Targeting the MAPK/ERK and PI3K/AKT Signaling Pathways Affects NRF2, Trx and GSH Antioxidant Systems in Leukemia Cells


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Inhibitors
2.2. Cell Culture
2.3. Cell Viability Assay
2.4. Drug Interaction Analysis
2.5. PI3K/MAPK Activity Assay
2.6. Apoptosis Assay
2.7. Oxidative Stress Assay
2.8. Determination of GSH/GSSG Ratio
2.9. Western Blotting
2.10. Statistical Analysis
3. Results
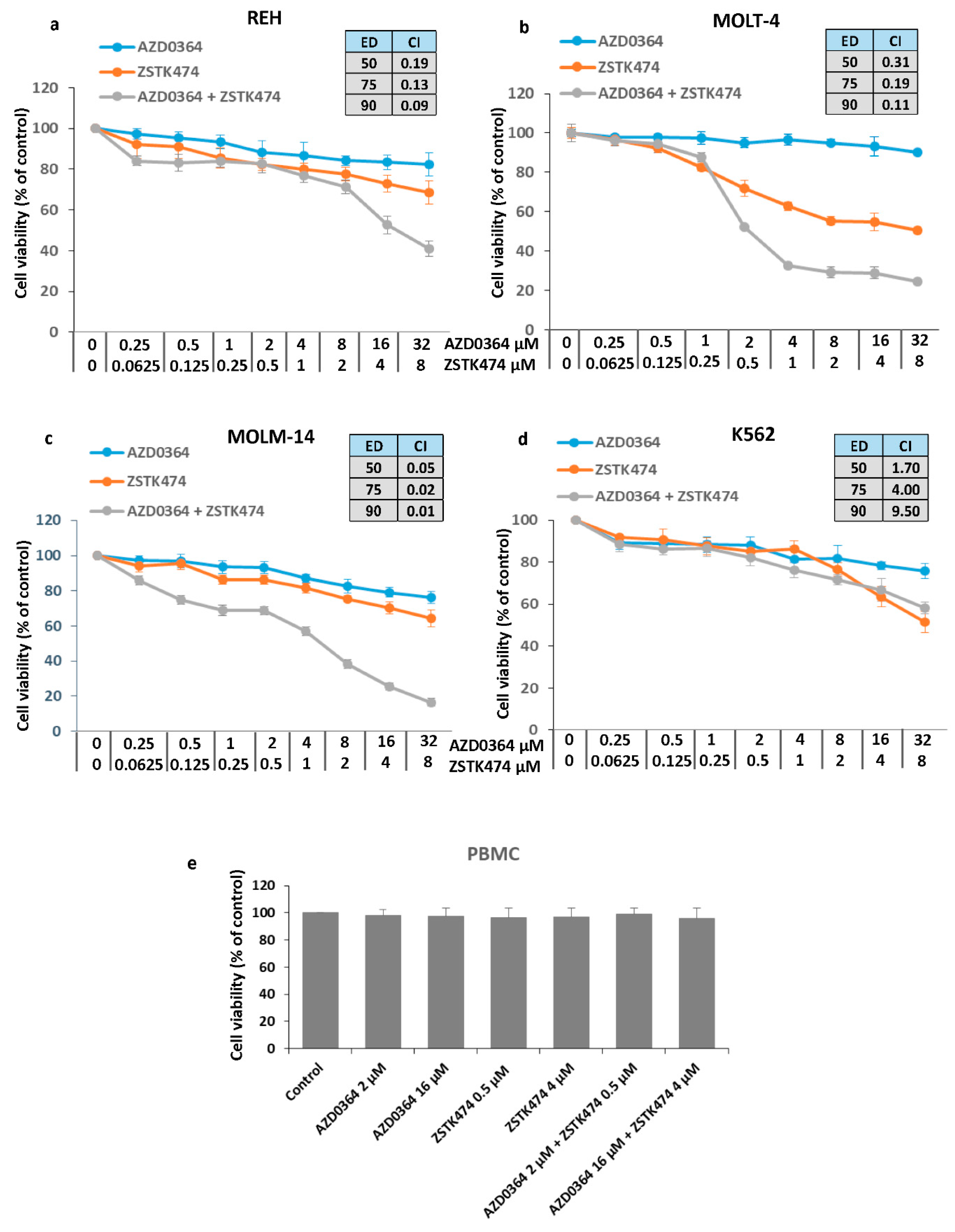
3.1. Synergistic Interaction between the ERK1/2 Inhibitor AZD034 and PI3K Inhibitor ZSTK474
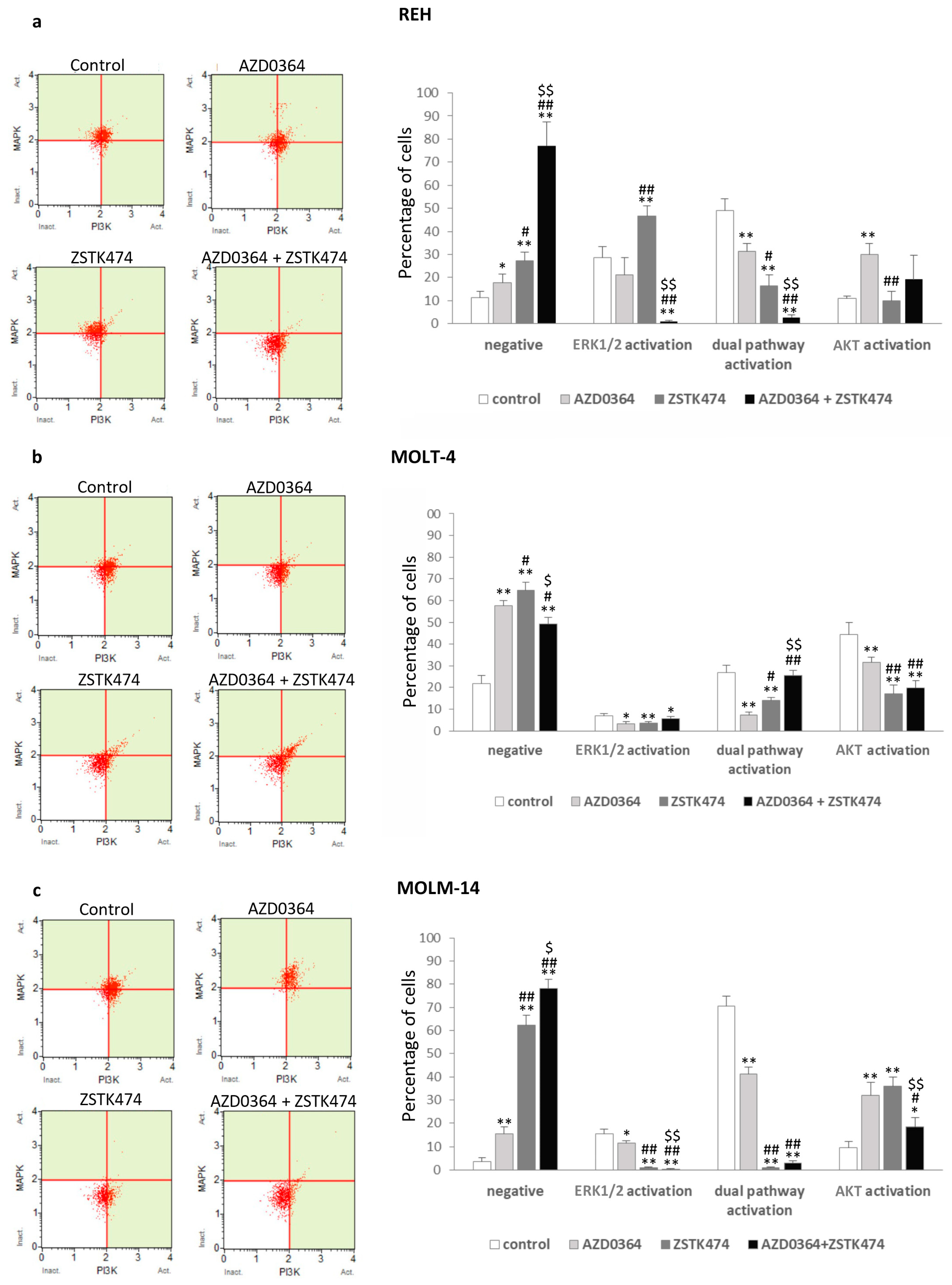
3.2. MAPK/ERK and PI3K/AKT Signaling Pathways are Modulated by AZD0364 and ZSTK474
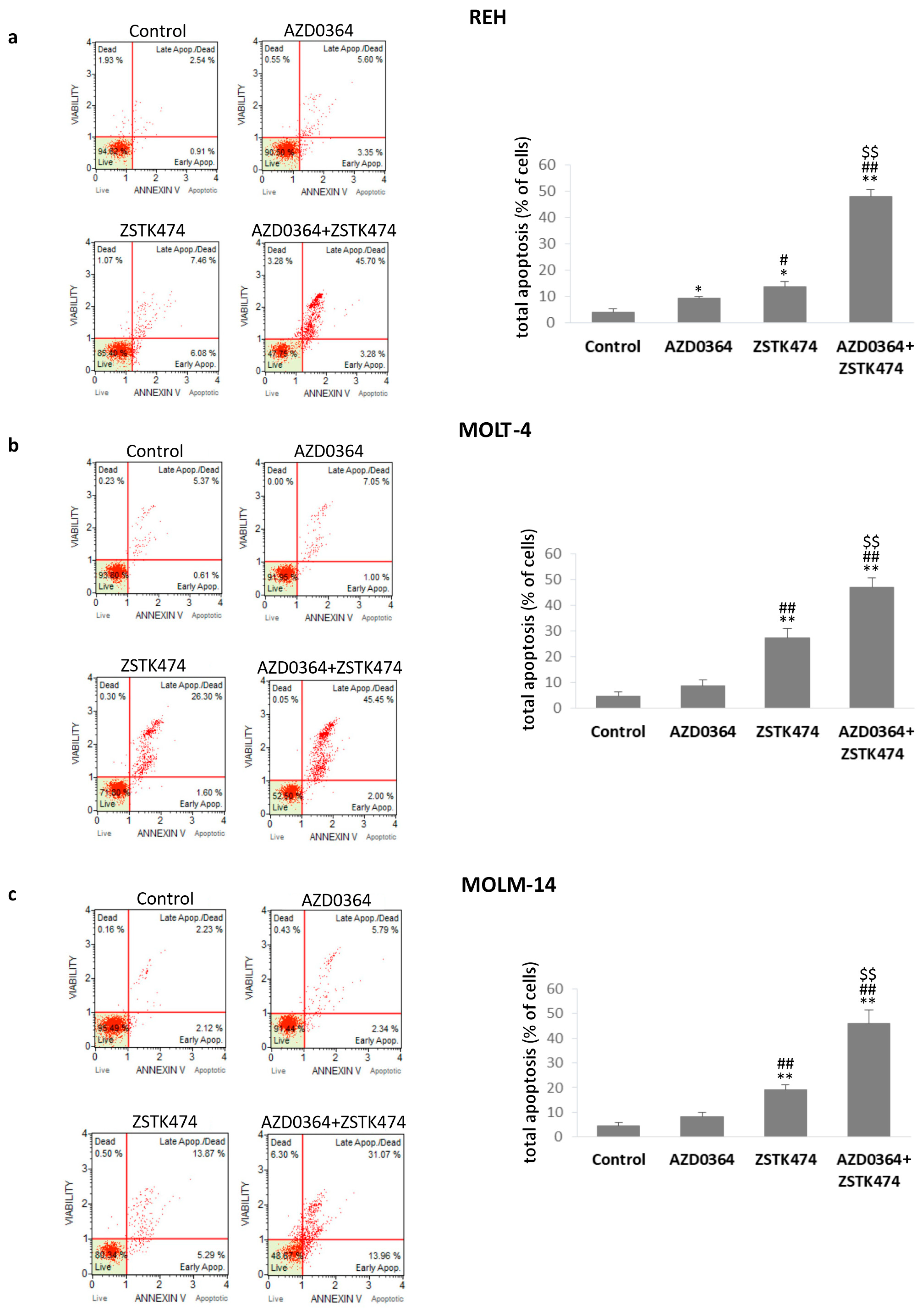
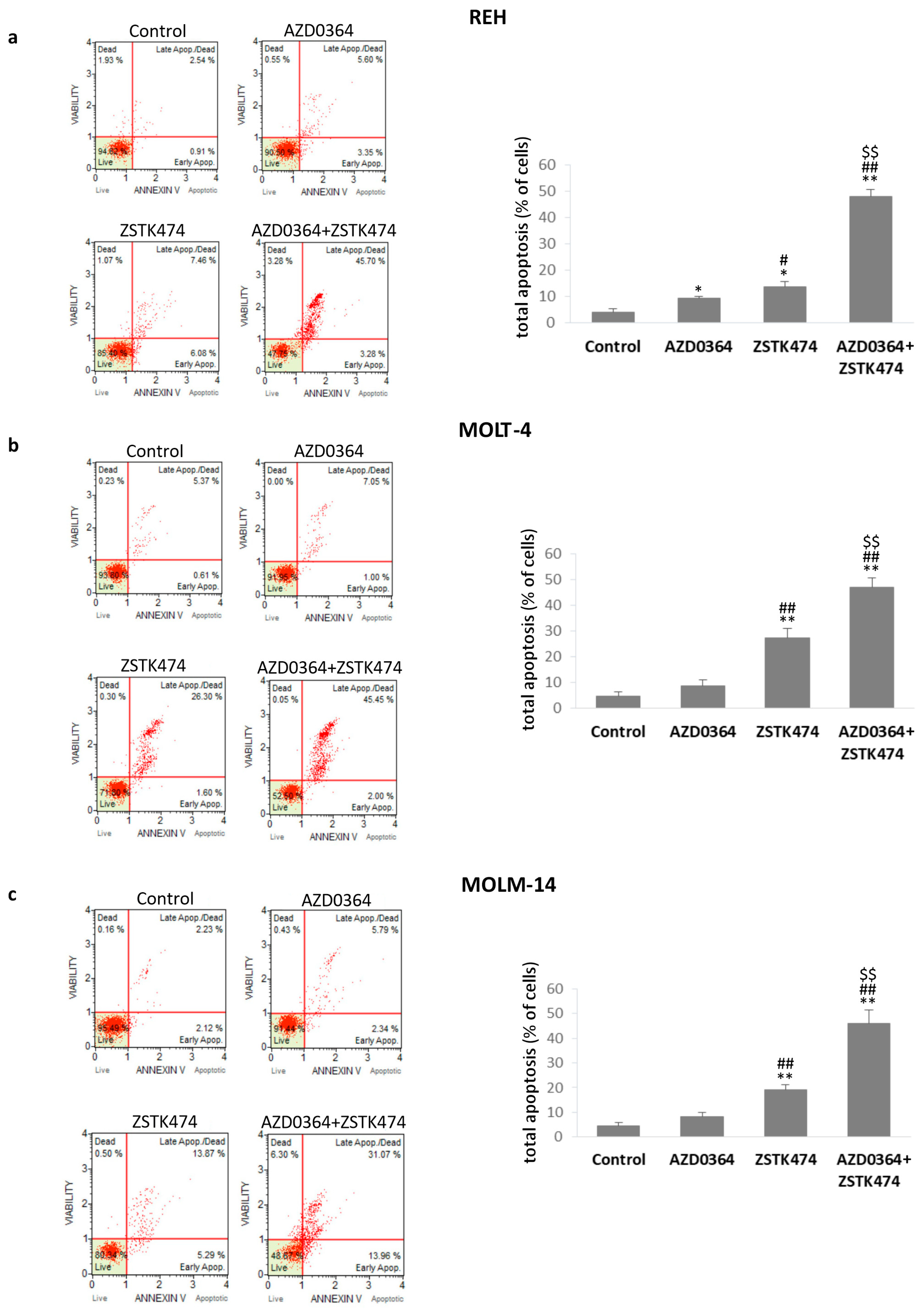
3.3. AZD034 in Combination with ZSTK474 Enhances Apoptosis in REH, MOLT-4 and MOLM-14 Cells
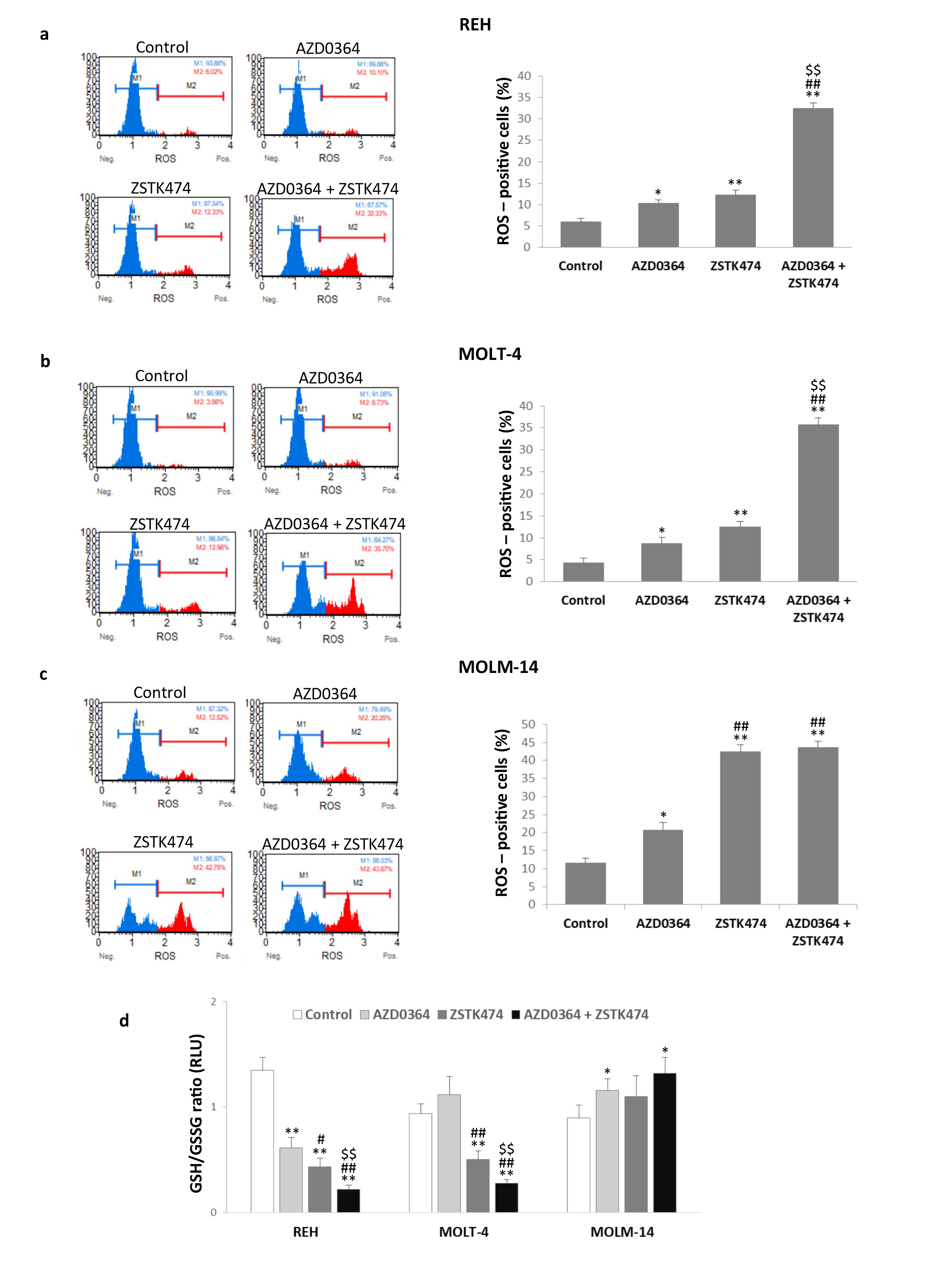
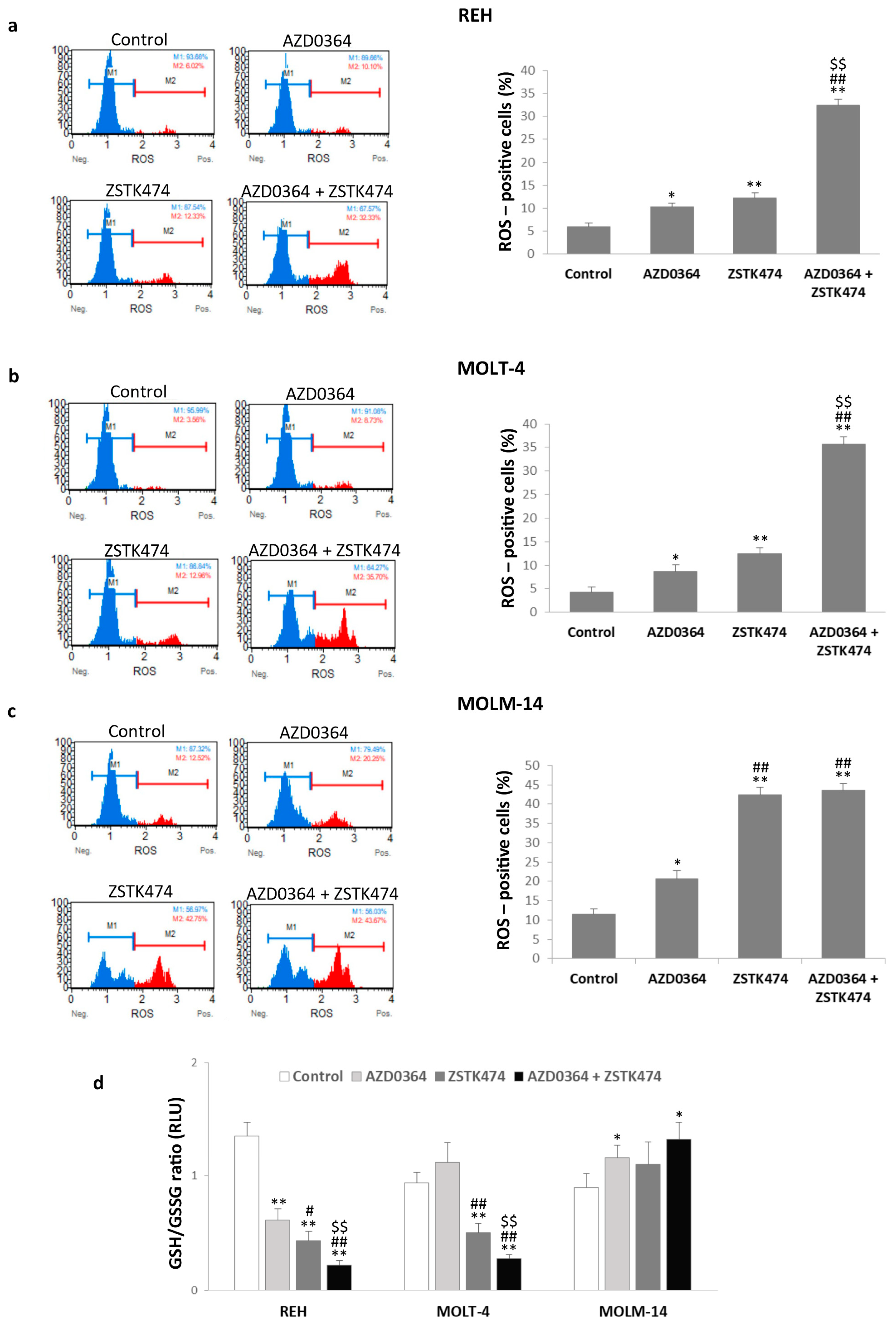
3.4. AZD0364 and ZSTK474 Increase ROS Production and Alter GSH/GSSG Ratio in Leukemia Cells
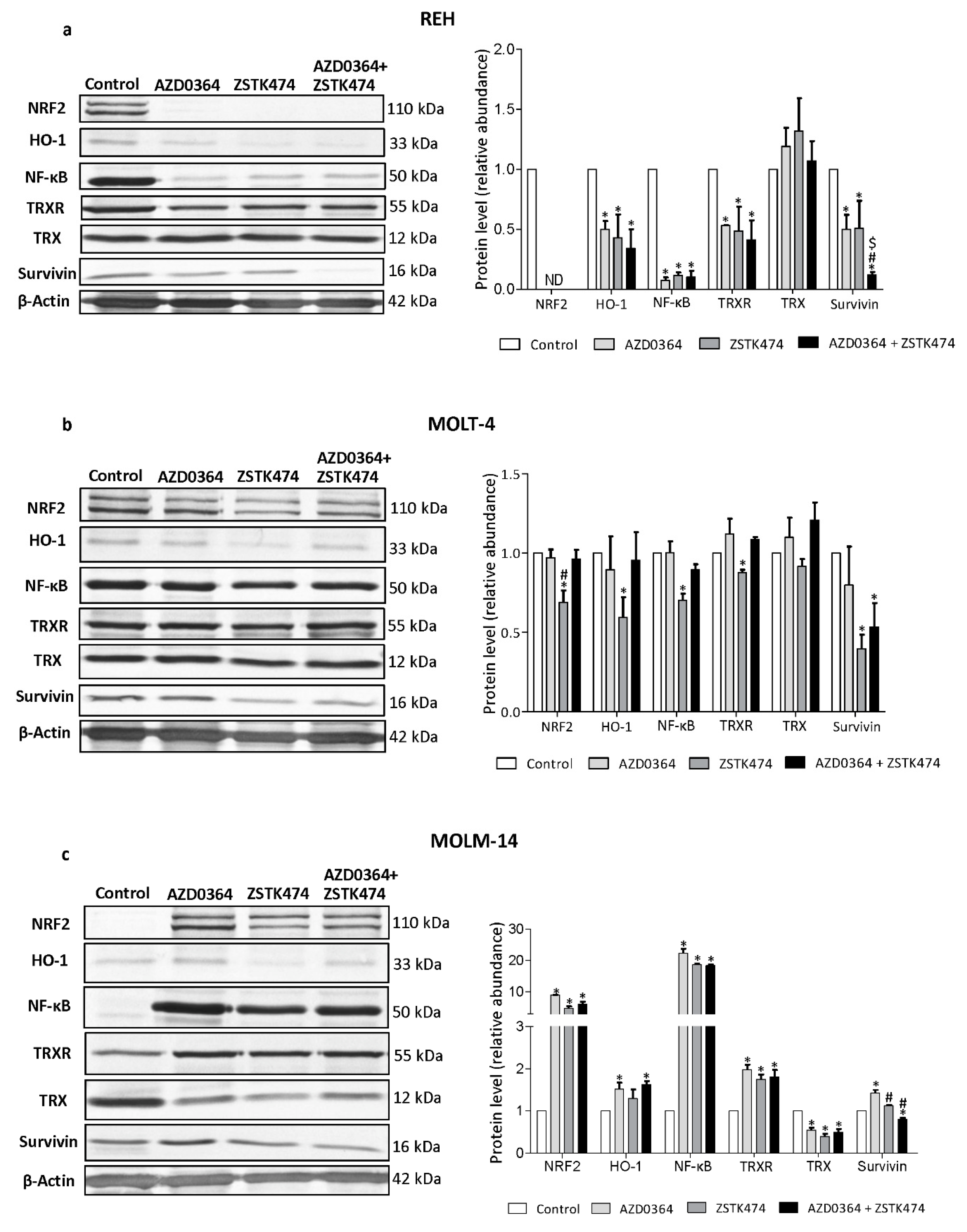
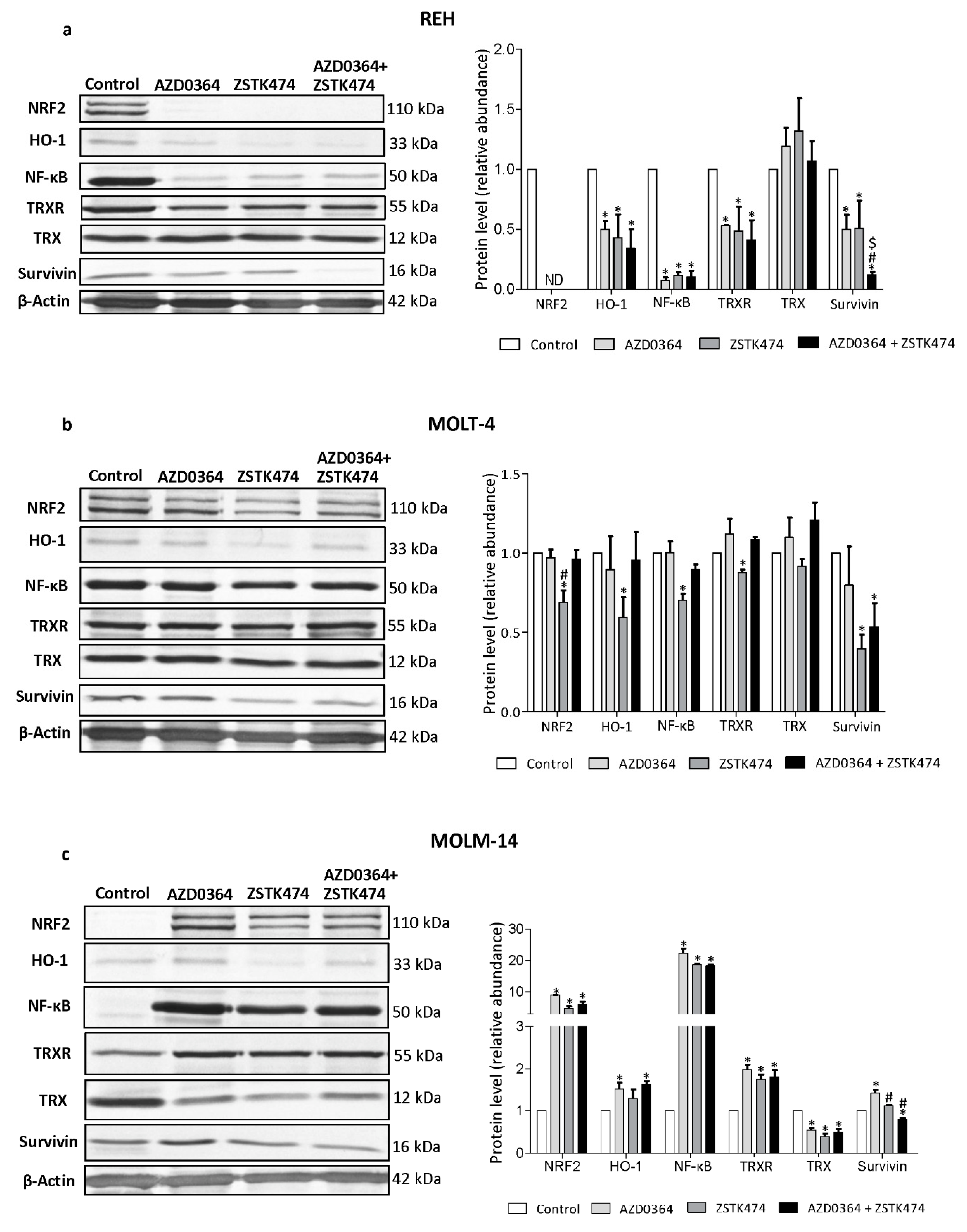
3.5. AZD034 and ZSTK474 Affect the Antioxidant and Apoptotic Protein Levels in Leukemia Cells
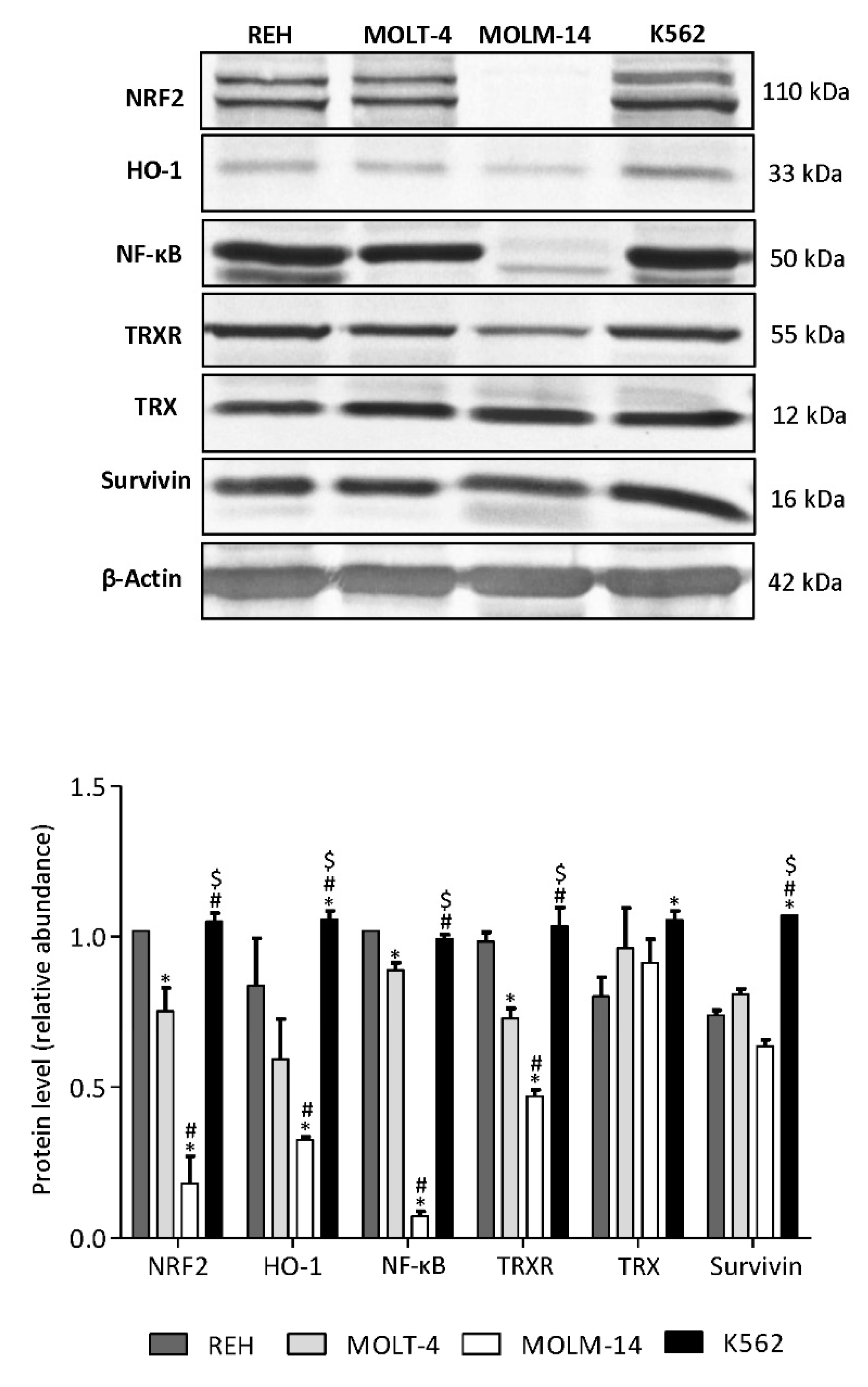
3.6. AML MOLM-14 Cell Line Demonstrates the Lowest Level of NF-κB, NRF2, HO-1 and TrxR Proteins, While CML K562 Cells Show the Highest Level of HO-1 and Survivin Compared with ALL and AML Cell Lines
4. Discussion
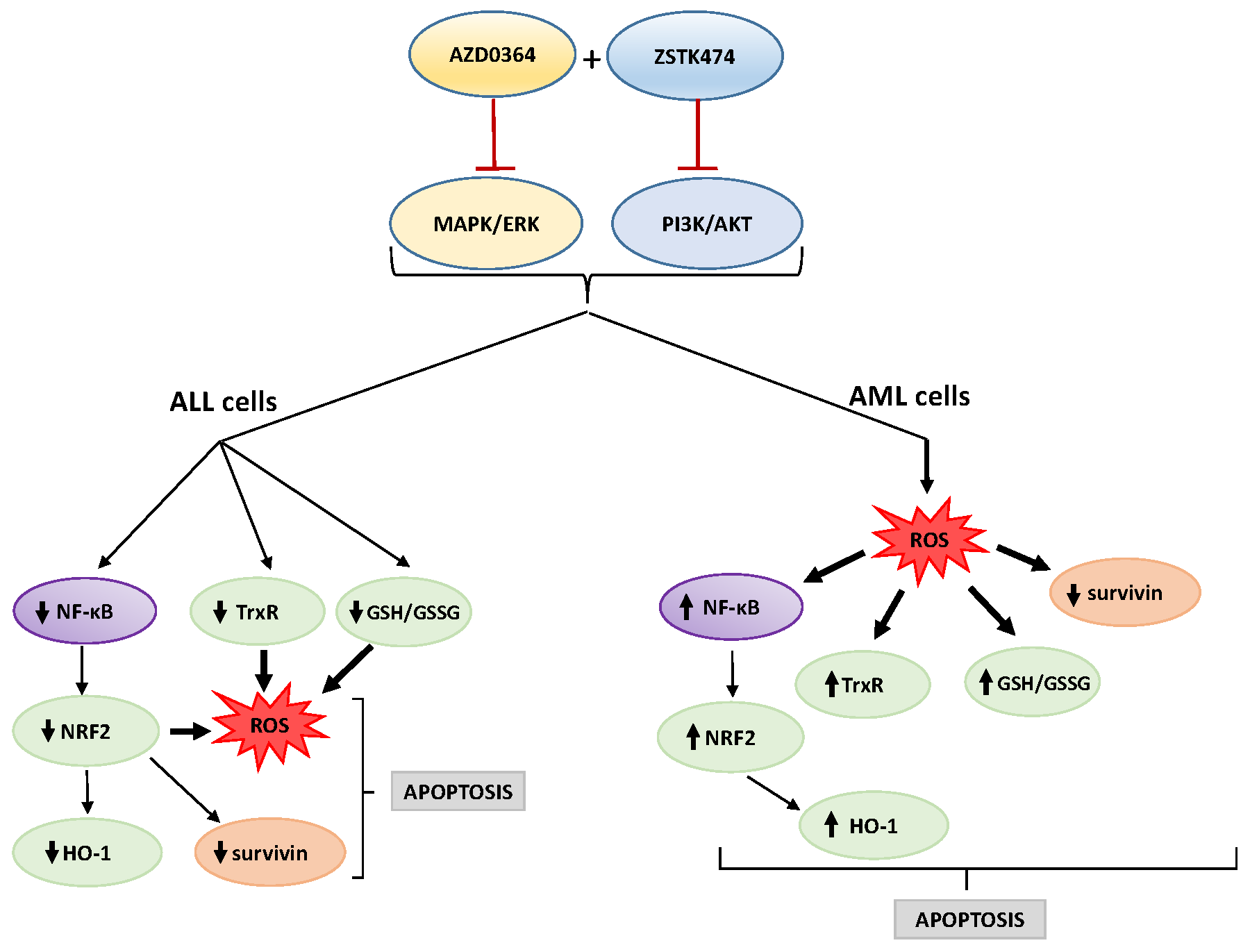
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Steelman, L.S.; Franklin, R.A.; Abrams, S.L.; Chappell, W.; Kempf, C.R.; Bäsecke, J.; Stivala, F.; Donia, M.; Fagone, P.; Nicoletti, F.; et al. Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia 2011, 25, 1080–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandage, V.L.; Gale, R.E.; Linch, D.C.; Khwaja, A. PI3-kinase/Akt is constitutively active in primary acute myeloid leukaemia cells and regulates survival and chemoresistance via NF-kappaB, Mapkinase and p53 pathways. Leukemia 2005, 19, 586–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martelli, A.M.; Nyåkern, M.; Tabellini, G.; Bortul, R.; Tazzari, P.L.; Evangelisti, C.; Cocco, L. Phosphoinositide 