Royal Jelly as an Intelligent Anti-Aging Agent—A Focus on Cognitive Aging and Alzheimer’s Disease: A Review



Abstract

1. An Overview of Cognitive Aging
2. Overview of Alzheimer’s Disease
3. The Mechanism Underlying AD Development
3.1. Role of the Immune System in Alzheimer’s Disease
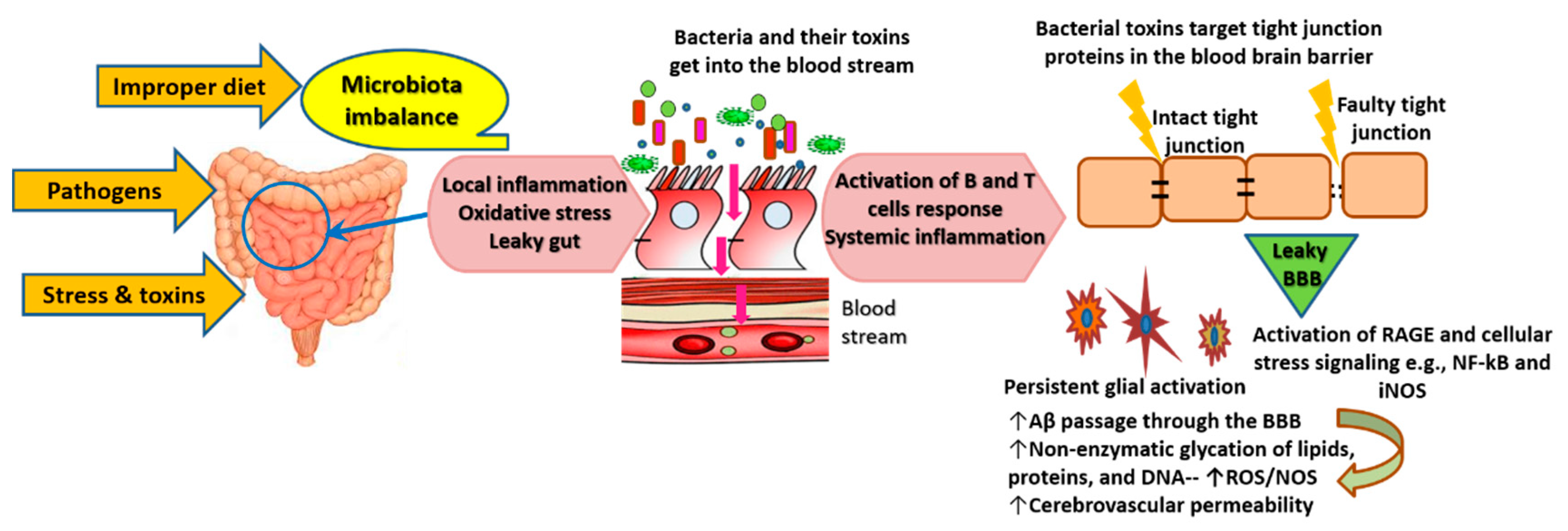
3.2. Role of the Gut-Brain Axis in AD Pathogenesis
3.3. Multiple Medical Conditions Contribute to AD by Enhancing Neuroinflammation
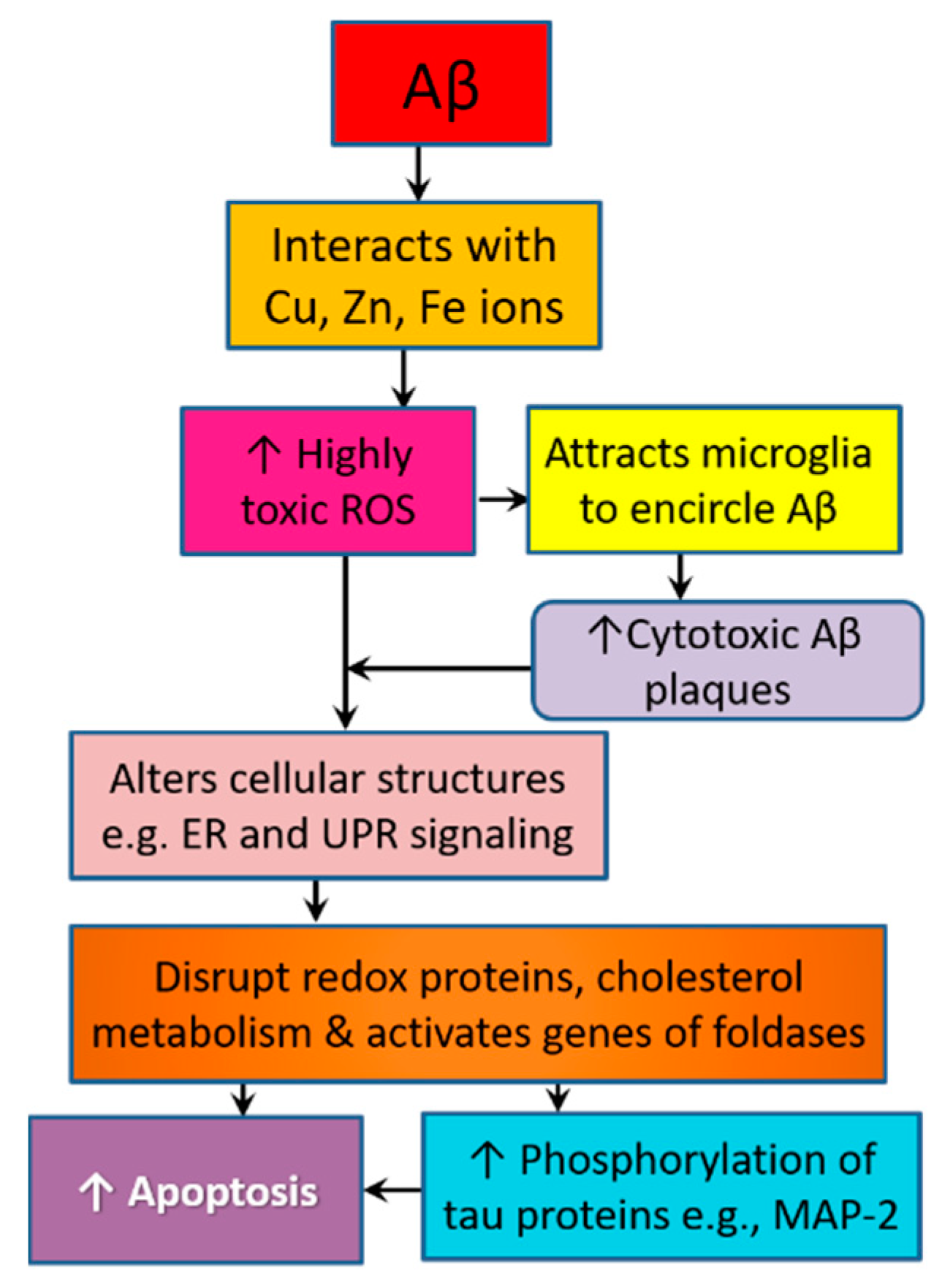
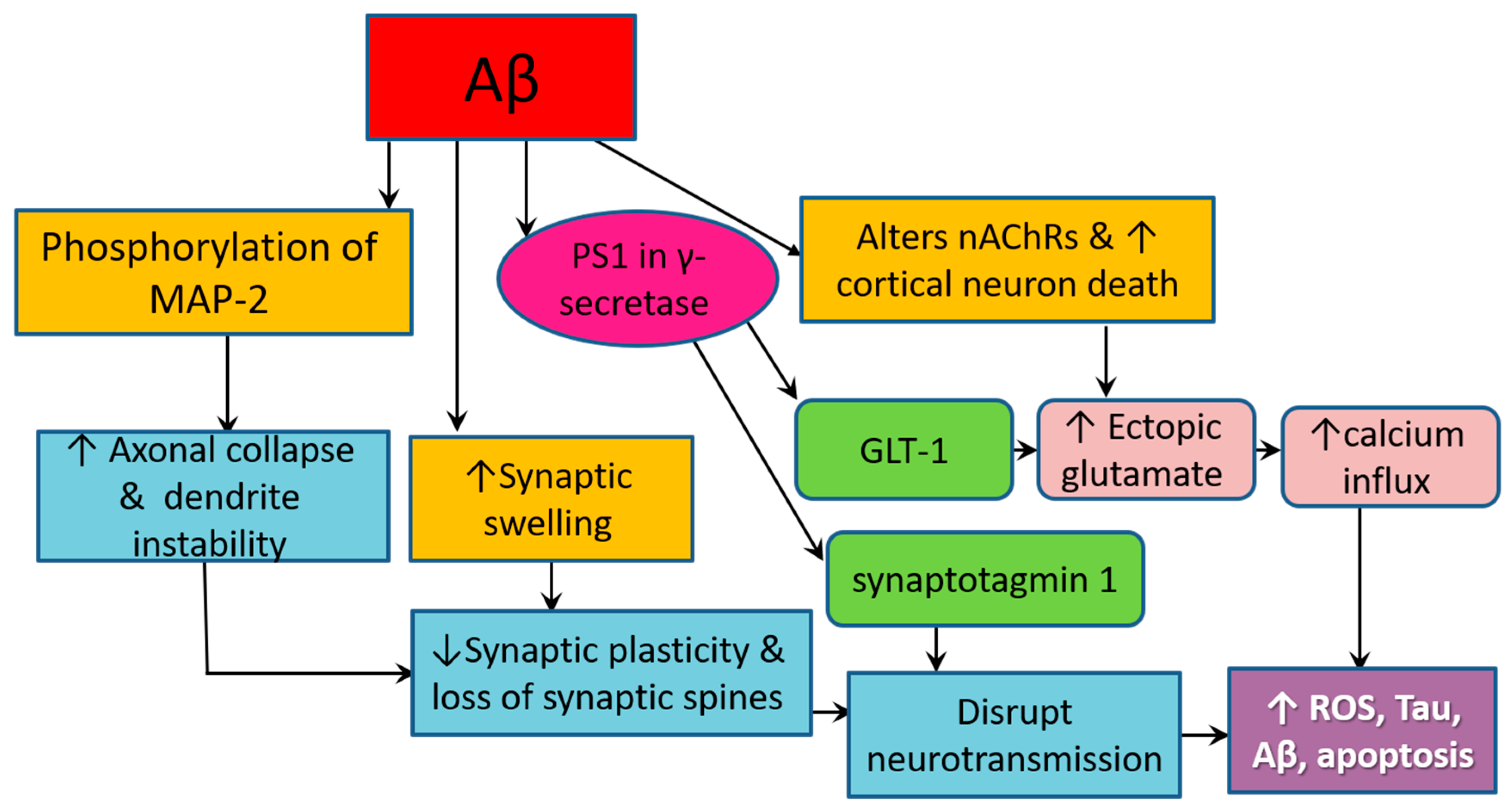
3.4. Amyloid Pathology Promotes Neurodegeneration by Altering Cellular Structure and Function
4. Current Treatments of AD
5. Royal Jelly, Its Ingredients and Pharmacological Properties
6. Effect of Royal Jelly on Cognitive Performance and Related Biological Markers
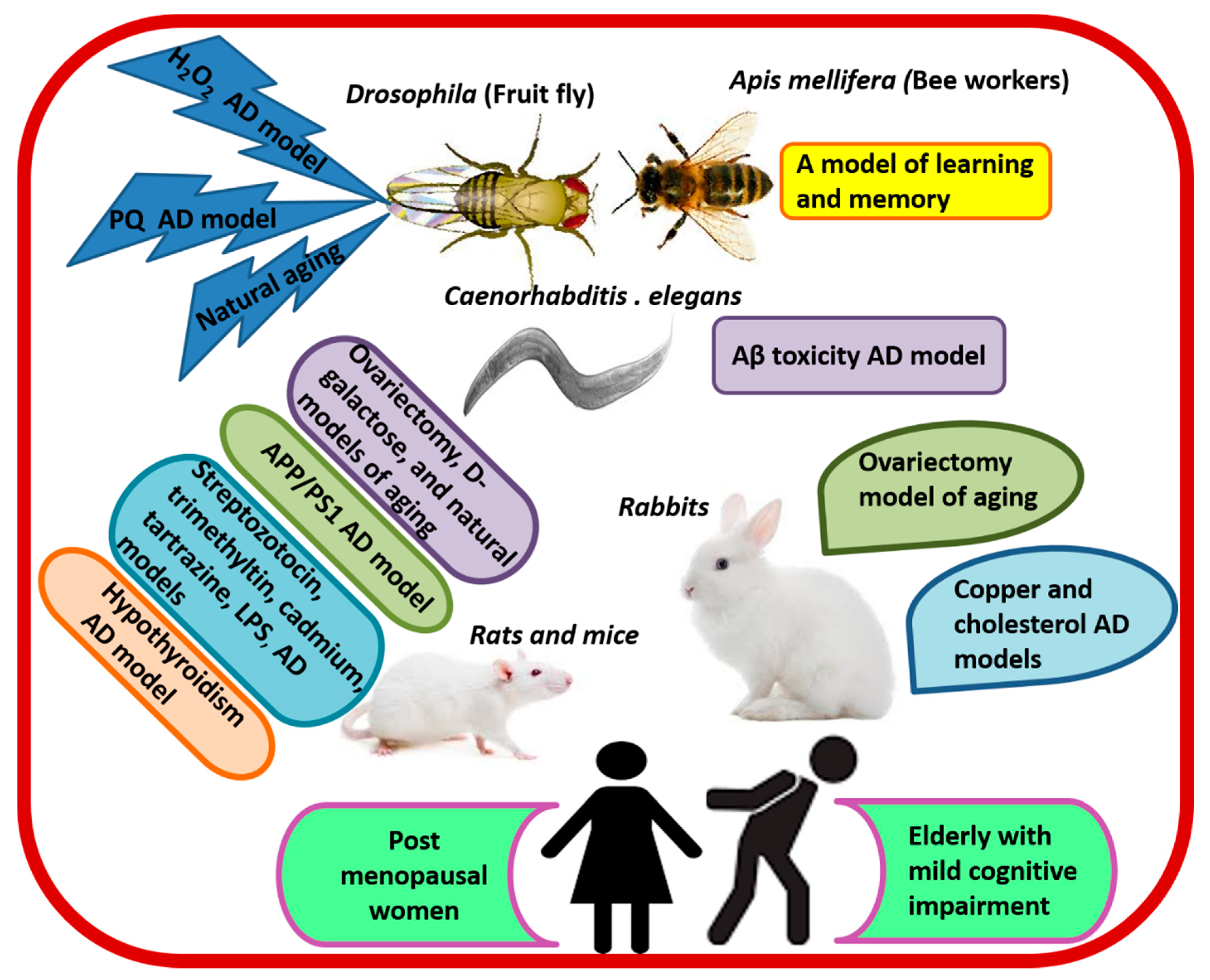
6.1. Evidence from Preclinical Studies
6.2. Evidence from Clinical Trials
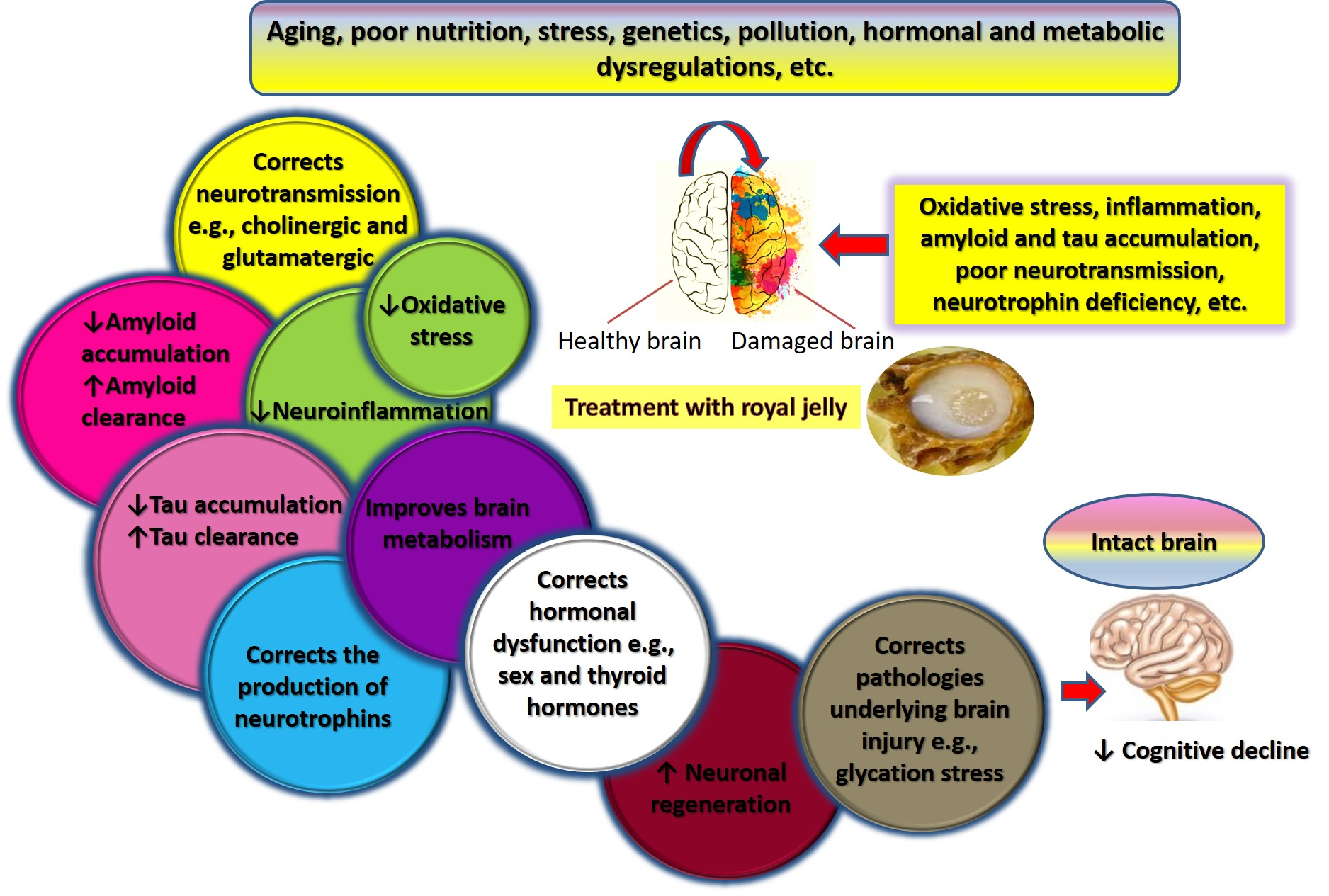
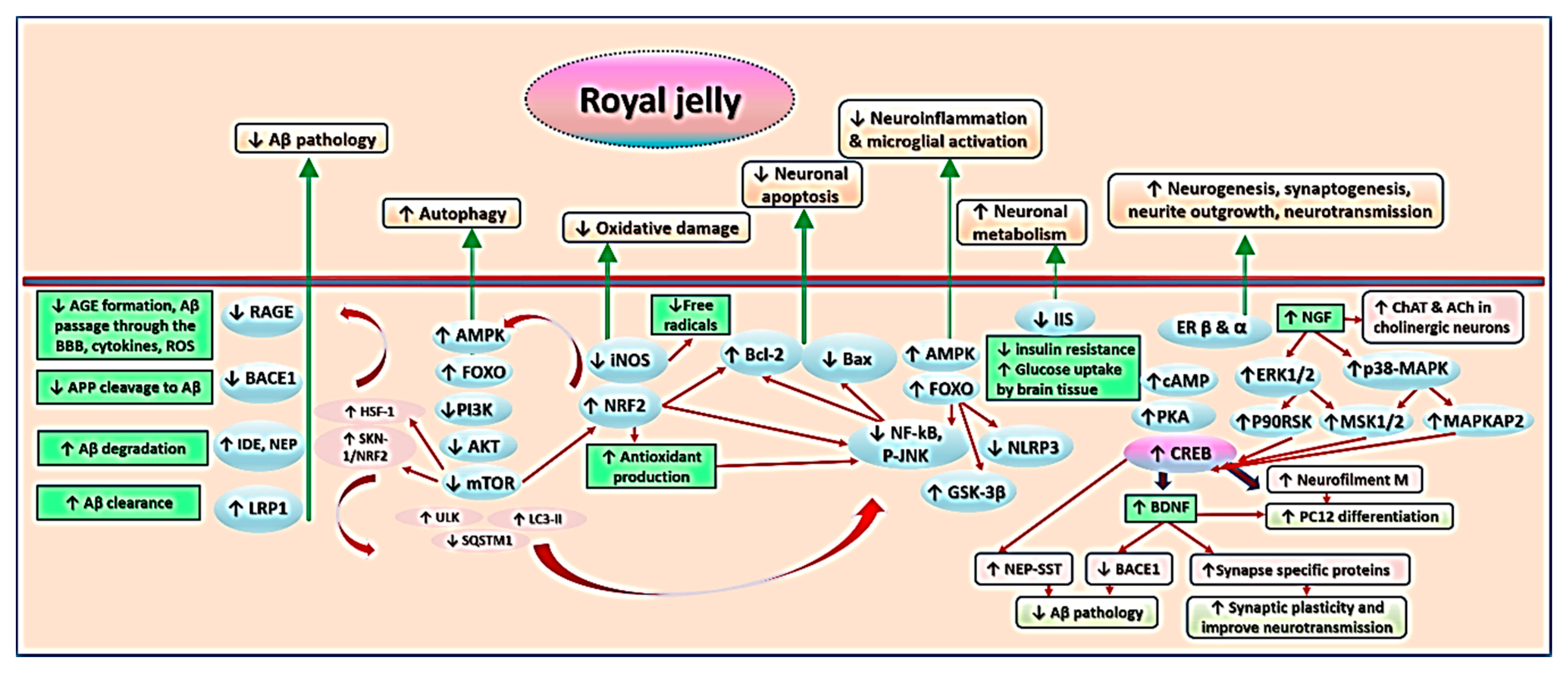
7. Mechanisms Underlying Effects of RJ on Cognition and AD-Related Pathology
7.1. Royal Jelly-Related Neuroprotection Is Mediated by Regulating the Production of Neurotrophins
7.2. Royal Jelly Regulates Neurotransmission in Models of Advanced Aging and Alzheimer’s Disease
7.3. Royal Jelly Regulates Energy Metabolism in the Brain
7.4. Royal Jelly Protects Against Neuroinflammation
7.5. Royal Jelly Protects Against Oxidative Stress
7.6. Royal Jelly Promotes Neuronal Regeneration and Attenuates Apoptosis
7.7. Royal Jelly Mitigates Amyloid-Related Neurotoxicity
7.8. Royal Jelly Alleviates Hormonal and Metabolic Abnormalities Underlying Cognitive Impairment
8. Discussion
8.1. Royal Jelly May Improve Health and Extend Lifespan in Cognitively Impaired Subjects
8.2. Tips for Identifying Treatment Targets
8.3. Issues of Concern Regarding the Use of Royal Jelly in Research Related to Cognitive Aging
8.4. Safety of Prolonged Consumption of Royal Jelly in Old Age
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5-HT | 5-hydroxytryptamine |
| 5-HTT | Serotonin transporter |
| 5HIAA | 5-hydroxyindoleacetic acid |
| 10-HDA | Trans-10-hydroxy-2-decenoic acid |
| Aβ | Beta-amyloid peptide |
| β-secretase/BACE1 | Beta-site APP cleaving enzyme 1 |
| ACh | Acetylcholine |
| AChE | Acetylcholinesterase |
| AChRs | Acetylcholine receptors |
| ADL | Activities of daily living |
| AGEs | Advanced glycation end products |
| AD | Alzheimer’s disease |
| AP-1 | Activator protein |
| AMPK | Adenosine monophosphate activated protein kinase |
| APP | Amyloid precursor protein |
| ATF4 | Activating transcription factor 4 |
| ATP | Adenosine triphosphate |
| APOE | Apolipoprotein E |
| BBB | Blood brain barrier |
| Caspase | Cysteine-aspartic acid protease |
| CaSR | Ca2+-sensing receptor |
| cAMP | cyclic adenosine mono phosphate |
| ChAT | Choline acetyltransferase |
| CHOP | CCAAT enhancer-binding (C/EBP) protein homologous protein |
| CNS | Central nervous system |
| COX | Cyclooxygenase |
| CREB | cAMP-response element (CRE)-binding protein |
| DAEE | Trans-2-decenoic acid ethyl ester |
| DG | Dentate gyrus |
| DOPAC | 3,4-dihydroxyphenylacetic acid |
| EAAT | Excitatory amino acid transporter |
| eIF2α | Eukaryotic initiation factor 2 alpha |
| eNOS | Endothelial NO synthase |
| ER | Endoplasmic reticulum |
| EREs | Estrogen Response Elements |
| ERK | Extracellular signal-regulated kinase |
| FOXO | Forkhead Box O transcription factor |
| fT4 | Free thyroxine |
| GABA | Gamma-aminobutyric acid |
| GAD | Glutamate decarboxylase |
| GABA-T | GABA-transaminase |
| GI | Gastrointestinal |
| GLT-1 | Glutamate transporter 1 |
| GLUT4 | Glucose transporter |
| GSH-Px | Glutathione peroxidase |
| GSK-3β | Glycogen synthase kinase-3β |
| H2O2 | Hydrogen peroxide |
| H2S | Hydrogen sulfide |
| HBMECs | Human brain microvascular endothelial cells |
| HIF-1α | Hypoxia inducible factor-1α |
| HO-1 | Heme oxygenase-1 |
| HPO-DAEE | 4-Hydroperoxy-2- decenoic acid ethyl ester |
| hSAA1 | Human Serum Amyloid A1 |
| HSF-1 | Heat shock transcription factor 1 |
| IDE | Insulin-degrading enzyme |
| IKK | Inhibitor of kappa B kinase |
| IIS | Insulin/insulin-like growth factor |
| IκB | Inhibitor of κB |
| iNOS | Inducible nitric oxide synthase |
| JNK | c-Jun NH2-terminal kinases |
| K63-Ub | K63-linked polyubiquitin |
| LC3-II | Microtubule-associated protein 1 light chain 3-II |
| LRP-1 | Low-density lipoprotein receptor-related protein 1 |
| M1-Ub | M1 position |
| MAP-2 | Microtubule-associated protein 2 |
| MAPK | Mitogen-activated protein kinase |
| MBP | Myelin basic protein |
| MCI | Mild cognitive impairment |
| MDA | Malonaldehyde |
| MOMP | Mitochondrial outer membrane permeabilization |
| mTOR | Mammalian target of rapamycin |
| NAD+ | Nucleotide nicotinamide adenine dinucleotide |
| NA | Nicotinic acid |
| NaMN | Nicotinic acid mononucleotide |
| NEP | Neprilysin |
| NF-κB | Nuclear factor-kappa B |
| NFTs | Neurofibrillary tangles |
| NO | Nitric oxide |
| NOS | Nitric oxide species |
| nNOS | Neuronal NO synthase |
| NMDAR | N-methyl-d-aspartate receptor |
| NLRP3 | Nucleotide-binding domain and leucine-rich repeat containing protein 3 |
| NRF2 | Nuclear factor-erythroid 2-related factor 2 |
| NS/NPCs | Neural stem/progenitor cells |
| p90RSK | pp90 ribosomal S6 kinase |
| PERK | Protein kinase RNA like ER kinase |
| PKA | cAMP-dependent protein kinase |
| PS1 | Presenilin 1 |
| RIPK1 | Receptor-interacting serine/threonine-protein kinase 1 |
| RAGE | Receptor for Advanced Glycation End products |
| RJ | Royal jelly |
| RJPs | Royal jelly peptides |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SPs | Senile plaques |
| SQSTM1/p62 | Sequestosome 1 |
| STAT1 | Signal transducer and activator of transcription 1 |
| T3 | Triiodothyronine |
| T4 | Thyroxine |
| TGF-β | Transforming growth factor-β |
| TAK | TGF-β-associated kinase |
| TAB | TAK binding protein |
| TLR | Toll-like receptor |
| TrkA | Tropomyosine-related kinase A |
| TNF-α | Tumor necrosis factor-α |
| TRAF | TNF receptor-associated factor 1 |
| UCBMCs | Umbilical cord blood mononuclear cells |
| ULK | Unc-51-like autophagy activating kinase |
| UPR | Unfolded protein response |
| VEGF | Vascular Endothelial Growth Factor |
| Zn | Zinc |
References
- Tobore, T.O. On the etiopathogenesis and pathophysiology of Alzheimer’s disease: A comprehensive theoretical review. J. Alzheimers Dis. 2019, 68, 417–437. [Google Scholar] [CrossRef]
- Miljkovic, N.; Lim, J.Y.; Miljkovic, I.; Frontera, W.R. Aging of skeletal muscle fibers. Ann. Rehabil. Med. 2015, 39, 155–162. [Google Scholar] [CrossRef]
- Niraula, A.; Sheridan, J.F.; Godbout, J.P. Microglia priming with aging and stress. Neuropsychopharmacology 2017, 42, 318–333. [Google Scholar] [CrossRef] [PubMed]
- De Nobrega, A.K.; Lyons, L.C. Aging and the clock: Perspective from flies to humans. Eur. J. Neurosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Budni, J.; Bellettini-Santos, T.; Mina, F.; Garcez, M.L.; Zugno, A.I. The involvement of BDNF, NGF and GDNF in aging and Alzheimer’s disease. Aging Dis. 2015, 6, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Salthouse, T.A. Trajectories of normal cognitive aging. Psychol. Aging 2019, 34, 17–24. [Google Scholar] [CrossRef]
- Gefen, T.; Kim, G.; Bolbolan, K.; Geoly, A.; Ohm, D.; Oboudiyat, C.; Shahidehpour, R.; Rademaker, A.; Weintraub, S.; Bigio, E.H.; et al. Activated microglia in cortical white matter across cognitive aging trajectories. Front. Aging Neurosci. 2019, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Raj, D.; Yin, Z.; Breur, M.; Doorduin, J.; Holtman, I.R.; Olah, M.; Mantingh-Otter, I.J.; Van Dam, D.; De Deyn, P.P.; den Dunnen, W.; et al. Increased white matter inflammation in aging-and Alzheimer’s disease brain. Front. Mol. Neurosci. 2017, 10, 206. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.H.; Low, K.A.; Chiarelli, A.M.; Fletcher, M.A.; Navarra, R.; Burzynska, A.Z.; Kong, T.S.; Zimmerman, B.; Maclin, E.L.; Sutton, B.P.; et al. Optical measures of cerebral arterial stiffness are associated with white matter signal abnormalities and cognitive performance in normal aging. Neurobiol. Aging 2019. [Google Scholar] [CrossRef] [PubMed]
