Impact of the APE1 Redox Function Inhibitor E3330 in Non-Small Cell Lung Cancer Cells Exposed to Cisplatin: Increased Cytotoxicity and Impairment of Cell Migration and Invasion


 ,
,  ,
,  ,
,  , ,
, ,  and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
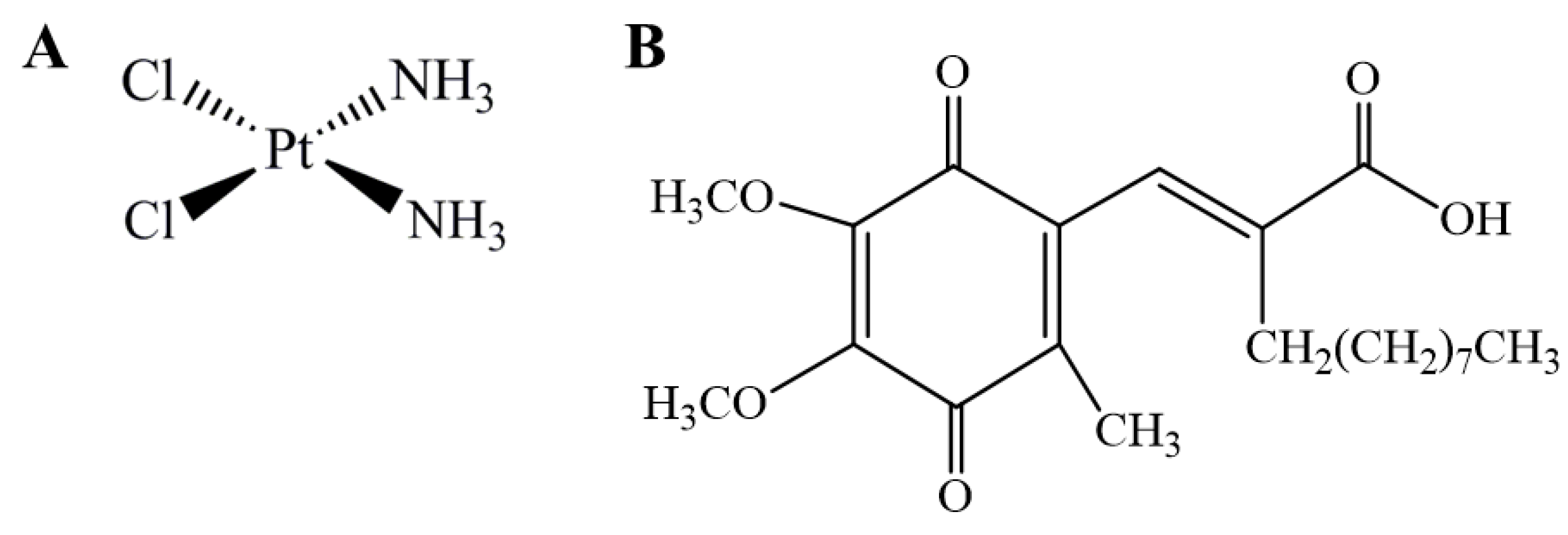
2.1. Chemicals
2.2. Cell Culture
2.3. Crystal Violet (CV) Staining Assay
2.4. MTS Reduction Assay
2.5. Cell DNA Content Analysis
2.6. Apoptosis Assay
2.7. Selection of Cisplatin and E3330 Concentrations for the Migration/Invasion Assays
2.8. In Vitro Wound-Healing Assay
2.9. Chemotaxis and Chemoinvasion Assays
3. Results
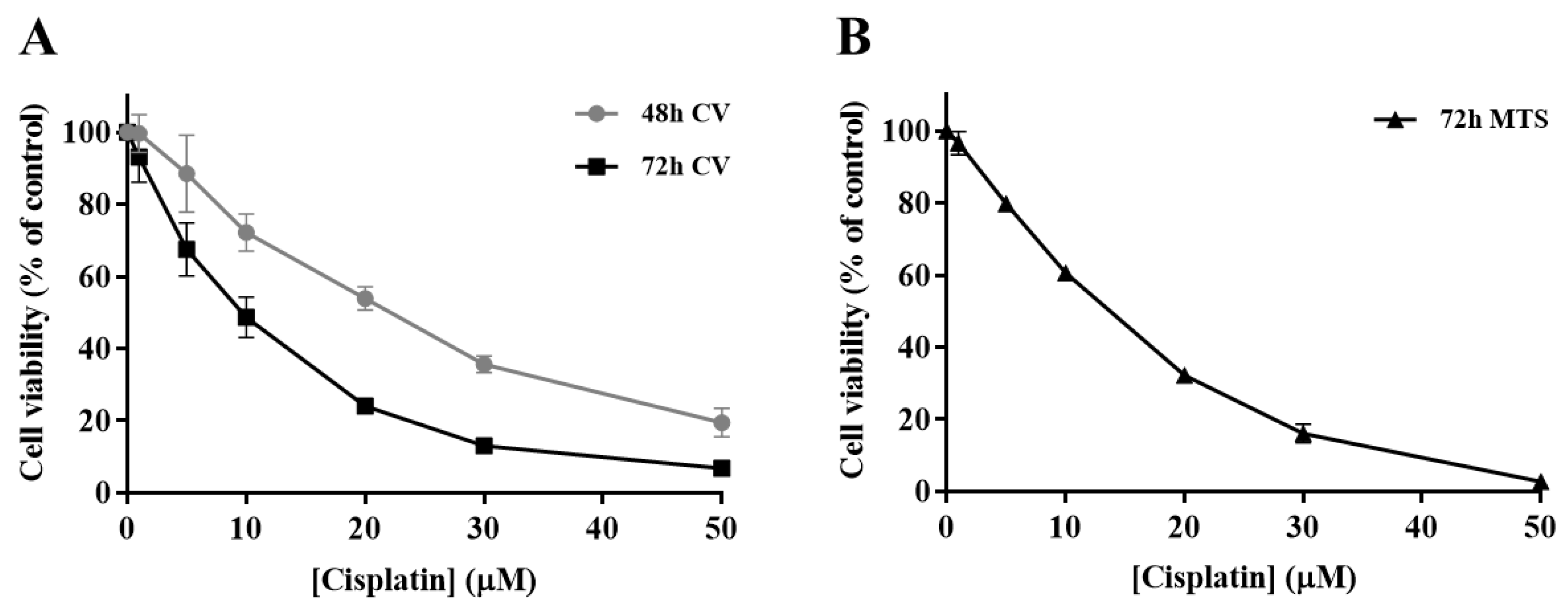
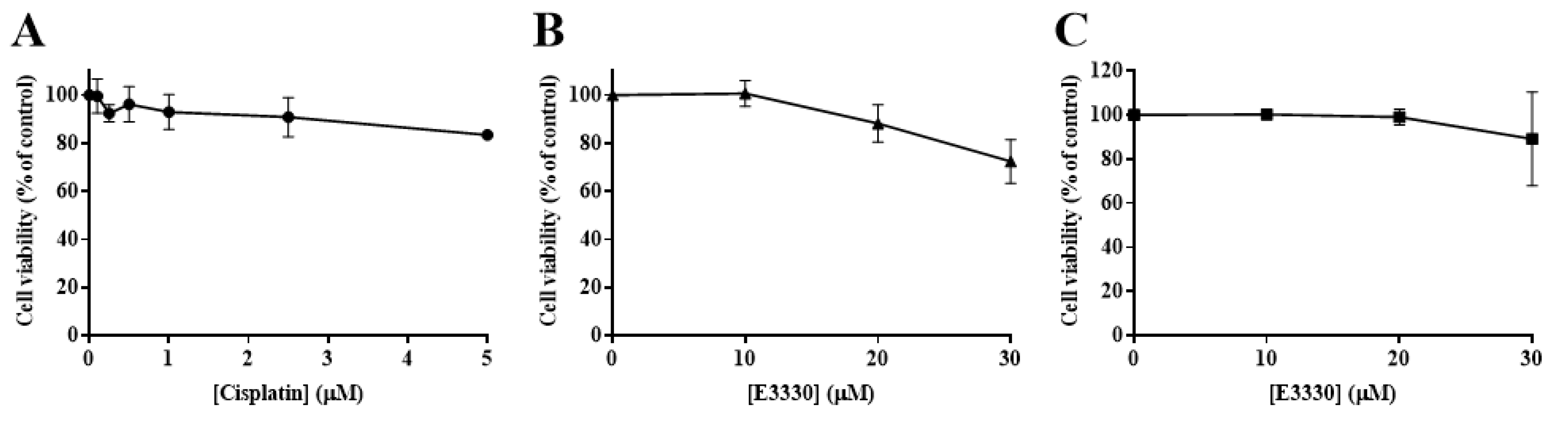
3.1. Cytotoxicity Profile of Cisplatin in H1975 Cells
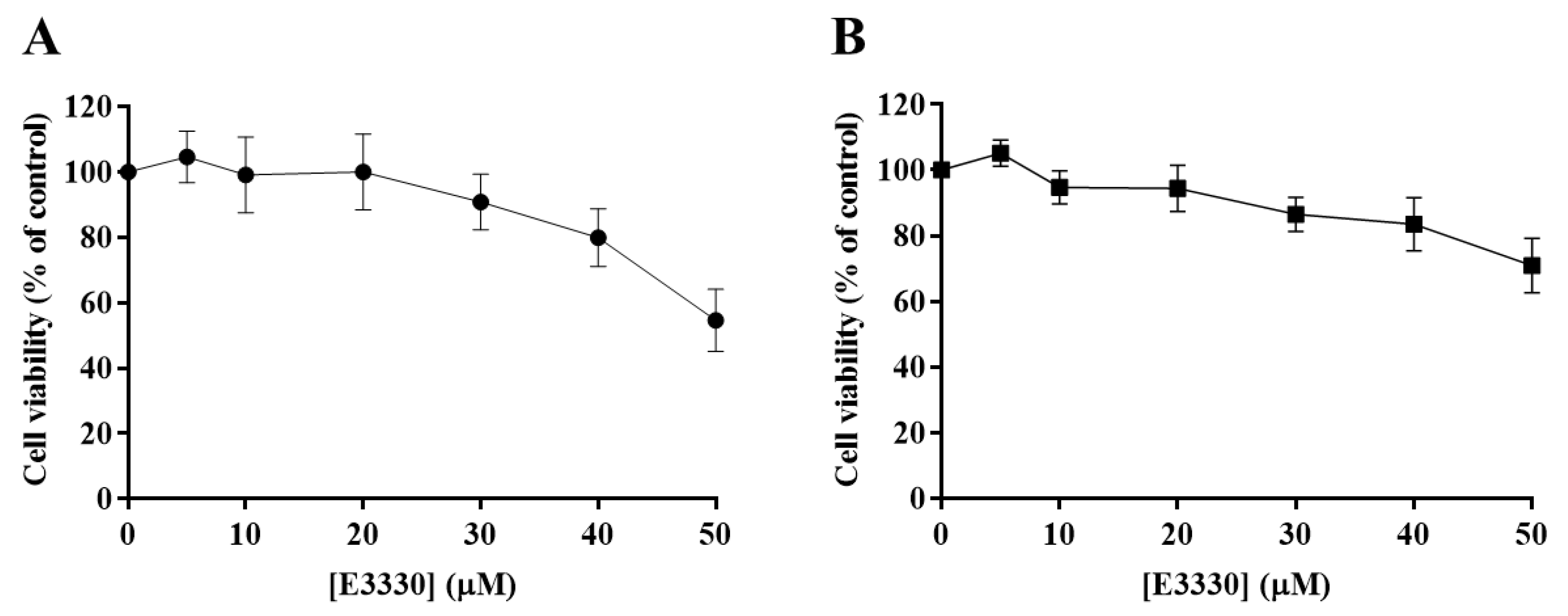
3.2. Impact of E3330 in the Viability of H1975 Cells
3.3. The Combination of E3330 and Cisplatin Displays a Synergistic Effect in Cell Viability
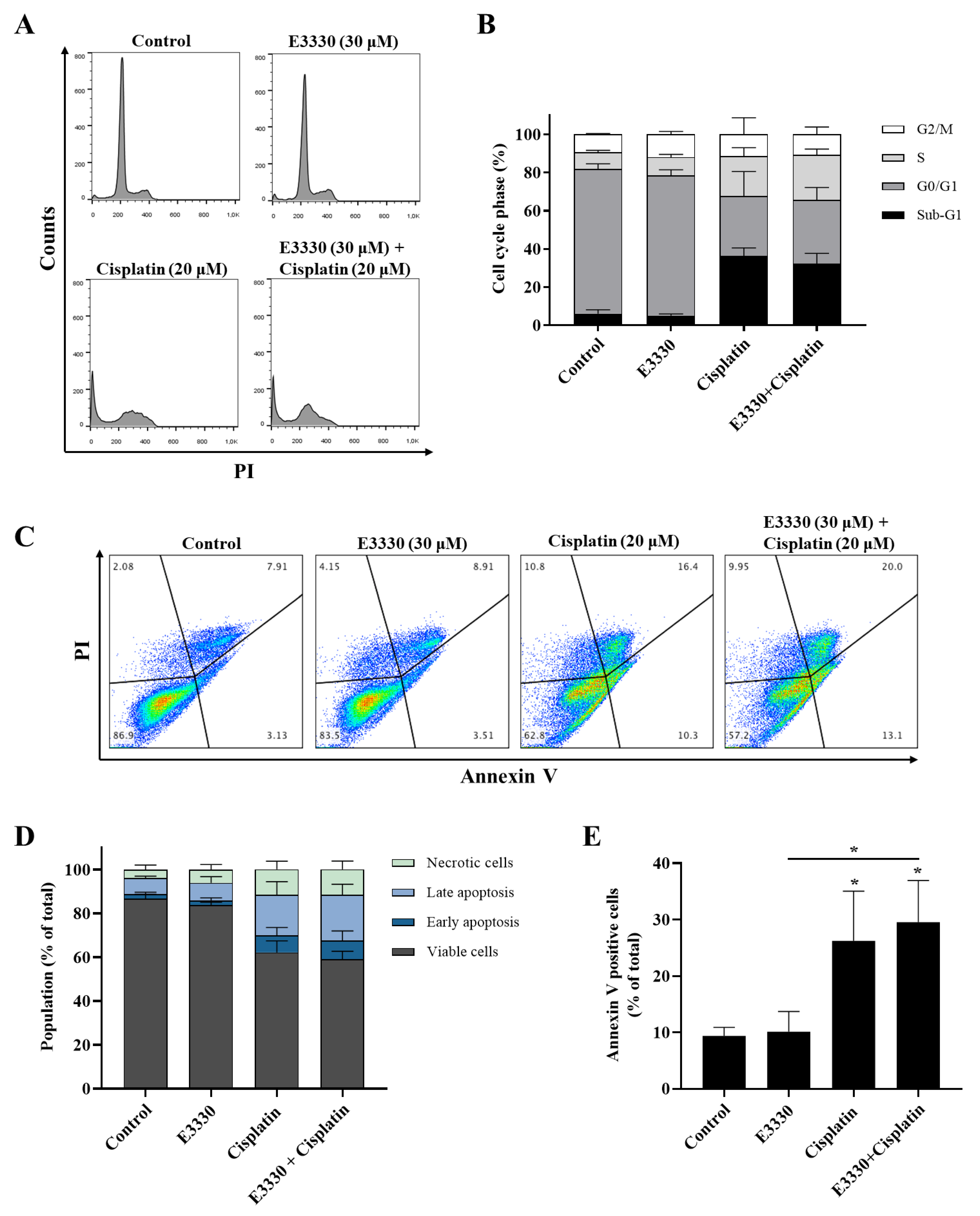
3.4. Effect of the Combination of E3330 and Cisplatin in Cell Cycle Distribution and Cell Death
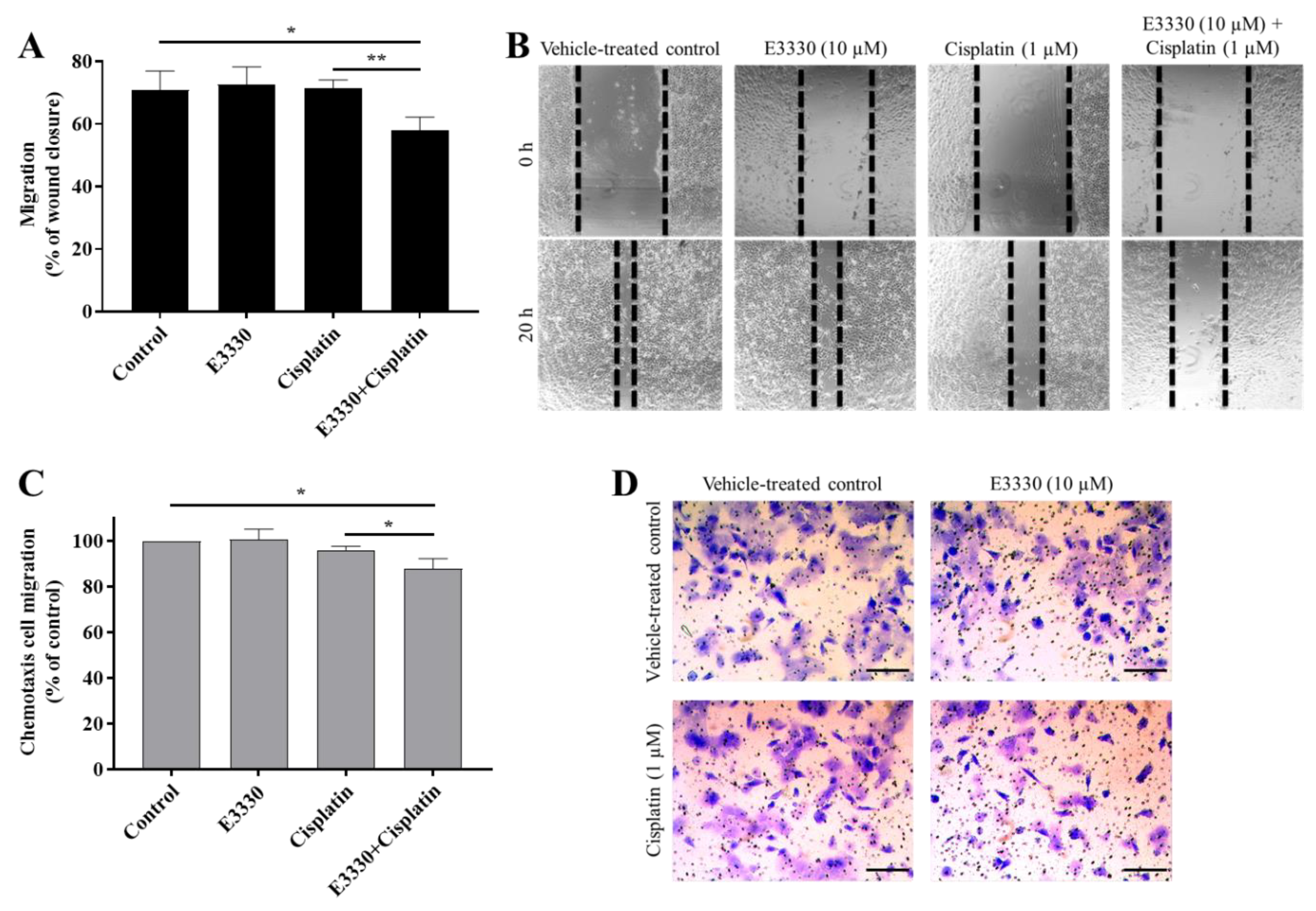
3.5. E3330 in Combination with Cisplatin Reduces Both Collective and Chemotactic Cell Migration
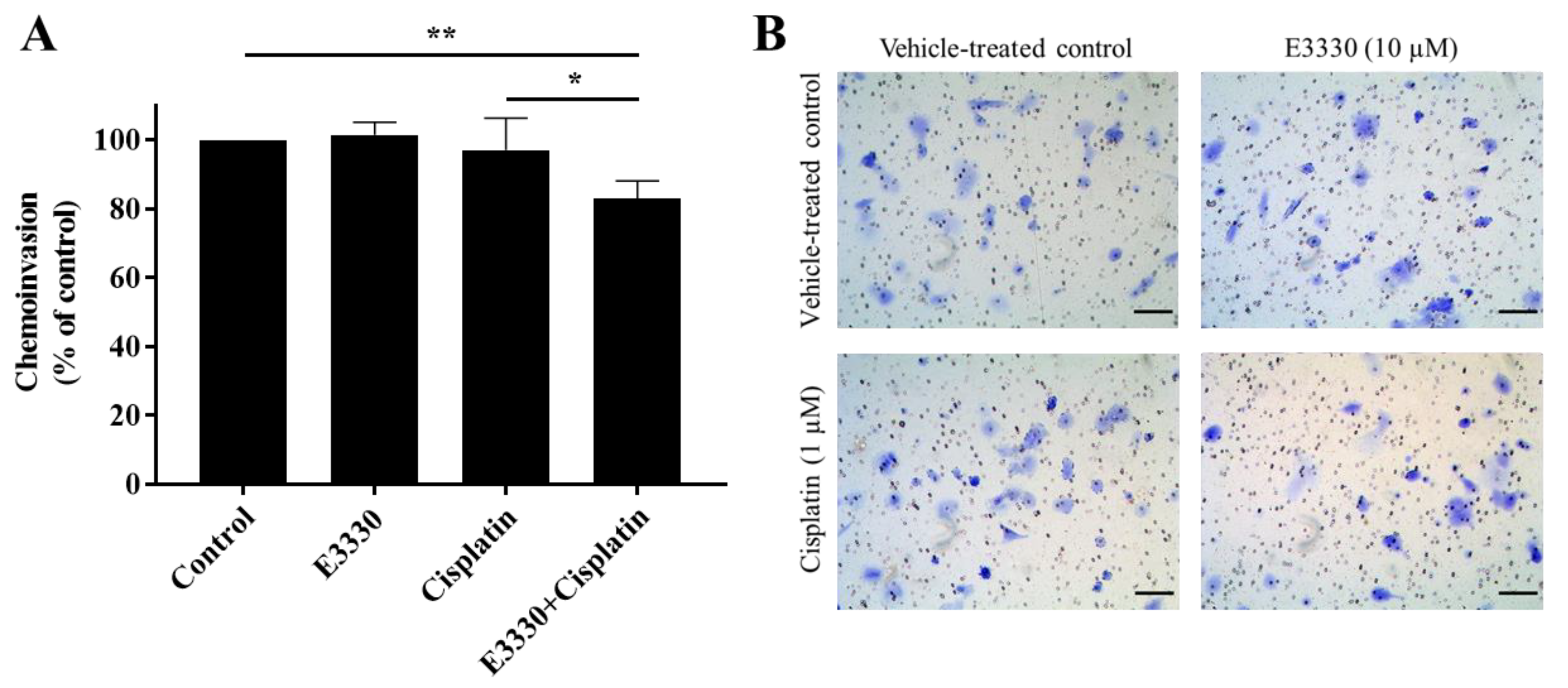
3.6. The Combination of E3330 and Cisplatin Decreases Invasion of H1975 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [Green Version]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Reed, E.; Li, Q.Q. Molecular basis of cellular response to cisplatin chemotherapy in non-small cell lung cancer (Review). Oncol. Rep. 2004, 12, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Rajeswaran, A.; Trojan, A.; Burnand, B.; Giannelli, M. Efficacy and side effects of cisplatin- and carboplatin-based doublet chemotherapeutic regimens versus non-platinum-based doublet chemotherapeutic regimens as first line treatment of metastatic non-small cell lung carcinoma: A systematic review of randomi. Lung Cancer 2008, 59, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kamdar, O.; Le, W.; Rosen, G.D.; Upadhyay, D. Nicotine induces resistance to chemotherapy by modulating mitochondrial signaling in lung cancer. Am. J. Respir. Cell Mol. Biol. 2009, 40, 135–146. [Google Scholar] [CrossRef]
