Abstract
Hypoxia is a common form of intrauterine stress characterized by exposure to low oxygen concentrations. Gestational hypoxia is associated with the generation of reactive oxygen species. Increase in oxidative stress is responsible for damage to proteins, lipids and DNA with consequent impairment of normal cellular functions. The purpose of this review is to propose a summary of preclinical and clinical evidences designed to outline the correlation between fetal hypoxia and oxidative stress. The results of the studies described show that increases of oxidative stress in the placenta is responsible for changes in fetal development. Specifically, oxidative stress plays a key role in vascular, cardiac and neurological disease and reproductive function dysfunctions. Moreover, the different finding suggests that the prenatal hypoxia-induced oxidative stress is associated with pregnancy complications, responsible for changes in fetal programming. In this way, fetal hypoxia predisposes the offspring to congenital anomalies and chronic diseases in future life. Several antioxidant agents, such as melatonin, erythropoietin, vitamin C, resveratrol and hydrogen, shown potential protective effects in prenatal hypoxia. However, future investigations will be needed to allow the implementation of these antioxidants in clinical practice for the promotion of health in early intrauterine life, in fetuses and children.
1. Introduction
Fetal hypoxia is a condition characterized by a reduction of oxygen responsible for impairing the fetus development and increases the risk of perinatal and infant mortality [1]. Indeed, fetal hypoxia represents 23% of neonatal deaths worldwide [2]. The most common risk factors causing fetal hypoxia are placental insufficiency, preclampsia, umbilical cord damage and maternal factors such as smoking, heart, kidney or lung dysfunction [3].
Prenatal hypoxia can be classified into three patterns: pre-placental; uteroplacental and post-placental. The pre-placental hypoxia affects the fetus and the mother, unlike post-placental hypoxia that induces only fetal damage. Instead, uteroplacental hypoxia is hallmarked by an altered uteroplacental circulation [1]. We will focus on the last two patterns that are mainly related to the direct impact of hypoxia on the fetus.
Fetal hypoxia is responsible for the production of reactive oxygen species (ROS). ROS are essential to perform several cellular functions. However, excessive production of ROS cannot be countered by the antioxidant defenses; in this way, ROS are responsible for cellular oxidative stress. In the placenta, DNA damage, protein denaturation and lipid peroxidation induced by ROS can modify placental function leading to a reduction in oxygen and nutrients in the fetus [4]. This cascade of events induces limited fetal growth leading to a decrease in weight and a body shape alteration of the offspring [5]. Moreover, growth restriction is often correlated to abnormal organ development. This involves the birth of offspring with organs that may not support proper functioning during future life [6].
Specifically, exposure to short periods of fetal hypoxia leads to a redistribution of blood flow towards the essential circulation to protect the brain and heart [7]. However, prolonged periods of intrauterine hypoxia could entail alterations of the initial homeostatic defenses [8,9]. This implicates a high strain on the developing heart and principal vessels, inducing impaired fetal heart function. In this way, the bodyweight/heart weight ratio increases and occur changes in cardiac gene expressions [10]. Therefore, fetal hypoxia, due to alterations of fetal programming, represents an immediate danger to fetal life, but also to the future life [9]. Indeed, the exposure of the fetus to hypoxic conditions predisposes the offspring to congenital anomalies and chronic diseases, such as cardiovascular, renal and metabolic disorders, in later life [3,11,12,13,14,15,16].
Therefore, therapeutic interventions during pregnancy represent an interesting opportunity aimed to reduce the harmful effects of fetal hypoxia-induced oxidative stress.
In this review, we want to provide an overview of preclinical and clinical evidences aimed to illustrate the correlation between fetal hypoxia and oxidative stress. The prevention of the oxidative process of a long fetal life has to be considered to promote healthcare in children.
2. Methodology
In order to write the paragraph “4. Animal models of prenatal hypoxia”, the bibliography research in PubMed was performed using the following keywords: “fetal hypoxia”, “prenatal hypoxia”, “placental hypoxia”, “oxidative stress”, “mice”, “rats”, “pigs”, “rabbits”, “antioxidants”, “neuroprotection”. In this review the articles published from 2000 to 2020 were taken into consideration. In this way, 269 articles were found, as shown in the Prisma flow diagram (Figure 1). In this manuscript were considered the preclinical studies evaluating the role of oxidative stress induced by prenatal hypoxia on the fetus were described. Moreover, in order to write the subparagraph “4.1. New antioxidant treatments for prenatal hypoxia” were also taken into consideration preclinical studies evaluating the effectiveness of new antioxidant agents.
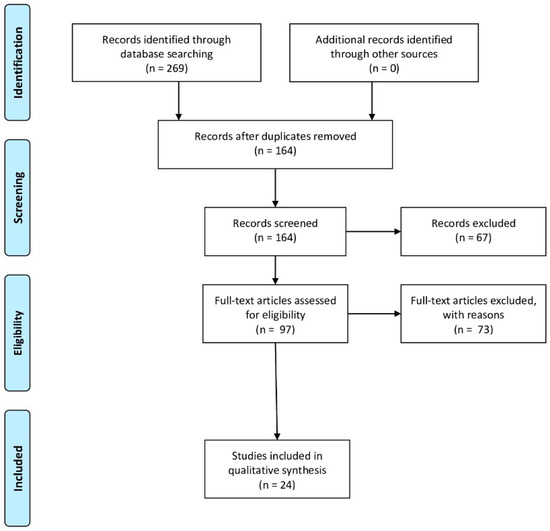
Figure 1.
Prisma flow diagram illustrating the selection methodology of the preclinical studies used for the writing of the review. Duplicate articles were excluded from the total of the studies recorded. Instead, were considered articles that evaluate the damage of oxidative stress induced by fetal hypoxia (The PRISMA Statement was published in: [17]).
3. Prenatal Oxidative Stress
Prenatal hypoxia is a condition responsible for the disease and fetal death or newborn [18]. The placenta is an organ important for communication between the pregnant woman and the fetus. The proper functioning of this organ is important for fetal development. Hypoxia is defined as a decrease in O2 necessary for the physiological functions of tissue [19]. An increase of ROS levels, generated from an incomplete reduction of O2, is one of the most common mechanisms induced to hypoxia. Normally the placenta produces ROS such as superoxide anion (O2−), hydroxyl radical (HO−) and hydrogen peroxide (H2O2) [20]. These molecules are highly unstable and possess a strong chemical reactivity due to the presence of unpaired electrons in the external orbital [21]. Due to this instability, ROS are inclined to yield or acquire an electron from other electrically unstable molecules, in order to achieve a stable energy state. In this way their lead a series of redox reactions that are important for the survival cellular. Normal ROS production is ensured by a balance between the production of these molecules and the antioxidant defense system. The main antioxidant system is provided to the activity of antioxidant enzymes such as superoxide dismutase (SOD), glutathione peroxidase (GPx) and catalase (CAT). Non-enzymatic antioxidants such as thiols (e.g., glutathione, GSH), protein thiols; vitamins A, B6, B12, C and E; selenium; folic acid; and the β-carotenoids, bilirubin and uric acid, represent another defense mechanism able to reduce the excessive ROS production [22]. During pregnancy, normal ROS levels can be involved in trophoblast proliferation and differentiation and in the modulation of the vascular responses of the placenta [19]. However, an increase in ROS levels is responsible for placental functional changes. Following fetal hypoxia, low levels of O2 lead to a reduction in the activity of the mitochondrial electron transport chain. Thereby, this reduction promoting an increase in the percentage of O2 incompletely reduced with consequent production of ROS, such as O2− [20,23]. The mitochondrial electron transport chain represents a major ROS producer. Another source of ROS is represented by NADPH oxidase, responsible for the endogenous production of O2− [24]. In the vascular endothelium, cytochrome P450 is another enzyme responsible for the production of OH− and O2− [25]. The metalloflavoprotein xanthine oxidase is another enzyme that following the oxidation of hypoxanthine to xanthine and uric acid, leads to the production of O2− [26]. In the placenta, in hypoxic conditions, the mitochondrial oxygen consumption is downregulated. This leads to a reduction in the reserves of high energy phosphates generating high levels of xanthine, hypoxanthine, NADH, FADH, hydrogen ions (H+) and lactic acid [27].