- Casaletto, K.B.; Elahi, F.M.; Staffaroni, A.M.; Walters, S.; Contreras, W.R.; Wolf, A.; Dubal, D.; Miller, B.; Yaffe, K.; Kramer, J.H. Cognitive aging is not created equally: Differentiating unique cognitive phenotypes in “normal” adults. Neurobiol. Aging 2019, 77, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, R.; Ji, E.; Kim, S.Y. Phytochemicals that regulate neurodegenerative disease by targeting neurotrophins: A comprehensive review. Biomed. Res. Int. 2015, 2015, 814068. [Google Scholar] [CrossRef] [PubMed]
- Zamani, Z.; Reisi, P.; Alaei, H.; Asghar Pilehvarian, A. Effect of royal jelly on spatial learning and memory in rat model of streptozotocin-induced sporadic Alzheimer’s disease. Adv. Biomed. Res. 2012, 1, 1–10. [Google Scholar] [CrossRef]
- G.B.D. Neurological Disorders Collaborator Group. Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the global burden of disease study 2015. Lancet. Neurol. 2017, 16, 877–897. [Google Scholar] [CrossRef]
- Anand, A.; Patience, A.A.; Sharma, N.; Khurana, N. The present and future of pharmacotherapy of Alzheimer’s disease: A comprehensive review. Eur. J. Pharmacol. 2017, 815, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Di Battista, A.M.; Heinsinger, N.M.; Rebeck, G.W. Alzheimer’s disease genetic risk factor APOE-epsilon4 also affects normal brain function. Curr. Alzheimer Res. 2016, 13, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Vermunt, L.; Sikkes, S.A.M.; van den Hout, A.; Handels, R.; Bos, I.; van der Flier, W.M.; Kern, S.; Ousset, P.J.; Maruff, P.; Skoog, I.; et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimers Dement. 2019, 15, 888–898. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef]
- Ali, A.M.; Ahmed, A.H.; Smail, L. Psychological climacteric symptoms and attitudes toward menopause among Emirati women. Int. J. Env. Res. Public Health 2020, 17, 5028. [Google Scholar] [CrossRef]
- Wise, E.A.; Rosenberg, P.B.; Lyketsos, C.G.; Leoutsakos, J.M. Time course of neuropsychiatric symptoms and cognitive diagnosis in national Alzheimer’s coordinating centers volunteers. Alzheimers Dement. 2019, 11, 333–339. [Google Scholar] [CrossRef]
- Kumar, A.; Nisha, C.M.; Silakari, C.; Sharma, I.; Anusha, K.; Gupta, N.; Nair, P.; Tripathi, T.; Kumar, A. Current and novel therapeutic molecules and targets in Alzheimer’s disease. J. Formos. Med. Assoc. 2016, 115, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, J.R.M.; Marques, D.F.F.; Baptista, S.J.; Pereira, C.M.F.; Moreira, P.I.; Dinis, T.C.P.; Santos, A.E.; Salvador, J.A.R. Highlights in BACE1 inhibitors for Alzheimer’s disease treatment. Front. Chem. 2018, 6, 178. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Pan, Y.; Liu, Y.; Chen, Y.; Wu, Y.; Si, J.; Wang, K.; Hu, F. Royal jelly alleviates cognitive deficits and β-amyloid accumulation in APP/PS1 mouse model via activation of the cAMP/PKA/CREB/BDNF pathway and inhibition of neuronal apoptosis. Front. Aging Neurosci. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.J.; Fulton-Howard, B.; Goate, A. Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet Neurol. 2020. [Google Scholar] [CrossRef]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hagg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Finneran, D.J.; Nash, K.R. Neuroinflammation and fractalkine signaling in Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 30. [Google Scholar] [CrossRef]
- Ali, A.M.; Kunugi, H. Bee honey protects astrocytes against oxidative stress: A preliminary in vitro investigation. Neuropsychopharmacol. Rep. 2019, 39, 312–314. [Google Scholar] [CrossRef]
- Sharman, M.J.; Verdile, G.; Kirubakaran, S.; Münch, G. Inflammation in Alzheimer’s disease, and prevention with antioxidants and phenolic compounds—What are the most promising candidates? In Neurodegeneration and Alzheimer’s Disease; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2019; pp. 233–266. [Google Scholar] [CrossRef]
- Arranz, A.M.; De Strooper, B. The role of astroglia in Alzheimer’s disease: Pathophysiology and clinical implications. Lancet Neurol. 2019. [Google Scholar] [CrossRef]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J.; et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993, 43, 1467–1472. [Google Scholar] [CrossRef]
- Zoltowska, K.M.; Maesako, M.; Meier, J.; Berezovska, O. Novel interaction between Alzheimer’s disease-related protein presenilin 1 and glutamate transporter 1. Sci. Rep. 2018, 8, 8718. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Kabir, M.T.; Al Mamun, A.; Abdel-Daim, M.M.; Barreto, G.E.; Ashraf, G.M. APOE and Alzheimer’s disease: Evidence mounts that targeting APOE4 may combat Alzheimer’s pathogenesis. Mol. Neurobiol. 2019, 56, 2450–2465. [Google Scholar] [CrossRef] [PubMed]
- Sasayama, D.; Hattori, K.; Yokota, Y.; Matsumura, R.; Teraishi, T.; Yoshida, S.; Kunugi, H. Increased apolipoprotein E and decreased TNF-α in the cerebrospinal fluid of nondemented APOE-ε4 carriers. Neuropsychopharmacol. Rep. 2020, 40, 201–205. [Google Scholar] [CrossRef]
- Pretorius, L.; Kell, D.B.; Pretorius, E. Iron dysregulation and dormant microbes as causative agents for impaired blood rheology and pathological clotting in Alzheimer’s type dementia. Front. Neurosci. 2018, 12, 851. [Google Scholar] [CrossRef]
- Jang, S.E.; Lim, S.M.; Jeong, J.J.; Jang, H.M.; Lee, H.J.; Han, M.J.; Kim, D.H. Gastrointestinal inflammation by gut microbiota disturbance induces memory impairment in mice. Mucosal Immunol. 2018, 11, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jaber, V.; Lukiw, W.J. Secretory products of the human GI tract microbiome and their potential impact on Alzheimer’s disease (AD): Detection of lipopolysaccharide (LPS) in AD hippocampus. Front. Cell Infect. Microbiol. 2017, 7, 318. [Google Scholar] [CrossRef]
- Ali, A.M.; Kunugi, H. Apitherapy for Parkinson’s disease: A focus on the effects of propolis and royal jelly. Oxid. Med. Cell Longev. 2020. accepted. [Google Scholar]
- Gubiani, D.; Fabbretti, E.; Cestnik, B.; Lavrač, N.; Urbančič, T. Outlier based literature exploration for cross-domain linking of Alzheimer’s disease and gut microbiota. Expert Syst. Appl. 2017, 85, 386–396. [Google Scholar] [CrossRef]
- Peterson, C.P.; Sauer, C.; Chatfield, C.H. The extracellular polymeric substances of Legionella pneumophila biofilms contain amyloid structures. Curr. Microbiol. 2018, 75, 736–744. [Google Scholar] [CrossRef]
- Erskine, E.; MacPhee, C.E.; Stanley-Wall, N.R. Functional amyloid and other protein fibers in the biofilm matrix. J. Mol. Biol. 2018, 430, 3642–3656. [Google Scholar] [CrossRef]
- Santoro, N.; Epperson, C.N.; Mathews, S.B. Menopausal symptoms and their management. Endocrinol. Metab. Clin. N. Am. 2015, 44, 497–515. [Google Scholar] [CrossRef] [PubMed]
- Burgess, E.J.; Hoyt, L.R.; Randall, M.J.; Mank, M.M.; Bivona, J.J., 3rd; Eisenhauer, P.L.; Botten, J.W.; Ballif, B.A.; Lam, Y.W.; Wargo, M.J.; et al. Bacterial lipoproteins constitute the TLR2-stimulating activity of serum amyloid A. J. Immunol. 2018, 201, 2377–2384. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.M.; Hendawy, A.O. So, antidepressant drugs have serious adverse effects, but what are the alternatives? Nov. Appro. Drug Des. Dev. 2018, 4, 555636. [Google Scholar] [CrossRef]
- Zhan, X.; Stamova, B.; Jin, L.W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016, 87, 2324–2332. [Google Scholar] [CrossRef]
- Doncker, W.D.; Dantzer, R.; Ormstad, H.; Kuppuswamy, A. Mechanisms of poststroke fatigue. J. Neurol. Neurosurg. Psychiatry 2017, 1–7. [Google Scholar] [CrossRef]
- Converso, D.; Viotti, S.; Sottimano, I.; Loera, B.; Molinengo, G.; Guidetti, G. The relationship between menopausal symptoms and burnout. A cross-sectional study among nurses. BMC Women’s Health 2019, 19. [Google Scholar] [CrossRef]
- Reed-Geaghan, E.G.; Savage, J.C.; Hise, A.G.; Landreth, G.E. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J. Neurosci. 2009, 29, 11982–11992. [Google Scholar] [CrossRef]
- Frederiksen, H.R.; Haukedal, H.; Freude, K. Cell type specific expression of toll-like receptors in human brains and implications in Alzheimer’s disease. Biomed. Res. Int. 2019, 2019, 7420189. [Google Scholar] [CrossRef]