- Nishioka, T.; Luo, L.Y.; Shen, L.; He, H.; Mariyannis, A.; Dai, W.; Chen, C. Nicotine increases the resistance of lung cancer cells to cisplatin through enhancing Bcl-2 stability. Br. J. Cancer 2014, 110, 1785–1792. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Ganapathy, S.; Avraham, H.; Nishioka, T.; Chen, C. Nicotine exposure potentiates lung tumorigenesis by perturbing cellular surveillance. Br. J. Cancer 2020, 122, 904–911. [Google Scholar] [CrossRef]
- Goldkorn, T.; Filosto, S.; Chung, S. Lung injury and lung cancer caused by cigarette smoke-induced oxidative stress: Molecular mechanisms and therapeutic opportunities involving the ceramide-generating machinery and epidermal growth factor receptor. Antioxid. Redox Signal. 2014, 21, 2149–2174. [Google Scholar] [CrossRef] [Green Version]
- Tell, G.; Fantini, D.; Quadrifoglio, F. Understanding different functions of mammalian AP endonuclease (APE1) as a promising tool for cancer treatment. Cell. Mol. Life Sci. 2010, 67, 3589–3608. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.R.; Georgiadis, M.M.; Fishel, M.L. APE1/Ref-1 Role in Redox Signaling: Translational Applications of Targeting the Redox Function of the DNA Repair/Redox Protein APE1/Ref-1. Curr. Mol. Pharmacol. 2012, 5, 36–53. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, P.S.; Corvacho, E.; Costa, J.G.; Saraiva, N.; Fernandes, A.S.; Castro, M.; Miranda, J.P.; Oliveira, N.G. The APE1 redox inhibitor E3330 reduces collective cell migration of human breast cancer cells and decreases chemoinvasion and colony formation when combined with docetaxel. Chem. Biol. Drug Des. 2017, 90, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, D.P.; Grimard, M.; Luo, M.; Shahda, S.; Jiang, Y.; Tong, Y.; Yu, Z.; Zyromski, N.; Schipani, E.; Carta, F.; et al. Regulation of HIF1α under Hypoxia by APE1/Ref-1 Impacts CA9 Expression: Dual Targeting in Patient-Derived 3D Pancreatic Cancer Models. Mol. Cancer Ther. 2016, 15, 2722–2732. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, A.A.; Jiang, Y.; Luo, M.; Reed, A.M.; Shahda, S.; He, Y.; Maitra, A.; Kelley, M.R.; Fishel, M.L. APE1/Ref-1 Regulates STAT3 Transcriptional Activity and APE1/Ref-1-STAT3 Dual-Targeting Effectively Inhibits Pancreatic Cancer Cell Survival. PLoS ONE 2012, 7, e47462. [Google Scholar] [CrossRef]
