, I., John, A., Eds.; Elsevier Health Sciences: New York, NY, USA, 2018; pp. 497–513. [Google Scholar]
- Abraham, N.G.; Kappas, A. Heme oxygenase and the cardiovascular-renal system. Free Radic. Biol. Med. 2005, 39, 1–25. [Google Scholar] [CrossRef]
- Poss, K.D.; Tonegawa, S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924. [Google Scholar] [CrossRef]
- Poss, K.D.; Tonegawa, S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10925–10930. [Google Scholar] [CrossRef]
- Tracz, M.J.; Juncos, J.P.; Croatt, A.J.; Ackerman, A.W.; Grande, J.P.; Knutson, K.L.; Kane, G.C.; Terzic, A.; Griffin, M.D.; Nath, K.A. Deficiency of heme oxygenase-1 impairs renal hemodynamics and exaggerates systemic inflammatory responses to renal ischemia. Kidney Int. 2007, 72, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, A.; Oda, Y.; Yachie, A.; Koizumi, S.; Nakanishi, I. Heme oxygenase-1 deficiency: The first autopsy case. Hum. Pathol. 2002, 33, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Ohta, K.; Yachie, A.; Fujimoto, K.; Kaneda, H.; Wada, T.; Toma, T.; Seno, A.; Kasahara, Y.; Yokoyama, H.; Seki, H.; et al. Tubular injury as a cardinal pathologic feature in human heme oxygenase-1 deficiency. Am. J. Kidney Dis. 2000, 35, 863–870. [Google Scholar] [CrossRef]
- Kartikasari, A.E.; Wagener, F.A.; Yachie, A.; Wiegerinck, E.T.; Kemna, E.H.; Swinkels, D.W. Hepcidin suppression and defective iron recycling account for dysregulation of iron homeostasis in heme oxygenase-1 deficiency. J. Cell. Mol. Med. 2009, 13, 3091–3102. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, N.; Yadav, S.P.; Sachdeva, A.; Pruthi, P.K.; Sawhney, S.; Piplani, T.; Wada, T.; Yachie, A. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J. Pediatr. Hematol. Oncol. 2011, 33, 74–78. [Google Scholar] [CrossRef]
- Rendic, S.; Di Carlo, F.J. Human cytochrome P450 enzymes: A status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab. Rev. 1997, 29, 413–580. [Google Scholar] [CrossRef]
- Manikandan, P.; Nagini, S. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Curr. Drug Targets 2018, 19, 38–54. [Google Scholar] [CrossRef]
- Cavezzi, A.; Troiani, E.; Corrao, S. COVID-19: Hemoglobin, iron, and hypoxia beyond inflammation. A narrative review. Clin. Pract. 2020, 10, 1271. [Google Scholar] [CrossRef]
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Investig. 1999, 103, 129–135. [Google Scholar] [CrossRef]
- Zhang, Q.; Shan, K.S.; Minalyan, A.; O’Sullivan, C.; Nace, T. A Rare Presentation of Coronavirus Disease 2019 (COVID-19) Induced Viral Myositis with Subsequent Rhabdomyolysis. Cureus 2020, 12, e8074. [Google Scholar] [CrossRef]
- Chan, K.H.; Farouji, I.; Abu Hanoud, A.; Slim, J. Weakness and elevated creatinine kinase as the initial presentation of coronavirus disease 2019 (COVID-19). Am. J. Emerg. Med. 2020, 38, 1548.e1–1548.e3. [Google Scholar] [CrossRef] [PubMed]
- Samies, N.L.; Pinninti, S.; James, S.H. Rhabdomyolysis and Acute Renal Failure in an Adolescent with COVID-19. J. Pediatric Infect. Dis. Soc. 2020. [Google Scholar] [CrossRef]
- Valente-Acosta, B.; Moreno-Sanchez, F.; Fueyo-Rodriguez, O.; Palomar-Lever, A. Rhabdomyolysis as an initial presentation in a patient diagnosed with COVID-19. BMJ Case Rep. 2020, 13. [Google Scholar] [CrossRef]
- Nath, K.A.; Balla, G.; Vercellotti, G.M.; Balla, J.; Jacob, H.S.; Levitt, M.D.; Rosenberg, M.E. Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J. Clin. Investig. 1992, 90, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Michaca, L.; Farrugia, G.; Croatt, A.J.; Alam, J.; Nath, K.A. Heme: A determinant of life and death in renal tubular epithelial cells. Am. J. Physiol. Ren. Physiol. 2004, 286, F370–F377. [Google Scholar] [CrossRef] [PubMed]
- Balla, J.; Jacob, H.S.; Balla, G.; Nath, K.; Eaton, J.W.; Vercellotti, G.M. Endothelial-cell heme uptake from heme proteins: Induction of sensitization and desensitization to oxidant damage. Proc. Natl. Acad. Sci. USA 1993, 90, 9285–9289. [Google Scholar] [CrossRef] [PubMed]
- Martel, J.; Ko, Y.F.; Young, J.D.; Ojcius, D.M. Could nasal nitric oxide help to mitigate the severity of COVID-19? Microbes Infect. 2020, 22, 168–171. [Google Scholar] [CrossRef]