Hypoxia induces a decrease in the enzymatic activity of the pumps of ATP-asi dependent membranes, reduction of the membrane potential and an increase in the flow of cytosolic calcium (Ca2+) levels. In hypoxic conditions, the increase in intracellular Ca2+, due to the activation of voltage-dependent channels and release by the mitochondria and the endoplasmic reticulum, establishes a loop that triggers the mechanism of apoptosis and neuronal necrosis [28]. Especially in the neuronal cells, the entry of Ca2+ favors the accumulation of glutamate. Glutamate, interacting with N-methyl-D aspartate (NMDA) receptors, intensifies the intracellular current of Ca2+, further contributing to neuronal damage [29].
Moreover, Ca2+ is responsible for activating nitric oxide synthase (NOS), involved in the production of nitric oxide (NO). Among the three known NOS isoforms, endothelial nitric oxide synthase (eNOS) is a Ca2+-dependent flavoenzyme that generates NO [30]. In this process Ca2+ plays an important role in the activation of eNOS, regulating the binding of eNOS with calmodulin [31] NO is a powerful endothelial vasodilator involved in the regulation of vascular tone, in the control of blood flow in the tissues and in the aggregation of platelets. In the placental, NO plays a key role in vasodilatation of the uteroplacental arteries, an important mechanism that determines the invasion of the trophoblasts and the remodeling of the endothelium [32]. Therefore, the altered balance of NO and ROS play a critical role in modulating the umbilical-placental vascular function in different prenatal conditions.
Therefore, high levels of ROS are responsible for damage to several cellular components such as DNA, proteins and lipids with consequent impairment of normal cellular functions [33]. During pregnancy, within 10–12 weeks of gestation, there is an increase in the flow of maternal blood into the placenta, which leads to a local increase in oxygen and consequently an increase in the activity of antioxidant enzymes. However, an excessive increase in ROS that cannot be countered by the antioxidant response, induce oxidative stress conditions. In the placenta oxidative stress, especially in this stage of pregnancy, is responsible for reducing the invasion of trophoblasts. This cascade of events induces different conditions that can be linked to alterations of fetal development and in serious cases even to early pregnancy failure [34,35].
The brain is more sensitive to changes in O2 levels. Oxidative stress is the main factor that induces neuronal cell death in the immature brain [36]. During embryogenesis, hypoxic damage also delays neuronal migration and alters the expression of numerous neurotransmitters [37,38]. These mechanisms increase the risk of neural birth defects, brain damage and long-term cognitive impairment in learning and memory [39,40]. Moreover, it can also predispose offspring to the future onset of epileptic conditions [41].
4. Animal Models of Prenatal Hypoxia
Several studies evaluate the effects of oxidative stress induced by prenatal hypoxia on the development of the fetus and the future consequences on offspring.
Matheson H. et al. have evaluated the chronic hypoxia in placental development. The authors explained about a reduction in fetal weight and an increase in placental weight due to hypoxic conditions by the Akt-mTOR signal and increased expression of heat shock protein 70 (HSP70) and HSP27, protecting the placentas from damage. Additionally, the authors reported a different gender-specific placental response to fetal hypoxia. They showed that the placentas of female fetuses were heavier and more sensitive to hypoxia-induced oxidative stress than those of males, although the mechanisms should be better clarified [42]. This finding was also supported by Song H. et al. in a model of pregnant guinea pigs and they demonstrated that chronic hypoxia inhibits mitochondrial function in placental tissues via increased peroxynitrite. This mechanism thus justifying the reduction of placental efficiency and fetal growth. Even in this study, the placental response exhibiting sexual dimorphism [43].
In order to clarify the role of oxidative stress in the prenatal hypoxia, Rueda-Clausen C. et al. used mice lacking the eNOS (eNOS−/−) and mice deficient in the enzyme catechol-O-methyl transferase (COMT−/−). Fetal hypoxia has led to a high mortality rate in eNOS−/− fetuses compared to COMT−/−. Instead, was recorded an increase of NO in the placentas of COMT−/− compare to eNOS−/−. This result would justify the increased tolerance to hypoxic stress in COMT−/− mice. However, this hypothesis should be investigated further in order to enhance the understanding of placental responses to hypoxic insult [44].
Other studies focused in the role of oxidative stress induced by fetal hypoxia in the embryonal development of the brain in the offspring. Baud O. et al., proved that the brain of the offspring exposed to fetal hypoxia presented of cysts and extracellular matrix abnormalities. These results were justified with an increase in the activity of lipid peroxidation, presence of macrophages and consequent neuronal death. Thereby, chronic fetal hypoxia, through an increase of oxidative stress, may be the main cause of neurological impairment [45]. Instead, Sab I. et al. evaluated the involvement of fetal hypoxic damage in behavioral abnormalities and long-term disturbances in the learning and memory of offspring. In the offspring of 3-months exposed to prenatal hypoxia, the authors showed a reduction in exploratory capacity and locomotor activity and an increase in anxious behavior through a reduction in the activity of GPx and an increase in NO. However, would be necessary to better clarify the role of fetal damage-induced oxidative stress in the long-term disturbances in learning and memory in the offspring [40].
Prenatal hypoxia is also responsible of the fetal cardiovascular dysfunctions. Zhu X. et al. proven that fetal hypoxia induced a decrease in the partial pressure of O2 and the saturation of O2, in the fetal thoracic aortas. These results were correlated with an increase in response to angiotensin II (Ang II) an increase of O2−, due to activation of NADPH oxidase 4 and reduction of SOD [46]. Instead, Chen L. et al. wanted to evaluate the effects of maternal exposure to chronic intermittent hypoxia (CIH). In fetal hearts, the hypoxic conditions induced hypertrophy, high contractility of the left ventricle associated with high arterial stiffness. The authors correlated these cardiovascular abnormalities with a significant increase of lipid peroxidation and SOD1. Furthermore, was observed a major increase of lipid peroxidation in male offspring compared to that female [47]. The mechanisms underlying sex-dependent programming for offspring in pregnancy need further studies. Moreover, Figueroa H. et al., using New Zealand rabbits exposed to fetal hypoxia, showed an increased dilation of the vessels in fetal hearts. The authors justified this results with an increase in the activity of eNOS, inducible nitric oxide synthase (iNOS) and of the levels of carbonyl proteins, suggesting that oxidative stress is an important stimulus to initiate adverse effects in fetal hearts [48].
Other researchers focused attention in the alteration programming cardiovascular function induced by fetal hypoxia in adult offspring. In this regard, Giussani D.A. et al. proven that fetal hypoxia induced an aortic thickening in the fetus correlated with an increase in HSP70 and nitrotyrosine. Moreover, in the adult offspring (4 months) were also observed an increase in myocardial contractility and an alteration in the peripheral resistance vessels eNOS-dependent [49]. Instead Chen X. et al. compared the effects of prenatal hypoxia between 5-month and 20-month offspring. The authors showed vasodilation in the mesenteric arteries only in the 20-month offspring. These results were correlated with a decrease in eNOS activity and the antioxidants levels such as SOD and CAT. Moreover, was also observed an increase in ROS and malondialdehyde levels in both plasma and mesenteric arteries. This study has identified aging, as a postnatal factor that improves vascular dysfunction induced by fetal hypoxia [50]. In line with these results, Rueda-Clausen C. et al. demonstrated that the 12-month-old offspring exposed to chronic fetal hypoxia, shown high levels of total GSH. Additionally, in the male offspring, these events were responsible for the presence of multiple isolated areas of intra-ventricular myocyte death [51].
Furthermore, cardiovascular dysfunctions are also induced by epigenetic modifications due to altered ROS levels following fetal hypoxia [52,53]. Patterson A.J. et al. showed that hypoxic damage induced an increase of ROS responsible of epigenetic repression of the PKCε gene, in the hearts of rat offspring. This epigenetic mutation compromises the cardioprotective role of the protein encoded by PKCε. In this way, hypoxia-mediated ROS production in the developing heart is involved in the susceptibility of offspring to heart disease [54].