- Cesaratto, L.; Codarin, E.; Vascotto, C.; Leonardi, A.; Kelley, M.R.; Tiribelli, C.; Tell, G. Specific Inhibition of the Redox Activity of Ape1/Ref-1 by E3330 Blocks Tnf-A-Induced Activation of Il-8 Production in Liver Cancer Cell Lines. PLoS ONE 2013, 8, e70909. [Google Scholar] [CrossRef]
- Biswas, A.; Khanna, S.; Roy, S.; Pan, X.; Sen, C.K.; Gordillo, G.M. Endothelial cell tumor growth is ape/ref-1 dependent. Am. J. Physiol. Cell Physiol. 2015, 309, C296–C307. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Zhou, S.; Sandusky, G.E.; Kelley, M.R.; Fishel, M.L. Reduced Expression of DNA Repair and Redox Signaling Protein APE1/Ref-1 Impairs Human Pancreatic Cancer Cell Survival, Proliferation, and Cell Cycle Progression. Cancer Investig. 2010, 28, 885–895. [Google Scholar] [CrossRef]
- Fishel, M.L.; Wu, X.; Devlin, C.M.; Logsdon, D.P.; Jiang, Y.; Luo, M.; He, Y.; Yu, Z.; Tong, Y.; Lipking, K.P.; et al. Apurinic/apyrimidinic endonuclease/redox factor-1 (APE1/Ref-1) redox function negatively regulates NRF2. J. Biol. Chem. 2015, 290, 3057–3068. [Google Scholar] [CrossRef] [Green Version]
- Lando, D.; Pongratz, I.; Poellinger, L.; Whitelaw, M.L. A redox mechanism controls differential DNA binding activities of hypoxia-inducible factor (HIF) 1α and the HIF-like factor. J. Biol. Chem. 2000, 275, 4618–4627. [Google Scholar] [CrossRef] [Green Version]
- Shan, J.L.; He, H.T.; Li, M.X.; Zhu, J.W.; Cheng, Y.; Hu, N.; Wang, G.; Wang, D.; Yang, X.Q.; He, Y.; et al. APE1 promotes antioxidant capacity by regulating Nrf-2 function through a redox-dependent mechanism. Free Radic. Biol. Med. 2015, 78, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.G.; Song, Y.J.; Cho, E.J.; Lee, S.K.; Park, J.B.; Yu, J.H.; Lim, S.P.; Kim, J.M.; Jeon, B.H. Alteration of APE1/ref-1 expression in non-small cell lung cancer: The implications of impaired extracellular superoxide dismutase and catalase antioxidant systems. Lung Cancer 2008, 60, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Tell, G.; Quadrifoglio, F.; Tiribelli, C.; Kelley, M.R. The many functions of APE1/Ref-1: Not only a DNA repair enzyme. Antioxid. Redox Signal. 2009, 11, 601–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Xiang, D.B.; Yang, X.Q.; Chen, L.S.; Li, M.X.; Zhong, Z.Y.; Zhang, Y.S. APE1 overexpression is associated with cisplatin resistance in non-small cell lung cancer and targeted inhibition of APE1 enhances the activity of cisplatin in A549 cells. Lung Cancer 2009, 66, 298–304. [Google Scholar] [CrossRef]