- Lei, C.; Su, B.; Dong, H.; Fakhr, B.S.; Grassi, L.G.; Di Fenza, R.; Gianni, S.; Pinciroli, R.; Vassena, E.; Morais, C.C.A.; et al. Protocol for a randomized controlled trial testing inhaled nitric oxide therapy in spontaneously breathing patients with COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Hedenstierna, G.; Chen, L.; Hedenstierna, M.; Lieberman, R.; Fine, D.H. Nitric oxide dosed in short bursts at high concentrations may protect against Covid 19. Nitric Oxide 2020, 103, 1–3. [Google Scholar] [CrossRef]
- Fang, L.; Karakiulakis, G.; Roth, M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir. Med. 2020, 8, e21. [Google Scholar] [CrossRef]
- Mount, P.F.; Power, D.A. Nitric oxide in the kidney: Functions and regulation of synthesis. Acta Physiol. (Oxf.) 2006, 187, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Szilagyi, J.T.; Mishin, V.; Heck, D.E.; Jan, Y.H.; Aleksunes, L.M.; Richardson, J.R.; Heindel, N.D.; Laskin, D.L.; Laskin, J.D. Selective Targeting of Heme Protein in Cytochrome P450 and Nitric Oxide Synthase by Diphenyleneiodonium. Toxicol. Sci. 2016, 151, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R.; Gao, Y.; DeLeon, E.R.; Arif, M.; Arif, F.; Arora, N.; Straub, K.D. Catalase as a sulfide-sulfur oxido-reductase: An ancient (and modern?) regulator of reactive sulfur species (RSS). Redox Biol. 2017, 12, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Ow, Y.P.; Green, D.R.; Hao, Z.; Mak, T.W. Cytochrome c: Functions beyond respiration. Nat. Rev. Mol. Cell Biol. 2008, 9, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Pechlaner, R.; Willeit, P.; Summerer, M.; Santer, P.; Egger, G.; Kronenberg, F.; Demetz, E.; Weiss, G.; Tsimikas, S.; Witztum, J.L.; et al. Heme oxygenase-1 gene promoter microsatellite polymorphism is associated with progressive atherosclerosis and incident cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 229–236. [Google Scholar] [CrossRef]
- Yamada, N.; Yamaya, M.; Okinaga, S.; Nakayama, K.; Sekizawa, K.; Shibahara, S.; Sasaki, H. Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to emphysema. Am. J. Hum. Genet. 2000, 66, 187–195. [Google Scholar] [CrossRef]
- Okamoto, I.; Krogler, J.; Endler, G.; Kaufmann, S.; Mustafa, S.; Exner, M.; Mannhalter, C.; Wagner, O.; Pehamberger, H. A microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with risk for melanoma. Int. J. Cancer 2006, 119, 1312–1315. [Google Scholar] [CrossRef]
- Hirai, H.; Kubo, H.; Yamaya, M.; Nakayama, K.; Numasaki, M.; Kobayashi, S.; Suzuki, S.; Shibahara, S.; Sasaki, H. Microsatellite polymorphism in heme oxygenase-1 gene promoter is associated with susceptibility to oxidant-induced apoptosis in lymphoblastoid cell lines. Blood 2003, 102, 1619–1621. [Google Scholar] [CrossRef]
- Guenegou, A.; Leynaert, B.; Benessiano, J.; Pin, I.; Demoly, P.; Neukirch, F.; Boczkowski, J.; Aubier, M. Association of lung function decline with the heme oxygenase-1 gene promoter microsatellite polymorphism in a general population sample. Results from the European Community Respiratory Health Survey (ECRHS), France. J. Med. Genet. 2006, 43, e43. [Google Scholar] [CrossRef]
- Exner, M.; Schillinger, M.; Minar, E.; Mlekusch, W.; Schlerka, G.; Haumer, M.; Mannhalter, C.; Wagner, O. Heme oxygenase-1 gene promoter microsatellite polymorphism is associated with restenosis after percutaneous transluminal angioplasty. J. Endovasc. Ther. 2001, 8, 433–440. [Google Scholar] [CrossRef]
- Bao, W.; Song, F.; Li, X.; Rong, S.; Yang, W.; Wang, D.; Xu, J.; Fu, J.; Zhao, Y.; Liu, L. Association between heme oxygenase-1 gene promoter polymorphisms and type 2 diabetes mellitus: A HuGE review and meta-analysis. Am. J. Epidemiol. 2010, 172, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.W.; Yeung, V.T.; Benzie, I.F. Heme oxygenase microsatellite polymorphism, oxidative stress, glycemic control, and complication development in type 2 diabetes patients. Free Radic. Biol. Med. 2012, 53, 60–63. [Google Scholar] [CrossRef]
- Endler, G.; Exner, M.; Schillinger, M.; Marculescu, R.; Sunder-Plassmann, R.; Raith, M.; Jordanova, N.; Wojta, J.; Mannhalter, C.; Wagner, O.F.; et al. A microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with increased bilirubin and HDL levels but not with coronary artery disease. Thromb. Haemost. 2004, 91, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Abraham, N.G.; Junge, J.M.; Drummond, G.S. Translational Significance of Heme Oxygenase in Obesity and Metabolic Syndrome. Trends Pharmacol. Sci. 2016, 37, 17–36. [Google Scholar] [CrossRef] [PubMed]
- Vitek, L.; Jirsa, M.; Brodanova, M.; Kalab, M.; Marecek, Z.; Danzig, V.; Novotny, L.; Kotal, P. Gilbert syndrome and ischemic heart disease: A protective effect of elevated bilirubin levels. Atherosclerosis 2002, 160, 449–456. [Google Scholar] [CrossRef]
