Glutamine Cooperatively Upregulates Lipopolysaccharide-Induced Nitric Oxide Production in BV2 Microglial Cells through the ERK and Nrf-2/HO-1 Signaling Pathway



 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Cell Viability
2.4. Flow Cytometry Analysis
2.5. NO Assay
2.6. Reverse Transcription Polymerase Chain Reactions (RT–PCR)
2.7. Western Blot Analysis
2.8. Electrophoretic Mobility Shift Assay (EMSA)
2.9. Transfection of siNrf2 and siERK
2.10. Statistical Analysis
3. Results
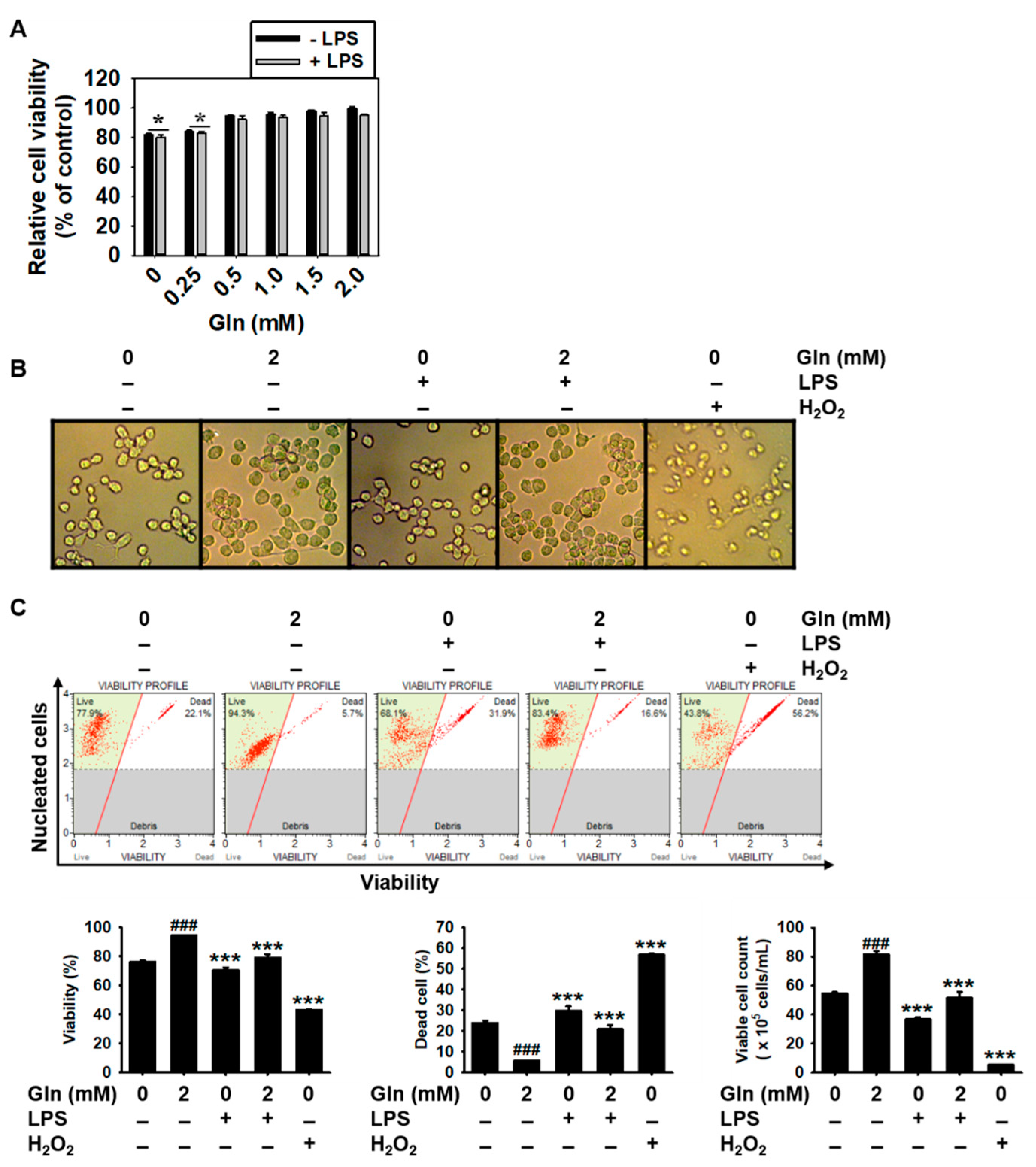
3.1. Gln Depletion Causes Mild Cytotoxicity in BV2 Microglial Cells
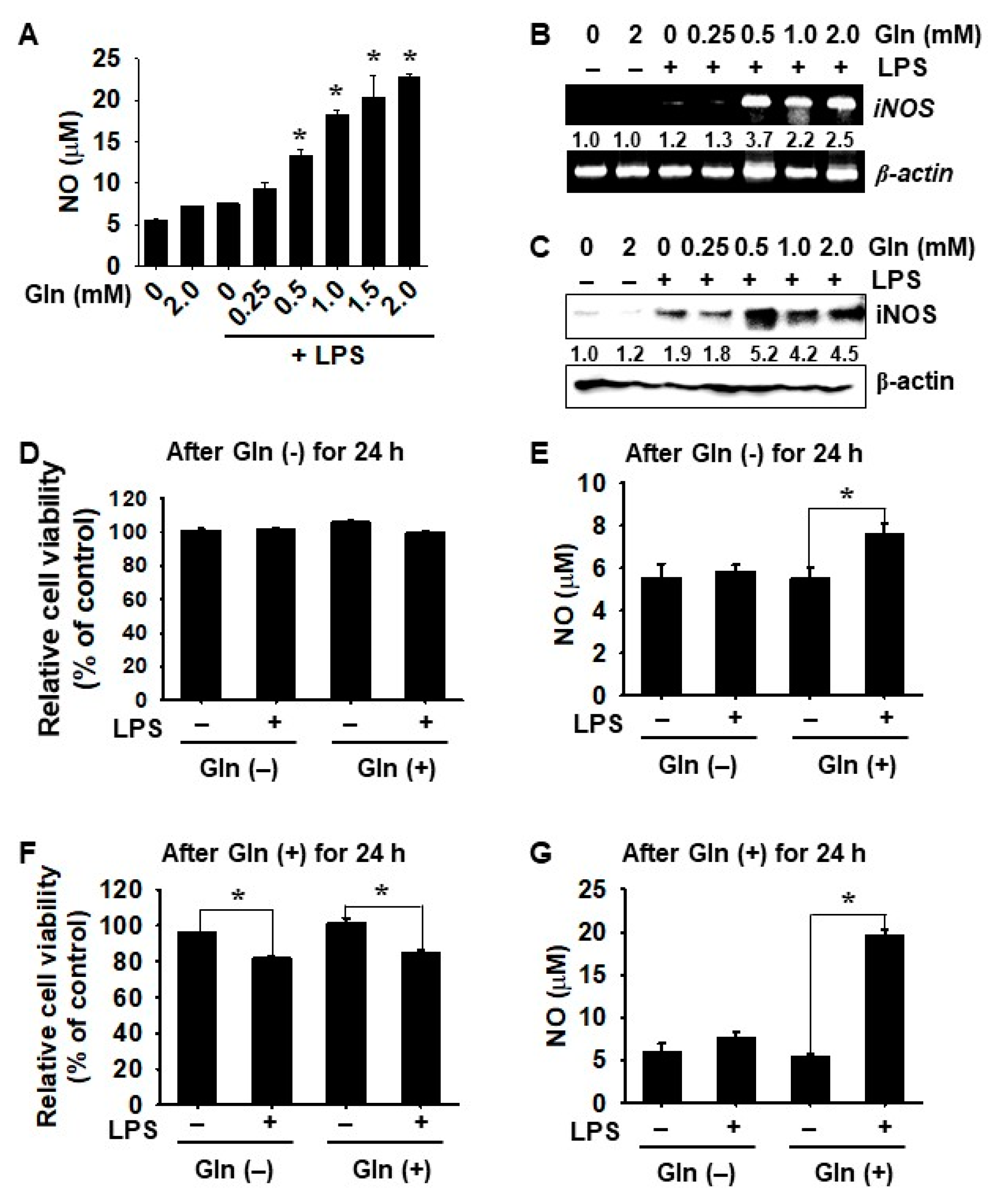
3.2. LPS-mediated iNOS Expression and NO Production Depend on the Presence of Gln
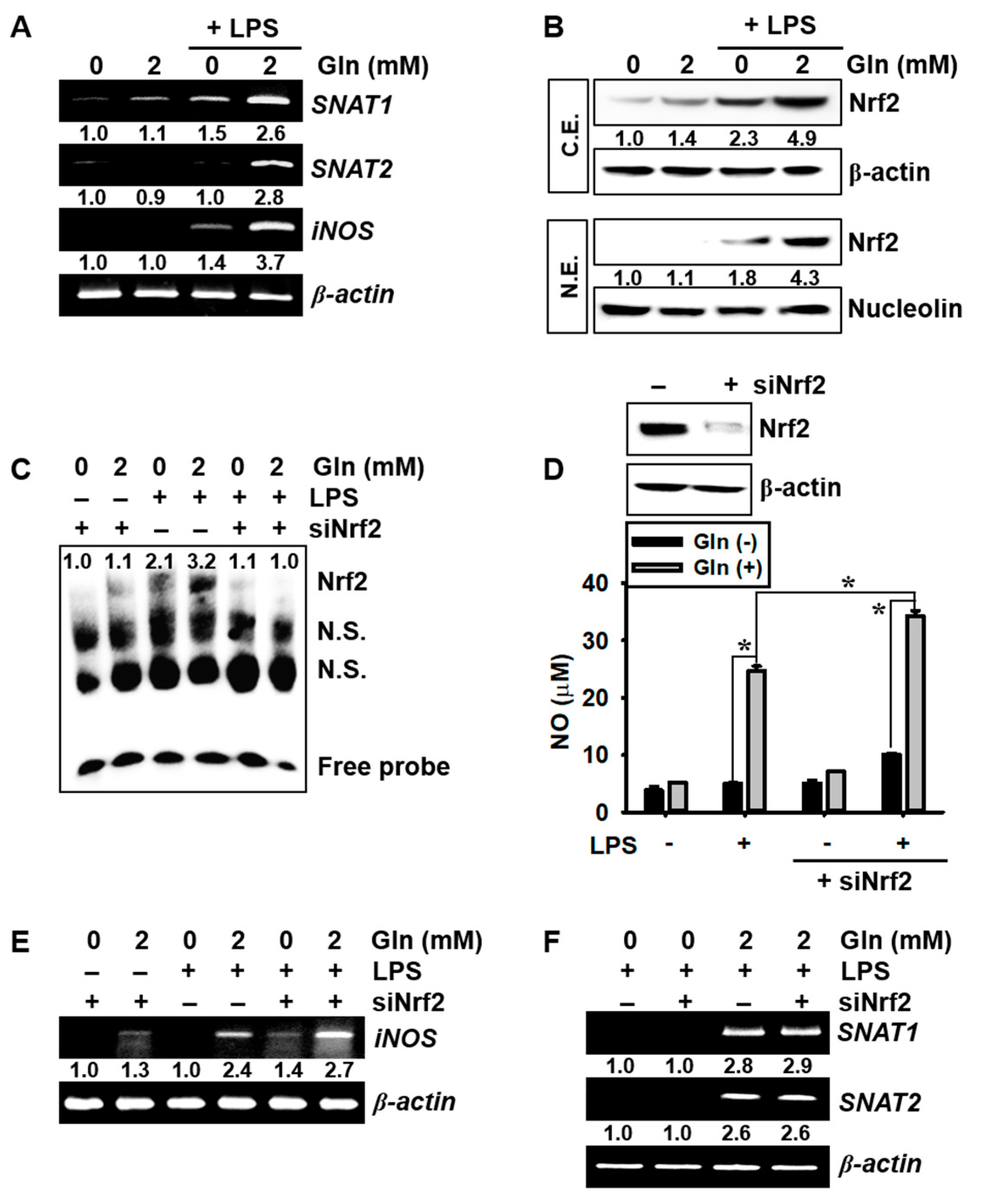
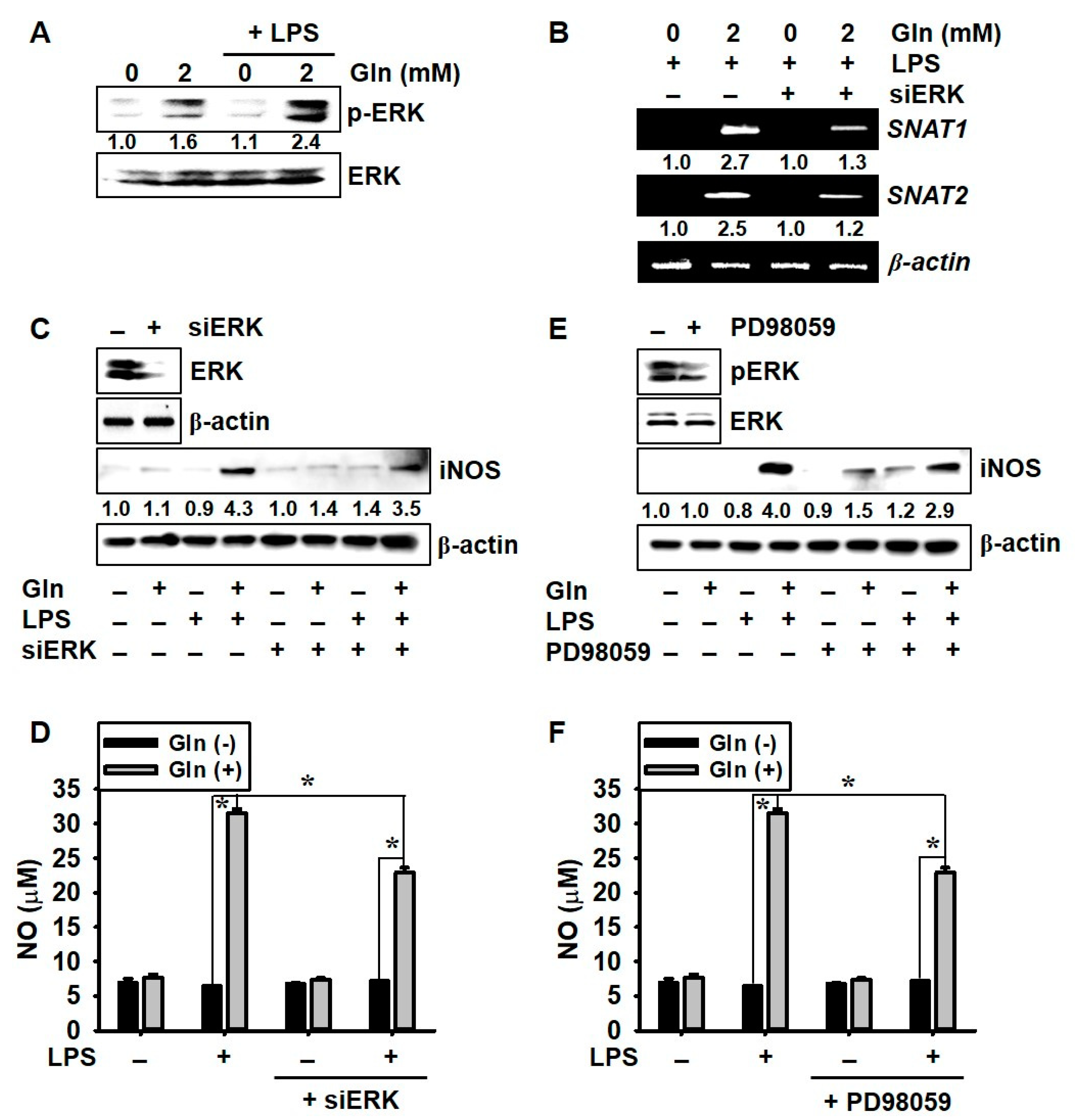
3.3. LPS Upregulates SNAT and iNOS Expression in the Presence of Gln and Nrf2 Activation Inhibits iNOS Expression and NO Production, but not SNAT Expression
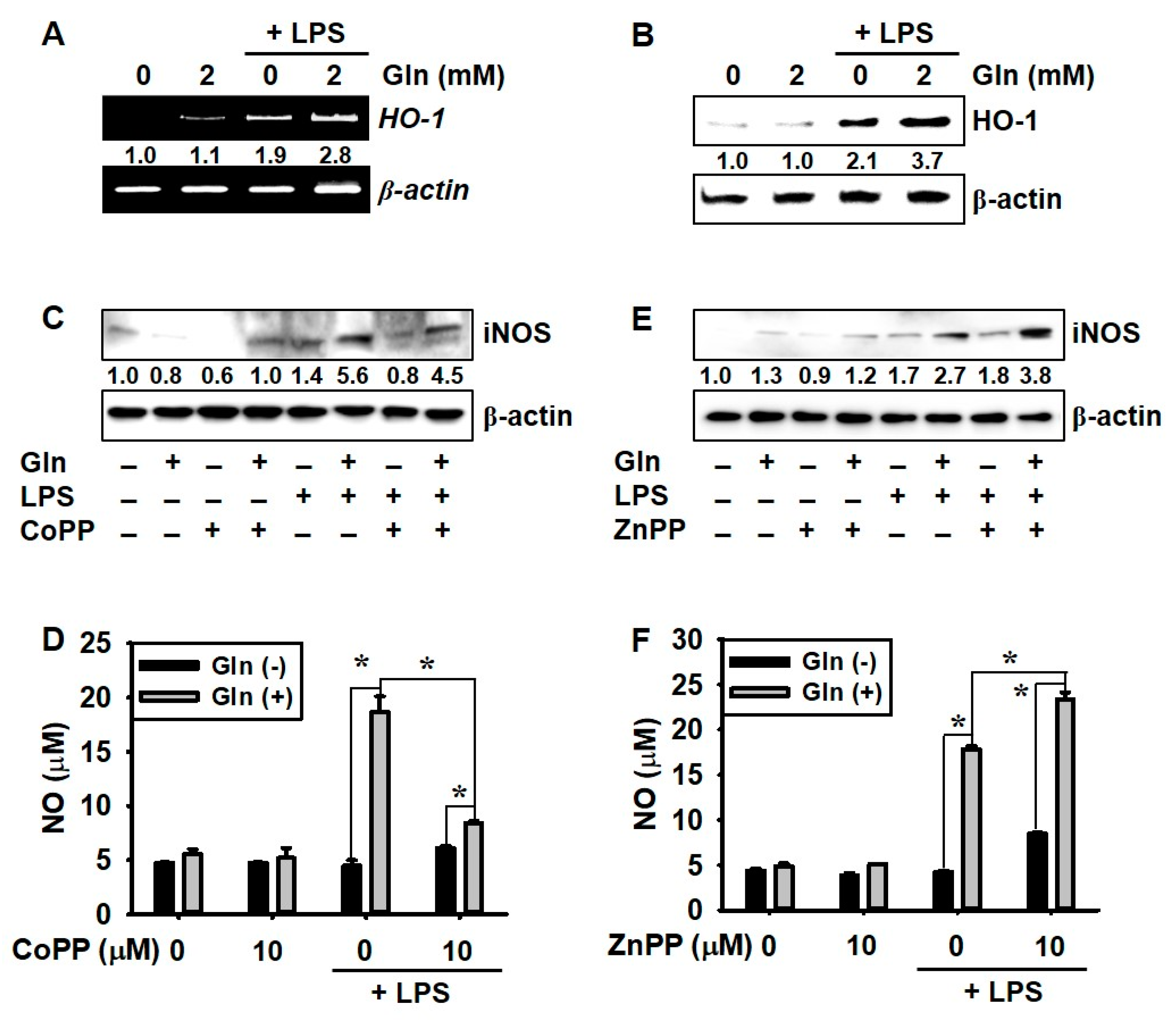
3.4. LPS Promotes HO-1 Expression Regardless of the Presence of Gln, which Decreases NO Production
3.5. ERK Phosphorylation Upregulates LPS-Induced NO Production in the Presence of Gln
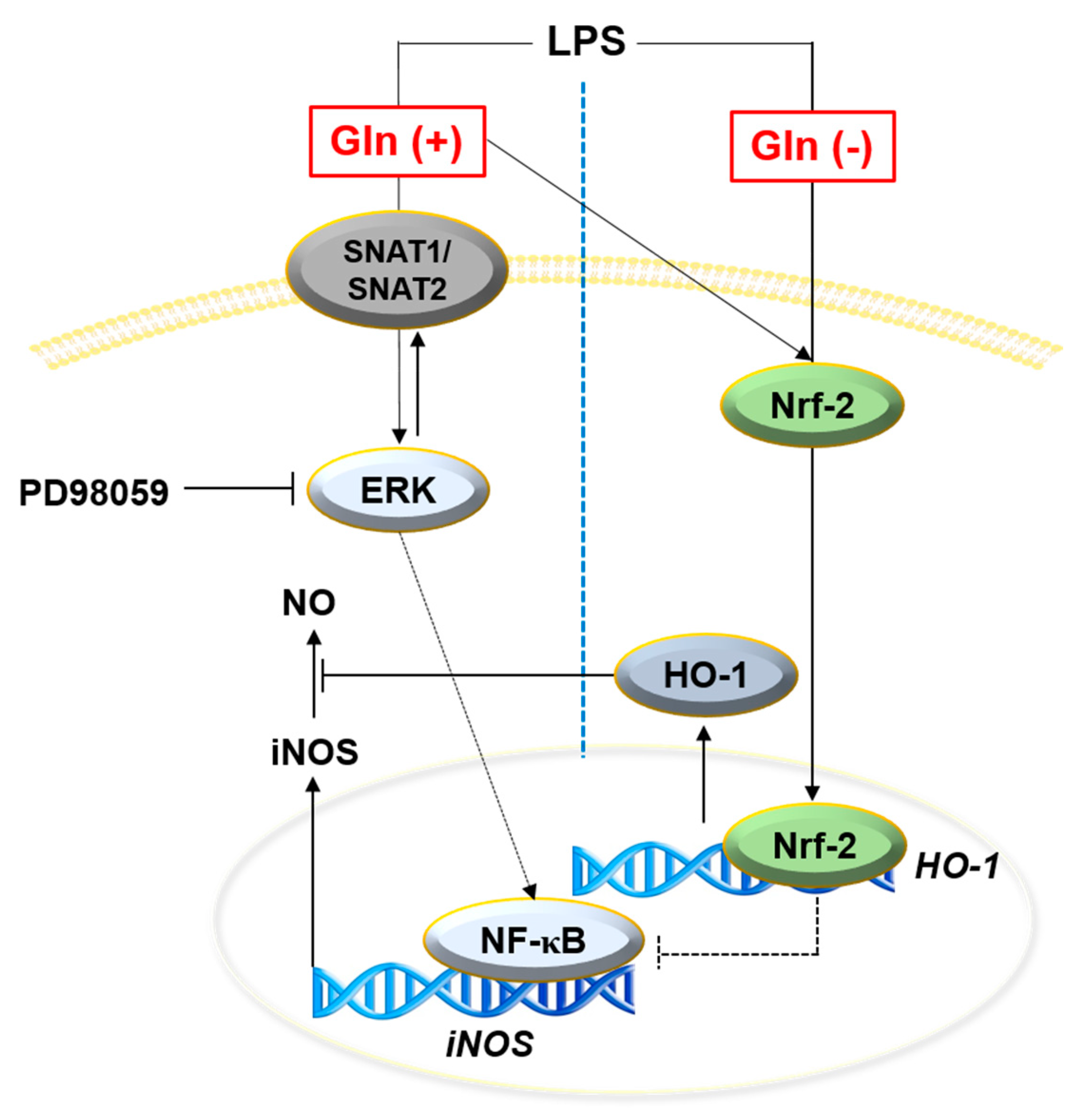
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Cowan, M.; Petri, W.A.J. Microglia: Immune Regulators of Neurodevelopment. Front. Immunol. 2018, 9, 2576. [Google Scholar] [CrossRef] [PubMed]