However, fetal hypoxia promotes damage also in the reproductive activity of the offspring. In this contest, Aiken C.E.et al. showed that hypoxic damage during the intrauterine period inducing aging oviducts in the 4-month female rats. These data were justified with increased markers of cell cycle such as p21 and p53 in the oviducts, two important regulators of the cell cycle. Parallelly, the oxidative stress negatively influences the functionality of the oviducts through alteration of the biogenesis of mitochondrial DNA [55]. Moreover, the authors in a later job reported that the offspring, shown a reduction in the number of primordial follicles available and an increase in oxidative stress markers such as Gp91phox and P22phox. Therefore, prolonged exposure to fetal hypoxia causes a reduction in fertility that accelerates with the aging of the ovary [56]. However, an understanding of these molecular pathways would be necessary to develop effective interventions to protect the female offspring of a high-risk pregnancy.
The results of all these studies showed that fetal hypoxia and the consequent increase in oxidative stress cause immediate adverse events on the fetus such as the limitation of fetal growth and the reduction of the weight of the unborn child. However, fetal hypoxia is also responsible for long-term damage to the offspring, such as an increased risk of subsequent cardiovascular dysfunction, cognitive delay and impaired reproductive function (Table 1).

Table 1.
Synthesis of the studies that evaluate the role of fetal hypoxia-induced oxidative stress in several animal models.
New Antioxidant Treatments for Prenatal Hypoxia
Complications of pregnancy such as fetal hypoxia activate physiological survival processes such as ROS production; therefore, clinical strategies such as the use of antioxidants can be helpful in managing fetal problems as well as long-term consequences on offspring (Table 2).
Table 2.
Synthesis of the studies aimed at testing the antioxidant properties of some substances such as melatonin, vitamin C, resveratrol, nMitoQ, hydrogen and erythropoietin. Specifically, the table shows the animal models used in the studies and the type of hypoxic damage induced. Additionally, the type of treatment, the dosage, the route of administration and the therapeutic effects obtained are described.
Okatani Y. et al. studied the effects of melatonin in improving the oxidative stress damage induced by ischemia/reperfusion of mitochondria in the rat placenta. Melatonin is an important scavenger secreted by the pineal gland that showed broad antioxidant, anti-inflammatory and antiapoptotic effects. The results showed that the administration of melatonin induced an increase in the markers of mitochondrial activity and a decrease in the concentration of reactive substances with thiobarbituric acid. Therefore, exogenous melatonin exhibited an antioxidant action and could be useful as a treatment for and fetal hypoxia [57].
Vitamin C is another antioxidant that may be used as a possible therapeutic treatment during fetal hypoxia. Vitamin C in mammals is one of the most important endogenous antioxidants. Richter H.G. et al. showed that maternal treatment with vitamin C prevented placental oxidative stress associated with maternal exposure to hypoxia [58].
Fetal hypoxia makes the offspring susceptible to the development of metabolic and cardiovascular disorders [59]. Resveratrol is a natural polyphenol that carries out its cardio-protective effects reducing ROS production, through the activation of molecules such as Adenosine Monophosphate Kinase Cardiac (AMPK) and the up-regulation of antioxidant enzymes such as SOD in endothelial cells of the arteries and smooth muscle cells [60,61]. Shah A. et al. evaluated the beneficial action of resveratrol in preventing alterations of metabolism and cardiac dysfunctions. The study showed that at 12 months of age, fetal hypoxia and the fatty acid diet have induced metabolic alteration in offspring, especially in male offspring. Resveratrol treatment has proven effective in preventing cardiovascular disease, probably through increasing cardiac SOD [62]. However, as in the previous study [62], even after 21 weeks, resveratrol treatment promotes an improvement in diastolic function and increases the ability to recover after an ischemia/reperfusion injury. The cardiovascular beneficial effects of resveratrol can be explained with a significant increase of the cardiac p-AMPK protein levels. Therefore, resveratrol could be a therapeutic opportunity used to counteract also the long-term oxidative damage induced by fetal hypoxia [63].
The use of mitochondrial antioxidants, such as MitoQ, could prevent the secretion of factors that cause DNA damage. Tom J. Phillips et al. assessed the ability of MitoQ to prevent DNA damage mediated by the release of harmful particles from the placenta induced following prenatal hypoxia. In order to prevent MitoQ from crossing the placenta, MitoQ was bonded with nanoparticles (nMitoQ), which accumulate in bilayered trophoblast barrier and do not cross through it, as previously demonstrated [64,65]. Administration of nMitoQQ appears to normalize alterations of microRNA, bone morphogenetic proteins and amino acids in placental secretions and plasma is capable of preventing molecular changes induced by fetal hypoxia, reducing oxidative stress in the placenta [66]. The same research team also evaluated the effects of placental treatment with nMitoQ in reducing the risk of developing cardiovascular disease in offspring exposed to prenatal hypoxia. The results of the study showed that hypoxia-induced changes in heart function that occurred following adulthood were attenuated by treatment with nMitoQ [67]. Additionally, the administration of nMitoQ reduced the O2− levels in both the placenta and the fetus and the nitrotyrosine levels in the placenta. In this way, nMitoQ reduces the oxidative stress levels, highlighting that the efficacy of the treatment is targeted to the placenta. Moreover, treatment with nMitoQ resulted in an increase of vascular endothelial growth factor A (VEGFA) and insulin-like growth factor 2 (IGF-2). In conclusion, nMitoQ, reducing oxidative stress, improves oxygenation, angiogenesis and placental morphology, especially in the placenta of female offspring [68].
During the first week after birth, negative geotropism is important, both for rats and mice, to develop a sense of adaptation to the environment; just as straightening reflex is a neuromuscular response aimed at bringing the body into the normal vertical position [69,70]. Liu W. et al. observed that offspring subjected to hypoxic damage showed an alteration of these sensory responses. However, treatment with hydrogen during pregnancy restoring the anomalies of sensory responses and therefore preventing neurological damage induced by hypoxia [71].
Fetal hypoxia is also responsible for the alteration of neurological development and consequent cognitive delays, behavioral deficits, cerebral paralysis and other complications [72]. The erythropoietin shows neuroprotective properties [73] through several mechanisms of action including the reduction of oxidative stress [74] and damage induced by NO [75]. In this regard, Mazur M. et al. have proven that the administration of neonatal endogenous erythropoietin, 4 days later to hypoxic damage, has promoted the survival of oligodendrocytes and neurons. Additionally, improving histological damage was also observed, even after 24 days of treatment. In conclusion, erythropoietin improved the neurological insult induced by hypoxia and allow the correct development of the nervous system [76]. In view of these findings, Jantzi L.L. et al. assessed the effects of erythropoietin on calpain activated following fetal hypoxia. Calpain is a protein involved in cellular homeostasis during the development of the central nervous system and in the degradation of proteins in the mature central nervous system. Therefore, its alteration may be responsible for the cognitive delay [77]. The hypoxic damage induced high calpain activity postnatal in the cortex. The erythropoietin treatment reduced cortical calpain activity and promoted positive modulation of markers of neurological development, such as neuronal potassium-chloride co-transporter (KCC2), Myelin Basic Protein (MBP) and phosphorylated-neurofilament (p-NF). Thereby, erythropoietin can be valid therapeutic tools aimed at protecting the offspring from possible alterations in the development of the central nervous system [78].
5. Intrauterine Hypoxia and Human Evidence of Fetal Developing and Programming
The antioxidant defenses in the mother and placenta during fetal life and in the newborn born at term, under physiologic conditions, are capable of neutralizing ROS thus avoiding negative health consequences [79]. Increased production of ROS and oxidative stress during organogenesis, a period in which cells continue to differentiate, act as teratogenic agents; through disrupting critical signaling events causing structural abnormalities, loss of cellular function or spontaneous abortion of the developing fetus [79,80]. The relationship between oxidative stress and congenital malformations is not fully elucidated; however, oxidative stress may play a significant causal role in birth defects.
The prevalence of structural birth defects varies globally, ranging from approximately 3% to 6% of all live births [81]. These birth defects are a leading cause of infant mortality.
Most structural birth defects develop early in embryogenesis, during the first 10 weeks gestation, and the vast majority of birth defects are “nonsyndromic” and rare, not associated with multi-organ syndromes. A complex interaction between genes and environmental factors has been described [82].
For instance, the incidence of Congenital Heart Diseases (CHDs) varies from 4/1000 to 50/1000 live births [83] and only 15% of CHDs can be attributed to a genetic cause.
All other cases result from a complex interaction between genetic susceptibility and environmental factors (maternal drugs and alcohol assumption, nutrient availability, cigarette smoking, diabetic pregnancy, exposure to industrial chemicals, infectious agents) whose common embryotoxic effect may be related to oxidant-or redox misregulation with increased ROS production and alteration of developmental signals [83]. In particular, significantly lower concentrations of GSH and significantly higher concentrations of glutathione disulfide (GSSG) has been described [84,85,86].
