Neuroprotection by Phytoestrogens in the Model of Deprivation and Resupply of Oxygen and Glucose In Vitro: The Contribution of Autophagy and Related Signaling Mechanisms

 , ,
, ,  ,
, 

Abstract
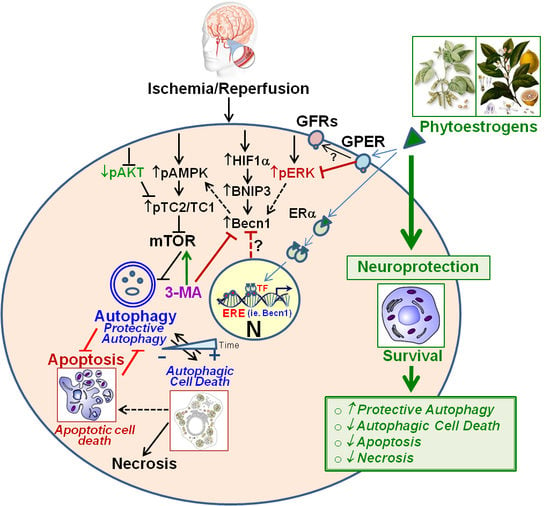
1. Introduction
2. Materials and Methods
2.1. Primary Cortical Neuron Cultures
2.2. Oxygen and Glucose Deprivation (OGD) and Resupply (OGDR)
2.3. Western Blotting
2.4. Immunocytochemistry
2.5. RT-PCR Analysis
2.5.1. RNA Extraction
2.5.2. Real-Time q-PCR
2.6. Cell Viability Test XTT
2.7. Caspase 3 Activity Test
2.8. Statistics
3. Results
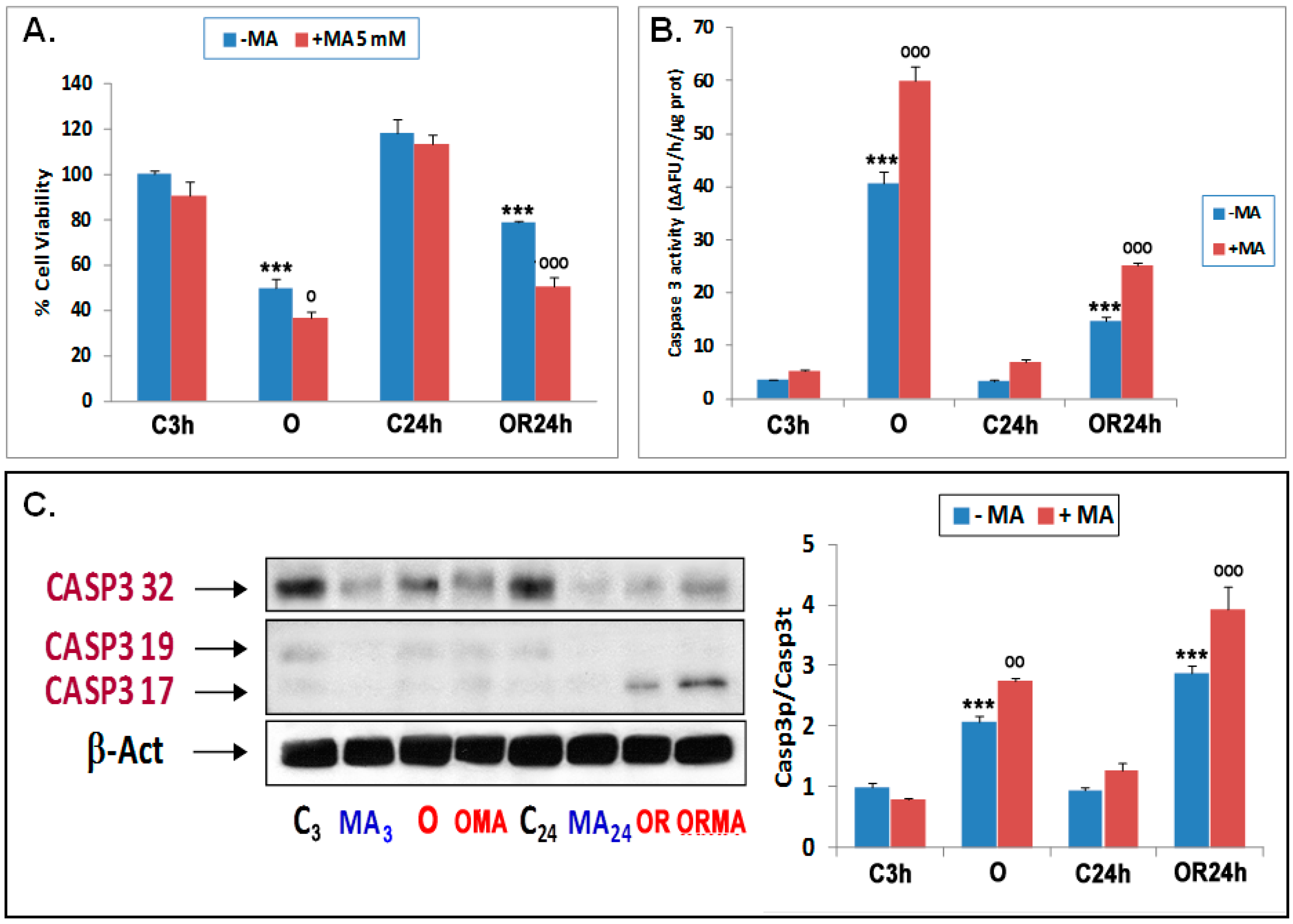
3.1. Early Autophagy Activation Plays a Protective Role in Primary Cortical Neurons upon Experimental OGD and OGDR
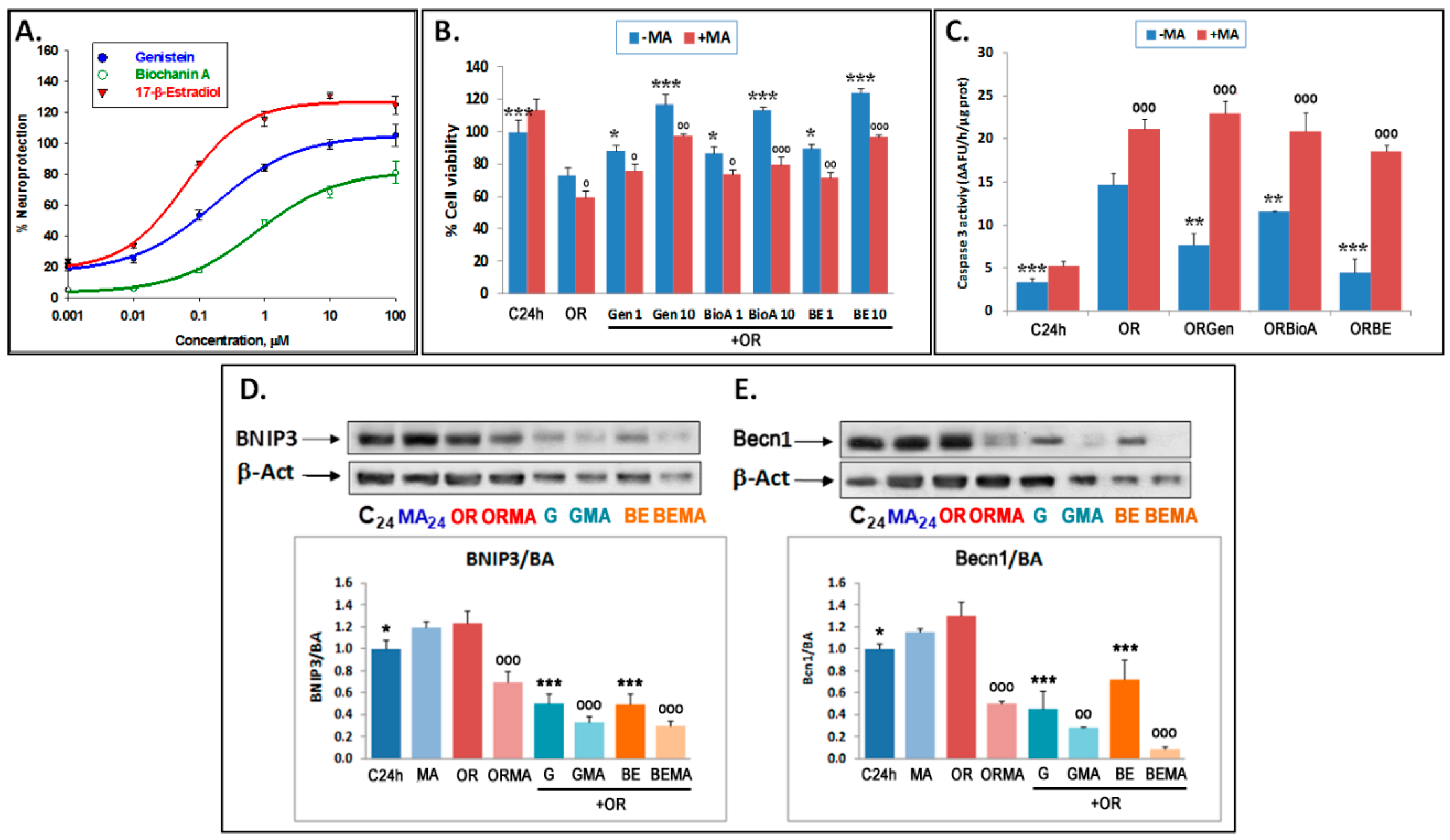
3.2. Phytoestrogens Afford Neuroprotection by Modulating Autophagy
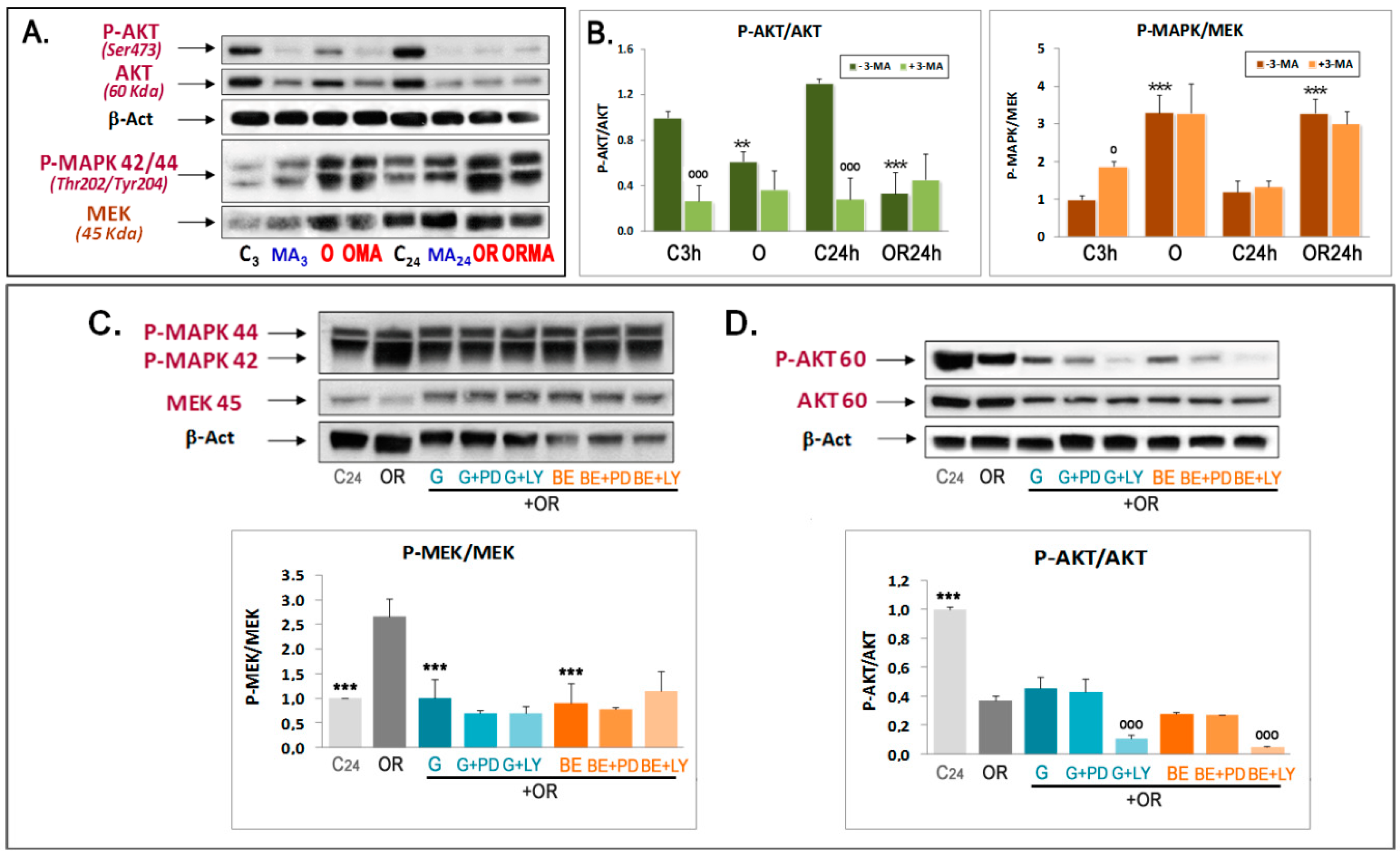
3.3. Phytoestrogens Activate mTOR and Act as MEK Inhibitors in Advanced Stages of OGDR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Gen | Genistein |
| Becn1 | Beclin 1 |
| BioA | Biochanin A |
| Daid | Daidzein |
| BE | 17β-estradiol |
| ER | Estrogen receptors |
| 3-MA | 3-Methyladenine |
| OGD | Oxygen and Glucose Deprivation |
| OGDR | Oxygen and Glucose Deprivation and Resupply |
| LY294002 | AKT inhibitor |
| PD98059 | MEK inhibitor |
References
- Castillo, J.; Alvarez-Sabin, J.; Dávalos, A.; Díez-Tejedor, E.; Lizasoain, I.; Martínez-Vila, E.; Vivancos, J.; Zarranz, J.J. Consensus review. Pharmacological neuroprotection in cerebral ischemia: Is it still a therapeutic option? Neurologia 2003, 18, 368–384. [Google Scholar]
- Pluta, R.; Salínska, E.; Puka, M.; Stafiej, A.; Lazarewicz, J.W. Early changes in extracellular amino acids and calcium concentrations in rabbit hippocampus following complete 15-min cerebral ischemia. Resuscitation 1988, 16, 193–210. [Google Scholar] [CrossRef]
- Hossmann, K.A. Pathophysiology and therapy of experimental stroke. Cell. Mol. Neurobiol. 2006, 26, 1057–1083. [Google Scholar] [CrossRef]
- Rami, A.; Kögel, D. Apoptosis meets Autophagy-Like cell death in the ischemic penumbra: Two sides of the same coin? Autophagy 2008, 4, 422–426. [Google Scholar] [CrossRef]