3-kinase/Akt signaling pathway and its therapeutical implications for human acute myeloid leukemia. Leukemia 2006, 20, 911–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, R.A.; Bethel, P.; Cook, C.; Davies, E.; Debreczeni, J.E.; Fairley, G.; Feron, L.; Flemington, V.; Graham, M.A.; Greenwood, R.; et al. Structure-guided discovery of potent and selective inhibitors of ERK1/2 from a modestly active and promiscuous chemical start point. J. Med. Chem. 2017, 60, 3438–3450. [Google Scholar] [CrossRef] [PubMed]
- Jasek-Gajda, E.; Gajda, M.; Jasińska, M.; Litwin, J.A.; Lis, G.J. TAK-733, a selective MEK inhibitor, enhances voreloxin-induced apoptosis in myeloid leukemia cells. Anticancer Res. 2018, 38, 6147–6156. [Google Scholar] [CrossRef]
- Jasek-Gajda, E.; Jurkowska, H.; Jasińska, M.; Litwin, J.A.; Lis, G.J. Combination of ERK2 inhibitor VX-11e and voreloxin synergistically enhances anti-proliferative and pro-apoptotic effects in leukemia cells. Apoptosis 2019, 24, 849–861. [Google Scholar] [CrossRef] [Green Version]
- Lonetti, A.; Cappellini, A.; Bertaina, A.; Locatelli, F.; Pession, A.; Buontempo, F.; Evangelisti, C.; Evangelisti, C.; Orsini, E.; Zambonin, L.; et al. Improving nelarabine efficacy in T cell acute lymphoblastic leukemia by targeting aberrant PI3K/AKT/mTOR signaling pathway. J. Hematol. Oncol. 2016, 9, 114. [Google Scholar] [CrossRef] [Green Version]
- Lonetti, A.; Cappellini, A.; Spartà, A.M.; Chiarini, F.; Buontempo, F.; Evangelisti, C.; Evangelisti, C.; Orsini, E.; McCubrey, J.A.; Martelli, A.M. PI3K pan-inhibition impairs more efficiently proliferation and survival of T-cell acute lymphoblastic leukemia cell lines when compared with isoform-selective PI3K inhibitors. Oncotarget 2015, 6, 10399–10414. [Google Scholar] [CrossRef] [Green Version]
- Evangelisti, C.; Chiarini, F.; Cappellini, A.; Paganelli, F.; Fini, M.; Santi, S.; Martelli, A.M.; Neri, L.M.; Evangelisti, C. Targeting Wnt/β-catenin and PI3K/Akt/mTOR pathways in T-cell acute lymphoblastic leukemia. J. Cell. Physiol. 2020, 235, 5413–5428. [Google Scholar] [CrossRef]