In the same way the human brain is particularly vulnerable to the damaging effects of reactive oxygen intermediates due to both its complexity and the long period of development. Several studies to delineate the mechanism underlying maternal diabetic embryopathy demonstrated that oxidative stress is a major contributor in neural tube defects (NTDs) and other malformation such as Holoprosencephaly (HPE) [87].
NTDs result from a failure of the neural tube to close during the fourth week of embryogenesis. NTDs are among the most common of human birth defects, with an overall prevalence of around 0.5–2/1000 pregnancies and frequently result in infant mortality or major health problems in surviving children [81,88,89]. Most human NTDs occur sporadically, with recurrences tending to fit a multifactorial polygenic or oligogenic pattern, rather than either dominant or recessive single gene inheritance with reduced penetrance. In addition to the mutation of the coding sequence, altered transcriptional regulation of these genes has the potential to cause NTDs. A consideration of potential causative factors in human NTDs should, therefore, account for the possibility of deregulation of redox signals [79,90,91]. A direct relationship between antioxidant enzymes and the development of the neural tube is reported. GPx, glutathione S-transferases (GSTs) and copper-zinc super-SOD enzymes are the most important protective systems in humans for NTDs. An impaired responsiveness of the antioxidant enzymes, such as CAT, SOD, GPx, GSTs and glutathione reductase (GR) that play an active role in the detoxification of hydrogen peroxidase, has crucial effects in oxygen-induced embryopathy and may result in neural-tube fails [92,93,94].
HPE is characterized by midline defects of the brain, facial and oral structures. It affects 1 in every 5000–10,000 live births [95]. Many cases of human HPE occur following fetal alcohol exposure or as a result of maternal diabetes both associated with elevated levels of ROS [96,97,98,99]. In other congenital malformations, such as urological and sex disorders, the role of oxidative stress-mediated by ROS has been also proposed [100,101,102,103].
In adverse pre- and perinatal conditions, oxidative stress can also predispose the newborn to a variety of health issues in later life [104]. The fetal programming concept suggests that the intrauterine environment to which a fetus is exposed can have a long-term impact on health after birth. Although the exact mechanisms of fetal programming have not yet been examined, the correlation between intrauterine stress and adverse effects in offspring has been confirmed for diseases such as metabolic dysfunction, type 2 diabetes, cardiovascular disease, neurological disorders, obesity [49,105,106,107,108,109,110,111].
Finally, increased activity of some important antioxidant enzymes (SOD1, CAT and GR) together with decreased GSH levels and higher levels of biomarkers of oxidative damage, such as protein carbonyls, malondialdehyde, allantoin or 8-hydroxydeoxyguanosine has been reported in chromosomal disorders, such as Down’s Syndrome (DS) [112,113,114,115,116,117,118,119,120,121]. In particular, a relationship between oxidative stress and DS clinical expression has been proposed [79].
6. Areas for Future Research
Several drugs with antioxidant properties, such as melatonin, vitamin C, resveratrol, nMitoQ, hydrogen and erythropoietin. These antioxidants exhibit potential beneficial effects in fetal hypoxia. Some of these substances, such as vitamin C and melatonin, are administered during the gestational period, while nMitoQ represents a treatment targeting the placenta. Instead, resveratrol and erythropoietin are used as postnatal treatments. Furthermore, the release of antioxidants through nanoparticles in the placenta may represent a new therapeutic approach that, having as a target exclusively the placenta, can avoid adverse effects on developing offspring. Future studies will be needed before validating the use of these antioxidant treatments in the routine clinical application.
Moreover, the microbiota profile and contaminants, provide a key interface between the mother and the fetal development in pregnancy [122]. The environmental role on the target tissue, including the placenta, fetus and neonate is not fully elucidated and is a target for investigating mechanisms of fetal programming [123].
The role of the endocrine receptor, such as glucocorticoid receptor isoforms, on the response to stress, has been proposed [124]. However, the potential role of these receptors in adapting to the maternal environment and regulating fetal growth is still to be explored in future research. Recently, the effect on the immune response, inflammation and oxidative stress of stem cells, such as Mesenchymal Stromal Cells (MSCs) has been reported also during pregnancy conditions [125]. Further experimental and clinical investigations are mandatory to define the emerging therapeutic potential of MSCs on the prevention of placental oxidative stress and/or treatment of prenatal hypoxia.
Furthermore, early detection of oxidative stress markers may be also considered as a crucial point for a better knowledge of the regulation of gene expression under hypoxic conditions, in order to identify new targets and possible treatments [126].
Epigenetic processes act in a cell-specific, temporally regulated manner to direct development, differentiation, organogenesis and related processes [127]. The hypoxia-induced epigenetic modifications may be implicated in fetal metabolic programming, congenital malformations and chronic diseases in future life [128]. It is hoped that future research will provide insights at the molecular level into these hypoxia-induced epigenetic mechanisms and clinical problems, in order to detect the development of preventive and intervention strategies to combat the epigenetic disorders and associated disease [129,130].
Progress in this area of research could benefit from integrated analyses combining knowledge gained from studies of human cohorts, animal models and cell systems, to promote an understanding of children’s health and illness.
7. Conclusions
This review provides an overview of animal models commonly used in order to evaluate the effects of oxidative stress induced by placental hypoxia on the development of the fetus. The in vivo models employed the exposure of pregnant animals to different concentrations of O2 in hypoxic chambers and the vascular clamping of the uterine artery. These animal models showed increased ROS levels induced by fetal hypoxia with consequent predispose the newborn to congenital malformation and/or pathological conditions in later life.
Additionally, we considered the efficacy of novel therapeutic approaches in preclinical studies. To date, the most used antioxidant agents are melatonin, vitamin C, resveratrol, hydrogen, erythropoietin, MitoQ. Specifically, the release of antioxidants through nanoparticles in the placenta may represent a new therapeutic approach that could avoid adverse effects on developing offspring.
Author Contributions
Conceptualization, V.C., G.P. and E.M.; writing—original draft preparation, S.S. and V.C.; writing—review and editing, P.B. and E.M.; funding acquisition, P.B. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by a Current Research Fund 2020, Ministry of Health, Italy.
