Mechanisms of Mitochondrial ROS Production in Assisted Reproduction: The Known, the Unknown, and the Intriguing

Abstract
1. Mitochondrial ROS, Oxidative Stress, and Assisted Reproduction: An Introduction
2. Mechanisms of Mitochondrial ROS Production: The Known
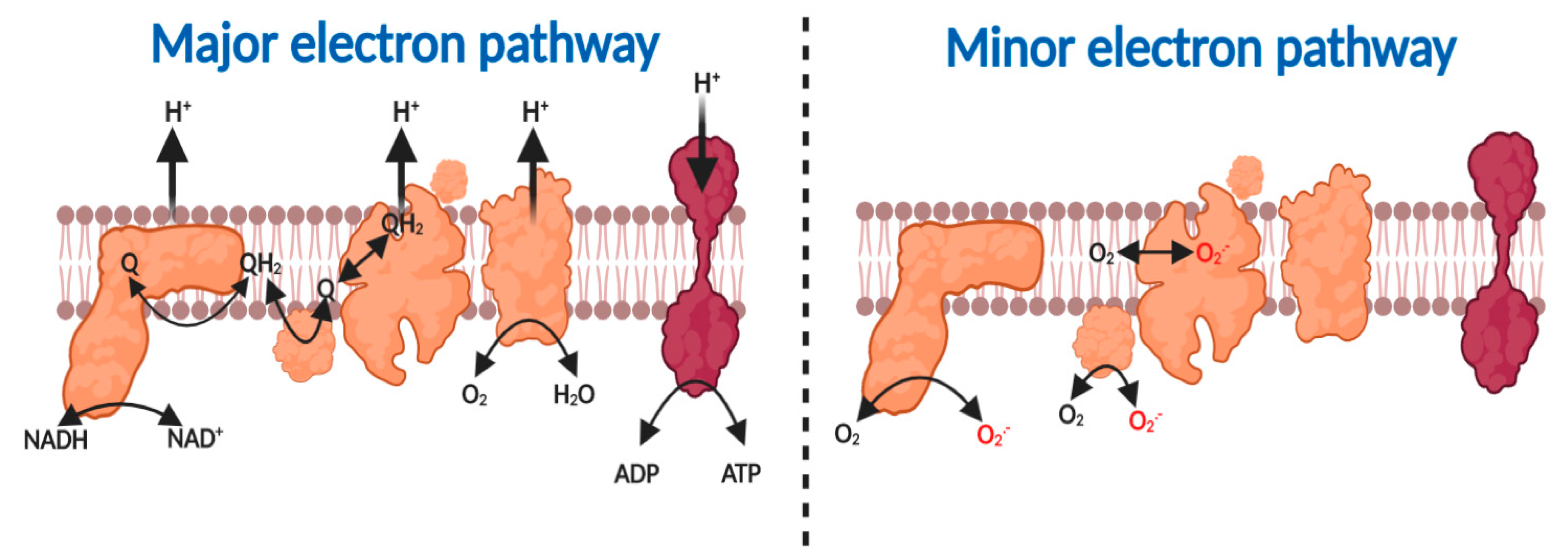
2.1. The Major and Minor Mitochondiral Electron Pathways
2.2. How Mitochondria Produce Superoxide
2.3. Complex I: Forward Mode
2.4. Complex I: Reverse Electron Transfer
2.5. Complex II
2.6. Complex III
2.7. Key Superoxide Production Modes
3. How Mitochondria Produce Superoxide in ART: The Unknown
3.1. General Considerations
3.2. Site and Mode-defined Mechanisms of Superoxide Production in ART
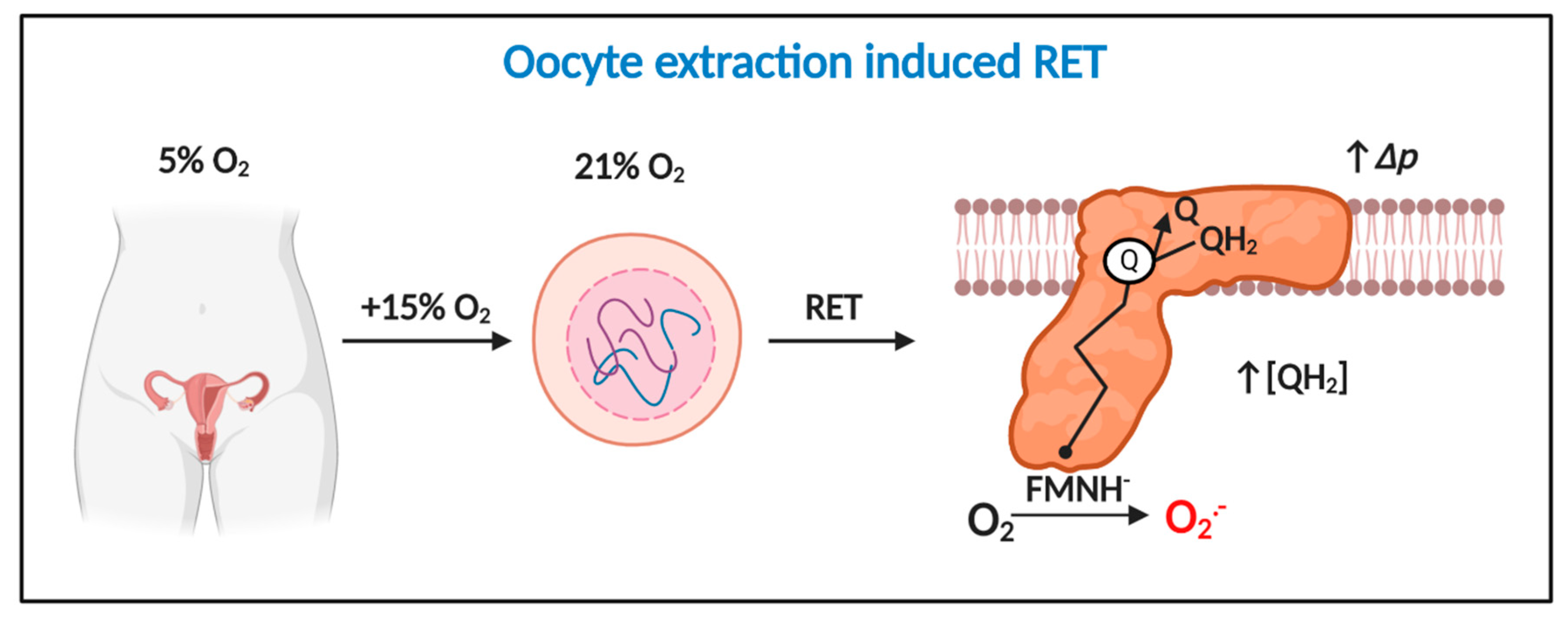
3.3. How Oocyte/Zygote Mitochondria Produce Superoxide is Unknown
4. A Framework for Interpreting the Role of Mitochondrial ROS in ART: The Intriguing
4.1. Interpreting Mitochondrial Superoxide Production
4.2. A Two-Step Bifurcation Model to Interpret ART-induced Oxidative Stress
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jensen, P. Antimycin-insensitive oxidation of succinate and reduced nicotinamide-adenine dinucleotide in electron-transport particles I. pH dependency and hydrogen peroxide formation. Biochim. Biophys. Acta 1966, 122, 157–166. [Google Scholar] [CrossRef]
- Steptoe, P.; Edwards, R. Birth after the reimplantation of a human embryo. Lancet 1978, 312, 366. [Google Scholar] [CrossRef]
- O’Flaherty, C. Reactive oxygen species and male fertility. Antioxidants 2020, 9, 287. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Stress: Introductory Remarks. In Oxidative Stress; Academic Press: Cambridge, MA, USA, 1985; pp. 1–5. [Google Scholar]
- Sawyer, D.T.; Valentine, J.S. How super is superoxide? Acc. Chem. Res. 1981, 14, 393–400. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Ferguson, S.J. Bioenergetics 4, 4th ed.; Academic Press: London, UK, 2013. [Google Scholar]
- Mailloux, R.J. An update on mitochondrial reactive oxygen species production. Antioxidants 2020, 9, 472. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology & Medicine, 5th ed.; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Johnson, M.H.; Nasresfahani, M.H. Radical solutions and cultural problems: Could free oxygen radicals be responsible for the impaired development of preimplantation mammalian embryos invitro? BioEssays 1994, 16, 31–38. [Google Scholar] [CrossRef]
- Guerin, P.; El Mouatassim, S.; Menezo, Y. Oxidative stress and protection against reactive oxygen species in the pre-implantation embryo and its surroundings. Hum. Reprod. Update 2001, 7, 175–189. [Google Scholar] [CrossRef]
- Agarwal, A.; Gupta, S.; Sekhon, L.; Shah, R. Redox considerations in female reproductive function and assisted reproduction: From molecular mechanisms to health implications. Antioxid. Redox Sign. 2008, 10, 1375–1404. [Google Scholar] [CrossRef]