- Blalock, W.L.; Navolanic, P.M.; Steelman, L.S.; Shelton, J.G.; Moye, P.W.; Lee, J.T.; Franklin, R.A.; Mirza, A.; McMahon, M.; White, M.K.; et al. Requirement for the PI3K/Akt pathway in MEK1-mediated growth and prevention of apoptosis: Identification of an Achilles heel in leukemia. Leukemia 2003, 17, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Maachani, U.B.; Schweitzer, M.; Singh, R.; Wang, M.; Chang, R.; Souweidane, M.M. Dual inhibition of PI3K/AKT and MEK/ERK pathways induces synergistic antitumor effects in diffuse intrinsic pontine glioma cells. Transl. Oncol. 2017, 10, 221–228. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Abrams, S.L.; Bertrand, F.E.; Ludwig, D.E.; Bäsecke, J.; Libra, M.; Stivala, F.; Milella, M.; Tafuri, A. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia 2008, 22, 708–722. [Google Scholar] [CrossRef] [Green Version]
- Irwin, M.E.; Rivera-Del Valle, N.; Chandra, J. Redox control of leukemia: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 1349–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sillar, J.R.; Germon, Z.P.; DeIuliis, G.N.; Dun, M.D. The role of reactive oxygen species in acute myeloid leukaemia. Int. J. Mol. Sci. 2019, 20, 6003. [Google Scholar] [CrossRef] [Green Version]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Roh, J.L.; Jang, H.; Kim, E.H.; Shin, D. Targeting of the glutathione, thioredoxin, and Nrf2 antioxidant systems in head and neck cancer. Antioxid. Redox Signal. 2017, 27, 106–114. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sánchez-Pérez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Zimta, A.A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The role of Nrf2 activity in cancer development and progression. Cancers (Basel) 2019, 11, 1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Aziz, A.; MacEwan, D.J.; Bowles, K.M.; Rushworth, S.A. Oxidative stress responses and NRF2 in human leukaemia. Oxid. Med. Cell. Longev. 