Acknowledgments
The authors would like to thank the Ministry of Health, Italy.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kingdom, J.C.; Kaufmann, P. Oxygen and placental villous development: Origins of fetal hypoxia. Placenta 1997, 18, 613–621. [Google Scholar] [CrossRef]
- Lawn, J.E.; Cousens, S.; Zupan, J.; Team, L.N.S.S. 4 million neonatal deaths: When? Where? Why? Lancet 2005, 365, 891–900. [Google Scholar] [CrossRef]
- Hutter, D.; Jaeggi, E. Causes and mechanisms of intrauterine hypoxia and its impact on the fetal cardiovascular system: A review. Int. J. Pediatrics 2010, 2010, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.J.; Bartho, L.A.; Perkins, A.V.; Holland, O.J. Placental mitochondria and reactive oxygen species in the physiology and pathophysiology of pregnancy. Clin. Exp. Pharmacol. Physiol. 2020, 47, 176–184. [Google Scholar] [CrossRef]
- Giussani, D.A.; Phillips, P.S.; Anstee, S.; Barker, D.J. Effects of altitude versus economic status on birth weight and body shape at birth. Pediatric Res. 2001, 49, 490–494. [Google Scholar] [CrossRef]
- Meyer, K.; Zhang, L. Fetal programming of cardiac function and disease. Reprod. Sci. 2007, 14, 209–216. [Google Scholar] [CrossRef]
- Giussani, D.; Spencer, J.; Moore, P.; Bennet, L.; Hanson, M. Afferent and efferent components of the cardiovascular reflex responses to acute hypoxia in term fetal sheep. J. Physiol. 1993, 461, 431–449. [Google Scholar] [CrossRef]
- Richardson, B.S.; Bocking, A.D. Metabolic and circulatory adaptations to chronic hypoxia in the fetus. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 1998, 119, 717–723. [Google Scholar] [CrossRef]
- Morrison, J.L. Sheep models of intrauterine growth restriction: Fetal adaptations and consequences. Clin. Exp. Pharmacol. Physiol. 2008, 35, 730–743. [Google Scholar] [CrossRef]
- Zhang, L. Prenatal hypoxia and cardiac programming. J. Soc. Gynecol. Investig. 2005, 12, 2–13. [Google Scholar] [CrossRef]
- Moritz, K.M.; Singh, R.R.; Probyn, M.E.; Denton, K.M. Developmental programming of a reduced nephron endowment: More than just a baby’s birth weight. Am. J. Physiol. -Ren. Physiol. 2009, 296, F1–F9. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.; Shiell, A.W.; Barker, M.E.; Law, C.M. Growth in utero and blood pressure levels in the next generation. J. Hypertens. 2000, 18, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.J.; Watson, M.S.; Prescott, G.J.; Sunderland, S.; Campbell, D.M.; Hannaford, P.; Smith, W.C. Hypertensive diseases of pregnancy and risk of hypertension and stroke in later life: Results from cohort study. BMJ 2003, 326, 845. [Google Scholar] [CrossRef] [PubMed]
- Cargill, K.R.; Chiba, T.; Murali, A.; Mukherjee, E.; Crinzi, E.; Sims-Lucas, S. Prenatal hypoxia increases susceptibility to kidney injury. PLoS ONE 2020, 15, e0229618. [Google Scholar] [CrossRef]
- Barker, D.J.; Osmond, C.; Golding, J.; Kuh, D.; Wadsworth, M.E. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 1989, 298, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Ray, J.G.; Vermeulen, M.J.; Schull, M.J.; Redelmeier, D.A. Cardiovascular health after maternal placental syndromes (champs): Population-based retrospective cohort study. Lancet 2005, 366, 1797–1803. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. The PRISMA Group. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med 2009, 6, 7. [Google Scholar] [CrossRef]
- Gunn, A.J.; Bennet, L. Fetal hypoxia insults and patterns of brain injury: Insights from animal models. Clin. Perinatol. 2009, 36, 579–593. [Google Scholar] [CrossRef]
- Herrera, E.A.; Krause, B.; Ebensperger, G.; Reyes, R.V.; Casanello, P.; Parra-Cordero, M.; Llanos, A.J. The placental pursuit for an adequate oxidant balance between the mother and the fetus. Front. Pharmacol. 2014, 5, 149. [Google Scholar] [CrossRef]
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex iii: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive oxygen species in living systems: Source, biochemistry, and role in human disease. Am. J. Med. 1991, 91, S14–S22. [Google Scholar] [CrossRef]
- Williamson, R.D.; McCarthy, C.; McCarthy, F.P.; Kenny, L.C. Oxidative stress in pre-eclampsia; have we been looking in the wrong place? Pregnancy Hypertens. Int. J. Womens Cardiovasc. Health 2017, 8, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex iii is required for hypoxia-induced ros production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-M.; Shah, A.M. Endothelial cell superoxide generation: Regulation and relevance for cardiovascular pathophysiology. Am. J. Physiol. -Regul. Integr. Comp. Physiol. 2004, 287, R1014–R1030. [Google Scholar] [CrossRef]
- Fleming, I. Cytochrome p450 and vascular homeostasis. Circ. Res. 2001, 89, 753–762. [Google Scholar] [CrossRef]
- Hewinson, J.; Stevens, C.R.; Millar, T.M. Vascular physiology and pathology of circulating xanthine oxidoreductase: From nucleotide sequence to functional enzyme. Redox Rep. 2004, 9, 71–79. [Google Scholar] [CrossRef]
- Tissot van Patot, M.C.; Murray, A.J.; Beckey, V.; Cindrova-Davies, T.; Johns, J.; Zwerdlinger, L.; Jauniaux, E.; Burton, G.J.; Serkova, N.J. Human placental metabolic adaptation to chronic hypoxia, high altitude: Hypoxic preconditioning. Am. J. Physiol. -Regul. Integr. Comp. Physiol. 2010, 298, R166–R172. [Google Scholar] [CrossRef]
- Wang, H.; Joseph, J.A. Mechanisms of hydrogen peroxide-induced calcium dysregulation in pc12 cells. Free Radic. Biol. Med. 2000, 28, 1222–1231. [Google Scholar] [CrossRef]
- Gallin, W.J.; Greenberg, M.E. Calcium regulation of gene expression in neurons: The mode of entry matters. Curr. Opin. Neurobiol. 1995, 5, 367–374. [Google Scholar] [CrossRef]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef]
- Stuehr, D.J. Mammalian nitric oxide synthases. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 1999, 1411, 217–230. [Google Scholar] [CrossRef]
- Kaufmann, P.; Black, S.; Huppertz, B. Endovascular trophoblast invasion: Implications for the pathogenesis of intrauterine growth retardation and preeclampsia. Biol. Reprod. 2003, 69, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gamaley, I.A.; Klyubin, I.V. Roles of reactive oxygen species: Signaling and regulation of cellular functions. Int. Rev. Cytol. 1999, 188, 203–255. [Google Scholar] [PubMed]
- Raijmakers, M.T.; Dechend, R.; Poston, L. Oxidative stress and preeclampsia: Rationale for antioxidant clinical trials. Hypertension 2004, 44, 374–380. [Google Scholar] [CrossRef]
- Jauniaux, E.; Watson, A.L.; Hempstock, J.; Bao, Y.-P.; Skepper, J.N.; Burton, G.J. Onset of maternal arterial blood flow and placental oxidative stress: A possible factor in human early pregnancy failure. Am. J. Pathol. 2000, 157, 2111–2122. [Google Scholar] [CrossRef]
- Buonocore, G.; Perrone, S.; Muraca, M. Free radicals and brain damage in newborns with hypoxic-ischemic lesion. Ann. Dell’istituto Super. Di Sanita 2001, 37, 527–535. [Google Scholar]
- Golan, M.H.; Mane, R.; Molczadzki, G.; Zuckerman, M.; Kaplan-Louson, V.; Huleihel, M.; Perez-Polo, J.R. Impaired migration signaling in the hippocampus following prenatal hypoxia. Neuropharmacology 2009, 57, 511–522. [Google Scholar] [CrossRef]