- Yun, J.; Yeo, I.J.; Hwang, C.J.; Choi, D.Y.; Im, H.S.; Kim, J.Y.; Choi, W.R.; Jung, M.H.; Han, S.B.; Hong, J.T. Estrogen deficiency exacerbates Abeta-induced memory impairment through enhancement of neuroinflammation, amyloidogenesis and NF-κB activation in ovariectomized mice. Brain Behav. Immun. 2018, 73, 282–293. [Google Scholar] [CrossRef]
- Shin, B.K.; Kang, S.; Kim, D.S.; Park, S. Intermittent fasting protects against the deterioration of cognitive function, energy metabolism and dyslipidemia in Alzheimer’s disease-induced estrogen deficient rats. Exp. Biol. Med. 2018, 243, 334–343. [Google Scholar] [CrossRef]
- Yang, J.T.; Wang, Z.J.; Cai, H.Y.; Yuan, L.; Hu, M.M.; Wu, M.N.; Qi, J.S. Sex differences in neuropathology and cognitive behavior in APP/PS1/tau triple-transgenic mouse model of Alzheimer’s disease. Neurosci. Bull. 2018, 34, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.A.; Jayakody, N.; Johnston, D.A.; Bechmann, I.; Carare, R.O. Failure of perivascular drainage of beta-amyloid in cerebral amyloid angiopathy. Brain Pathol. 2014, 24, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Merlini, M.; Wanner, D.; Nitsch, R.M. Tau pathology-dependent remodelling of cerebral arteries precedes Alzheimer’s disease-related microvascular cerebral amyloid angiopathy. Acta Neuropathol. 2016, 131, 737–752. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.M.; Verjee, M.A.; Bener, A.; Gerber, L.M. The hopeless age? A qualitative exploration of the experience of menopause in Arab women in Qatar. Climacteric 2013, 16, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Chornenkyy, Y.; Wang, W.X.; Wei, A.; Nelson, P.T. Alzheimer’s disease and type 2 diabetes mellitus are distinct diseases with potential overlapping metabolic dysfunction upstream of observed cognitive decline. Brain Pathol. 2019, 29, 3–17. [Google Scholar] [CrossRef]
- Bredesen, D.E. Metabolic profiling distinguishes three subtypes of Alzheimer’s disease. Aging 2015, 7, 595–600. [Google Scholar] [CrossRef]
- Holscher, C. Insulin signaling impairment in the brain as a risk factor in Alzheimer’s disease. Front. Aging Neurosci. 2019, 11, 88. [Google Scholar] [CrossRef]
- Kunugi, H.; Ali, A.M. Royal jelly and its components promote healthy aging and longevity: From animal models to humans. Int. J. Mol. Sci. 2019, 20, 4662. [Google Scholar] [CrossRef]
- MacLean, M.; Derk, J.; Ruiz, H.H.; Juranek, J.K.; Ramasamy, R.; Schmidt, A.M. The receptor for advanced glycation end products (RAGE) and DIAPH1: Implications for vascular and neuroinflammatory dysfunction in disorders of the central nervous system. Neurochem. Int. 2019, 126, 154–164. [Google Scholar] [CrossRef]
- Jeon, S.Y.; Byun, M.S.; Yi, D.; Lee, J.H.; Choe, Y.M.; Ko, K.; Sohn, B.K.; Choi, H.J.; Lee, J.Y.; Lee, D.Y. Influence of hypertension on brain amyloid deposition and Alzheimer’s disease signature neurodegeneration. Neurobiol. Aging 2019, 75, 62–70. [Google Scholar] [CrossRef]
- Ding, J.; Davis-Plourde, K.L.; Sedaghat, S.; Tully, P.J.; Wang, W.; Phillips, C.; Pase, M.P.; Himali, J.J.; Gwen Windham, B.; Griswold, M.; et al. Antihypertensive medications and risk for incident dementia and Alzheimer’s disease: A meta-analysis of individual participant data from prospective cohort studies. Lancet Neurol. 2019. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, L.M.; Chen, Z.H.; Zhao, Z.P.; Li, Y.C.; Deng, Q.; Huang, Z.J.; Zhang, X.; Li, C.; Zhou, M.G.; et al. Multilevel logistic regression analysis on hypercholesterolemia related risk factors among adults in China. Zhonghua Yu Fang Yi Xue Za Zhi 2018, 52, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Fabiani, C.; Antollini, S.S. Alzheimer’s disease as a membrane disorder: Spatial cross-talk among beta-amyloid peptides, nicotinic acetylcholine receptors and lipid rafts. Front. Cell Neurosci. 2019, 13, 309. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Mukherjee, S. Cholesterol: A key in the pathogenesis of Alzheimer’s disease. ChemMedChem 2018, 13, 1742–1743. [Google Scholar] [CrossRef]
- Pan, Y.; Xu, J.; Chen, C.; Chen, F.; Jin, P.; Zhu, K.; Hu, C.W.; You, M.; Chen, M.; Hu, F. Royal jelly reduces cholesterol levels, ameliorates Aβ pathology and enhances neuronal metabolic activities in a rabbit model of Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 50. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox. Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef]
- Seymen, C.M.; Cakir Gundogdu, A.; Bulut, D.I.; Yilmaz Demirtas, C.; Elmas, C. Royal jelly increased map-2 expression in hippocampal neurons of hypothyroid rats: An immunohistochemical study. Biotech. Histochem. 2019, 1–9. [Google Scholar] [CrossRef]
- Strodel, B.; Coskuner-Weber, O. Transition metal ion interactions with disordered amyloid-beta peptides in the pathogenesis of Alzheimer’s disease: Insights from computational chemistry studies. J. Chem. Inf. Model. 2019, 59, 1782–1805. [Google Scholar] [CrossRef]
- Lee, M.C.; Yu, W.C.; Shih, Y.H.; Chen, C.Y.; Guo, Z.H.; Huang, S.J.; Chan, J.C.C.; Chen, Y.R. Zinc ion rapidly induces toxic, off-pathway amyloid-beta oligomers distinct from amyloid-beta derived diffusible ligands in Alzheimer’s disease. Sci. Rep. 2018, 8, 4772. [Google Scholar] [CrossRef]
- Barnham, K.J.; Haeffner, F.; Ciccotosto, G.D.; Curtain, C.C.; Tew, D.; Mavros, C.; Beyreuther, K.; Carrington, D.; Masters, C.L.; Cherny, R.A.; et al. Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer’s disease beta-amyloid. FASEB J. 2004, 18, 1427–1429. [Google Scholar] [CrossRef] [PubMed]
- Milton, N.G. Role of hydrogen peroxide in the aetiology of Alzheimer’s disease: Implications for treatment. Drugs Aging 2004, 21, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, J.; Liang, S.H.; Xu, Y.; Moore, A.; Ran, C. Imaging hydrogen peroxide in Alzheimer’s disease via cascade signal amplification. Sci. Rep. 2016, 6, 35613. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Archana, A.; Jan, A.T.; Minakshi, R. Dissecting endoplasmic reticulum unfolded protein response (UPR(ER)) in managing clandestine modus operandi of Alzheimer’s disease. Front. Aging Neurosci 2018, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.C.; Jiang, T.; Yang, A.F.; Du, Y.J.; Wu, M.; Kong, L.H. Epigenetic modulation on Tau phosphorylation in Alzheimer’s disease. Neural Plast. 2019, 2019, 6856327. [Google Scholar] [CrossRef] [PubMed]
- Ovsepian, S.V.; O’Leary, V.B.; Zaborszky, L.; Ntziachristos, V.; Dolly, J.O. Amyloid plaques of Alzheimer’s disease as hotspots of glutamatergic activity. Neuroscientist 2019, 25, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Khanegheini, A.; Meftahi, G.H.; Zarrindast, M.R.; Afarinesh, M.R.; Sahraei, H.; Jahromi, G.P.; Shahyad, S. Involvement of CA1 GABAA Receptors in ketamine-induced impairment of spatial and non-spatial novelty detection in mice. Neurochem. J. 2019, 13, 81–89. [Google Scholar] [CrossRef]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef]
- Zhang, X.; Lao, K.; Qiu, Z.; Rahman, M.S.; Zhang, Y.; Gou, X. Potential astrocytic receptors and transporters in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 2019, 67, 1109–1122. [Google Scholar] [CrossRef]
- Hussain, A.; Tabrez, E.S.; Muhammad, A.; Peela, J.R. The mechanisms of dietary phytoestrogen as a potential treatment and prevention agent against Alzheimer’s disease. Crit. Rev. Eukaryot. Gene Expr. 2018, 28, 321–327. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Minami, A.; Matsushita, H.; Ieno, D.; Matsuda, Y.; Horii, Y.; Ishii, A.; Takahashi, T.; Kanazawa, H.; Wakatsuki, A.; Suzuki, T. Improvement of neurological disorders in postmenopausal model rats by administration of royal jelly. Climacteric 2016, 19, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Blake, M.G.; Boccia, M.M. Basal forebrain cholinergic system and memory. Curr. Top. Behav. Neurosci. 2018, 37, 253–273. [Google Scholar] [CrossRef] [PubMed]
- Polverino, A.; Grimaldi, M.; Sorrentino, P.; Jacini, F.; D’Ursi, A.M.; Sorrentino, G. Effects of acetylcholine on beta-amyloid-induced cPLA2 activation in the TB neuroectodermal cell line: Implications for the pathogenesis of Alzheimer’s disease. Cell Mol. Neurobiol. 2018, 38, 817–826. [Google Scholar] [CrossRef]
- Kruthika, K.R.; Rajeswari; Maheshappa, H.D. Multistage classifier-based approach for Alzheimer’s disease prediction and retrieval. IMU 2019, 14, 34–42. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement. 2019, 5, 272–293. [Google Scholar] [CrossRef]