- Pacifico, F.; Mauro, C.; Barone, C.; Crescenzi, E.; Mellone, S.; Monaco, M.; Chiappetta, G.; Terrazzano, G.; Liguoro, D.; Vito, P.; et al. Oncogenic and anti-apoptotic activity of NF-κB in human thyroid carcinomas. J. Biol. Chem. 2004, 279, 54610–54619. [Google Scholar] [CrossRef] [Green Version]
- Yeh, P.Y.; Chuang, S.E.; Yeh, K.H.; Song, Y.C.; Ea, C.K.; Cheng, A.L. Increase of the resistance of human cervical carcinoma cells to cisplatin by inhibition of the MEK to ERK signaling pathway partly via enhancement of anticancer drug-induced NFκB activation. Biochem. Pharmacol. 2002, 63, 1423–1430. [Google Scholar] [CrossRef]
- Deben, C.; Deschoolmeester, V.; De Waele, J.; Jacobs, J.; Van Den Bossche, J.; Wouters, A.; Peeters, M.; Rolfo, C.; Smits, E.; Lardon, F.; et al. Hypoxia-induced cisplatin resistance in non-small cell lung cancer cells is mediated by HIF-1α and mutant p53 and can be overcome by induction of oxidative stress. Cancers 2018, 10, 126. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, N.; Sugimoto, K.; Tang, J.; Nishi, T.; Sato, I.; Hiramoto, M.; Aizawa, S.; Hatakeyama, M.; Ohba, R.; Hatori, H.; et al. High-performance affinity beads for identifying drug receptors. Nat. Biotechnol. 2000, 18, 877–881. [Google Scholar] [CrossRef]
- Shah, F.; Logsdon, D.; Messmann, R.A.; Fehrenbacher, J.C.; Fishel, M.L.; Kelley, M.R. Exploiting the Ref-1-APE1 node in cancer signaling and other diseases: From bench to clinic. NPJ Precis. Oncol. 2017, 1, 1–19. [Google Scholar] [CrossRef] [Green Version]
- McIlwain, D.W.; Fishel, M.L.; Boos, A.; Kelley, M.R.; Jerde, T.J. APE1/Ref-1 redox-specific inhibition decreases survivin protein levels and induces cell cycle arrest in prostate cancer cells. Oncotarget 2018, 9, 10962–10977. [Google Scholar] [CrossRef] [Green Version]
- Fishel, M.L.; Jiang, Y.; Rajeshkumar, N.V.; Scandura, G.; Sinn, A.L.; He, Y.; Shen, C.; Jones, D.R.; Pollok, K.E.; Ivan, M.; et al. Impact of APE1/Ref-1 Redox Inhibition on Pancreatic Tumor Growth. Mol. Cancer Ther. 2012, 10, 1698–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, J.G.; Saraiva, N.; Guerreiro, P.S.; Louro, H.; Silva, M.J.; Miranda, J.P.; Castro, M.; Batinic-Haberle, I.; Fernandes, A.S.; Oliveira, N.G. Ochratoxin A-induced cytotoxicity, genotoxicity and reactive oxygen species in kidney cells: An integrative approach of complementary endpoints. Food Chem. Toxicol. 