- Fernández-Gómez, F.J.; Hernández, F.; Argandoña, L.; Galindo, M.F.; Segura, T.; Jordán, J. Farmacología de la neuroprotección en el ictus isquémico agudo. Rev. Neurol. 2008, 47, 253–260. [Google Scholar] [CrossRef]
- Murrow, L.; Debnath, J. Autophagy as a Stress-Response and Quality-Control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. 2013, 8, 105–137. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Adhami, F.; Liao, G.; Morozov, Y.M.; Schloemer, A.; Schmithorst, V.J.; Lorenz, J.N.; Dunn, R.S.; Vorhees, C.V.; Wills-Karp, M.; Degen, J.L.; et al. Cerebral Ischemia-Hypoxia induces intravascular coagulation and autophagy. Am. J. Pathol. 2006, 169, 566–583. [Google Scholar] [CrossRef] [PubMed]
- Gabryel, B.; Kost, A.; Kasprowska, D. Neuronal autophagy in cerebral ischemia–a potential target for neuroprotective strategies? Pharmacol. Rep. 2012, 64, 1–15. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, J.; Li, D.; Zhang, C.; Liu, M. Calcium antagonists for acute ischemic stroke. Cochrane Database Syst. Rev. 2019, 13, CD001928. [Google Scholar] [CrossRef]
- Sarkar, S.; Chakraborty, D.; Bhowmik, A.; Ghosh, M.K. Cerebral ischemic stroke: Cellular fate and therapeutic opportunities. Front. Biosci. (Landmark. Ed.) 2019, 24, 435–450. [Google Scholar] [PubMed]
- Ginsberg, M.D. Neuroprotection for ischemic stroke: Past, present and future. Neuropharmacology 2008, 55, 363–389. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, Á.; Dirnagl, U.; Urra, X.; Planas, A.M. Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016, 15, 869–881. [Google Scholar] [CrossRef]
- Radak, D.; Katsiki, N.; Resanovic, I.; Jovanovic, A.; Sudar-Milovanovic, E.; Zafirovic, S.; Mousad, S.A.; Isenovic, E.R. Apoptosis and Acute Brain Ischemia in Ischemic Stroke. Curr. Vasc. Pharmacol. 2017, 15, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Ferrari, F.; Villa, R.F. Neuroprotection for ischaemic stroke: Current status and challenges. Pharmacol. Ther. 2015, 146, 23–34. [Google Scholar] [CrossRef]