2015, 2015, 454659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basak, P.; Sadhukhan, P.; Sarkar, P.; Sil, P.C. Perspectives of the Nrf-2 signaling pathway in cancer progression and therapy. Toxicol. Rep. 2017, 4, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Mense, S.M.; Zhang, L. Heme: A versatile signaling molecule controlling the activities of diverse regulators ranging from transcription factors to MAP kinases. Cell Res. 2006, 16, 681–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, T.; Kirino, Y.; Takeno, M.; Samukawa, S.; Hama, M.; Tanaka, M.; Yamaji, S.; Ueda, A.; Tomita, N.; Fujita, H.; et al. Expression of heme oxygenase-1 in human leukemic cells and its regulation by transcriptional repressor Bach1. Cancer Sci. 2010, 101, 1409–1416. [Google Scholar] [CrossRef]
- Rushworth, S.A.; MacEwan, D.J. HO-1 underlies resistance of AML cells to TNF-induced apoptosis. Blood 2008, 111, 3793–3801. [Google Scholar] [CrossRef] [Green Version]
- Fidyt, K.; Pastorczak, A.; Goral, A.; Szczygiel, K.; Fendler, W.; Muchowicz, A.; Bartlomiejczyk, M.A.; Madzio, J.; Cyran, J.; Graczyk-Jarzynka, A.; et al. Targeting the thioredoxin system as a novel strategy against B-cell acute lymphoblastic leukemia. Mol. Oncol. 2019, 13, 1180–1195. [Google Scholar] [CrossRef] [Green Version]
- Arnér, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef]
- Meuillet, E.J.; Mahadevan, D.; Berggren, M.; Coon, A.; Powis, G. Thioredoxin-1 binds to the C2 domain of PTEN inhibiting PTEN’s lipid phosphatase activity and membrane binding: A mechanism for the functional loss of PTEN’s tumor suppressor activity. Arch. Biochem. Biophys. 2004, 429, 123–133. [Google Scholar] [CrossRef]
- Zhou, F.L.; Zhang, W.G.; Wei, Y.C.; Meng, S.; Bai, G.G.; Wang, B.Y.; Yang, H.Y.; Tian, W.; Meng, X.; Zhang, H.; et al. Involvement of oxidative stress in the relapse of acute myeloid leukemia. J. Biol. Chem. 2010, 285, 15010–15015. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.J.; Liu, Q.; Wei, H.L.; Yi, J.; Zhao, H.S.; Gao, L.P. Inhibition of thioredoxin reductase by auranofin induces apoptosis in adriamycin-resistant human K562 chronic myeloid leukemia cells. Pharmazie 2011, 66, 440–444. [Google Scholar] [PubMed]
- Rahman, I.; Biswas, S.K.; Jimenez, L.A.; Torres, M.; Forman, H.J. Glutathione, stress responses, and redox signaling in lung inflammation. Antioxid. Redox Signal. 2005, 7, 42–59. [Google Scholar] [CrossRef]