- Saijo, K.; Crotti, A.; Glass, C.K. Regulation of microglia activation and deactivation by nuclear receptors. Glia 2012, 61, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Perry, V.H. Microglial Physiology: Unique Stimuli, Specialized Responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Kittl, M.; Dobias, H.; Beyreis, M.; Kiesslich, T.; Mayr, C.; Gaisberger, M.; Ritter, M.; Kerschbaum, H.H.; Jakab, M. Glycine Induces Migration of Microglial BV-2 Cells via SNAT-Mediated Cell Swelling. Cell. Physiol. Biochem. 2018, 50, 1460–1473. [Google Scholar] [CrossRef] [PubMed]
- De Simone, R.; Vissicchio, F.; Mingarelli, C.; De Nuccio, C.; Visentin, S.; Ajmone-Cat, M.A.; Minghetti, L. Branched-chain amino acids influence the immune properties of microglial cells and their responsiveness to pro-inflammatory signals. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Struzynska, L.; Sulkowski, G. Relationships between glutamine, glutamate, and GABA in nerve endings under Pb-toxicity conditions. J. Inorg. Biochem. 2004, 98, 951–958. [Google Scholar] [CrossRef]
- Nakajima, K.; Kanamatsu, T.; Takezawa, Y.; Kohsaka, S. Up-regulation of glutamine synthesis in microglia activated with endotoxin. Neurosci. Lett. 2015, 591, 99–104. [Google Scholar] [CrossRef]