- Gibson, C.L.; Gray, L.J.; Murphy, S.P.; Bath, P.M. Estrogens and experimental ischemic stroke: A systematic review. J. Cereb. Blood Flow Metab. 2006, 26, 1103–1113. [Google Scholar] [CrossRef]
- Etgen, A.M.; Jover-Mengual, T.; Zukin, R.S. Neuroprotective Actions of Estradiol and Novel Estrogen Analogs in Ischemia: Translational Implications. Front. Neuroendocr. 2011, 32, 336–352. [Google Scholar] [CrossRef]
- Bramlett, H.M.; Dietrich, W.D. Neuropathological protection after traumatic brain injury in intact female rats versus males or ovariectomized females. J. Neurotrauma 2001, 18, 891–900. [Google Scholar] [CrossRef]
- Schreihofer, D.A.; Oppong-Gyebi, A. Genistein: Mechanisms of action for a pleiotropic neuroprotective agent in stroke. Nutr. Neurosci. 2019, 22, 375–391. [Google Scholar] [CrossRef]
- Shambayati, M.; Patel, M.; Ma, Y.; Cunningham, R.L.; Schreihofer, D.A. Central inflammatory response to experimental stroke is inhibited by a neuroprotective dose of dietary soy. Brain Res. 2014, 1593, 76–82. [Google Scholar] [CrossRef]
- Castelló-Ruiz, M.; Torregrosa, G.; Burguete, M.C.; Salom, J.B.; Gil, J.V.; Miranda, F.J.; Jover-Mengual, T.; Marrachelli, V.G.; Alborch, E. Soy-Derived phytoestrogens as preventive and acute neuroprotectors in experimental ischemic stroke: Influence of rat strain. Phytomedicine 2011, 18, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Cortina, B.; Torregrosa, G.; Castelló-Ruiz, M.; Burguete, M.C.; Moscardó, A.; Latorre, A.; Salom, J.B.; Vallés, J.; Santos, M.T.; Alborch, E. Improvement of the circulatory function partially accounts for the neuroprotective action of the phytoestrogen genistein in experimental ischemic stroke. Eur. J. Pharmacol. 2013, 708, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Schreihofer, D.A.; Do, K.D.; Schreihofer, A.M. High-Soy diet decreases infarct size after permanent middle cerebral artery occlusion in female rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R103–R108. [Google Scholar] [CrossRef] [PubMed]
- Schreihofer, D.A. Phytoestrogens as neuroprotectants. Drugs Today (Barc.) 2009, 45, 609–627. [Google Scholar] [CrossRef]