- Wang, X.; Cao, M.; Dong, Y. Royal jelly promotes DAF-16-mediated proteostasis to tolerate β-amyloid toxicity in C. elegans model of Alzheimer’s disease. Oncotarget 2016, 7, 54183–54193. [Google Scholar] [CrossRef]
- Fujiwara, H.; Kogure, A.; Sakamoto, M.; Yamakuni, T.; Mimaki, Y.; Murata, K.; Hitomi, N.; Yamaguchi, K.; Ohizumi, Y. Honeybee royal jelly and nobiletin stimulate CRE-mediated transcription in ERK-independent and -dependent fashions, respectively, in PC12D cells. J. Pharmacol. Sci. 2011, 116, 384–387. [Google Scholar] [CrossRef]
- Ramadan, M.F.; Al-Ghamdi, A. Bioactive compounds and health-promoting properties of royal jelly: A review. J. Funct. Foods 2012, 4, 39–52. [Google Scholar] [CrossRef]
- Pan, Y.; Xu, J.; Jin, P.; Yang, Q.; Zhu, K.; You, M.; Chen, M.; Hu, F. Royal jelly ameliorates behavioral deficits, cholinergic system deficiency, and autonomic nervous dysfunction in ovariectomized cholesterol-fed rabbits. Molecules 2019, 24, 1149. [Google Scholar] [CrossRef]
- Qiu, W.; Chen, X.; Tian, Y.; Wu, D.; Du, M.; Wang, S. Protection against oxidative stress and anti-aging effect in drosophila of royal jelly-collagen peptide. Food Chem. Toxicol. 2020, 135, 110881. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, A.N.K.G.; Nair, A.J.; Sugunan, V.S. A review on royal jelly proteins and peptides. J. Funct. Foods 2018, 44, 255–264. [Google Scholar] [CrossRef]
- Alu’datt, M.H.; Rababah, T.; Sakandar, H.A.; Imran, M.; Mustafa, N.; Alhamad, M.N.; Mhaidat, N.; Kubow, S.; Tranchant, C.; Al-Tawaha, A.R.; et al. Fermented food-derived bioactive compounds with anticarcinogenic properties: Fermented royal jelly as a novel source for compounds with health benefits. In Anticancer Plants: Properties and Application; Akhtar, M., Swamy, M., Eds.; Springer: Singapore, 2018. [Google Scholar]
- Ali, A.M.; Kunugi, H. Apitherapy for age-related skeletal muscle dysfunction (sarcopenia): A review on the effects of royal jelly, propolis, and bee pollen. Foods 2020, in press. [Google Scholar]
- Fratini, F.; Cilia, G.; Mancini, S.; Felicioli, A. Royal jelly: An ancient remedy with remarkable antibacterial properties. Microbiol. Res. 2016, 192, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Hossen, M.S.; Nahar, T.; Gan, S.H.; Khalil, M.I. Bioinformatics and therapeutic insights on proteins in royal jelly. Curr. Proteom. 2019, 16, 84–101. [Google Scholar] [CrossRef]
- Kocot, J.; Kielczykowska, M.; Luchowska-Kocot, D.; Kurzepa, J.; Musik, I. Antioxidant potential of propolis, bee pollen, and royal jelly: Possible medical application. Oxid. Med. Cell. Longev. 2018, 2018, 7074209. [Google Scholar] [CrossRef]
- Xue, X.; Wu, L.; Wang, K. Chemical composition of royal jelly. In Bee Products—Chemical and Biological Properties; Alvarez-Suarez, J.M., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 181–190. [Google Scholar] [CrossRef]
- Virgiliou, C.; Kanelis, D.; Pina, A.; Gika, H.; Tananaki, C.; Zotou, A.; Theodoridis, G. A targeted approach for studying the effect of sugar bee feeding on the metabolic profile of Royal Jelly. J. Chromatogr. A 2019, 460783. [Google Scholar] [CrossRef]
- Gong, Z.; Tan, K.; Nieh, J.C. First demonstration of olfactory learning and long-term memory in honey bee queens. J. Exp. Biol. 2018, 221. [Google Scholar] [CrossRef]
- Wang, C.; Gong, Z.W.; Li, X.Y.; He, S.Y.; Tan, K. Effect of royal jelly to worker bees olfactory learning. Apic. China 2015, 66, 27–28. [Google Scholar]
- Shi, J.-l.; Liao, C.-h.; Wang, Z.-l.; Wu, X.-b. Effect of royal jelly on longevity and memory-related traits of Apis mellifera workers. J. Asia-Pac. Entomol. 2018, 21, 1430–1433. [Google Scholar] [CrossRef]
- Pyrzanowska, J.; Piechal, A.; Blecharz-Klin, K.; Joniec-Maciejak, I.; Graikou, K.; Chinou, I.; Widy-Tyszkiewicz, E. Long-term administration of Greek royal jelly improves spatial memory and influences the concentration of brain neurotransmitters in naturally aged Wistar male rats. J. Ethnopharmacol. 2014, 155, 343–351. [Google Scholar] [CrossRef]
- Zamani, Z.; Reisi, P.; Alaei, H.; Pilehvarian, A. Effect of royal jelly on improving passive avoidance learning and spatial learning and memory in rats. J. Shahid Sadoughi Univ. Med. Sci. 2012, 20, 211–219. [Google Scholar]
- Chen, D.; Liu, F.; Wan, J.-B.; Lai, C.-Q.; Shen, L.-r. Effect of major royal jelly proteins on spatial memory in aged rats: Metabolomics analysis in urine. J. Agric. Food Chem. 2017, 65, 3151–3159. [Google Scholar] [CrossRef] [PubMed]
- Pyrzanowska, J.; Piechal, A.; Blecharz-Klin, K.; Graikou, K.; Widy-Tyszkiewicz, E.; Chinou, I. Chemical analysis of Greek royal jelly—Its influence of the long-term administration on spatial memory in aged rats. Planta Med. 2012, 78. [Google Scholar] [CrossRef]
- Guardia de Souza, E.S.T.; do Val de Paulo, M.E.F.; da Silva, J.R.M.; da Silva Alves, A.; Britto, L.R.G.; Xavier, G.F.; Lopes Sandoval, M.R. Oral treatment with royal jelly improves memory and presents neuroprotective effects on icv-STZ rat model of sporadic Alzheimer’s disease. Heliyon 2020, 6, e03281. [Google Scholar] [CrossRef]
- Hattori, N.; Ohta, S.; Sakamoto, T.; Mishima, S.; Furukawa, S. Royal jelly facilitates restoration of the cognitive ability in trimethyltin-intoxicated mice. Evid. Based Complement. Altern. Med. 2011, 2011, 165968. [Google Scholar] [CrossRef]
- You, M.M.; Chen, Y.F.; Pan, Y.M.; Liu, Y.C.; Tu, J.; Wang, K.; Hu, F.L. Royal jelly attenuates LPS-induced inflammation in BV-2 microglial cells through modulating NF-kappaB and p38/JNK signaling pathways. Mediat. Inflamm. 2018, 2018, 7834381. [Google Scholar] [CrossRef]
- You, M.; Miao, Z.; Pan, Y.; Hu, F. Trans-10-hydroxy-2-decenoic acid alleviates LPS-induced blood-brain barrier dysfunction by activating the AMPK/PI3K/AKT pathway. Eur. J. Pharm. 2019, 865, 172736. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, Y.; Sun, P.; Fan, Z.; Zhang, W.; Feng, C. Royal jelly peptides: Potential inhibitors of β-secretase in N2a/APP695swe cells. Sci. Rep. 2019, 9, 168. [Google Scholar] [CrossRef]
- Kawahata, I.; Xu, H.; Takahashi, M.; Murata, K.; Han, W.; Yamaguchi, Y.; Fujii, A.; Yamaguchi, K.; Yamakuni, T. Royal jelly coordinately enhances hippocampal neuronal expression of somatostatin and neprilysin genes conferring neuronal protection against toxic soluble amyloid-β oligomers implicated in Alzheimer’s disease pathogenesis. J. Funct. Foods 2018, 51, 28–38. [Google Scholar] [CrossRef]
- You, M.; Miao, Z.; Tian, J.; Hu, F. Trans-10-hydroxy-2-decenoic acid protects against LPS-induced neuroinflammation through FOXO1-mediated activation of autophagy. Eur. J. Nutr. 2019. [Google Scholar] [CrossRef]
- Cheraghi, O.; Abdollahpourasl, M.; Rezabakhsh, A.; Rahbarghazi, R. Distinct effects of royal jelly on human endothelial cells under high glucose condition. Iran. J. Pharm. Res. 2018, 17, 1361–1370. [Google Scholar] [PubMed]
- Almeer, R.S.; Kassab, R.B.; AlBasher, G.I.; Alarifi, S.; Alkahtani, S.; Ali, D.; Abdel Moneim, A.E. Royal jelly mitigates cadmium-induced neuronal damage in mouse cortex. Mol. Biol. Rep. 2019, 46, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.A.-R.; Galal, A.A.A.; Elewa, Y.H.A. Comparative protective effects of royal jelly and cod liver oil against neurotoxic impact of tartrazine on male rat pups brain. Acta Histochem. 2015, 117, 649–658. [Google Scholar] [CrossRef]
- Pyrzanowska, J.; Piechal, A.; Blecharz-Klin, K.; Joniec-Maciejak, I.; Graikou, K.; Widy-Tyszkiewicz, E.; Chinou, I. Administration of Greek royal jelly produces fast response in neurotransmission of aged wistar male rats. J. Pre-Clin. Clin. Res. 2015, 9, 151–157. [Google Scholar] [CrossRef][Green Version]
- Pyrzanowska, J.; Wawer, A.; Joniec-Maciejak, I.; Piechal, A.; Blecharz-Klin, K.; Graikou, K.; Chinou, I.; Widy-Tyszkiewicz, E. Long-term administration of Greek royal jelly decreases GABA concentration in the striatum and hypothalamus of naturally aged wistar male rats. Neurosci. Lett. 2018, 675, 17–22. [Google Scholar] [CrossRef]
- Ji, W.Z.; Zhang, C.P.; Wei, W.T.; Hu, F.L. The in vivo antiaging effect of enzymatic hydrolysate from royal jelly in d-galactose induced aging mouse. J. Chin. Inst. Food Sci. Technol. 2016, 16, 18–25. [Google Scholar]