- Kalousova, M.; Novotny, L.; Zima, T.; Braun, M.; Vitek, L. Decreased levels of advanced glycation end-products in patients with Gilbert syndrome. Cell. Mol. Biol. (Noisy-le-Grand) 2005, 51, 387–392. [Google Scholar]
- Maruhashi, T.; Soga, J.; Fujimura, N.; Idei, N.; Mikami, S.; Iwamoto, Y.; Kajikawa, M.; Matsumoto, T.; Kihara, Y.; Chayama, K.; et al. Hyperbilirubinemia, augmentation of endothelial function, and decrease in oxidative stress in Gilbert syndrome. Circulation 2012, 126, 598–603. [Google Scholar] [CrossRef]
- Zhou, Y.; Rui, L. Leptin signaling and leptin resistance. Front. Med. 2013, 7, 207–222. [Google Scholar] [CrossRef]
- Peterson, S.J.; Dave, N.; Kothari, J. The Effects of Heme Oxygenase Upregulation on Obesity and the Metabolic Syndrome. Antioxid. Redox Signal. 2020, 32, 1061–1070. [Google Scholar] [CrossRef]
- Zhang, Y.; Scarpace, P.J. The role of leptin in leptin resistance and obesity. Physiol. Behav. 2006, 88, 249–256. [Google Scholar] [CrossRef]
- LaPensee, C.R.; Hugo, E.R.; Ben-Jonathan, N. Insulin stimulates interleukin-6 expression and release in LS14 human adipocytes through multiple signaling pathways. Endocrinology 2008, 149, 5415–5422. [Google Scholar] [CrossRef] [PubMed]
- Zammit, V.A.; Waterman, I.J.; Topping, D.; McKay, G. Insulin stimulation of hepatic triacylglycerol secretion and the etiology of insulin resistance. J. Nutr. 2001, 131, 2074–2077. [Google Scholar] [CrossRef]
- Peterson, S.J.; Shapiro, J.I.; Thompson, E.; Singh, S.; Liu, L.; Weingarten, J.A.; O’Hanlon, K.; Bialczak, A.; Bhesania, S.R.; Abraham, N.G. Oxidized HDL, Adipokines, and Endothelial Dysfunction: A Potential Biomarker Profile for Cardiovascular Risk in Women with Obesity. Obesity (Silver Spring) 2019, 27, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.J.; Vanella, L.; Bialczak, A.; Schragenheim, J.; Li, M.; Bellner, L.; Shapiro, J.I.; Abraham, N.G. Oxidized HDL and Isoprostane Exert a Potent Adipogenic Effect on Stem Cells: Where in the Lineage? Cell Stem Cells Regen. Med. 2016, 2, 2472–6990. [Google Scholar] [CrossRef] [PubMed]
- Aghagoli, G.; Gallo Marin, B.; Soliman, L.B.; Sellke, F.W. Cardiac involvement in COVID-19 patients: Risk factors, predictors, and complications: A review. J. Card. Surg. 2020, 35, 1302–1305. [Google Scholar] [CrossRef]
- Kruger, A.L.; Peterson, S.; Turkseven, S.; Kaminski, P.M.; Zhang, F.F.; Quan, S.; Wolin, M.S.; Abraham, N.G. D-4F induces heme oxygenase-1 and extracellular superoxide dismutase, decreases endothelial cell sloughing, and improves vascular reactivity in rat model of diabetes. Circulation 2005, 111, 3126–3134. [Google Scholar] [CrossRef]
- Singh, S.; McClung, J.; Thompson, E.; Glick, Y.; Greenberg, M.; Acosta-Baez, G.; Edris, B.; Shapiro, J.; Abraham, N.G. Cardioprotective heme oxygenase-1-PGC-1α signaling in epicardial fat attenuates cardiovascular risk in humans as in obese mice. Obesity (Silver Spring) 2019. [Google Scholar] [CrossRef]
- Peterson, S.J.; Yadav, R.; Iacobellis, G. Cardioprotective Heme Oxygenase 1-PGC1alpha Signaling in Epicardial Fat Attenuates Cardiovascular Risk in Humans as in Obese Mice. Obesity (Silver Spring) 2019, 27, 1560–1561. [Google Scholar] [CrossRef]
- Peterson, S.J.; Rubinstein, R.; Faroqui, M.; Raza, A.; Boumaza, I.; Zhang, Y.; Stec, D.; Abraham, N.G. Positive Effects of Heme Oxygenase Upregulation on Adiposity and Vascular Dysfunction: Gene Targeting vs. Pharmacologic Therapy. Int. J. Mol. Sci. 2019, 20, 2514. [Google Scholar] [CrossRef]
- Goossens, G.H. The Metabolic Phenotype in Obesity: Fat Mass, Body Fat Distribution, and Adipose Tissue Function. Obes. Facts 2017, 10, 207–215. [Google Scholar] [CrossRef]
- Trayhurn, P.; Wood, I.S. Adipokines: Inflammation and the pleiotropic role of white adipose tissue. Br. J. Nutr. 2004, 92, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M. The Cell: A Molecular Approach, 2nd ed.; Sinauer Associate: Sunderland, UK, 2000. [Google Scholar]
- Smeitink, J.; van den Heuvel, L.; DiMauro, S. The genetics and pathology of oxidative phosphorylation. Nat. Rev. Genet. 2001, 2, 342–352. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Apostolova, N.; Victor, V.M. Molecular strategies for targeting antioxidants to mitochondria: Therapeutic implications. Antioxid. Redox Signal. 2015, 22, 686–729. [Google Scholar] [CrossRef]