- Schreihofer, D.A.; Redmond, L. Soy phytoestrogens are neuroprotective against Stroke-Like injury in vitro. Neuroscience 2009, 158, 602–609. [Google Scholar] [CrossRef]
- Ma, T.C.; Campana, A.; Lange, P.S.; Lee, H.H.; Banerjee, K.; Bryson, J.B.; Mahishi, L.; Alam, S.; Giger, R.J.; Barnes, S.; et al. A Large-Scale chemical screen for regulators of the arginase 1 promoter identifies the soy isoflavone Daidzein as a clinically approved small molecule that can promote neuronal protection or regeneration via a cAMP-Independent pathway. J. Neurosci. 2010, 30, 739–748. [Google Scholar] [CrossRef]
- Ma, Y.; Sullivan, J.C.; Schreihofer, D.A. Dietary Genistein and equol (4,7 isoflavandiol) reduce oxidative stress and protect rats against focal cerebral ischemia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, 871–877. [Google Scholar] [CrossRef]
- Kim, J.; Fann, D.Y.; Seet, R.C.; Jo, D.G.; Mattson, M.P.; Arumugam, T.V. Phytochemicals in ischemic stroke. Neuromol. Med. 2016, 18, 283–305. [Google Scholar] [CrossRef]
- Arce, C.; Arteaga, J.L.; Sánchez-Mendoza, E.; Oset-Gasque, M.J.; Cañadas, S.; González, M.P. Added after Anoxia-Reoxigenation stress, genistein rescues from death the rat embryo cortical neurons. Neurosci. Med. 2010, 1, 50–59. [Google Scholar]
- Hurtado, O.; Ballesteros, I.; Cuartero, M.I.; Moraga, A.; Pradillo, J.M.; Ramírez-Franco, J.; Bartolomé-Martín, D.; Pascual, D.; Torres, M.; Sánchez-Prieto, J.; et al. Daidzein has neuroprotective effects through ligand-binding-independent PPAR γ activation. Neurochem. Int. 2012, 61, 119–127. [Google Scholar] [CrossRef]
- Aras, A.B.; Guven, M.; Akman, T.; Alacam, H.; Kalkan, Y.; Silan, C.; Cosar, M. Genistein exerts neuroprotective effect on focal cerebral ischemia injury in rats. Inflammation 2015, 38, 1311–1321. [Google Scholar] [CrossRef]
- Guo, M.; Lu, H.; Qin, J.; Qu, S.; Wang, W.; Guo, Y.; Liao, W.; Song, M.; Chen, J.; Wang, Y. Biochanin a provides neuroprotection against cerebral Ischemia-Reperfusion injury by Nrf2-Mediated inhibition of oxidative stress and inflammation signaling pathway in rats. Med. Sci. Monit. 2019, 25, 8975–8983. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, S.; Oset-Gasque, M.J.; Arce, C.; Martínez-Honduvilla, C.; González, M.P. Michocondrial involvement in nitric Oxide-Induced cellular death in cortical neurons in culture. J. Neurosc. Res. 2006, 83, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Ryou, M.G.; Mallet, R.T. An in vitro Oxygen-Glucose deprivation model for studying ischemia-reperfusion injury of neuronal cells. Methods Mol. Biol. 2018, 1717, 229–235. [Google Scholar] [PubMed]
- Wu, Y.T.; Tan, H.L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Tavares, M.R.; Pavan, I.C.; Amaral, C.L.; Meneguello, L.; Luchessi, A.D.; Simabuco, F.M. The S6K protein family in health and disease. Life Sci. 2015, 131, 1–10. [Google Scholar] [CrossRef]
- Lu, N.; Li, X.; Tan, R.; An, J.; Cai, Z.; Hu, X.; Wang, F.; Wang, H.; Lu, C.; Lu, H. HIF-1α/Beclin1-Mediated autophagy is involved in neuroprotection induced by hypoxic preconditioning. Mol. Neurosci. 2018, 66, 238–250. [Google Scholar] [CrossRef]
- Sale, E.M.; Sale, G.J. Protein kinase B: Signalling roles and therapeutic targeting. Cell. Mol. Life Sci. 2008, 65, 113–127. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Xu, F.; Gu, J.H.; Qin, Z.H. Neuronal autophagy in cerebral ischemia. Neurosci. Bull. 2012, 28, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Sun, Y.Y.; Liu, K.Y. Autophagy and inflammation in ischemic stroke. Neural Regen. Res. 2020, 15, 1388–1396. [Google Scholar]
- Hou, K.; Xu, D.; Li, F.; Chen, S.; Li, Y. The progress of neuronal autophagy in cerebral ischemia stroke: Mechanisms, roles and research methods. J. Neurol. Sci. 2019, 400, 72–82. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Sureda, A.; Sanches-Silva, A.; Pandima Devi, K.; Ahmed, T.; Shahid, M.; Sobarzo-Sánchez, E.; Dacrema, M.; Daglia, M.; Braidy, N.; et al. Novel therapeutic strategies for stroke: The role of autophagy. Crit. Rev. Clin. Lab. Sci. 2019, 56, 182–199. [Google Scholar] [CrossRef] [PubMed]
- Altavilla, D.; Crisafulli, A.; Marini, H.; Espósito, M.; D’Anna, R.; Corrado, F.; Bitto, A.; Squadrito, F. Cardiovascular effects of the phytoestrogen genistein. Med. Chem. Cardiovasc. Hematol. Agents 2004, 2, 179–186. [Google Scholar] [CrossRef]