- Palmieri, E.M.; Menga, A.; Lebrun, A.; Hooper, D.C.; Butterfield, D.A.; Mazzone, M.; Castegna, A. Blockade of Glutamine Synthetase Enhances Inflammatory Response in Microglial Cells. Antioxidants Redox Signal. 2016, 26, 351–363. [Google Scholar] [CrossRef]
- Jin, L.-W.; Horiuchi, M.; Wulff, H.; Liu, X.-B.; Cortopassi, G.A.; Erickson, J.D.; Wulff, H. Dysregulation of glutamine transporter SNAT1 in Rett syndrome microglia: A mechanism for mitochondrial dysfunction and neurotoxicity. J. Neurosci. 2015, 35, 2516–2529. [Google Scholar] [CrossRef]
- Grewal, S.; Defamie, N.; Zhang, X.; De Gois, S.; Shawki, A.; MacKenzie, B.; Chen, C.; Varoqui, H.; Erickson, J.D. SNAT2 Amino Acid Transporter Is Regulated by Amino Acids of the SLC6 γ-Aminobutyric Acid Transporter Subfamily in Neocortical Neurons and May Play No Role in Delivering Glutamine for Glutamatergic Transmission. J. Boil. Chem. 2009, 284, 11224–11236. [Google Scholar] [CrossRef]
- MacKenzie, B.; Schäfer, M.K.-H.; Erickson, J.D.; Hediger, M.A.; Weihe, E.; Varoqui, H. Functional Properties and Cellular Distribution of the System A Glutamine Transporter SNAT1 Support Specialized Roles in Central Neurons. J. Boil. Chem. 2003, 278, 23720–23730. [Google Scholar] [CrossRef] [PubMed]