2016, 87, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.G.; Saraiva, N.; Batinic-Haberle, I.; Castro, M.; Oliveira, N.G.; Fernandes, A.S. The SOD mimic MnTnHex-2-PyP5+ reduces the viability and migration of 786-O human renal cancer cells. Antioxidants 2019, 8, 490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, A.S.; Flórido, A.; Saraiva, N.; Cerqueira, S.; Ramalhete, S.; Cipriano, M.; Cabral, M.F.; Miranda, J.P.; Castro, M.; Costa, J.; et al. Role of the Copper(II) Complex Cu[15]pyN5 in Intracellular ROS and Breast Cancer Cell Motility and Invasion. Chem. Biol. Drug Des. 2015, 86, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.G.; Keser, V.; Jackson, C.; Saraiva, N.; Guerreiro, Í.; Almeida, N.; Camões, S.P.; Manguinhas, R.; Castro, M.; Miranda, J.P.; et al. A multiple endpoint approach reveals potential in vitro anticancer properties of thymoquinone in human renal carcinoma cells. Food Chem. Toxicol. 2020, 136, 111076. [Google Scholar] [CrossRef] [PubMed]
- Flórido, A.; Saraiva, N.; Cerqueira, S.; Almeida, N.; Parsons, M.; Batinic-Haberle, I.; Miranda, J.P.; Costa, J.G.; Carrara, G.; Castro, M.; et al. The manganese(III) porphyrin MnTnHex-2-PyP5+ modulates intracellular ROS and breast cancer cell migration: Impact on doxorubicin-treated cells. Redox Biol. 2019, 20, 367–378. [Google Scholar] [CrossRef]
- Karachaliou, N.; Moreno, M.D.L.L.G.; Sosa, A.E.; Santarpia, M.; Lazzari, C.; Capote, A.R.; Massuti, B.; Rosell, R. Using genetics to predict patient response to platinum-based chemotherapy. Expert Rev. Precis. Med. Drug Dev. 2017, 2, 21–32. [Google Scholar] [CrossRef]
- Rosell, R.; Karachaliou, N.; Arrieta, O. Novel molecular targets for the treatment of lung cancer. Curr. Opin. Oncol. 2020, 32, 37–43. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, D.F.; Rui, Y.; Jiang, A.B.; Liu, Z.L.; Huang, L.N. Prognosis value of HIF-1α expression in patients with non-small cell lung cancer. Gene 2014, 541, 69–74. [Google Scholar] [CrossRef]