- Herlenius, E.; Lagercrantz, H. Neurotransmitters and neuromodulators during early human development. Early Hum. Dev. 2001, 65, 21–37. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, P.J.; Xiong, F.; Li, Y.; Zhou, J.; Zhang, L. Fetal hypoxia increases vulnerability of hypoxic–ischemic brain injury in neonatal rats: Role of glucocorticoid receptors. Neurobiol. Dis. 2014, 65, 172–179. [Google Scholar] [CrossRef]
- Sab, I.; Ferraz, M.; Amaral, T.; Resende, A.; Ferraz, M.; Matsuura, C.; Brunini, T.; Mendes-Ribeiro, A. Prenatal hypoxia, habituation memory and oxidative stress. Pharmacol. Biochem. Behav. 2013, 107, 24–28. [Google Scholar] [CrossRef]
- Louzoun-Kaplan, V.; Zuckerman, M.; Perez-Polo, J.R.; Golan, H.M. Prenatal hypoxia down regulates the gaba pathway in newborn mice cerebral cortex; partial protection by mgso4. Int. J. Dev. Neurosci. 2008, 26, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Matheson, H.; Veerbeek, J.H.; Charnock-Jones, D.S.; Burton, G.J.; Yung, H.W. Morphological and molecular changes in the murine placenta exposed to normobaric hypoxia throughout pregnancy. J. Physiol. 2016, 594, 1371–1388. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Telugu, B.P.; Thompson, L.P. Sexual dimorphism of mitochondrial function in the hypoxic guinea pig placenta. Biol. Reprod. 2019, 100, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Rueda-Clausen, C.; Stanley, J.; Thambiraj, D.; Poudel, R.; Davidge, S.; Baker, P. Effect of prenatal hypoxia in transgenic mouse models of preeclampsia and fetal growth restriction. Reprod. Sci. 2014, 21, 492–502. [Google Scholar] [CrossRef]
- Baud, O.; Daire, J.L.; Dalmaz, Y.; Fontaine, R.H.; Krueger, R.C.; Sebag, G.; Evrard, P.; Gressens, P.; Verney, C. Gestational hypoxia induces white matter damage in neonatal rats: A new model of periventricular leukomalacia. Brain Pathol. 2004, 14, 1–10. [Google Scholar] [CrossRef]
- Zhu, X.; Gao, Q.; Tu, Q.; Zhong, Y.; Zhu, D.; Mao, C.; Xu, Z. Prenatal hypoxia enhanced angiotensin ii-mediated vasoconstriction via increased oxidative signaling in fetal rats. Reprod. Toxicol. 2016, 60, 21–28. [Google Scholar] [CrossRef]
- Chen, L.; Zadi, Z.H.; Zhang, J.; Scharf, S.M.; Pae, E.-K. Intermittent hypoxia in utero damages postnatal growth and cardiovascular function in rats. J. Appl. Physiol. 2018, 124, 821–830. [Google Scholar] [CrossRef]
- Figueroa, H.; Alvarado, C.; Cifuentes, J.; Lozano, M.; Rocco, J.; Cabezas, C.; Illanes, S.E.; Eixarch, E.; Hernandez-Andrade, E.; Gratacos, E.; et al. Oxidative damage and nitric oxide synthase induction by surgical uteroplacental circulation restriction in the rabbit fetal heart. Prenat. Diagn. 2017, 37, 453–459. [Google Scholar] [CrossRef]
- Giussani, D.A.; Camm, E.J.; Niu, Y.; Richter, H.G.; Blanco, C.E.; Gottschalk, R.; Blake, E.Z.; Horder, K.A.; Thakor, A.S.; Hansell, J.A. Developmental programming of cardiovascular dysfunction by prenatal hypoxia and oxidative stress. PLoS ONE 2012, 7, e31017. [Google Scholar] [CrossRef]
- Chen, X.; Qi, L.; Fan, X.; Tao, H.; Zhang, M.; Gao, Q.; Liu, Y.; Xu, T.; Zhang, P.; Su, H. Prenatal hypoxia affected endothelium-dependent vasodilation in mesenteric arteries of aged offspring via increased oxidative stress. Hypertens. Res. 2019, 42, 863–875. [Google Scholar] [CrossRef]
- Rueda-Clausen, C.; Morton, J.; Oudit, G.; Kassiri, Z.; Jiang, Y.; Davidge, S. Effects of hypoxia-induced intrauterine growth restriction on cardiac siderosis and oxidative stress. J. Dev. Orig. Health Dis. 2012, 3, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Xiao, Y.; Estrella, J.L.; Ducsay, C.A.; Gilbert, R.D.; Zhang, L. Effect of fetal hypoxia on heart susceptibility to ischemia and reperfusion injury in the adult rat. J. Soc. Gynecol. Investig. 2003, 10, 265–274. [Google Scholar] [CrossRef]
- Xue, Q.; Zhang, L. Prenatal hypoxia causes a sex-dependent increase in heart susceptibility to ischemia and reperfusion injury in adult male offspring: Role of protein kinase cϵ. J. Pharmacol. Exp. Ther. 2009, 330, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.J.; Xiao, D.; Xiong, F.; Dixon, B.; Zhang, L. Hypoxia-derived oxidative stress mediates epigenetic repression of pkcɛ gene in foetal rat hearts. Cardiovasc. Res. 2012, 93, 302–310. [Google Scholar] [CrossRef]
- Aiken, C.E.; Tarry-Adkins, J.L.; Spiroski, A.M.; Nuzzo, A.M.; Ashmore, T.J.; Rolfo, A.; Sutherland, M.J.; Camm, E.J.; Giussani, D.A.; Ozanne, S.E. Chronic fetal hypoxia disrupts the peri-conceptual environment in next-generation adult female rats. J. Physiol. 2019, 597, 2391–2401. [Google Scholar] [CrossRef]
- Aiken, C.E.; Tarry-Adkins, J.L.; Spiroski, A.-M.; Nuzzo, A.M.; Ashmore, T.J.; Rolfo, A.; Sutherland, M.J.; Camm, E.J.; Giussani, D.A.; Ozanne, S.E. Chronic gestational hypoxia accelerates ovarian aging and lowers ovarian reserve in next-generation adult rats. FASEB J. 2019, 33, 7758–7766. [Google Scholar] [CrossRef]
- Okatani, Y.; Wakatsuki, A.; Shinohara, K.; Taniguchi, K.; Fukaya, T. Melatonin protects against oxidative mitochondrial damage induced in rat placenta by ischemia and reperfusion. J. Pineal Res. 2001, 31, 173–178. [Google Scholar] [CrossRef]
- Richter, H.; Camm, E.; Modi, B.; Naeem, F.; Cross, C.; Cindrova-Davies, T.; Spasic-Boskovic, O.; Dunster, C.; Mudway, I.; Kelly, F. Ascorbate prevents placental oxidative stress and enhances birth weight in hypoxic pregnancy in rats. J. Physiol. 2012, 590, 1377–1387. [Google Scholar] [CrossRef]
- Giussani, D.; Davidge, S. Developmental programming of cardiovascular disease by prenatal hypoxia. J. Dev. Orig. Health Dis. 2013, 4, 328–337. [Google Scholar] [CrossRef]
- Ungvari, Z.; Labinskyy, N.; Mukhopadhyay, P.; Pinto, J.T.; Bagi, Z.; Ballabh, P.; Zhang, C.; Pacher, P.; Csiszar, A. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1876–H1881. [Google Scholar] [CrossRef]
- Li, Y.; Cao, Z.; Zhu, H. Upregulation of endogenous antioxidants and phase 2 enzymes by the red wine polyphenol, resveratrol in cultured aortic smooth muscle cells leads to cytoprotection against oxidative and electrophilic stress. Pharmacol. Res. 2006, 53, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Reyes, L.M.; Morton, J.S.; Fung, D.; Schneider, J.; Davidge, S.T. Effect of resveratrol on metabolic and cardiovascular function in male and female adult offspring exposed to prenatal hypoxia and a high-fat diet. J. Physiol. 2016, 594, 1465–1482. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Quon, A.; Morton, J.S.; Davidge, S.T. Postnatal resveratrol supplementation improves cardiovascular function in male and female intrauterine growth restricted offspring. Physiol. Rep. 2017, 5, e13109. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.; Salih, S.; Roh, D.; Lacharme-Lora, L.; Parry, M.; Hardiman, B.; Keehan, R.; Grummer, R.; Winterhager, E.; Gokhale, P. Signalling of DNA damage and cytokines across cell barriers exposed to nanoparticles depends on barrier thickness. Nat. Nanotechnol. 2011, 6, 824. [Google Scholar] [CrossRef]
- Jones, A.J.; Gokhale, P.J.; Allison, T.F.; Sampson, B.; Athwal, S.; Grant, S.; Andrews, P.W.; Allen, N.D.; Case, C.P. Evidence for bystander signalling between human trophoblast cells and human embryonic stem cells. Sci. Rep. 2015, 5, 1–14. [Google Scholar] [CrossRef]