- Lord, T.; Nixon, B.; Jones, K.T.; Aitken, R.J. Melatonin prevents postovulatory oocyte aging in the mouse and extends the window for optimal fertilization in vitro. Biol. Reprod. 2013, 88, 67. [Google Scholar] [CrossRef]
- Lord, T.; Aitken, R.J. Oxidative stress and ageing of the post-ovulatory oocyte. Reproduction 2013, 146, R217–R227. [Google Scholar] [CrossRef]
- Zhang, M.; Shiyang, X.; Zhang, Y.; Miao, Y.; Chen, Y.; Cui, Z.; Xiong, B. Coenzyme Q10 ameliorates the quality of postovulatory aged oocytes by suppressing DNA damage and apoptosis. Free. Radic. Biol. Med. 2019, 143, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-G.; Park, H.-J.; Kim, J.-W.; Jung, J.-M.; Kim, M.-J.; Jegal, H.-G.; Kim, I.-S.; Kang, M.-J.; Wee, G.; Yang, H.-Y.; et al. Mito-TEMPO improves development competence by reducing superoxide in preimplantation porcine embryos. Sci. Rep. 2018, 8, 10130. [Google Scholar] [CrossRef] [PubMed]
- Bontekoe, S.; Mantikou, E.; Van Wely, M.; Seshadri, S.; Repping, S.; Mastenbroek, S. Low oxygen concentrations for embryo culture in assisted reproductive technologies. Cochrane Database Syst. Rev. 2012, 19, 209. [Google Scholar] [CrossRef] [PubMed]
- Rampon, C.; Volovitch, M.; Joliot, A.; Vriz, S. Hydrogen Peroxide and Redox Regulation of Developments. Antioxidants 2018, 7, 159. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Aitken, R.J.; Drevet, J.R. The importance of oxidative stress in determining the functionality of mammalian spermatozoa: A two-edged sword. Antioxidants 2020, 9, 111. [Google Scholar] [CrossRef]
- Lane, N. Oxygen: The Molecule that Made the World; Oxford University Press: Oxford, UK, 2002. [Google Scholar]
- Fridovich, I. Oxygen: How do we stand it? Med. Princ. Pract. 2012, 22, 131–137. [Google Scholar] [CrossRef]
- Abele, D. Toxic oxygen: The radical life-giver. Nature 2002, 420, 2002. [Google Scholar] [CrossRef]
- Ogilby, P.R. Singlet oxygen: There is indeed something new under the sun. Chem. Soc. Rev. 2010, 39, 3181–3209. [Google Scholar] [CrossRef]
- Kaila, V.R.I.; Verkhovsky, M.I.; Wikström, M. Proton-coupled electron transfer in cytochrome oxidase. Chem. Rev. 2010, 110, 7062–7081. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, B.C.; Chang, C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 2011, 7, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. The biology of oxygen radicals. Science 1978, 201, 875–880. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Ann. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Stress: Concept and some Practical Aspects. Antioxidants 2020, 9, 852. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Stress: Eustress and Distress; Academic Press: London, UK, 2020. [Google Scholar]