- Okabe, T.; Okamoto, I.; Tamura, K.; Terashima, M.; Yoshida, T.; Satoh, T.; Takada, M.; Fukuoka, M.; Nakagawa, K. Differential constitutive activation of the epidermal growth factor receptor in non-small cell lung cancer cells bearing EGFR gene mutation and amplification. Cancer Res. 2007, 67, 2046–2053. [Google Scholar] [CrossRef] [Green Version]
- Umelo, I.A.; De Wever, O.; Kronenberger, P.; Noor, A.; Teugels, E.; Chen, G.; Bracke, M.; De Grève, J. Combined inhibition of rho-associated protein kinase and EGFR suppresses the invasive phenotype in EGFR-dependent lung cancer cells. Lung Cancer 2015, 90, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Yang, Y.; Katz, S.I. Dexamethasone pretreatment impairs the thymidylate synthase inhibition mediated flare in thymidine salvage pathway activity in non-small cell lung cancer. PLoS ONE 2018, 13, e0202384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.C.; Liang, X.; Liu, Z.Y.; Cui, J.; Liu, Y.; Jing, L.; Jiang, L.L.; Ma, J.Q.; Han, L.L.; Guo, Q.Q.; et al. In vitro synergistic antitumor efficacy of sequentially combined chemotherapy/icotinib in non-small cell lung cancer cell lines. Oncol. Rep. 2015, 33, 239–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Xu, P.; Liu, Z.; Zhen, Y.; Chen, Y.; Liu, Y.; Fu, Q.; Deng, X.; Liang, Z.; Li, Y.; et al. Dual roles of miR-374a by modulated c-Jun respectively targets CCND1-inducing PI3K/AKT signal and PTEN-suppressing Wnt/β-catenin signaling in non-small-cell lung cancer article. Cell Death Dis. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Miao, H.; Ma, S.; Zhang, L.; You, C.; Tang, F.; Yang, C.; Tian, X.; Wang, F.; Luo, Y.; et al. FePt-Cys nanoparticles induce ROS-dependent cell toxicity, and enhance chemo-radiation sensitivity of NSCLC cells in vivo and in vitro. Cancer Lett. 2018, 418, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.F.; Hanke, N.T.; Sands, B.J.; Carbajal, L.; Anderl, J.L.; Garland, L.L. Carfilzomib demonstrates broad anti-tumor activity in pre-clinical non-small cell and small cell lung cancer models. J. Exp. Clin. Cancer Res. 2014, 33, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Delaplane, S.; Jiang, A.; Reed, A.; He, Y.; Fishel, M.; Nyland, R.L.; Borch, R.F.; Qiao, X.; Georgiadis, M.M.; et al. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: Small-molecule inhibition of the redox function of Ape1. Antioxid. Redox Signal. 2008, 10, 1853–1867. [Google Scholar] [CrossRef]