- Peng, Y.-r.; Zhong, F.-x.; Yang, B.; Rong-jing, G.; Su, F. Effect of royal jelly on learning and memory performance of aged rats. Food Sci. 2011, 15, 269–272. [Google Scholar]
- Yakoot, M.; Salem, A.; Helmy, S. Effect of Memo®, a natural formula combination, on mini-mental state examination scores in patients with mild cognitive impairment. Clin. Interv. Aging 2013, 8, 975–981. [Google Scholar] [CrossRef]
- Georgiev, D.B.; Metka, M.; Huber, J.C.; Goudev, A.R.; Manassiev, N. Effects of an herbal medication containing bee products on menopausal symptoms and cardiovascular risk markers: Results of a pilot open-uncontrolled trial. MedGenMed 2004, 6, 46. [Google Scholar]
- Yakoot, M.; Salem, A.; Omar, A.M. Effectiveness of a herbal formula in women with menopausal syndrome. Complement. Med. Res. 2011, 18, 264–268. [Google Scholar] [CrossRef]
- Szanto, E.; Gruber, D.; Sator, M.; Knogler, W.; Huber, J.C. Placebo-controlled study of melbrosia in treatment of climacteric symptoms. Wien. Med. Wochenschr. 1994, 144, 130–133. [Google Scholar] [PubMed]
- Cattaneo, A.; Capsoni, S. Painless Nerve Growth Factor: A TrkA biased agonist mediating a broad neuroprotection via its actions on microglia cells. Pharmacol. Res. 2019, 139, 17–25. [Google Scholar] [CrossRef]
- Hattori, N.; Nomoto, H.; Fukumitsu, H.; Mishima, S.; Furukawa, S. AMP N1-oxide, a unique compound of royal jelly, induces neurite outgrowth from PC12 vells via signaling by protein kinase a independent of that by mitogen-activated protein kinase. Evid.-Based Complement. Altern. Med. 2010, 7, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Nomoto, H.; Mishima, S.; Inagaki, S.; Goto, M.; Sako, M.; Furukawa, S. Identification of AMP N1-oxide in royal jelly as a component neurotrophic toward cultured rat pheochromocytoma PC12 cells. Biosci. Biotechnol. Biochem. 2006, 70, 897–906. [Google Scholar] [CrossRef]
- Cragnolini, A.B.; Montenegro, G.; Friedman, W.J.; Mascó, D.H. Brain-region specific responses of astrocytes to an in vitro injury and neurotrophins. Mol. Cell. Neurosci. 2018, 88, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Rafii, M.S.; Tuszynski, M.H.; Thomas, R.G.; Barba, D.; Brewer, J.B.; Rissman, R.A.; Siffert, J.; Aisen, P.S. Adeno-associated viral vector (serotype 2)-nerve growth factor for patients with Alzheimer disease: A randomized clinical trial. JAMA Neurol. 2018, 75, 834–841. [Google Scholar] [CrossRef] [PubMed]
- De Bellis, A.; de Bellis, M.; Aloe, L. Long-term non-invasive treatment via intranasal administration of nerve growth factor protects the human brain in frontotemporal dementia associated with corticobasal syndrome: A pilot study. J. Alzheimers Dis. Rep. 2018, 2, 67–77. [Google Scholar] [CrossRef]
- Xu, D.; Wu, D.; Qin, M.; Nih, L.R.; Liu, C.; Cao, Z.; Ren, J.; Chen, X.; He, Z.; Yu, W.; et al. Efficient delivery of nerve growth factors to the central nervous system for neural regeneration. Adv. Mater. 2019, 31, e1900727. [Google Scholar] [CrossRef]
- Hattori, N.; Nomoto, H.; Fukumitsu, H.; Mishima, S.; Furukawa, S. Royal jelly-induced neurite outgrowth from rat pheochromocytoma PC12 cells requires integrin signal independent of activation of extracellular signalregulated kinases. Biomed. Res. 2007, 28, 139–146. [Google Scholar] [CrossRef]
- Skaper, S.D. The neurotrophin family of neurotrophic factors: An overview. Methods Mol. Biol. 2012, 846, 1–12. [Google Scholar] [CrossRef]
- Koga, Y.; Tsurumaki, H.; Aoki-Saito, H.; Sato, M.; Yatomi, M.; Takehara, K.; Hisada, T. Roles of cyclic AMP response element binding activation in the ERK1/2 and p38 MAPK signalling pathway in central nervous system, cardiovascular system, osteoclast differentiation and mucin and cytokine production. Int. J. Mol. Sci. 2019, 20, 1346. [Google Scholar] [CrossRef]
- Ohashi, E.; Kohno, K.; Arai, N.; Harashima, A.; Ariyasu, T.; Ushio, S. Adenosine N1-oxide exerts anti-inflammatory effects through the PI3K/Akt/GSK-3beta signaling pathway and promotes osteogenic and adipocyte differentiation. Biol. Pharm. Bull. 2019, 42, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, N.; Fujii, T.; Combarros, O.; Kamboh, M.I.; Tsai, S.J.; Matsushita, S.; Nacmias, B.; Comings, D.E.; Arboleda, H.; Ingelsson, M.; et al. Sexually dimorphic effect of the Val66Met polymorphism of BDNF on susceptibility to Alzheimer’s disease: New data and meta-analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153, 235–242. [Google Scholar] [CrossRef]
- Kunugi, H.; Ueki, A.; Otsuka, M.; Isse, K.; Hirasawa, H.; Kato, N.; Nabika, T.; Kobayashi, S.; Nanko, S. A novel polymorphism of the brain-derived neurotrophic factor (BDNF) gene associated with late-onset Alzheimer’s disease. Mol. Psychiatry 2001, 6, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Nomoto, H.; Fukumitsu, H.; Mishima, S.; Furukawa, S. Royal jelly and its unique fatty acid, 10-hydroxy-trans-2-decenoic acid, promote neurogenesis by neural stem/progenitor cells in vitro. Biomed. Res. 2007, 28, 261–266. [Google Scholar] [CrossRef]
- Ali, A.M.; Hendawy, A.O. Royal jelly acid, 10-hydroxy-trans-2-decenoic acid, for psychiatric and neurological disorders: How helpful could it be?! Edelweiss J. Food Sci. Technol. 2019, 1, 1–4. [Google Scholar]
- Makino, A.; Iinuma, M.; Fukumitsu, H.; Soumiya, H.; Furukawa, Y.; Furukawa, S. 2-Decenoic acid ethyl ester possesses neurotrophin-like activities to facilitate intracellular signals and increase synapse-specific proteins in neurons cultured from embryonic rat brain. Biomed. Res. 2010, 31, 379–386. [Google Scholar] [CrossRef]
- Petukhova, E.O.; Mukhamedshina, Y.O.; Salafutdinov, I.I.; Garanina, E.E.; Kaligin, M.S.; Leushina, A.V.; Rizvanov, A.A.; Reis, H.J.; Palotas, A.; Zefirov, A.L.; et al. Effects of transplanted umbilical cord blood mononuclear cells overexpressing GDNF on spatial memory and hippocampal synaptic proteins in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2019, 69, 443–453. [Google Scholar] [CrossRef]
- Hashimoto, M.; Kanda, M.; Ikeno, K.; Hayashi, Y.; Nakamura, T.; Ogawa, Y.; Fukumitsu, H.; Nomoto, H.; Furukawa, S. Oral administration of royal jelly facilitates mRNA expression of glial cell line-derived neurotrophic factor and neurofilament H in the hippocampus of the adult mouse brain. Biosci. Biotechnol. Biochem. 2005, 69, 800–805. [Google Scholar] [CrossRef]
- Ebrahimi-Ghiri, M.; Rostampour, M.; Jamshidi-Mehr, M.; Nasehi, M.; Zarrindast, M.R. Role of CA1 GABAA and GABAB receptors on learning deficit induced by D-AP5 in passive avoidance step-through task. Brain Res. 2018, 1678, 164–173. [Google Scholar] [CrossRef]
- Noda, M.; Ifuku, M.; Hossain, M.S.; Katafuchi, T. Glial activation and expression of the serotonin transporter in chronic fatigue syndrome. Front. Psychiatry 2018, 9, 589. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, T.A.; Nguyen, J.C.; Polglaze, K.E.; Bertrand, P.P. Influence of tryptophan and serotonin on mood and cognition with a possible role of the gut-brain axis. Nutrients 2016, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Beggs, K.T.; Mercer, A.R. Dopamine receptor activation by honey bee queen pheromone. Curr. Biol. 2009, 19, 1206–1209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Xue, X.F.; Zhou, J.H.; Chen, F.; Wu, L.M.; Li, Y.; Zhao, J. Determination of tryptophan in bee pollen and royal jelly by high-performance liquid chromatography with fluorescence detection. Biomed. Chromatogr. 2009, 23, 994–998. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K. Nutrition and dopamine: An intake of tyrosine in royal jelly can affect the brain levels of dopamine in male honeybees (Apis mellifera L.). J. Insect Physiol. 2016, 87, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Nagao, T.; Sasaki, K. Consumption of tyrosine in royal jelly increases brain levels of dopamine and tyramine and promotes transition from normal to reproductive workers in queenless honey bee colonies. Gen. Comp. Endocrinol. 2015, 211, 1–8. [Google Scholar] [CrossRef]
- Inoue, Y.; Hara, H.; Mitsugi, Y.; Yamaguchi, E.; Kamiya, T.; Itoh, A.; Adachi, T. 4-hydroperoxy-2-decenoic acid ethyl ester protects against 6-hydroxydopamine-induced cell death via activation of Nrf2-ARE and eIF2α-ATF4 pathways. Neurochem. Int. 2018, 112, 288–296. [Google Scholar] [CrossRef]