- Burguete, M.C.; Torregrosa, G.; Pérez-Asensio, F.J.; Castelló-Ruiz, M.; Salom, J.B.; Gil, J.V.; Alborch, E. Dietary phytoestrogens improve stroke outcome after transient focal cerebral ischemia in rats. Eur. J. Neurosci. 2006, 23, 703–710. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef]
- Shi, R.Y.; Zhu, S.H.; Li, V.; Gibson, S.B.; Xu, X.S.; Kong, J.M. BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci. Ther. 2014, 20, 1045–1055. [Google Scholar] [CrossRef]
- Ułamek-Kozioł, M.; Kocki, J.; Bogucka-Kocka, A.; Januszewski, S.; Bogucki, J.; Czuczwar, S.J.; Pluta, R. Autophagy, mitophagy and apoptotic gene changes in the hippocampal CA1 area in a rat ischemic model of Alzheimer’s disease. Pharmacol. Rep. 2017, 69, 1289–1294. [Google Scholar]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Kocki, J.; Januszewski, S.; Bogucki, J.; Bogucka-Kocka, A.; Pluta, R. Dysregulation of Autophagy, Mitophagy, and apoptosis genes in the CA3 region of the hippocampus in the ischemic model of Alzheimer’s disease in the rat. J. Alzheimer’s Dis. 2019, 72, 1279–1286. [Google Scholar]
- Ułamek-Kozioł, M.; Kocki, J.; Bogucka-Kocka, A.; Petniak, A.; Gil-Kulik, P.; Januszewski, S.; Bogucki, J.; Jabłoński, M.; Furmaga-Jabłońska, W.; Brzozowska, J.; et al. Dysregulation of autophagy, Mitophagy, and apoptotic genes in the medial temporal lobe cortex in an ischemic model of Alzheimer’s disease. J. Alzheimer’s Dis. 2016, 54, 113–121. [Google Scholar]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell. Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Tamargo-Gómez, I.; Mariño, G. AMPK: Regulation of metabolic dynamics in the context of autophagy. Int. J. Mol. Sci. 2018, 19, 3812. [Google Scholar] [CrossRef]
- Wang, J.; Whiteman, M.W.; Lian, H.; Wang, G.; Singh, A.; Huang, D.; Denmark, T.A. Non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J. Biol. Chem. 2009, 284, 21412–21424. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Adhami, F.; Schloemer, A.; Kuan, C.Y. The roles of autophagy in cerebral ischemia. Autophagy 2007, 3, 42–44. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef]