- Zou, G.-M.; Maitra, A. Small-molecule inhibitor of the AP endonuclease 1/REF-1 E3330 inhibits pancreatic cancer cell growth and migration. Mol. Cancer Ther. 2008, 7, 2012–2021. [Google Scholar] [CrossRef] [Green Version]
- Singh-Gupta, V.; Joiner, M.C.; Runyan, L.; Yunker, C.K.; Sarkar, F.H.; Miller, S.; Gadgeel, S.M.; Konski, A.A.; Hillman, G.G. Soy isoflavones augment radiation effect by inhibiting APE1/ref-1 DNA repair activity in non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 688–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, G.I.; Edwards, C.D.; Ewen, M.E.; Rollins, B.J. p16INK4A Participates in a G1 Arrest Checkpoint in Response to DNA Damage. Mol. Cell. Biol. 1998, 18, 378–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorenson, C.M.; Barry, M.A.; Eastman, A. Analysis of events associated with cell cycle arrest at G2 phase and cell death induced by cisplatin. J. Natl. Cancer Inst. 1990, 82, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Sarin, N.; Engel, F.; Kalayda, G.V.; Mannewitz, M.; Cinatl, J.; Rothweiler, F.; Michaelis, M.; Saafan, H.; Ritter, C.A.; Jaehde, U.; et al. Cisplatin resistance in non-small cell lung cancer cells is associated with an abrogation of cisplatin-induced G2/M cell cycle arrest. PLoS ONE 2017, 12, e0181081. [Google Scholar] [CrossRef] [PubMed]
- Nyland, R.L.; Luo, M.; Kelley, M.R.; Borch, R.F. Design and Synthesis of Novel Quinone Inhibitors Targeted to the Redox Function of Apurinic/Apyrimidinic Endonuclease 1/Redox Enhancing Factor-1 (Ape1/Ref-1). J. Med. Chem. 2010, 53, 1200–1210. [Google Scholar] [CrossRef] [Green Version]
- Jiang, A.; Gao, H.; Kelley, M.R.; Qiao, X. Inhibition of APE1/Ref-1 Redox Activity with APX3330 Blocks Retinal Angiogenesis in vitro and in vivo. Vis. Res. 2011, 51, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Ahmed, F.; Ali, S.; Philip, P.A.; Kucuk, O.; Sarkar, F.H. Inactivation of Nuclear Factor kB by Soy Isoflavone Genistein Contributes to Increased Apoptosis Induced by Chemotherapeutic Agents in Human Cancer Cells. Cancer Res. 2005, 65, 6934–6942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Gao, Y.; Cui, Y.; Zhang, T.; Cui, R.; Jiang, Y.; Shi, J. Overexpression of CD44 is associated with the occurrence and migration of non-small cell lung cancer. Mol. Med. Rep. 2016, 14, 3159–3167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauvois, B. New facets of matrix metalloproteinases MMP-2 and MMP-9 as cell surface transducers: Outside-in signaling and relationship to tumor progression. Biochim. Biophys. Acta 2012, 1825, 29–36. [Google Scholar] [CrossRef]