- Gil-Iturbe, E.; Solas, M.; Cuadrado-Tejedo, M.; García-Osta, A.; Escoté, X.; Ramírez, M.J.; Lostao, M.P. GLUT12 expression in brain of mouse models of Alzheimer’s disease. Mol. Neurobiol. 2020, 57, 798–805. [Google Scholar] [CrossRef]
- Smiljanic, K.; Todorovic, S.; Mladenovic Djordjevic, A.; Vanmierlo, T.; Lütjohann, D.; Ivkovic, S.; Kanazir, S. Limited daily feeding and intermittent feeding have different effects on regional brain energy homeostasis during aging. Biogerontology 2018, 19, 121–132. [Google Scholar] [CrossRef]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef]
- Castellano, C.A.; Nugent, S.; Paquet, N.; Tremblay, S.; Bocti, C.; Lacombe, G.; Imbeault, H.; Turcotte, É.; Fulop, T.; Cunnane, S.C. Lower brain 18F-fluorodeoxyglucose uptake but normal 11C-acetoacetate metabolism in mild Alzheimer’s disease dementia. J. Alzheimers Dis. 2015, 43, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Scavello, F.; Zeni, F.; Tedesco, C.C.; Mensa, E.; Veglia, F.; Procopio, A.D.; Bonfigli, A.R.; Olivieri, F.; Raucci, A. Modulation of soluble receptor for advanced glycation end-products (RAGE) isoforms and their ligands in healthy aging. Aging 2019, 11, 1648–1663. [Google Scholar] [CrossRef] [PubMed]
- Placais, P.Y.; de Tredern, E.; Scheunemann, L.; Trannoy, S.; Goguel, V.; Han, K.A.; Isabel, G.; Preat, T. Upregulated energy metabolism in the drosophila mushroom body is the trigger for long-term memory. Nat. Commun. 2017, 8, 15510. [Google Scholar] [CrossRef]
- Tubbs, E.; Chanon, S.; Robert, M.; Bendridi, N.; Bidaux, G.; Chauvin, M.-A.; Ji-Cao, J.; Durand, C.; Gauvrit-Ramette, D.; Vidal, H.; et al. Disruption of mitochondria-associated endoplasmic reticulum membrane (MAM) integrity contributes to muscle insulin resistance in mice and humans. Diabetes 2018, 67, 636–650. [Google Scholar] [CrossRef]
- Omer, K.; Gelkopf, M.J.; Newton, G. Effectiveness of royal jelly supplementation in glycemic regulation: A systematic review. World J. Diabetes 2019, 10, 96–113. [Google Scholar] [CrossRef]
- Ota, M.; Matsuo, J.; Ishida, I.; Hattori, K.; Teraishi, T.; Tonouchi, H.; Ashida, K.; Takahashi, T.; Kunugi, H. Effect of a ketogenic meal on cognitive function in elderly adults: Potential for cognitive enhancement. Psychopharmacology 2016, 233, 3797–3802. [Google Scholar] [CrossRef] [PubMed]
- Ota, M.; Matsuo, J.; Ishida, I.; Takano, H.; Yokoi, Y.; Hori, H.; Yoshida, S.; Ashida, K.; Nakamura, K.; Takahashi, T.; et al. Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neurosci. Lett. 2019, 690, 232–236. [Google Scholar] [CrossRef]
- Honda, Y.; Araki, Y.; Hata, T.; Ichihara, K.; Ito, M.; Tanaka, M.; Honda, S. 10-Hydroxy-2-decenoic acid, the major lipid component of royal jelly, extends the lifespan of caenorhabditis elegans through dietary restriction and target of rapamycin signaling. J. Aging Res. 2015, 2015, 425261. [Google Scholar] [CrossRef]
- Hindupur, S.K.; González, A.; Hall, M.N. The opposing actions of target of rapamycin and AMP-activated protein kinase in cell growth control. Cold Spring Harb. Perspect. Biol. 2015, 7, a019141. [Google Scholar] [CrossRef]
- Streit, W.J.; Khoshbouei, H.; Bechmann, I. Dystrophic microglia in late-onset Alzheimer’s disease. Glia 2020. [Google Scholar] [CrossRef]
- Frank, P.G.; Lisanti, M.P. ICAM-1: Role in inflammation and in the regulation of vascular permeability. Am. J. Physiol Heart Circ. Physiol 2008, 295, H926–H927. [Google Scholar] [CrossRef] [PubMed]
- Ramos, T.N.; Bullard, D.C.; Barnum, S.R. ICAM-1: Isoforms and phenotypes. J. Immunol. 2014, 192, 4469–4474. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Nagai, H.; Miyamoto, M.; Kimura, M.; Yonekura, M. Identification of a royal jelly glycoprotein that carries unique complex-type N-glycans harboring the T-antigen (Galβ1-3GalNAc) unit. Biosci. Biotechnol. Biochem. 2010, 74, 2148–2150. [Google Scholar] [CrossRef] [PubMed]
- Edilova, M.I.; Abdul-Sater, A.A.; Watts, T.H. TRAF1 signaling in human health and disease. Front. Immunol. 2018, 9, 2969. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, M.M.; Lei, X.Y.; Yu, X.J. SFTS phlebovirus promotes LC3-II accumulation and nonstructural protein of SFTS phlebovirus co-localizes with autophagy proteins. Sci. Rep. 2018, 8, 5287. [Google Scholar] [CrossRef] [PubMed]
- Francois, A.; Rioux Bilan, A.; Quellard, N.; Fernandez, B.; Janet, T.; Chassaing, D.; Paccalin, M.; Terro, F.; Page, G. Longitudinal follow-up of autophagy and inflammation in brain of APPswePS1dE9 transgenic mice. J. Neuroinflamm. 2014, 11, 139. [Google Scholar] [CrossRef]
- Feng, Y.; Kang, H.H.; Wong, P.M.; Gao, M.; Wang, P.; Jiang, X. Unc-51-like kinase (ULK) complex-independent autophagy induced by hypoxia. Protein Cell 2019, 10, 376–381. [Google Scholar] [CrossRef]
- Sebastiani, A.; Golz, C.; Sebastiani, P.G.; Bobkiewicz, W.; Behl, C.; Mittmann, T.; Thal, S.C.; Engelhard, K. Sequestosome 1 deficiency delays, but does not prevent brain damage formation following acute brain injury in adult mice. Front. Neurosci. 2017, 11, 678. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. AMPK activation inhibits the functions of myeloid-derived suppressor cells (MDSC): Impact on cancer and aging. J. Mol. Med. 2019, 97, 1049–1064. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Yoshimori, T. Autophagy and longevity. Mol. Cells 2018, 41, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Partridge, L. Promoting health and longevity through diet: From model organisms to humans. Cell 2015, 161, 106–118. [Google Scholar] [CrossRef]
- Martins, R.; Lithgow, G.J.; Link, W. Long live FOXO: Unraveling the role of FOXO proteins in aging and longevity. Aging Cell 2016, 15, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T. mTOR as regulator of lifespan, aging, and cellular senescence: A mini-review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Kroncke, K.D.; Fehsel, K.; Kolb-Bachofen, V. Inducible nitric oxide synthase in human diseases. Clin. Exp. Immunol. 1998, 113, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Adamiak, M.; Abdelbaset-Ismail, A.; Moore, J.B.T.; Zhao, J.; Abdel-Latif, A.; Wysoczynski, M.; Ratajczak, M.Z. Inducible nitric oxide synthase (iNOS) is a novel negative regulator of hematopoietic stem/progenitor cell trafficking. Stem Cell Rev. Rep. 2017, 13, 92–103. [Google Scholar] [CrossRef]
- Liu, Y.; Clement, J.; Grant, R.; Sachdev, P.; Braidy, N. Quantitation of NAD+: Why do we need to measure it? Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2527–2532. [Google Scholar] [CrossRef]
- Johnson, S.; Wozniak, D.F.; Imai, S. CA1 Nampt knockdown recapitulates hippocampal cognitive phenotypes in old mice which nicotinamide mononucleotide improves. NPJ Aging Mech. Dis. 2018, 4, 10. [Google Scholar] [CrossRef]
- Dou, X.; Shen, C.; Wang, Z.; Li, S.; Zhang, X.; Song, Z. Protection of nicotinic acid against oxidative stress-induced cell death in hepatocytes contributes to its beneficial effect on alcohol-induced liver injury in mice. J. Nutr. Biochem. 2013, 24, 1520–1528. [Google Scholar] [CrossRef]
- Hattori, N.; Nomoto, H.; Fukumitsu, H.; Mishima, S.; Furukawa, S. AMP N1-oxide potentiates astrogenesis by cultured neural stem/progenitor cells through STAT3 activation. Biomed. Res. 2007, 28, 295–299. [Google Scholar] [CrossRef] [PubMed]
- D’Orsi, B.; Mateyka, J.; Prehn, J.H.M. Control of mitochondrial physiology and cell death by the Bcl-2 family proteins Bax and Bok. Neurochem. Int. 2017, 109, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Henderson, L.E.; Abdelmegeed, M.A.; Yoo, S.H.; Rhee, S.G.; Zhu, X.; Smith, M.A.; Nguyen, R.Q.; Perry, G.; Song, B.J. Enhanced phosphorylation of Bax and its translocation into mitochondria in the brains of individuals affiliated with Alzheimer’s disease. Open Neurol. J. 2017, 11, 48–58. [Google Scholar] [CrossRef]
- McCarty, M.F.; O’Keefe, J.H.; DiNicolantonio, J.J. A diet rich in taurine, cysteine, folate, B12 and betaine may lessen risk for Alzheimer’s disease by boosting brain synthesis of hydrogen sulfide. Med. Hypotheses 2019, 132, 109356. [Google Scholar] [CrossRef]
- El Idrissi, A. Taurine regulation of neuroendocrine function. Adv. Exp. Med. Biol. 2019, 1155, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Gorgani, S.; Jahanshahi, M.; Elyasi, L. Taurine prevents passive avoidance memory impairment, accumulation of amyloid-β plaques, and neuronal loss in the hippocampus of scopolamine-treated rats. Neurophysiology 2019, 51, 171–179. [Google Scholar] [CrossRef]