- Ferraris, A.M.; Rolfo, M.; Mangerini, R.; Gaetani, G.F. Increased glutathione in chronic lymphocytic leukemia lymphocytes. Am. J. Hematol. 1994, 47, 237–238. [Google Scholar] [CrossRef] [PubMed]
- Singh Ghalaut, V.; Kharb, S.; Ghalaut, P.S.; Rawal, A. Lymphocyte glutathione levels in acute leukemia. Clin. Chim. Acta 1999, 285, 85–89. [Google Scholar] [CrossRef]
- Maung, Z.T.; Hogarth, L.; Reid, M.M.; Proctor, S.J.; Hamilton, P.J.; Hall, A.G. Raised intracellular glutathione levels correlate with in vitro resistance to cytotoxic drugs in leukaemic cells from patients with acute lymphoblastic leukemia. Leukemia 1994, 8, 1487–1491. [Google Scholar] [PubMed]
- Calvert, P.; Yao, K.S.; Hamilton, T.C.; O’Dwyer, P.J. Clinical studies of reversal of drug resistance based on glutathione. Chem. Biol. Interact 1998, 111–112, 213–324. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Fruehauf, J.P.; Meyskens, F.L., Jr. Reactive oxygen species: A breath of life or death? Clin. Cancer Res. 2007, 13, 789–794. [Google Scholar] [CrossRef] [Green Version]
- Heasman, S.A.; Zaitseva, L.; Bowles, K.M.; Rushworth, S.A.; Macewan, D.J. Protection of acute myeloid leukaemia cells from apoptosis induced by front-line chemotherapeutics is mediated by haem oxygenase-1. Oncotarget 2011, 2, 658–668. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Mobley, A.; Miller, C.; Boklan, J.; Chandra, J. Potentiation of reactive oxygen species is a marker for synergistic cytotoxicity of MS-275 and 5-azacytidine in leukemic cells. Leuk. Res. 2008, 32, 771–780. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, Z. Increased Oxidative Stress as a Selective Anticancer Therapy. Oxid. Med. Cell. Longev. 2015, 2015, 294303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgieva, E.; Ivanova, D.; Zhelev, Z.; Bakalova, R.; Gulubova, M.; Aoki, I. Mitochondrial dysfunction and redox imbalance as a diagnostic marker of “free radical diseases”. Anticancer Res. 2017, 37, 5373–5381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayerhofer, M.; Florian, S.; Krauth, M.T.; Aichberger, K.J.; Bilban, M.; Marculescu, R.; Printz, D.; Fritsch, G.; Wagner, O.; Selzer, E.; et al. Identification of heme oxygenase-1 as a novel BCR/ABL-dependent survival factor in chronic myeloid leukemia. Cancer Res. 2004, 64, 3148–3154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, H.; Zhen, C.; Liu, J.; Yang, P.; Hu, L.; Shang, P. Unraveling the potential role of glutathione in multiple forms of cell death in cancer therapy. Oxid. Med. Cell. Longev. 