- Elattar, S.; Satyanarayana, A. Can Brown Fat Win the Battle against White Fat? J. Cell. Physiol. 2015, 230, 2311–2317. [Google Scholar] [CrossRef] [PubMed]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef]
- Berry, B.J.; Trewin, A.J.; Amitrano, A.M.; Kim, M.; Wojtovich, A.P. Use the Protonmotive Force: Mitochondrial Uncoupling and Reactive Oxygen Species. J. Mol. Biol. 2018, 430, 3873–3891. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Schragenheim, J.; Cao, J.; Falck, J.R.; Abraham, N.G.; Bellner, L. PGC-1 alpha regulates HO-1 expression, mitochondrial dynamics and biogenesis: Role of epoxyeicosatrienoic acid. Prostaglandins Other Lipid Mediat. 2016, 125, 8–18. [Google Scholar] [CrossRef]
- Schragenheim, J.; Maayan, O.; Abraham, N.G. Chapter 4: HO-1-derived CO Is a Regulator of Vascular Function and Metabolic Syndrome. RSC Metallobiology 2018, 59–100. [Google Scholar] [CrossRef]
- Stuart, J.A.; Brindle, K.M.; Harper, J.A.; Brand, M.D. Mitochondrial proton leak and the uncoupling proteins. J. Bioenerg. Biomembr. 1999, 31, 517–525. [Google Scholar] [CrossRef]
- Kaczara, P.; Motterlini, R.; Rosen, G.M.; Augustynek, B.; Bednarczyk, P.; Szewczyk, A.; Foresti, R.; Chlopicki, S. Carbon monoxide released by CORM-401 uncouples mitochondrial respiration and inhibits glycolysis in endothelial cells: A role for mitoBKCa channels. Biochim. Biophys. Acta 2015, 1847, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Jezek, P.; Holendova, B.; Garlid, K.D.; Jaburek, M. Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling. Antioxid. Redox Signal. 2018, 29, 667–714. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R.; Foresti, R. Biological signaling by carbon monoxide and carbon monoxide-releasing molecules. Am. J. Physiol. Cell Physiol. 2017, 312, C302–C313. [Google Scholar] [CrossRef]
- Wilson, J.L.; Bouillaud, F.; Almeida, A.S.; Vieira, H.L.; Ouidja, M.O.; Dubois-Rande, J.L.; Foresti, R.; Motterlini, R. Carbon monoxide reverses the metabolic adaptation of microglia cells to an inflammatory stimulus. Free Radic. Biol. Med. 2017, 104, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Choi, A.M. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl. Res. 2016, 167, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Weaver, L.; Hamoud, A.R.; Stec, D.E.; Hinds, T.D., Jr. Biliverdin reductase and bilirubin in hepatic disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G668–G676. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Hosick, P.A.; Chen, S.; Tukey, R.H.; Hankins, M.W.; Nestor-Kalinoski, A.; Stec, D.E. Mice with hyperbilirubinemia due to Gilbert’s syndrome polymorphism are resistant to hepatic steatosis by decreased serine 73 phosphorylation of PPARalpha. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E244–E252. [Google Scholar] [CrossRef]
- Singh, S.P.; Greenberg, M.; Glick, Y.; Bellner, L.; Favero, G.; Rezzani, R.; Rodella, L.F.; Agostinucci, K.; Shapiro, J.I.; Abraham, N.G. Adipocyte Specific HO-1 Gene Therapy is Effective in Antioxidant Treatment of Insulin Resistance and Vascular Function in an Obese Mice Model. Antioxidants 2020, 9, 40. [Google Scholar] [CrossRef]
- Waldman, M.; Bellner, L.; Vanella, L.; Schragenheim, J.; Sodhi, K.; Singh, S.P.; Lin, D.; Lakhkar, A.; Li, J.; Hochhauser, E.; et al. Epoxyeicosatrienoic Acids Regulate Adipocyte Differentiation of Mouse 3T3 Cells, Via PGC-1alpha Activation, Which Is Required for HO-1 Expression and Increased Mitochondrial Function. Stem Cells Dev. 2016, 25, 1084–1094. [Google Scholar] [CrossRef]
- Zhu, J.; Ji, P.; Pang, J.; Zhong, Z.; Li, H.; He, C.; Zhang, J.; Zhao, C. Clinical characteristics of 3062 COVID-19 patients: A meta-analysis. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Slaats, J.; Ten Oever, J.; van de Veerdonk, F.L.; Netea, M.G. IL-1beta/IL-6/CRP and IL-18/ferritin: Distinct Inflammatory Programs in Infections. PLoS Pathog. 2016, 12, e1005973. [Google Scholar] [CrossRef] [PubMed]
- Pulivarthi, S.; Gurram, M.K. Effectiveness of d-dimer as a screening test for venous thromboembolism: An update. N. Am. J. Med. Sci. 2014, 6, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Kollias, A.; Kyriakoulis, K.G.; Dimakakos, E.; Poulakou, G.; Stergiou, G.S.; Syrigos, K. Thromboembolic risk and anticoagulant therapy in COVID-19 patients: Emerging evidence and call for action. Br. J. Haematol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pei, G.; Zhang, Z.; Peng, J.; Liu, L.; Zhang, C.; Yu, C.; Ma, Z.; Huang, Y.; Liu, W.; Yao, Y.; et al. Renal Involvement and Early Prognosis in Patients with COVID-19 Pneumonia. J. Am. Soc. Nephrol. 2020. [Google Scholar] [CrossRef]