- Perez, F.P.; Moinuddin, S.S.; ul ain Shamim, Q.; Joseph, D.J.; Morisaki, J.; Zhou, X. Longevity pathways: HSF1 and FoxO pathways, a new therapeutic target to prevent age-related diseases. Curr. Aging Sci. 2012, 5, 87–95. [Google Scholar] [CrossRef]
- Uchoa, M.F.; Moser, V.A.; Pike, C.J. Interactions between inflammation, sex steroids, and Alzheimer’s disease risk factors. Front. Neuroendocr. 2016, 43, 60–82. [Google Scholar] [CrossRef]
- Lv, W.; Du, N.; Liu, Y.; Fan, X.; Wang, Y.; Jia, X.; Hou, X.; Wang, B. Low testosterone level and risk of Alzheimer’s disease in the elderly men: A systematic review and meta-analysis. Mol. Neurobiol. 2016, 53, 2679–2684. [Google Scholar] [CrossRef]
- Morita, H.; Ikeda, T.; Kajita, K.; Fujioka, K.; Mori, I.; Okada, H.; Uno, Y.; Ishizuka, T. Effect of royal jelly ingestion for six months on healthy volunteers. Nutr. J. 2012, 11, 77. [Google Scholar] [CrossRef]
- Zárate, S.; Stevnsner, T.; Gredilla, R. Role of estrogen and other sex hormones in brain aging. Neuroprotection and DNA repair. Front. Aging Neurosci. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Ghanbari, E.; Khazaei, M.R.; Khazaei, M.; Nejati, V. Royal jelly promotes ovarian follicles growth and increases steroid hormones in immature rats. Int. J. Fertil. Steril. 2018, 11, 263–269. [Google Scholar] [CrossRef]
- Su, L.; Chen, S.; Zheng, C.; Wei, H.; Song, X. Meta-analysis of gene expression and identification of biological regulatory mechanisms in Alzheimer’s disease. Front. Neurosci. 2019, 13, 633. [Google Scholar] [CrossRef]
- Weiser, M.J.; Grimshaw, V.; Wynalda, K.M.; Mohajeri, M.H.; Butt, C.M. Long-term administration of queen bee acid (QBA) to rodents reduces anxiety-like behavior, promotes neuronal health and improves body composition. Nutrients 2017, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Bjornstrom, L.; Sjoberg, M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Gan, E.H.; Pearce, S.H. Clinical review: The thyroid in mind: Cognitive function and low thyrotropin in older people. J. Clin. Endocrinol. Metab. 2012, 97, 3438–3449. [Google Scholar] [CrossRef]
- Nomoto, S.; Kinno, R.; Ochiai, H.; Kubota, S.; Mori, Y.; Futamura, A.; Sugimoto, A.; Kuroda, T.; Yano, S.; Murakami, H.; et al. The relationship between thyroid function and cerebral blood flow in mild cognitive impairment and Alzheimer’s disease. PLoS ONE 2019, 14, e0214676. [Google Scholar] [CrossRef]
- Rivera, O.; McHan, L.; Konadu, B.; Patel, S.; Sint Jago, S.; Talbert, M.E. A high-fat diet impacts memory and gene expression of the head in mated female Drosophila melanogaster. J. Comp. Physiol. B 2019, 189, 179–198. [Google Scholar] [CrossRef]
- Pan, Y.; Rong, Y.; You, M.; Ma, Q.; Chen, M.; Hu, F. Royal jelly causes hypotension and vasodilation induced by increasing nitric oxide production. Food Sci. Nutr. 2019, 7, 1361–1370. [Google Scholar] [CrossRef]
- Takaki-Doi, S.; Hashimoto, K.; Yamamura, M.; Kamei, C. Antihypertensive activities of royal jelly protein hydrolysate and its fractions in spontaneously hypertensive rats. Acta Med. Okayama 2009, 63, 57–64. [Google Scholar] [CrossRef]
- Surgucheva, I.; Shestopalov, V.I.; Surguchov, A. Effect of γ-synuclein silencing on apoptotic pathways in retinal ganglion cells. J. Biol. Chem. 2008, 283, 36377–36385. [Google Scholar] [CrossRef] [PubMed]
- Higham, J.P.; Malik, B.R.; Buhl, E.; Dawson, J.M.; Ogier, A.S.; Lunnon, K.; Hodge, J.J.L. Alzheimer’s disease associated genes ankyrin and tau cause shortened lifespan and memory loss in drosophila. Front. Cell Neurosci. 2019, 13, 260. [Google Scholar] [CrossRef]
- Kaku, M.; Rocabado, J.M.R.; Kitami, M.; Ida, T.; Uoshima, K. Royal jelly affects collagen crosslinking in bone of ovariectomized rats. J. Funct. Foods 2014, 7, 398–406. [Google Scholar] [CrossRef]
- Hidaka, S.; Okamoto, Y.; Uchiyama, S.; Nakatsuma, A.; Hashimoto, K.; Ohnishi, S.T.; Yamaguchi, M. Royal jelly prevents osteoporosis in rats: Beneficial effects in ovariectomy model and in bone tissue culture model. Evid. Based Complement. Altern. Med. 2006, 3, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Nitta, Y.; Fukumitsu, H.; Soumiya, H.; Ikeno, K.; Nakamura, T.; Furukawa, S. Antidepressant-like activity of 10-hydroxy-trans-2-decenoic acid, a unique unsaturated fatty acid of royal jelly, in stress-inducible depression-like mouse model. Evid.-Based Complementary Altern. Med. 2012, 2012, 139140. [Google Scholar] [CrossRef]
- Santos, L.E.; Beckman, D.; Ferreira, S.T. Microglial dysfunction connects depression and Alzheimer’s disease. Brain Behav. Immun. 2016, 55, 151–165. [Google Scholar] [CrossRef]
- Alford, S.; Patel, D.; Perakakis, N.; Mantzoros, C.S. Obesity as a risk factor for Alzheimer’s disease: Weighing the evidence. Obes. Rev. 2018, 19, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Elsworthy, R.J.; Aldred, S. Depression in Alzheimer’s disease: An alternative role for selective serotonin reuptake inhibitors? J. Alzheimers Dis. 2019, 69, 651–661. [Google Scholar] [CrossRef]
- Ramos, O.Y.; Basualdo, M.; Libonatti, C.; Vega, M.F. Current status and application of lactic acid bacteria in animal production systems with a focus on bacteria from honey bee colonies. J. Appl. Microbiol. 2019. [Google Scholar] [CrossRef]
- Guldas, M. Effects of Royal Jelly and Bee Pollen on the Growth of Selected Probiotic Bacteria (Bf. animalis spp. Lactis, L. acidophilus and L. casei). J. Apic. Sci. 2016, 60, 129–140. [Google Scholar] [CrossRef]
- Kazemi, V.; Mojtahedzadeh, M.; Siadat, S.D.; Hadjiakhondi, A.; Ameri, A.; Ahmadi Badi, S.; Manayi, A.; Bagheri, M. Evaluation the effect of royal jelly on the growth of two members of gut microbiota; Bacteroides fragillis and Bacteroides thetaiotaomicron. J. Contemp. Med. Sci. 2019, 5, 20–23. [Google Scholar]
- Kaynar, L.; Cetin, A.; Hacioglu, S.K.; Eser, B.; Kocyigit, I.; Canoz, O.; Tasdemir, A.; Karadag, C.; Kurnaz, F.; Saraymen, R.; et al. Efficacy of royal jelly on methotrexate-induced systemic oxidative stress and damage to small intestine in rats. Afr. J. Tradit. Complement. Altern. Med. 2012, 9, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Guendouz, M.; Haddi, A.; Grar, H.; Kheroua, O.; Saidi, D.; Kaddouri, H. Preventive effects of royal jelly against anaphylactic response in a murine model of cow’s milk allergy. Pharm. Biol. 2017, 55, 2145–2152. [Google Scholar] [CrossRef] [PubMed]
- Karaca, T.; Bayiroglu, F.; Yoruk, M.; Kaya, M.S.; Uslu, S.; Comba, B.; Mis, L. Effect of royal jelly on experimental colitis Induced by acetic acid and alteration of mast cell distribution in the colon of rats. Eur. J. Histochem. 2010, 54, e35. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ho, L.; Faith, J.; Ono, K.; Janle, E.M.; Lachcik, P.J.; Cooper, B.R.; Jannasch, A.H.; D’Arcy, B.R.; Williams, B.A.; et al. Role of intestinal microbiota in the generation of polyphenol-derived phenolic acid mediated attenuation of Alzheimer’s disease beta-amyloid oligomerization. Mol. Nutr. Food Res. 2015, 59, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Kanelis, D.; Tananaki, C.; Liolios, V.; Dimou, M.; Goras, G.; Rodopoulou, M.A.; Karazafiris, E.; Thrasyvoulou, A. A suggestion for royal jelly specifications. Arh. Hig. Rada. Toksikol. 2015, 66, 275–284. [Google Scholar] [CrossRef]
- Munstedt, K.; Bargello, M.; Hauenschild, A. Royal jelly reduces the serum glucose levels in healthy subjects. J. Med. Food 2009, 12, 1170–1172. [Google Scholar] [CrossRef]
- Böhme, F.; Bischoff, G.; Zebitz, C.P.W.; Rosenkranz, P.; Wallner, K. From field to food—Will pesticide-contaminated pollen diet lead to a contamination of royal jelly? Apidologie 2018, 49, 112–119. [Google Scholar] [CrossRef]
- Shaha, A.; Mizuguchi, H.; Kitamura, Y.; Fujino, H.; Yabumoto, M.; Takeda, N.; Fukui, H. Effect of royal jelly and brazilian green propolis on the signaling for histamine H1 receptor and interleukin-9 gene expressions responsible for the pathogenesis of the allergic rhinitis. Biol. Pharm. Bull. 2018, 41, 1440–1447. [Google Scholar] [CrossRef]
- Gu, H.; Song, I.-B.; Han, H.-J.; Lee, N.-Y.; Cha, J.-Y.; Son, Y.-K.; Kwon, J. Antioxidant activity of royal jelly hydrolysates obtained by enzymatic treatment. Korean J. Food Sci. Animal Resour. 2018, 38, 135–142. [Google Scholar] [CrossRef]