- Yan, C.; Boyd, D.D. Regulation of matrix metalloproteinase gene expression. J. Cell. Physiol. 2007, 211, 19–26. [Google Scholar] [CrossRef]
- El-badrawy, M.K.; Yousef, A.M.; Shaalan, D.; Elsamanoudy, A.Z. Matrix Metalloproteinase-9 Expression in Lung Cancer Patients and Its Relation to Serum MMP-9 Activity, Pathologic Type, and Prognosis. J. Bronchol. Interv. Pulmonol. 2014, 21, 327–334. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manguinhas, R.; Fernandes, A.S.; Costa, J.G.; Saraiva, N.; Camões, S.P.; Gil, N.; Rosell, R.; Castro, M.; Miranda, J.P.; Oliveira, N.G. Impact of the APE1 Redox Function Inhibitor E3330 in Non-Small Cell Lung Cancer Cells Exposed to Cisplatin: Increased Cytotoxicity and Impairment of Cell Migration and Invasion. Antioxidants 2020, 9, 550. https://doi.org/10.3390/antiox9060550
Manguinhas R, Fernandes AS, Costa JG, Saraiva N, Camões SP, Gil N, Rosell R, Castro M, Miranda JP, Oliveira NG. Impact of the APE1 Redox Function Inhibitor E3330 in Non-Small Cell Lung Cancer Cells Exposed to Cisplatin: Increased Cytotoxicity and Impairment of Cell Migration and Invasion. Antioxidants. 2020; 9(6):550. https://doi.org/10.3390/antiox9060550
Chicago/Turabian StyleManguinhas, Rita, Ana S. Fernandes, João G. Costa, Nuno Saraiva, Sérgio P. Camões, Nuno Gil, Rafael Rosell, Matilde Castro, Joana P. Miranda, and Nuno G. Oliveira. 2020. "Impact of the APE1 Redox Function Inhibitor E3330 in Non-Small Cell Lung Cancer Cells Exposed to Cisplatin: Increased Cytotoxicity and Impairment of Cell Migration and Invasion" Antioxidants 9, no. 6: 550. https://doi.org/10.3390/antiox9060550
APA StyleManguinhas, R., Fernandes, A. S., Costa, J. G., Saraiva, N., Camões, S. P., Gil, N., Rosell, R., Castro, M., Miranda, J. P., & Oliveira, N. G. (2020). Impact of the APE1 Redox Function Inhibitor E3330 in Non-Small Cell Lung Cancer Cells Exposed to Cisplatin: Increased Cytotoxicity and Impairment of Cell Migration and Invasion. Antioxidants, 9(6), 550. https://doi.org/10.3390/antiox9060550