- Jones, D.P.; Sies, H. The Redox Code. Antioxid. Redox Sign. 2015, 23, 734–746. [Google Scholar] [CrossRef]
- Cobley, J.N. How Exercise Induces Oxidative Eustress. In Oxidative Stress; Sies, H., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 447–462. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Murphy, M.P.; Holmgren, A.; Larsson, N.-G.; Halliwell, B.; Chang, C.J.; Kalyanaraman, B.; Rhee, S.G.; Thornalley, P.J.; Partridge, L.; Gems, D.; et al. Unraveling the biological roles of reactive oxygen species. Cell Metab. 2011, 13, 361–366. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Gluud, C.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst. Rev. 2012, 3, CD007176. [Google Scholar] [CrossRef]
- Moser, C.C.; Farid, T.A.; Chobot, S.E.; Dutton, P.L. Electron tunneling chains of mitochondria. Biochim. Biophys. Acta 2006, 1757, 1096–1109. [Google Scholar] [CrossRef]
- Moser, C.C.; Page, C.C.; Dutton, P.L. Darwin at the molecular scale: Selection and variance in electron tunnelling proteins including cytochrome c oxidase. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Wikström, M.; Sharma, V.; Kaila, V.R.I.; Hosler, J.P.; Hummer, G. New perspectives on proton pumping in cellular respiration. Chem. Rev. 2015, 115, 2196–2221. [Google Scholar] [CrossRef]
- De Vault, D.; Chance, B. Studies of photosynthesis using a pulsed laser: I. temperature dependence of cytochrome oxidation rate in chromatium. evidence for tunneling. Biophys. J. 1966, 6, 825–847. [Google Scholar] [CrossRef]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol. 2015, 4, 381–398. [Google Scholar] [CrossRef]
- Saraste, M. Oxidative Phosphorylation at the fin de siècle. Science 1999, 283, 1488–1493. [Google Scholar] [CrossRef]
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 192, 452–454. [Google Scholar] [CrossRef]
- Walker, J.E. The ATP synthase: The understood, the uncertain and the unknown. Biochem. Soc. Trans. 2013, 41, 1–16. [Google Scholar] [CrossRef]
- Yoshida, M.; Muneyuki, E.; Hisabori, T. ATP synthase—A marvellous rotary engine of the cell. Nat. Rev. Mol. Cell Biol. 2001, 2, 669–677. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2008, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Moser, C.C.; Keske, J.M.; Warncke, K.; Farid, R.S.; Dutton, P.L. Nature of biological electron transfer. Nature 1992, 355, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A.; Sethu, R.; Rohaun, S.K. Evolutionary adaptations that enable enzymes to tolerate oxidative stress. Free. Radic. Biol. Med. 2019, 140, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef]
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 1973, 134, 707–716. [Google Scholar] [CrossRef]