- Danser, A.H.J.; Epstein, M.; Batlle, D. Renin-Angiotensin System Blockers and the COVID-19 Pandemic: At Present There Is No Evidence to Abandon Renin-Angiotensin System Blockers. Hypertension 2020, 75, 1382–1385. [Google Scholar] [CrossRef] [PubMed]
- Mancia, G.; Rea, F.; Ludergnani, M.; Apolone, G.; Corrao, G. Renin-Angiotensin-Aldosterone System Blockers and the Risk of Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Fosbol, E.L.; Butt, J.H.; Ostergaard, L.; Andersson, C.; Selmer, C.; Kragholm, K.; Schou, M.; Phelps, M.; Gislason, G.H.; Gerds, T.A.; et al. Association of Angiotensin-Converting Enzyme Inhibitor or Angiotensin Receptor Blocker Use with COVID-19 Diagnosis and Mortality. JAMA 2020. [Google Scholar] [CrossRef]
- Goyal, P.; Choi, J.J.; Pinheiro, L.C.; Schenck, E.J.; Chen, R.; Jabri, A.; Satlin, M.J.; Campion, T.R., Jr.; Nahid, M.; Ringel, J.B.; et al. Clinical Characteristics of Covid-19 in New York City. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, G.; Zangrillo, A.; Zanella, A.; Antonelli, M.; Cabrini, L.; Castelli, A.; Cereda, D.; Coluccello, A.; Foti, G.; Fumagalli, R.; et al. Baseline Characteristics and Outcomes of 1591 Patients Infected with SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA 2020. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients with 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020. [Google Scholar] [CrossRef]
- Savarino, A.; Di Trani, L.; Donatelli, I.; Cauda, R.; Cassone, A. New insights into the antiviral effects of chloroquine. Lancet Infect. Dis. 2006, 6, 67–69. [Google Scholar] [CrossRef]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): Clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 2005, 5, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Haertter, S. Recent examples on the clinical relevance of the CYP2D6 polymorphism and endogenous functionality of CYP2D6. Drug Metabol. Drug Interact. 2013, 28, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.J.; Poland, R.E.; Han, G.; Konishi, T.; Zheng, Y.P.; Berman, N.; Lin, K.M. Analysis of the CYP2D6 gene polymorphism and enzyme activity in African-Americans in southern California. Pharmacogenetics 2001, 11, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Yee, M.M.; Josephson, C.; Hill, C.E.; Harrington, R.; Castillejo, M.I.; Ramjit, R.; Osunkwo, I. Cytochrome P450 2D6 polymorphisms and predicted opioid metabolism in African American children with sickle cell disease. J. Pediatr. Hematol. Oncol. 2013, 35, e301–e305. [Google Scholar] [CrossRef] [PubMed]
- Gaedigk, A.; Bhathena, A.; Ndjountche, L.; Pearce, R.E.; Abdel-Rahman, S.M.; Alander, S.W.; Bradford, L.D.; Rogan, P.K.; Leeder, J.S. Identification and characterization of novel sequence variations in the cytochrome P4502D6 (CYP2D6) gene in African Americans. Pharmacogenomics J. 2005, 5, 173–182. [Google Scholar] [CrossRef]
- Lee, J.Y.; Vinayagamoorthy, N.; Han, K.; Kwok, S.K.; Ju, J.H.; Park, K.S.; Jung, S.H.; Park, S.W.; Chung, Y.J.; Park, S.H. Association of Polymorphisms of Cytochrome P450 2D6 with Blood Hydroxychloroquine Levels in Patients with Systemic Lupus Erythematosus. Arthritis Rheumatol. 2016, 68, 184–190. [Google Scholar] [CrossRef]
- Wahie, S.; Daly, A.K.; Cordell, H.J.; Goodfield, M.J.; Jones, S.K.; Lovell, C.R.; Carmichael, A.J.; Carr, M.M.; Drummond, A.; Natarajan, S.; et al. Clinical and pharmacogenetic influences on response to hydroxychloroquine in discoid lupus erythematosus: A retrospective cohort study. J. Investig. Dermatol. 2011, 131, 1981–1986. [Google Scholar] [CrossRef]
- He, X.; Pan, M.; Zeng, W.; Zou, C.; Pi, L.; Qin, Y.; Zhao, L.; Qin, P.; Lu, Y.; Baird, J.K.; et al. Multiple relapses of Plasmodium vivax malaria acquired from West Africa and association with poor metabolizer CYP2D6 variant: A case report. BMC Infect. Dis. 2019, 19, 704. [Google Scholar] [CrossRef]
- Haraya, K.; Kato, M.; Chiba, K.; Sugiyama, Y. Prediction of inter-individual variability on the pharmacokinetics of CYP2C8 substrates in human. Drug Metab. Pharmacokinet. 2017, 32, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Silvino, A.C.; Costa, G.L.; Araujo, F.C.; Ascher, D.B.; Pires, D.E.; Fontes, C.J.; Carvalho, L.H.; Brito, C.F.; Sousa, T.N. Variation in Human Cytochrome P-450 Drug-Metabolism Genes: A Gateway to the Understanding of Plasmodium vivax Relapses. PLoS ONE 2016, 11, e0160172. [Google Scholar] [CrossRef]