2019, 2019, 3150145. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Hu, C.; Li, H. Survivin as a novel target protein for reducing the proliferation of cancer cells. Biomed. Rep. 2018, 8, 399–406. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Lyu, H.; Wang, J.; Liu, B. Influence of survivin-targeted therapy on chemosensitivity in the treatment of acute myeloid leukemia. Cancer Lett. 2015, 366, 160–172. [Google Scholar] [CrossRef] [Green Version]
- Carter, B.Z.; Milella, M.; Altieri, D.C.; Andreeff, M. Cytokine-regulated expression of survivin in myeloid leukemia. Blood 2001, 97, 2784–2790. [Google Scholar] [CrossRef] [Green Version]
- Serrano-López, J.; Serrano, J.; Figueroa, V.; Torres-Gomez, A.; Tabares, S.; Casaño, J.; Fernandez-Escalada, N.; Sánchez-Garcia, J. Cytoplasmic localization of wild-type survivin is associated with constitutive activation of the PI3K/Akt signaling pathway and represents a favorable prognostic factor in patients with acute myeloid leukemia. Haematologica 2013, 98, 1877–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Ruvolo, V.R.; Gao, C.; Zhou, L.; Bornmann, W.; Tsao, T.; Schober, W.D.; Smith, P.; Guichard, S.; Konopleva, M.; et al. Evaluation of apoptosis induction by concomitant inhibition of MEK, mTOR, and Bcl-2 in human acute myelogenous leukemia cells. Mol. Cancer Ther. 2014, 13, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- Fan, R.; Wang, Y.; Wang, Y.; Wei, L.; Zheng, W. Mechanism of progestin resistance in endometrial precancer/cancer through Nrf2-survivin pathway. Am. J. Transl. Res. 2017, 9, 1483–1491. [Google Scholar] [PubMed]