- Komm, B.; Beyreis, M.; Kittl, M.; Jakab, M.; Ritter, M.; Kerschbaum, H.H. Glycine modulates membrane potential, cell volume, and phagocytosis in murine microglia. Amino Acids 2014, 46, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Jayasooriya, R.G.P.T.; Lee, K.-T.; Lee, H.-J.; Choi, Y.H.; Jeong, J.-W.; Kim, G.-Y. Anti-inflammatory effects of β-hydroxyisovalerylshikonin in BV2 microglia are mediated through suppression of the PI3K/Akt/NF-kB pathway and activation of the Nrf2/HO-1 pathway. Food Chem. Toxicol. 2014, 65, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Iizumi, T.; Takahashi, S.; Mashima, K.; Minami, K.; Izawa, Y.; Abe, T.; Hishiki, T.; Suematsu, M.; Kajimura, M.; Suzuki, N. A possible role of microglia-derived nitric oxide by lipopolysaccharide in activation of astroglial pentose-phosphate pathway via the Keap1/Nrf2 system. J. Neuroinflammation 2016, 13, 99. [Google Scholar] [CrossRef] [PubMed]
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 2010, 185, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Lawson, L.; Perry, V.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Boje, K.M.; Arora, P.K. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992, 587, 250–256. [Google Scholar] [CrossRef]
- Yuste, J.E.; Tarragon, E.; Campuzano, C.M.; Ros, F. Implications of glial nitric oxide in neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 322. [Google Scholar] [CrossRef]