- Wang, B.; Yang, L.P.; Zhang, X.Z.; Huang, S.Q.; Bartlam, M.; Zhou, S.F. New insights into the structural characteristics and functional relevance of the human cytochrome P450 2D6 enzyme. Drug Metab. Rev. 2009, 41, 573–643. [Google Scholar] [CrossRef] [PubMed]
- Mottet, F.; Vardeny, O.; de Denus, S. Pharmacogenomics of heart failure: A systematic review. Pharmacogenomics 2016, 17, 1817–1858. [Google Scholar] [CrossRef] [PubMed]
- Qaseem, A.; Yost, J.; Etxeandia-Ikobaltzeta, I.; Miller, M.C.; Abraham, G.M.; Obley, A.J.; Forciea, M.A.; Jokela, J.A.; Humphrey, L.L. Should Clinicians Use Chloroquine or Hydroxychloroquine Alone or in Combination with Azithromycin for the Prophylaxis or Treatment of COVID-19? Ann. Intern. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Geleris, J.; Sun, Y.; Platt, J.; Zucker, J.; Baldwin, M.; Hripcsak, G.; Labella, A.; Manson, D.K.; Kubin, C.; Barr, R.G.; et al. Observational Study of Hydroxychloroquine in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2020, 382, 2411–2418. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.L.; Andres, E.L.; Sims, A.C.; Graham, R.L.; Sheahan, T.P.; Lu, X.; Smith, E.C.; Case, J.B.; Feng, J.Y.; Jordan, R.; et al. Coronavirus Susceptibility to the Antiviral Remdesivir (GS-5734) Is Mediated by the Viral Polymerase and the Proofreading Exoribonuclease. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, T.P.; Sims, A.C.; Leist, S.R.; Schafer, A.; Won, J.; Brown, A.J.; Montgomery, S.A.; Hogg, A.; Babusis, D.; Clarke, M.O.; et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 2020, 11, 222. [Google Scholar] [CrossRef]
- de Wit, E.; Feldmann, F.; Cronin, J.; Jordan, R.; Okumura, A.; Thomas, T.; Scott, D.; Cihlar, T.; Feldmann, H. Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc. Natl. Acad. Sci. USA 2020, 117, 6771–6776. [Google Scholar] [CrossRef]
- Werk, A.N.; Cascorbi, I. Functional gene variants of CYP3A4. Clin. Pharmacol. Ther. 2014, 96, 340–348. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Du, G. Remdesivir in Adults with Severe COVID-19: A Randomized, Double-Blind, Placebo-Controlled, Multicenter Trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Du, Y.X.; Chen, X.P. Favipiravir: Pharmacokinetics and Concerns about Clinical Trials for 2019-nCoV Infection. Clin. Pharmacol. Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Yang, M.; Liu, D.; Chen, J.; Shu, D.; Xia, J.; Liao, X.; Gu, Y.; Cai, Q.; Yang, Y.; et al. Experimental Treatment with Favipiravir for COVID-19: An Open-Label Control Study. Engineering (Beijing) 2020. [Google Scholar] [CrossRef] [PubMed]
- Negahdaripour, M. The Battle against COVID-19: Where Do We Stand Now? Iran. J. Med. Sci. 2020, 45, 81–82. [Google Scholar] [CrossRef]
- Yazdanpanah, Y.; Guery, B. Antiretroviral drugs in severe acute respiratory syndrome. Presse Med. 2006, 35, 105–107. [Google Scholar] [CrossRef]
- Hui, D.S.; Wong, G.W. Advancements in the battle against severe acute respiratory syndrome. Expert Opin. Pharmacother. 2004, 5, 1687–1693. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- CN, C.C. Kaletra Capsules and Oral Solution. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2007/021226s018lbl.pdf (accessed on 30 June 2020).
- Martinez, M.A. Compounds with Therapeutic Potential against Novel Respiratory 2019 Coronavirus. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Fu, B.; Xu, X.; Wei, H. Why tocilizumab could be an effective treatment for severe COVID-19? J. Transl. Med. 2020, 18, 164. [Google Scholar] [CrossRef]
- Chaidos, A.; Katsarou, A.; Mustafa, C.; Milojkovic, D.; Karadimitris, A. Interleukin 6-blockade treatment for severe COVID-19 in two patients with multiple myeloma. Br. J. Haematol. 2020. [Google Scholar] [CrossRef]
- Zhang, X.; Peck, R. Clinical pharmacology of tocilizumab for the treatment of patients with rheumatoid arthritis. Expert Rev. Clin. Pharmacol. 2011, 4, 539–558. [Google Scholar] [CrossRef] [PubMed]
- Miao, M.; De Clercq, E.; Li, G. Clinical significance of chemokine receptor antagonists. Expert Opin. Drug Metab. Toxicol. 2020, 16, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Kotch, C.; Barrett, D.; Teachey, D.T. Tocilizumab for the treatment of chimeric antigen receptor T cell-induced cytokine release syndrome. Expert Rev. Clin. Immunol. 2019, 15, 813–822. [Google Scholar] [CrossRef]