- Phillips, T.J.; Scott, H.; Menassa, D.A.; Bignell, A.L.; Sood, A.; Morton, J.S.; Akagi, T.; Azuma, K.; Rogers, M.F.; Gilmore, C.E. Treating the placenta to prevent adverse effects of gestational hypoxia on fetal brain development. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]
- Aljunaidy, M.M.; Morton, J.S.; Kirschenman, R.; Phillips, T.; Case, C.P.; Cooke, C.-L.M.; Davidge, S.T. Maternal treatment with a placental-targeted antioxidant (mitoq) impacts offspring cardiovascular function in a rat model of prenatal hypoxia. Pharmacol. Res. 2018, 134, 332–342. [Google Scholar] [CrossRef]
- Ganguly, E.; Aljunaidy, M.; Kirschenman, R.; Spaans, F.; Morton, J.S.; Phillips, T.; Case, P.; Cooke, C.-L.; Davidge, S.T. Sex-specific effects of nanoparticle-encapsulated mitoq (nmitoq) delivery to the placenta in a rat model of fetal hypoxia. Front. Physiol. 2019, 10, 562. [Google Scholar] [CrossRef]
- Zhuravin, I.; Dubrovskaya, N.; Tumanova, N. Postnatal physiological development of rats after acute prenatal hypoxia. Neurosci. Behav. Physiol. 2004, 34, 809–816. [Google Scholar] [CrossRef]
- Golan, H.; Kashtutsky, I.; Hallak, M.; Sorokin, Y.; Huleihel, M. Maternal hypoxia during pregnancy delays the development of motor reflexes in newborn mice. Dev. Neurosci. 2004, 26, 24–29. [Google Scholar] [CrossRef]
- Liu, W.; Chen, O.; Chen, C.; Wu, B.; Tang, J.; Zhang, J.H. Protective effects of hydrogen on fetal brain injury during maternal hypoxia. In Intracerebral Hemorrhage Research; Springer: Berlin/Heidelberg, Germany, 2011; pp. 307–311. [Google Scholar]
- Kostović, I.; Jovanov-Milošević, N.; Radoš, M.; Sedmak, G.; Benjak, V.; Kostović-Srzentić, M.; Vasung, L.; Čuljat, M.; Radoš, M.; Hüppi, P. Perinatal and early postnatal reorganization of the subplate and related cellular compartments in the human cerebral wall as revealed by histological and mri approaches. Brain Struct. Funct. 2014, 219, 231–253. [Google Scholar] [CrossRef] [PubMed]
- McPherson, R.J.; Juul, S.E. Recent trends in erythropoietin-mediated neuroprotection. Int. J. Dev. Neurosci. 2008, 26, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Akisu, M.; Tuzun, S.; Arslanoglu, S.; Yalaz, M.; Kultursay, N. Effect of recombinant human erythropoietin administration on lipid peroxidation and antioxidant enzyme (s) activities in preterm infants. Acta Med. Okayama 2001, 55, 357–362. [Google Scholar] [PubMed]
- Calapai, G.; Marciano, M.C.; Corica, F.; Allegra, A.; Parisi, A.; Frisina, N.; Caputi, A.P.; Buemi, M. Erythropoietin protects against brain ischemic injury by inhibition of nitric oxide formation. Eur. J. Pharmacol. 2000, 401, 349–356. [Google Scholar] [CrossRef]
- Mazur, M.; Miller, R.H.; Robinson, S. Postnatal erythropoietin treatment mitigates neural cell loss after systemic prenatal hypoxic-ischemic injury. J. Neurosurg. Pediatrics 2010, 6, 206–221. [Google Scholar] [CrossRef]
- Zadran, S.; Akopian, G.; Zadran, H.; Walsh, J.; Baudry, M. Rvg-mediated calpain2 gene silencing in the brain impairs learning and memory. Neuromol. Med. 2013, 15, 74–81. [Google Scholar] [CrossRef]
- Jantzie, L.L.; Winer, J.L.; Corbett, C.J.; Robinson, S. Erythropoietin modulates cerebral and serum degradation products from excess calpain activation following prenatal hypoxia-ischemia. Dev. Neurosci. 2016, 38, 15–26. [Google Scholar] [CrossRef]
- Laforgia, N.; Di Mauro, A.; Favia Guarnieri, G.; Varvara, D.; De Cosmo, L.; Panza, R.; Capozza, M.; Baldassarre, M.E.; Resta, N. The role of oxidative stress in the pathomechanism of congenital malformations. Oxidative Med. Cell. Longev. 2018, 2018, 1–12. [Google Scholar] [CrossRef]
- Wu, F.; Tian, F.-J.; Lin, Y. Oxidative stress in placenta: Health and diseases. Biomed Res. Int. 2015, 2015, 1–15. [Google Scholar] [CrossRef]
- Christianson, A.; Howson, C.P.; Modell, B. March of Dimes: Global Report on Birth Defects, the Hidden Toll of Dying and Disabled Children; March of Dimes Birth Defects Foundation: White Plains, NY, USA, 2005. [Google Scholar]
- Hobbs, C.A.; Chowdhury, S.; Cleves, M.A.; Erickson, S.; MacLeod, S.L.; Shaw, G.M.; Shete, S.; Witte, J.S.; Tycko, B. Genetic epidemiology and nonsyndromic structural birth defects: From candidate genes to epigenetics. JAMA Pediatr. 2014, 168, 371–377. [Google Scholar] [CrossRef]
- Hoffman, J.I.; Kaplan, S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Rossi, R.; Colombo, R.; Giustarini, D.; Milzani, A. Biomarkers of oxidative damage in human disease. Clin. Chem. 2006, 52, 601–623. [Google Scholar] [CrossRef] [PubMed]
- Redón, J.; Oliva, M.R.; Tormos, C.; Giner, V.; Chaves, J.; Iradi, A.; Sáez, G.T. Antioxidant activities and oxidative stress byproducts in human hypertension. Hypertension 2003, 41, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, C.A.; Cleves, M.A.; Melnyk, S.; Zhao, W.; James, S.J. Congenital heart defects and abnormal maternal biomarkers of methionine and homocysteine metabolism. Am. J. Clin. Nutr. 2005, 81, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Dheen, S.T.; Tay, S.S.; Boran, J.; Ting, L.W.; Kumar, S.D.; Fu, J.; Ling, E.A. Recent studies on neural tube defects in embryos of diabetic pregnancy: An overview. Curr. Med. Chem. 2009, 16, 2345–2354. [Google Scholar] [CrossRef]
- Copp, A.J.; Stanier, P.; Greene, N.D. Neural tube defects: Recent advances, unsolved questions, and controversies. Lancet Neurol. 2013, 12, 799–810. [Google Scholar] [CrossRef]
- Wallingford, J.B.; Niswander, L.A.; Shaw, G.M.; Finnell, R.H. The continuing challenge of understanding, preventing, and treating neural tube defects. Science 2013, 339, 1222002. [Google Scholar] [CrossRef]
- Agopian, A.; Tinker, S.C.; Lupo, P.J.; Canfield, M.A.; Mitchell, L.E.; Study, N.B.D.P. Proportion of neural tube defects attributable to known risk factors. Birth Defects Res. Part A Clin. Mol. Teratol. 2013, 97, 42–46. [Google Scholar] [CrossRef]
- El-hage, S.; Singh, S.M. Temporal expression of genes encoding free radical–metabolizing enzymes is associated with higher mrna levels during in utero development in mice. Dev. Genet. 1990, 11, 149–159. [Google Scholar] [CrossRef]
- Graf, W.D.; Pippenger, C.; Shurtleff, D.B. Erythrocyte antioxidant enzyme activities in children with myelomeningocele. Dev. Med. Child Neurol. 1995, 37, 900–905. [Google Scholar] [CrossRef]
- Knapen, M.F.; Zusterzeel, P.L.; Peters, W.H.; Steegers, E.A. Glutathione and glutathione-related enzymes in reproduction: A review. Eur. J. Obstet. Gynecol. Reprod. Biol. 1999, 82, 171–184. [Google Scholar] [CrossRef]
- Gaeta, L.M.; Tozzi, G.; Pastore, A.; Federici, G.; Bertini, E.; Piemonte, F. Determination of superoxide dismutase and glutathione peroxidase activities in blood of healthy pediatric subjects. Clin. Chim. Acta 2002, 322, 117–120. [Google Scholar] [CrossRef]
- Cohen, M.M., Jr.; Shiota, K. Teratogenesis of holoprosencephaly. Am. J. Med Genet. 2002, 109, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Aoto, K.; Shikata, Y.; Higashiyama, D.; Shiota, K.; Motoyama, J. Fetal ethanol exposure activates protein kinase a and impairs shh expression in prechordal mesendoderm cells in the pathogenesis of holoprosencephaly. Birth Defects Res. Part A Clin. Mol. Teratol. 2008, 82, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Belloni, E.; Muenke, M.; Roessler, E.; Traverse, G.; Siegel-Bartelt, J.; Frumkin, A.; Mitchell, H.; Donis-Keller, H.; Helms, C.; Hing, A. Identification of sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat. Genet. 1996, 14, 353. [Google Scholar] [CrossRef] [PubMed]