- Sallmyr, A.; Fan, J.; Datta, K.; Kim, K.T.; Grosu, D.; Shapiro, P.; Small, D.; Rassool, F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: Implications for poor prognosis in AML. Blood 2008, 111, 3173–3182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grafone, T.; Palmisano, M.; Nicci, C.; Storti, S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: Biology and treatment. Oncol. Rev. 2012, 6, e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapidus, R.G.; Carter-Cooper, B.A.; Sadowska, M.; Choi, E.Y.; Wonodi, O.; Muvarak, N.; Natarajan, K.; Pidugu, L.S.; Jaiswal, A.; Toth, E.A.; et al. Hydroxylated dimeric naphthoquinones increase the generation of reactive oxygen species, induce apoptosis of acute myeloid leukemia cells and are not substrates of the multidrug resistance proteins ABCB1 and ABCG2. Pharmaceuticals (Basel) 2016, 9, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickinson, D.A.; Forman, H.J. Glutathione in defense and signaling: Lessons from a small thiol. Ann. N. Y. Acad. Sci. 2002, 973, 488–504. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, N. Multiple role of 3-mercaptopyruvate sulfurtransferase: Antioxidative function, H2S and polysulfide production and possible SOx production. Br. J. Pharmacol. 2018, 175, 577–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Yang, Z.; Xiao, X.; An, T.; Li, B.; Ouyang, J.; Li, H.; Wang, C.; Zhang, Y.; Zhang, H.; et al. A thioredoxin reductase inhibitor ethaselen induces growth inhibition and apoptosis in gastric cancer. J. Cancer 2020, 11, 3013–3019. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, S.; Singh, P.; Moh, A.; Abe, M.; Conway, E.M.; Boswell, H.S.; Yamaguchi, S.; Fu, X.Y.; Pelus, L.M. Survivin mediates aberrant hematopoietic progenitor cell proliferation and acute leukemia in mice induced by internal tandem duplication of Flt3. Blood 2009, 114, 394–403. [Google Scholar] [CrossRef] [Green Version]
- Ahamed, M.; Akhtar, M.J.; Raja, M.; Ahmad, I.; Siddiqui, M.K.; AlSalhi, M.S.; Alrokayan, S.A. ZnO nanorod-induced apoptosis in human alveolar adenocarcinoma cells via p53, survivin and bax/bcl-2 pathways: Role of oxidative stress. Nanomedicine 2011, 7, 904–913. [Google Scholar] [CrossRef]
- White-Gilbertson, S.J.; Kasman, L.; McKillop, J.; Tirodkar, T.; Lu, P.; Voelkel-Johnson, C. Oxidative stress sensitizes bladder cancer cells to TRAIL mediated apoptosis by down-regulating anti-apoptotic proteins. J. Urol. 2009, 182, 1178–1185. [Google Scholar] [CrossRef] [Green Version]