- Honda, Y.; Fujita, Y.; Maruyama, H.; Araki, Y.; Ichihara, K.; Sato, A.; Kojima, T.; Tanaka, M.; Nozawa, Y.; Ito, M.; et al. Lifespan-extending effects of royal jelly and its related substances on the nematode Caenorhabditis elegans. PLoS ONE 2011, 6, e23527. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Percentage | Examples | References |
|---|---|---|---|
| Moisture | 50–70% | Mainly water. | [96,98,99] |
| Carbohydrates | 7.5–16% | Sugars such as fructose, glucose, maltose, melibiose, and ribose. | [98,99] |
| Proteins | 9–18% | MRJPs (80% of protein content), minor proteins (e.g., aspimin, royalisin and jelleines), peptides (in the form of dipeptides or tripeptides, e.g., alanine-leucine, leucine- aspartic acid-arginine), and free amino acids (e.g., threonine, valine, glycine, isoleucine, leucine, proline, serine, methionine, and tryptophan). | [63,99,102] |
| Lipidsa | 3–6% | 10-HDA, sebacic acid, phenols (4–10%), waxes (5–6%), steroids (3–4%), and phospholipids (0.4–0.8%). | [63,98,99,101,102] |
| Vitamins | ? | B5 (52.8 mg/100 g), B6 (42.42 mg/100 g), niacin (42.42 mg/100 g), and traces of B1, B2, B8, B9, B12, ascorbic acid (vitamin C), vitamin E and A. | [63,102] |
| Minerals | ? | Potassium, sodium, magnesium, calcium, phosphor, sulfur, cupper, iron, zinc, selenium, barium, cobalt, manganese, etc. | [42,63,98] |
| Bioactive compounds | ? | ACh and nucleotides both as free bases (e.g., adenosine, guanosine, iridin, and cytidine) and as phosphates (e.g., adenosine 5′-monophosphate, adenosine 5′-diphosphate, and adenosine 5′-triphosphate). | [63,102] |
| Others | ? | Volatile organic compounds (e.g., esters, aldehydes, ketones, and alcohols), and minor heterocyclic compounds. | [63,98,102] |
| Animal/Cell Line Model | RJ Treatment | Summary of Effects and Mechanism | Reference |
|---|---|---|---|
| Hippocampal SST and NEP positive neurons | DRJ (100 mg/mL) | ↑SST and NEP gene expression and CREB-binding to CRE at the promoter region of SST. | [116] |
| N2a/APP695 cells | RJPs (1–9 μg/mL) | ↓Aβ1-40, Aβ1-42, and BACE1. | [115] |
| LPS-stimulated BV-2 microglia | RJ (0.3–3 mg/mL) | ↓IL-6, IL-1β, TNF-α, iNOS, and COX-2. | [113] |
| Apis mellifera workers as a model of learning | RJ (10% and 20%) in 50% sucrose solution | ↑Olfactory learning, memory, and expression of memory-related genes (GluRA and Nmdar1). | [105,106] |
| Naturally aged Drosophila and Drosophila treated with H2O2 and paraquat | eRJ (1–5 mg/mL) plus CP at a ratio of 2:3 | ↑T-SOD, GSH-Px, CAT, average life span, food consumption, weight gain, and exercise capacity. ↓MDA and protein carbonyl. | [96] |
| Aβ toxicity in CL2006 worm model of AD | RJ (2 mg/mL) and eRJ 1 mg/mL)/day/10 days at 20 °C | ↓Aβ species, Aβ-induced body paralysis, and IIS signaling. ↑Soluble proteins. | [92] |
| LPS-stimulated C57BL/6J mice and microglial BV-2 cells | Oral 10-HAD (100 mg/kg/day for 1 month) | ↓TNF-α, Tnfrsf8, Traf1, IL-1β, NF-κB and NLRP3 inflammasome-IL-1β signaling, and SQSTM1. ↑FOXO1-mediated autophagy, ULK, and LC3-II. | [117] |
| LPS-stimulated C57BL/6 mice and HBMECs | Oral 10-HAD (100 mg/kg/day for 1 month) | ↓ROS, CCL-2, CCL-3, ICAM-1, VCAM-1, MMP-2, and MMP-9, BBB permeability, and tight junction proteins degradation. ↑Expression of tight junction proteins, and AMPK/PI3K/AKT signaling. | [114] |
| OVX cholesterol-fed rabbit model of AD | Oral RJ (400 mg/kg/day/12 weeks) | ↓Behavioral cognitive deficits, body weight, blood lipid, BBB permeability, brain levels of MDA, Aβ, AchE, BACE1, and RAGE. ↑ChAT, SOD, LRP-1, heart rate variability, and Baroreflex sensitivity. | [95] |
| OVX rat model of aging | Oral eRJ (250 mg/mL tap water: 10 mL/kg/day/82 days) | ↓Cognitive and depressive-like behavioral deficits. ↑Brain weight and myelin galactolipids. | [87] |
| A rat model of AD induced by streptozotocin (icv) | Oral RJ (200 mg/kg/day/14 days) | ↓O2- (in the DG and hilus regions) and neurodegeneration (in the DG). ↑Working memory and neurogenesis in the DG. | [111] |
| Hypothyroidism rat model of cognitive impairment | Intragastric RJ (100 mg/kg/day/20 days) | ↓Brain vascular dilation, edema, and degeneration. ↑MAP-2 and fT4. | [73] |
| APP/PS1 transgenic mice model of AD | Intragastric RJ (300 mg/kg/day/3 months) | ↑Spatial learning and memory. ↓MDA, p-JNK and bax/bcl-2 ratio, caspase-3, BACE1, Aβ40 and Aβ42, and the total area and number of senile plaques in the cortex and hippocampus. ↑cAMP, p-PKA, p-CREB, BDNF, IDE, and LRP-1. | [28] |
| A rabbit model of AD induced by cholesterol diet and copper sulfate | Oral RJ (400 mg/kg/day/12 weeks) | ↓TC, LDL-C, MDA, ROS, RNS, Cho/Cr, mI/Cr, caspase-3, BACE1, Aβ1-40, Aβ1-42, Aβ plaque, and neuronal loss. ↑SOD, LRP-1, IDE, NAA/Cr, and glutamate/Cr. | [70] |
| A mouse model of streptozotocin-induced cognitive impairment | Dietary RJ (3% w/w/day/10 days) | ↓Streptozotocin-induced defects in learning and memory. | [16] |
| A mouse model of trimethyltin-induced hippocampal DG damage | Dietary RJ (1% or 5% w/w/day/6 days) | ↓Cognitive impairment and neuronal cell loss. ↑Number of hippocampal DG granule cells. | [112] |
| A mouse model of cadmium-induced cortical damage | Intragastric RJ (85 mg/kg/day/7 days) | ↑NRF2, GSH-Px, GSH-R, SOD, CAT, Bcl-2, norepinephrine, dopamine, and serotonin. ↓iNOS, ROS, NOS, TNF-α, IL-1β, Bax, caspase-3, and cadmium level in cortical neurons. | [119] |
| A mouse model of tartrazine-induced cortical damage | Ora RJ (300 mg/kg/day/30 days) | ↑ CAT, SOD, GSH, and brain levels of GABA, dopamine, and 5HT. ↓MDA, cortical pyknotic nuclei, and ssDNA positive apoptotic cells. | [120] |
| Naturally aged rats | Oral RJ (50 and 100 mg/kg/day/8 weeks) | ↑Memory and learning. | [107] |
| Naturally aged rats | Dietary RJ (3% w/w/day/10 days) | ↑Memory and learning. | [108] |
| Naturally aged rats | Intragastric MRJPs (125 mg/kg/day/14 weeks) | ↑Learning, memory, gluconeogenesis, brain glucose supply and ATP level, nicotinate and nicotinamide metabolism—NaMN, and cysteine-taurine metabolism. ↓ROS, AKT, and GABA. | [109] |
| Naturally aged rats | Oral/intragastric RJ (50 and 100 mg/kg/day/8 weeks) | ↑Learning, spatial memory, and motor performance. ↓5-HT, dopamine, MHPG and its turnover. ↑5HIAA, DOPAC and their turnover in the prefrontal cortex. ↑DOPAC and ↓5HIAA in the striatum. | [107,110] |
| Naturally aged rats | Subcutaneous RJ (100 and 500 mg/kg/day/6 days) | ↑ Serotonin activity in the hippocampus and prefrontal cortex. | [121] |
| Naturally aged rats | Intragastric RJ (50 and 100 mg/kg/day/8 weeks) | ↓GABA in the striatum and hypothalamus. | [122] |
| d-galactose induced mouse model of aging | Intragastric RJ and eRJ (0.7 and 1.4 mg/kg/day/90 days) | ↓ROS and body weight loss. ↑Memory, learning, muscular performance, and levels of internal antioxidant enzymes. | [123] |
| d-galactose induced mouse model of aging | Intragastric RJ (0.7 and 1.4 mg/kg/day/90 days) | ↑Spatial learning, memory, brain levels of norepinephrine, dopamine, and SOD. ↓MDA. | [124] |
| Elderly with MCI | RJ plus herbal extracts | ↑Scores of the Mini-Mental State Scale. | [125] |
| Postmenopausal women with menopausal complaints | RJ plus flower pollen | ↑Problem-solving ability, HDL, and TG. ↓Depression, menopausal symptoms, TC, and LDL. | [126] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.M.; Kunugi, H. Royal Jelly as an Intelligent Anti-Aging Agent—A Focus on Cognitive Aging and Alzheimer’s Disease: A Review. Antioxidants 2020, 9, 937. https://doi.org/10.3390/antiox9100937
Ali AM, Kunugi H. Royal Jelly as an Intelligent Anti-Aging Agent—A Focus on Cognitive Aging and Alzheimer’s Disease: A Review. Antioxidants. 2020; 9(10):937. https://doi.org/10.3390/antiox9100937
Chicago/Turabian StyleAli, Amira Mohammed, and Hiroshi Kunugi. 2020. "Royal Jelly as an Intelligent Anti-Aging Agent—A Focus on Cognitive Aging and Alzheimer’s Disease: A Review" Antioxidants 9, no. 10: 937. https://doi.org/10.3390/antiox9100937
APA StyleAli, A. M., & Kunugi, H. (2020). Royal Jelly as an Intelligent Anti-Aging Agent—A Focus on Cognitive Aging and Alzheimer’s Disease: A Review. Antioxidants, 9(10), 937. https://doi.org/10.3390/antiox9100937