- Tovar-y-Romo, L.B.; Penagos-Puig, A.; Ramírez-Jarquín, J.O. Endogenous recovery after brain damage: Molecular mechanisms that balance neuronal life/death fate. J. Neurochem. 2016, 136, 13–27. [Google Scholar] [CrossRef]
- Acaz-Fonseca, E.; Castelló-Ruiz, M.; Burguete, M.C.; Aliena-Valero, A.; Salom, J.B.; Torregrosa, G.; García-Segura, L.M. Insight into the molecular sex dimorphism of ischaemic stroke in rat cerebral cortex: Focus on neuroglobin, sex steroids and autophagy. Eur. J. Neurosci. 2020. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay Type | Primary Ab | Concentration | Producer | MW (kDa) | Secondary Ab | Concentration | Producer |
|---|---|---|---|---|---|---|---|
| (A) Western Blot | Rabbit-anti LC3B | 1/1000 | Cell Signalling | 16, 14 | HRP-conjugated goat anti-rabbit | 1/1000 | Sigma-Aldrich |
| Rabbit-anti caspase 3 Rabbit-anti-Cleaved-casp. 3 | 1/1000 | Cell Signalling | 32, 19, 17 | HRP-conjugated goat anti-rabbit | 1/1000 | Sigma-Aldrich | |
| Rabbit-anti mTOR Rabbit-anti Ser2448P-mTOR | 1/1000 | Cell Signalling | 285 | HRP-conjugated goat anti-rabbit | 1/1000 | Sigma-Aldrich | |
| Rabbit-anti AMPKα Rabbit-anti Thr172P-AMPK | 1/1000 | Cell Signalling | 62 | HRP-conjugated goat anti-rabbit | 1/1000 | Sigma-Aldrich | |
| Rabbit-anti p70S6K Rabbit-anti Thr389P-p70S6K | 1/1000 | Cell Signalling | 70, 85 | HRP-conjugated goat anti-rabbit | 1/1000 | Sigma-Aldrich | |
| Rabbit-anti AKT Rabbit-anti Ser473P-AKT | 1/1000 | Cell Signalling | 60 | HRP-conjugated goat anti-rabbit | 1/1000 | Sigma-Aldrich | |
| Rabbit-anti Thr202/Tyr204P-MAPK42,44 | 1/1000 | Cell Signalling | 42, 44 | HRP-conjugated goat anti-rabbit | 1/1000 | Sigma-Aldrich | |
| Rabbit-anti Beclin-1 | 1/1000 | Cell Signalling | 60 | HRP-conjugated goat anti-rabbit | 1/1000 | Sigma-Aldrich | |
| Rabbit-anti BNIP3 | 1/1000 | Cell Signalling | 30 | HRP-conjugated goat anti-rabbit | 1/1000 | Sigma-Aldrich | |
| Mouse-anti β-Actin | 1/6000 | Sigma-Aldrich | 42 | HRP-conjugated goat anti-mouse | 1/4000 | Sigma-Aldrich | |