- Pervin, S.; Tran, L.; Urman, R.; Braga, M.; Parveen, M.; Li, S.A.; Chaudhuri, G.; Singh, R. Oxidative stress specifically downregulates survivin to promote breast tumour formation. Br. J. Cancer 2013, 108, 848–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hare, T.; Eide, C.A.; Deininger, M.W. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007, 110, 2242–2249. [Google Scholar] [CrossRef] [PubMed]
- Carrà, G.; Torti, D.; Crivellaro, S.; Panuzzo, C.; Taulli, R.; Cilloni, D.; Guerrasio, A.; Saglio, G.; Morotti, A. The BCR-ABL/NF-κB signal transduction network: A long lasting relationship in Philadelphia positive Leukemias. Oncotarget 2016, 7, 66287–66298. [Google Scholar] [CrossRef] [Green Version]
- Conte, E.; Stagno, F.; Guglielmo, P.; Scuto, A.; Consoli, C.; Messina, A. Survivin expression in chronic myeloid leukemia. Cancer Lett. 2005, 225, 105–110. [Google Scholar] [CrossRef]
- Wang, Z.; Sampath, J.; Fukuda, S.; Pelus, L.M. Disruption of the inhibitor of apoptosis protein survivin sensitizes Bcr-abl-positive cells to STI571-induced apoptosis. Cancer Res. 2005, 65, 8224–8232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jasek-Gajda, E.; Jurkowska, H.; Jasińska, M.; Lis, G.J. Targeting the MAPK/ERK and PI3K/AKT Signaling Pathways Affects NRF2, Trx and GSH Antioxidant Systems in Leukemia Cells. Antioxidants 2020, 9, 633. https://doi.org/10.3390/antiox9070633
Jasek-Gajda E, Jurkowska H, Jasińska M, Lis GJ. Targeting the MAPK/ERK and PI3K/AKT Signaling Pathways Affects NRF2, Trx and GSH Antioxidant Systems in Leukemia Cells. Antioxidants. 2020; 9(7):633. https://doi.org/10.3390/antiox9070633
Chicago/Turabian StyleJasek-Gajda, Ewa, Halina Jurkowska, Małgorzata Jasińska, and Grzegorz J. Lis. 2020. "Targeting the MAPK/ERK and PI3K/AKT Signaling Pathways Affects NRF2, Trx and GSH Antioxidant Systems in Leukemia Cells" Antioxidants 9, no. 7: 633. https://doi.org/10.3390/antiox9070633
APA StyleJasek-Gajda, E., Jurkowska, H., Jasińska, M., & Lis, G. J. (2020). Targeting the MAPK/ERK and PI3K/AKT Signaling Pathways Affects NRF2, Trx and GSH Antioxidant Systems in Leukemia Cells. Antioxidants, 9(7), 633. https://doi.org/10.3390/antiox9070633