- Nishiyama, A.; Yokote, Y.; Sakagami, H. Changes in amino acid metabolism during activation of mouse macrophage-like cell lines. In Vivo 2010, 24, 857–860. [Google Scholar]
- Chen, J.; Herrup, K. Glutamine Acts as a Neuroprotectant against DNA Damage, Beta-Amyloid and H2O2-Induced Stress. PLoS ONE 2012, 7, e33177. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Y.; Vail, G.; Chow, H.-M.; Zhang, Y.; Louie, L.; Li, J.; Hart, R.P.; Plummer, M.R.; Herrup, K. The impact of glutamine supplementation on the symptoms of ataxia-telangiectasia: A preclinical assessment. Mol. Neurodegener. 2016, 11, 60. [Google Scholar] [CrossRef]
- Albrecht, J.; Sidoryk-Wegrzynowicz, M.; Zielińska, M.; Aschner, M. Roles of glutamine in neurotransmission. Neuron Glia Boil. 2010, 6, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Petersen, K.F.; Behar, K.L.; Brown, P.; Nixon, T.W.; Mason, G.F.; Petroff, O.A.C.; Shulman, G.I.; Shulman, R.G.; Rothman, D.L. Determination of the rate of the glutamate/glutamine cycle in the human brain by in vivo 13C NMR. Proc. Natl. Acad. Sci. USA 1999, 96, 8235–8240. [Google Scholar] [CrossRef] [PubMed]
- Raposo, B.; Vaartjes, D.; Ahlqvist, E.; Nandakumar, K.S.; Holmdahl, R. System A amino acid transporters regulate glutamine uptake and attenuate antibody-mediated arthritis. Immunology 2015, 146, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Yamada, D.; Kawabe, K.; Tosa, I.; Tsukamoto, S.; Nakazato, R.; Kou, M.; Fujikawa, K.; Nakamura, S.; Ono, M.; Oohashi, T.; et al. Inhibition of the glutamine transporter SNAT1 confers neuroprotection in mice by modulating the mTOR-autophagy system. Commun. Boil. 2019, 2, 346–411. [Google Scholar] [CrossRef]
- Solbu, T.T.; Bjørkmo, M.; Berghuis, P.; Harkany, T.; Chaudhry, F.A. SAT1, A Glutamine Transporter, is Preferentially Expressed in GABAergic Neurons. Front. Neuroanat. 2010, 4, 1. [Google Scholar] [CrossRef]
- Jenstad, M.; Quazi, A.Z.; Zilberter, M.; Haglerød, C.; Berghuis, P.; Saddique, N.; Goiny, M.; Buntup, D.; Davanger, S.; Haug, F.-M.S.; et al. System A Transporter SAT2 Mediates Replenishment of Dendritic Glutamate Pools Controlling Retrograde Signaling by Glutamate. Cereb. Cortex 2008, 19, 1092–1106. [Google Scholar] [CrossRef]
- Lister, A.; Bourgeois, S.; Silva, P.H.I.; Rubio-Aliaga, I.; Marbet, P.; Walsh, J.; Shelton, L.M.; Keller, B.; Verrey, F.; Devuyst, O.; et al. NRF2 regulates the glutamine transporter Slc38a3 (SNAT3) in kidney in response to metabolic acidosis. Sci. Rep. 2018, 8, 5629. [Google Scholar] [CrossRef]
- Łoboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef]
- Ryter, S.W. Heme oxygenase-1/carbon monoxide as modulators of autophagy and inflammation. Arch. Biochem. Biophys. 2019, 678, 108186. [Google Scholar] [CrossRef]