| (B) Fluorescence Immunocytochemistry | Rabbit-anti LC3B | 1/300 | Cell Signalling | 16, 14 | Donkey anti-rabbit Alexa Fluor 488 | 1/300 | Sigma-Aldrich |
| DAPI | 1/1000 | Sigma-Aldrich | 277.3 | - | - | - |
| Primer | Sequence |
|---|---|
| βAct forward | 5′-GCCAACCGTGAAAAGATGA-3′ |
| βAct reverse | 5′-TACGACCAGAGGCATACAGG-3′ |
| Becn1 forward | 5′-GGCTCCTATTCCATCAAAACC-3′ |
| Becn1 reverse | 5′-GGACACCCAAGCAAGACC-3′ |
| BNIP3 forward | 5′-GCTGAAATAGACACCCACAGC-3′ |
| BNIP3 reverse | 5′-GACTTGACCAATCCCATATCC-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abbruzzese, G.; Morón-Oset, J.; Díaz-Castroverde, S.; García-Font, N.; Roncero, C.; López-Muñoz, F.; Marco Contelles, J.L.; Oset-Gasque, M.J. Neuroprotection by Phytoestrogens in the Model of Deprivation and Resupply of Oxygen and Glucose In Vitro: The Contribution of Autophagy and Related Signaling Mechanisms. Antioxidants 2020, 9, 545. https://doi.org/10.3390/antiox9060545
Abbruzzese G, Morón-Oset J, Díaz-Castroverde S, García-Font N, Roncero C, López-Muñoz F, Marco Contelles JL, Oset-Gasque MJ. Neuroprotection by Phytoestrogens in the Model of Deprivation and Resupply of Oxygen and Glucose In Vitro: The Contribution of Autophagy and Related Signaling Mechanisms. Antioxidants. 2020; 9(6):545. https://doi.org/10.3390/antiox9060545
Chicago/Turabian StyleAbbruzzese, Giuseppe, Javier Morón-Oset, Sabela Díaz-Castroverde, Nuria García-Font, Cesáreo Roncero, Francisco López-Muñoz, José Luis Marco Contelles, and María Jesús Oset-Gasque. 2020. "Neuroprotection by Phytoestrogens in the Model of Deprivation and Resupply of Oxygen and Glucose In Vitro: The Contribution of Autophagy and Related Signaling Mechanisms" Antioxidants 9, no. 6: 545. https://doi.org/10.3390/antiox9060545
APA StyleAbbruzzese, G., Morón-Oset, J., Díaz-Castroverde, S., García-Font, N., Roncero, C., López-Muñoz, F., Marco Contelles, J. L., & Oset-Gasque, M. J. (2020). Neuroprotection by Phytoestrogens in the Model of Deprivation and Resupply of Oxygen and Glucose In Vitro: The Contribution of Autophagy and Related Signaling Mechanisms. Antioxidants, 9(6), 545. https://doi.org/10.3390/antiox9060545