- Roessler, E.; Belloni, E.; Gaudenz, K.; Jay, P.; Berta, P.; Scherer, S.W.; Tsui, L.-C.; Muenke, M. Mutations in the human sonic hedgehog gene cause holoprosencephaly. Nat. Genet. 1996, 14, 357–360. [Google Scholar] [CrossRef]
- Hammerschmidt, M.; Bitgood, M.J.; McMahon, A.P. Protein kinase a is a common negative regulator of hedgehog signaling in the vertebrate embryo. Genes Dev. 1996, 10, 647–658. [Google Scholar] [CrossRef]
- Avci, V.; Ayengin, K.; Alp, H.H. Oxidative DNA damage and nox4 levels in children with undescended testes. Eur. J. Pediatric Surg. 2019, 29, 545–550. [Google Scholar] [CrossRef]
- Froehlich, J.W.; Kostel, S.A.; Cho, P.S.; Briscoe, A.C.; Steen, H.; Vaezzadeh, A.R.; Lee, R.S. Urinary proteomics yield pathological insights for ureteropelvic junction obstruction. Mol. Cell. Proteom. 2016, 15, 2607–2615. [Google Scholar] [CrossRef]
- Ko, E.Y.; Sabanegh, E.S., Jr.; Agarwal, A. Male infertility testing: Reactive oxygen species and antioxidant capacity. Fertil. Steril. 2014, 102, 1518–1527. [Google Scholar] [CrossRef]
- Agarwal, A.; Sharma, R.K.; Desai, N.R.; Prabakaran, S.; Tavares, A.; Sabanegh, E. Role of oxidative stress in pathogenesis of varicocele and infertility. Urology 2009, 73, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.P. Fetal growth and adult disease. Br. J. Obstet. Gynaecol. 1992, 99, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Perrone, S.; Santacroce, A.; Picardi, A.; Buonocore, G. Fetal programming and early identification of newborns at high risk of free radical-mediated diseases. World J. Clin. Pediatrics 2016, 5, 172. [Google Scholar] [CrossRef] [PubMed]
- Menon, R. Oxidative stress damage as a detrimental factor in preterm birth pathology. Front. Immunol. 2014, 5, 567. [Google Scholar] [CrossRef] [PubMed]
- Giussani, D.A.; Niu, Y.; Herrera, E.A.; Richter, H.G.; Camm, E.J.; Thakor, A.S.; Kane, A.D.; Hansell, J.A.; Brain, K.L.; Skeffington, K.L. Heart disease link to fetal hypoxia and oxidative stress. In Advances in Fetal and Neonatal Physiology; Springer: Berlin/Heidelberg, Germany, 2014; pp. 77–87. [Google Scholar]
- Entringer, S.; Buss, C.; Wadhwa, P.D. Prenatal stress, telomere biology, and fetal programming of health and disease risk. Sci. Signal. 2012, 5, pt12. [Google Scholar] [CrossRef] [PubMed]
- Ghulmiyyah, L.M.; Costantine, M.M.; Yin, H.; Tamayo, E.; Clark, S.M.; Hankins, G.D.; Saade, G.R.; Longo, M. The role of oxidative stress in the developmental origin of adult hypertension. Am. J. Obstet. Gynecol. 2011, 205, 155.e7–155.e11. [Google Scholar] [CrossRef]
- Pinney, S.E.; Simmons, R.A. Epigenetic mechanisms in the development of type 2 diabetes. Trends Endocrinol. Metab. 2010, 21, 223–229. [Google Scholar] [CrossRef]
- Franco, M.C.P.; Dantas, A.P.V.; Akamine, E.H.; Kawamoto, E.M.; Fortes, Z.B.; Scavone, C.; Tostes, R.C.; Carvalho, M.H.C.; Nigro, D. Enhanced oxidative stress as a potential mechanism underlying the programming of hypertension in utero. J. Cardiovasc. Pharmacol. 2002, 40, 501–509. [Google Scholar] [CrossRef]
- Engidawork, E.; Lubec, G. Molecular changes in fetal down syndrome brain. J. Neurochem. 2003, 84, 895–904. [Google Scholar] [CrossRef]
- Sanchez-Font, M.; Sebastia, J.; Sanfeliu, C.; Cristofol, R.; Marfany, G.; Gonzàles-Duarte, R. Peroxiredoxin 2 (prdx2), an antioxidant enzyme, is underexpressed in down syndrome fetal brains. Cell. Mol. Life Sci. CMLS 2003, 60, 1513–1523. [Google Scholar] [CrossRef]
- Busciglio, J.; Yankner, B.A. Apoptosis and increased generation of reactive oxygen species in down’s syndrome neurons in vitro. Nature 1995, 378, 776–779. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Li, T.; Chen, J.; Liu, Y.; Xiong, F.; Yang, J.; Song, C. Plasma antioxidant enzymes and lipoperoxidation status in children with down syndrome. Clin. Biochem. 2016, 49, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D.; Iuliano, L.; Amerio, G.; Tang, L.X.; Rokach, J.; Sabatino, G.; Violi, F. Down’s syndrome is associated with increased 8, 12-iso-ipf2α-vi levels: Evidence for enhanced lipid peroxidation in vivo. Ann. Neurol. 2000, 48, 795–798. [Google Scholar] [CrossRef]
- Perluigi, M.; Di Domenico, F.; Buttterfield, D.A. Unraveling the complexity of neurodegeneration in brains of subjects with down syndrome: Insights from proteomics. Proteom. Clin. Appl. 2014, 8, 73–85. [Google Scholar] [CrossRef]
- Balcz, B.; Kirchner, L.; Cairns, N.; Fountoulakis, M.; Lubec, G. Increased brain protein levels of carbonyl reductase and alcohol dehydrogenase in down syndrome and alzheimer’s disease. J. Neural Transm. Suppl. 2001, 193–201. [Google Scholar] [CrossRef]
- Garcez, M.E.; Peres, W.; Salvador, M. Oxidative stress and hematologic and biochemical parameters in individuals with down syndrome. Mayo Clin. Proc. 2005, 80, 1607–1611. [Google Scholar] [CrossRef] [PubMed]
- Conti, A.; Fabbrini, F.; D’Agostino, P.; Negri, R.; Greco, D.; Genesio, R.; D’Armiento, M.; Olla, C.; Paladini, D.; Zannini, M.; et al. Altered expression of mitochondrial and extracellular matrix genes in the heart of human fetuses with chromosome 21 trisomy. BMC Genom. 2007, 8, 268. [Google Scholar] [CrossRef]
- Perluigi, M.; di Domenico, F.; Fiorini, A.; Cocciolo, A.; Giorgi, A.; Foppoli, C.; Butterfield, D.A.; Giorlandino, M.; Giorlandino, C.; Schinina, M.E.; et al. Oxidative stress occurs early in down syndrome pregnancy: A redox proteomics analysis of amniotic fluid. Proteom. Clin. Appl. 2011, 5, 167–178. [Google Scholar] [CrossRef]
- Chen, H.J.; Gur, T.L. Intrauterine microbiota: Missing, or the missing link? Trends Neurosci. 2019, 42, 402–413. [Google Scholar] [CrossRef]
- Singh, L.; Anand, M.; Singh, S.; Taneja, A. Environmental toxic metals in placenta and their effects on preterm delivery-current opinion. Drug Chem. Toxicol. 2018, 1–8. [Google Scholar] [CrossRef]
- Clifton, V.L.; Cuffe, J.; Moritz, K.M.; Cole, T.J.; Fuller, P.J.; Lu, N.Z.; Kumar, S.; Chong, S.; Saif, Z. Review: The role of multiple placental glucocorticoid receptor isoforms in adapting to the maternal environment and regulating fetal growth. Placenta 2017, 54, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Suvakov, S.; Richards, C.; Nikolic, V.; Simic, T.; McGrath, K.; Krasnodembskaya, A.; McClements, L. Emerging therapeutic potential of mesenchymal stem/stromal cells in preeclampsia. Curr. Hypertens. Rep. 2020, 22, 37. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Kataoka, N. Regulation of gene expression under hypoxic conditions. Int. J. Mol. Sci. 2019, 20, 3278. [Google Scholar] [CrossRef] [PubMed]
- Gheorghe, C.P.; Goyal, R.; Mittal, A.; Longo, L.D. Gene expression in the placenta: Maternal stress and epigenetic responses. Int. J. Dev. Biol. 2010, 54, 507–523. [Google Scholar] [CrossRef]
- Zhu, Z.; Cao, F.; Li, X. Epigenetic programming and fetal metabolic programming. Front. Endocrinol. 2019, 10, 764. [Google Scholar] [CrossRef]
- Kubota, T. Preemptive epigenetic medicine based on fetal programming. Adv. Exp. Med. Biol. 2018, 1012, 85–95. [Google Scholar]
- Bellanti, J.A. Epigenetic studies and pediatric research. Pediatr. Res. 2020, 87, 378–384. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).