- Jiao, X.; Nawab, O.; Patel, T.; Kossenkov, A.V.; Halama, N.; Jaeger, D.; Pestell, R.G. Recent Advances Targeting CCR5 for Cancer and Its Role in Immuno-Oncology. Cancer Res. 2019, 79, 4801–4807. [Google Scholar] [CrossRef] [PubMed]
- Lederman, M.M.; Penn-Nicholson, A.; Cho, M.; Mosier, D. Biology of CCR5 and its role in HIV infection and treatment. JAMA 2006, 296, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M. Cytokine storm and immunomodulatory therapy in COVID-19: Role of chloroquine and anti-IL-6 monoclonal antibodies. Int. J. Antimicrob. Agents 2020, 105982. [Google Scholar] [CrossRef]
- Chary, M.A.; Barbuto, A.F.; Izadmehr, S.; Hayes, B.D.; Burns, M.M. COVID-19: Therapeutics and Their Toxicities. J. Med. Toxicol. 2020. [Google Scholar] [CrossRef]
- Pandey, S.; Vyas, G.N. Adverse effects of plasma transfusion. Transfusion 2012, 52 (Suppl. 1), 65S–79S. [Google Scholar] [CrossRef]
- Shang, L.; Zhao, J.; Hu, Y.; Du, R.; Cao, B. On the use of corticosteroids for 2019-nCoV pneumonia. Lancet 2020, 395, 683–684. [Google Scholar] [CrossRef]
- Ledford, H. Coronavirus breakthrough: Dexamethasone is first drug shown to save lives. Nature 2020. [Google Scholar] [CrossRef]
- Lanteri, R.; Acquaviva, R.; Di, G.C.; Caltabiano, R.; Li, D.G.; Vanella, L.; Santangelo, M.; Lanzafame, S.; Di, C.A. Heme oxygenase 1 expression in postischemic reperfusion liver damage: Effect of L-arginine. Microsurgery 2006, 26, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Staudinger, R.; Abraham, N.G.; Levere, R.D.; Kappas, A. Inhibition of human immunodeficiency virus-1 reverse transcriptase by heme and synthetic heme analogs. Proc. Assoc. Am. Physicians 1996, 108, 47–54. [Google Scholar]
- Abraham, N.G.; Chertkov, J.L.; Staudinger, R.; Jiang, S.; Lutton, J.D.; Argani, I.; Levere, R.D.; Kappas, A. Long-term bone marrow stromal and hemopoietic toxicity to AZT: Protective role of heme and IL-1. Exp. Hematol. 1993, 21, 263–268. [Google Scholar]
- Levere, R.D.; Gong, Y.F.; Kappas, A.; Bucher, D.J.; Wormser, G.P.; Abraham, N.G. Heme inhibits human immunodeficiency virus 1 replication in cell cultures and enhances the antiviral effect of zidovudine. Proc. Natl. Acad. Sci. USA 1991, 88, 1756–1759. [Google Scholar] [CrossRef] [PubMed]
- Hill-Batorski, L.; Halfmann, P.; Neumann, G.; Kawaoka, Y. The cytoprotective enzyme heme oxygenase-1 suppresses Ebola virus replication. J. Virol. 2013, 87, 13795–13802. [Google Scholar] [CrossRef] [PubMed]
- Toledo, S.L.O.; Guedes, J.V.M.; Alpoim, P.N.; Rios, D.R.A.; Pinheiro, M.B. Sickle cell disease: Hemostatic and inflammatory changes, and their interrelation. Clin. Chim. Acta 2019, 493, 129–137. [Google Scholar] [CrossRef]
- Nath, K.A.; Grande, J.P.; Haggard, J.J.; Croatt, A.J.; Katusic, Z.S.; Solovey, A.; Hebbel, R.P. Oxidative stress and induction of heme oxygenase-1 in the kidney in sickle cell disease. Am. J. Pathol. 2001, 158, 893–903. [Google Scholar] [CrossRef]
- Balla, G.; Jacob, H.S.; Balla, J.; Rosenberg, M.; Nath, K.; Apple, F.; Eaton, J.W.; Vercellotti, G.M. Ferritin: A cytoprotective antioxidant strategem of endothelium. J. Biol. Chem. 1992, 267, 18148–18153. [Google Scholar]
- Eisenstein, R.S.; Garcia-Mayol, D.; Pettingell, W.; Munro, H.N. Regulation of ferritin and heme oxygenase synthesis in rat fibroblasts by different forms of iron. Proc. Natl. Acad. Sci. USA 1991, 88, 688–692. [Google Scholar] [CrossRef]
- Mancuso, C.; Bonsignore, A.; Di, S.E.; Mordente, A.; Motterlini, R. Bilirubin and S-nitrosothiols interaction: Evidence for a possible role of bilirubin as a scavenger of nitric oxide. Biochem. Pharmacol. 2003, 66, 2355–2363. [Google Scholar] [CrossRef]
- Schwartzman, M.L.; Martasek, P.; Rios, A.R.; Levere, R.D.; Solangi, K.; Goodman, A.I.; Abraham, N.G. Cytochrome P450-dependent arachidonic acid metabolism in human kidney. Kidney Int. 1990, 37, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Sacerdoti, D.; Escalante, B.; Abraham, N.G.; McGiff, J.C.; Levere, R.D.; Schwartzman, M.L. Treatment with tin prevents the development of hypertension in spontaneously hypertensive rats. Science 1989, 243, 388–390. [Google Scholar] [CrossRef] [PubMed]