- Chora, Â.A.; Fontoura, P.; Cunha, A.; Pais, T.F.; Cardoso, S.; Ho, P.P.; Lee, L.Y.; Sobel, R.A.; Steinman, L.; Soares, M.P. Heme oxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation. J. Clin. Investig. 2007, 117, 438–447. [Google Scholar] [CrossRef]
- Fagone, P.; Mangano, K.; Quattrocchi, C.; Motterlini, R.; Di Marco, R.; Magro, G.; Penacho, N.; Romão, C.C.C.; Nicoletti, F. Prevention of clinical and histological signs of proteolipid protein (PLP)-induced experimental allergic encephalomyelitis (EAE) in mice by the water-soluble carbon monoxide-releasing molecule (CORM)-A1. Clin. Exp. Immunol. 2011, 163, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Fagone, P.; Mangano, K.; Coco, M.; Perciavalle, V.; Garotta, G.; Romão, C.C.C.; Nicoletti, F. Therapeutic potential of carbon monoxide in multiple sclerosis. Clin. Exp. Immunol. 2012, 167, 179–187. [Google Scholar] [CrossRef]
- Džoljić, E.; Grabatinić, I.; Kostić, V. Why is nitric oxide important for our brain? Funct. Neurol. 2015, 30, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Mancuso, C.; Calvani, M.; Rizzarelli, E.; Butterfield, D.A.; Stella, A.M.G. Nitric oxide in the central nervous system: Neuroprotection versus neurotoxicity. Nat. Rev. Neurosci. 2007, 8, 766–775. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jayasooriya, R.G.P.T.; Molagoda, I.M.N.; Dilshara, M.G.; Choi, Y.H.; Kim, G.-Y. Glutamine Cooperatively Upregulates Lipopolysaccharide-Induced Nitric Oxide Production in BV2 Microglial Cells through the ERK and Nrf-2/HO-1 Signaling Pathway. Antioxidants 2020, 9, 536. https://doi.org/10.3390/antiox9060536
Jayasooriya RGPT, Molagoda IMN, Dilshara MG, Choi YH, Kim G-Y. Glutamine Cooperatively Upregulates Lipopolysaccharide-Induced Nitric Oxide Production in BV2 Microglial Cells through the ERK and Nrf-2/HO-1 Signaling Pathway. Antioxidants. 2020; 9(6):536. https://doi.org/10.3390/antiox9060536
Chicago/Turabian StyleJayasooriya, Rajapaksha Gedara Prasad Tharanga, Ilandarage Menu Neelaka Molagoda, Matharage Gayani Dilshara, Yung Hyun Choi, and Gi-Young Kim. 2020. "Glutamine Cooperatively Upregulates Lipopolysaccharide-Induced Nitric Oxide Production in BV2 Microglial Cells through the ERK and Nrf-2/HO-1 Signaling Pathway" Antioxidants 9, no. 6: 536. https://doi.org/10.3390/antiox9060536
APA StyleJayasooriya, R. G. P. T., Molagoda, I. M. N., Dilshara, M. G., Choi, Y. H., & Kim, G.-Y. (2020). Glutamine Cooperatively Upregulates Lipopolysaccharide-Induced Nitric Oxide Production in BV2 Microglial Cells through the ERK and Nrf-2/HO-1 Signaling Pathway. Antioxidants, 9(6), 536. https://doi.org/10.3390/antiox9060536