- Puri, N.; Arefiev, Y.; Chao, R.; Sacerdoti, D.; Chaudry, H.; Nichols, A.; Srikanthan, K.; Nawab, A.; Sharma, D.; Lakhani, V.H.; et al. Heme Oxygenase Induction Suppresses Hepatic Hepcidin and Rescues Ferroportin and Ferritin Expression in Obese Mice. J. Nutr. Metab. 2017, 2017, 4964571. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.H.; Peterson, S.J.; Bellner, L.; Choudhary, A.; Levy, L.; Gancz, L.; Sasson, A.; Trainer, J.; Rezzani, R.; Resnick, A.; et al. Cold-Pressed Nigella Sativa Oil Standardized to 3% Thymoquinone Potentiates Omega-3 Protection against Obesity-Induced Oxidative Stress, Inflammation, and Markers of Insulin Resistance Accompanied with Conversion of White to Beige Fat in Mice. Antioxidants 2020, 9, 489. [Google Scholar] [CrossRef]
- Raffaelle, M.L.; Amin, S.; Alex, R.; Shen, H.H.; Singh, S.; Vanella, L.; Rezzani, R.; Bonomini, F.; Peterson, S.J.; Abraham, N.G. Dietary Supplementation of Cold Press Pomegranate Seed Oil Attenuates Hepatic Steatosis Fibrosis through Antioxidant and Mitochondrial Pathways in Obese Mice. Int. J. Mol. Sci. 2020. under review. [Google Scholar]
- Sayed-Tabatabaei, F.A.; Oostra, B.A.; Isaacs, A.; van Duijn, C.M.; Witteman, J.C. ACE polymorphisms. Circ. Res. 2006, 98, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Cassis, L.A.; Kooi, C.W.; Daugherty, A. Structure and functions of angiotensinogen. Hypertens. Res. 2016, 39, 492–500. [Google Scholar] [CrossRef]
- Wu, C.; Lu, H.; Cassis, L.A.; Daugherty, A. Molecular and Pathophysiological Features of Angiotensinogen: A Mini Review. N. Am. J. Med. Sci. (Boston) 2011, 4, 183–190. [Google Scholar] [CrossRef]
- Yiannikouris, F.; Gupte, M.; Putnam, K.; Thatcher, S.; Charnigo, R.; Rateri, D.L.; Daugherty, A.; Cassis, L.A. Adipocyte deficiency of angiotensinogen prevents obesity-induced hypertension in male mice. Hypertension 2012, 60, 1524–1530. [Google Scholar] [CrossRef]
- Slamkova, M.; Zorad, S.; Krskova, K. Alternative renin-angiotensin system pathways in adipose tissue and their role in the pathogenesis of obesity. Endocr. Regul. 2016, 50, 229–240. [Google Scholar] [CrossRef][Green Version]
- Li, M.Y.; Li, L.; Zhang, Y.; Wang, X.S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Kassir, R. Risk of COVID-19 for patients with obesity. Obes. Rev. 2020, 21, e13034. [Google Scholar] [CrossRef] [PubMed]
- Kaklamani, V.G.; Wisinski, K.B.; Sadim, M.; Gulden, C.; Do, A.; Offit, K.; Baron, J.A.; Ahsan, H.; Mantzoros, C.; Pasche, B. Variants of the adiponectin (ADIPOQ) and adiponectin receptor 1 (ADIPOR1) genes and colorectal cancer risk. JAMA 2008, 300, 1523–1531. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Consequences of Hemoprotein Malfunction/Deficiency | |||
|---|---|---|---|
| Hemoprotein | Deficiency Consequence | Clinical Manifestation | Main Organs Affected |
| Catalase | ↑ ROS formation | Tissue damage | High [ACE-2R] organs |
| Cytochrome of the ETC | ↓ Aerobic respiration ↑ Apoptosis | Tissue damage | High [ACE-2R] organs |
| NO synthetase | ↓ vessel hemostasis ↓ smooth M. relaxation ↑ cardiac contractility ↓ regulation of renal hemodynamics, sodium regulation, and tubuloglomerular feedback | Prothrombotic state Vasoconstriction CHF/tachycardia AKI/ARF | High [ACE-2R] organs |
| Myoglobin | ↑ Rhabdomyolysis | Rhabdomyolysis Acute kidney injury Electrolyte derangement | Skeletal muscle |
| Hemoglobin | ↓ O2 and CO2 affinity ↓ Production | Prolonged and increasing O2 requirements Anemia (AOCI) | RBCs |
| All cells with porphyrin in membrane | No deficiency. Allows COVID-19 to bind with high affinity leading to infection | Progressive tissue damage, especially as virus continues to replicate within the cell | Low [ACE-2R] organs |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fakhouri, E.W.; Peterson, S.J.; Kothari, J.; Alex, R.; Shapiro, J.I.; Abraham, N.G. Genetic Polymorphisms Complicate COVID-19 Therapy: Pivotal Role of HO-1 in Cytokine Storm. Antioxidants 2020, 9, 636. https://doi.org/10.3390/antiox9070636
Fakhouri EW, Peterson SJ, Kothari J, Alex R, Shapiro JI, Abraham NG. Genetic Polymorphisms Complicate COVID-19 Therapy: Pivotal Role of HO-1 in Cytokine Storm. Antioxidants. 2020; 9(7):636. https://doi.org/10.3390/antiox9070636
Chicago/Turabian StyleFakhouri, Eddie W., Stephen J. Peterson, Janish Kothari, Ragin Alex, Joseph I. Shapiro, and Nader G. Abraham. 2020. "Genetic Polymorphisms Complicate COVID-19 Therapy: Pivotal Role of HO-1 in Cytokine Storm" Antioxidants 9, no. 7: 636. https://doi.org/10.3390/antiox9070636
APA StyleFakhouri, E. W., Peterson, S. J., Kothari, J., Alex, R., Shapiro, J. I., & Abraham, N. G. (2020). Genetic Polymorphisms Complicate COVID-19 Therapy: Pivotal Role of HO-1 in Cytokine Storm. Antioxidants, 9(7), 636. https://doi.org/10.3390/antiox9070636