- Barja, G. Updating the mitochondrial free radical theory of aging: An integrated view, key aspects, and confounding concepts. Antioxid. Redox Sign. 2013, 19, 1420–1445. [Google Scholar] [CrossRef]
- Mccord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar]
- Keele, B.B.; Mccord, J.M.; Fridovich, I. Superoxide dismutase from escherichia coli B. A new manganese-containing enzyme. J. Biol. Chem. 1970, 245, 6176–6181. [Google Scholar]
- Loschen, G.; Azzi, A.; Richter, C.; Flohé, L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974, 42, 68–72. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J. Mitochondrial Complex I. Ann. Rev. Biochem. 2013, 82, 551–575. [Google Scholar] [CrossRef] [PubMed]
- Vinothkumar, K.R.; Zhu, J.; Hirst, J. Architecture of mammalian respiratory complex I. Nature 2014, 515, 80–84. [Google Scholar] [CrossRef]
- Brandt, U. A two-state stabilization-change mechanism for proton-pumping complex I. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 1364–1369. [Google Scholar] [CrossRef]
- Kaila, V.R.I. Long-range proton-coupled electron transfer in biological energy conversion: Towards mechanistic understanding of respiratory complex I. J. R. Soc. Interface 2018, 15. [Google Scholar] [CrossRef]
- Parey, K.; Wirth, C.; Vonck, J.; Zickermann, V. Respiratory complex I—structure, mechanism and evolution. Curr. Opin. Struct. Biol. 2020, 63, 1–9. [Google Scholar] [CrossRef]
- Zickermann, V.; Wirth, C.; Nasiri, H.; Siegmund, K.; Schwalbe, H.; Hunte, C.; Brandt, U. Mechanistic insight from the crystal structure of mitochondrial complex I. Science 2015, 347, 44–49. [Google Scholar] [CrossRef]
- Kussmaul, L.; Hirst, J. The mechanism of superoxide production by NADH: Ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef]
- Pryde, K.R.; Hirst, J. Superoxide is produced by the reduced flavin in mitochondrial complex I. J. Biol. Chem. 2011, 286, 18056–18065. [Google Scholar] [CrossRef]
- Birrell, J.A.; Morina, K.; Bridges, H.R.; Friedrich, T.; Hirst, J. Investigating the function of [2Fe–2S] cluster N1a, the off-pathway cluster in complex I, by manipulating its reduction potential. Biochem. J. 2013, 456, 139–146. [Google Scholar] [CrossRef]
- Schulte, M.; Frick, K.; Gnandt, E.; Jurkovic, S.; Burschel, S.; Labatzke, R.; Aierstock, K.; Fiegen, D.; Wohlwend, D.; Gerhardt, S.; et al. A mechanism to prevent production of reactive oxygen species by Escherichia coli respiratory complex I. Nat. Commun. 2019, 10, 2551. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free. Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Hinkle, P.C.; Butow, R.A.; Rackers, E.; Chance, B. Partial resolution of the enzymes catalyzing oxidative phosphorylation. XV. Reverse electron transfer in the flavin-cytochrome β region of the respiratory chain of beef heart submitochondrial particles. J. Biol. Chem. 1967, 242, 5169–5173. [Google Scholar]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cochemé, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Martin, J.L.; Costa, A.S.H.; Gruszczyk, A.V.; Beach, T.E.; Allen, F.M.; Prag, H.A.; Hinchy, E.C.; Mahbubani, K.T.; Hamed, M.; Tronci, L.; et al. Succinate accumulation drives ischaemia-reperfusion injury during organ transplantation. Nat. Metab. 2019, 1, 966–974. [Google Scholar] [CrossRef]
- Scialo’, F.; Sriram, A.; Fernández-Ayala, D.J.M.; Gubina, N.; Lõhmus, M.; Nelson, G.; Logan, A.; Cooper, H.M.; Navas, P.; Enriquez, J.A.; et al. Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. 2016, 23, 725–734. [Google Scholar] [CrossRef]
- Onukwufor, J.O.; Berry, B.J.; Wojtovich, A.P. Physiologic implications of reactive oxygen species production by mitochondrial complex I reverse electron transport. Antioxidants 2019, 8, 285. [Google Scholar] [CrossRef]
- Fernández-Agüera, M.C.; Gao, L.; González-Rodríguez, P.; Pintado, C.O.; Arias-Mayenco, I.; García-Flores, P.; García-Pergañeda, A.; Pascual, A.; Ortega-Sáenz, P.; López-Barneo, J. Oxygen sensing by arterial chemoreceptors depends on mitochondrial complex I signaling. Cell Metab. 2015, 22, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Arias-Mayenco, I.; González-Rodríguez, P.; Torres-Torrelo, H.; Gao, L.; Fernández-Agüera, M.C.; Bonilla-Henao, V.; Ortega-Sáenz, P.; López-Barneo, J. Acute O2 sensing: Role of coenzyme QH2/Q ratio and mitochondrial ROS compartmentalization. Cell Metab. 2018, 28, 145–158. [Google Scholar] [CrossRef]
- Guarás, A.; Perales-Clemente, E.; Calvo, E.; Acín-Pérez, R.; Loureiro-López, M.; Pujol, C.; Martínez-Carrascoso, I.; Núñez, E.; García-Marqués, F.; Rodríguez-Hernández, A.; et al. The CoQH2/CoQ ratio serves as a sensor of respiratory chain efficiency. Cell Rep. 2016, 15, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Kempf, A.; Song, S.M.; Talbot, C.B.; Miesenböck, G. A potassium channel β-subunit couples mitochondrial electron transport to sleep. Nature 2019, 568, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, S.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 2018, 293, 9869–9879. [Google Scholar] [CrossRef]
- Yankovskaya, V.; Horsefield, R.; Törnroth-Horsefield, S.; Luna-Chavez, C.; Miyoshi, H.; Léger, C.; Byrne, B.; Cecchini, G.; Iwata, S. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 2003, 299, 700–704. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef]
- Siebels, I.; Dröse, S. Q-site inhibitor induced ROS production of mitochondrial complex II is attenuated by TCA cycle dicarboxylates. Biochim. Biophys. Acta Bioenerg. 2013, 1827, 1156–1164. [Google Scholar] [CrossRef]
- Messner, K.; Imlay, J.A. Mechanism of superoxide and hydrogen peroxide formation by fumarate reductase, succinate dehydrogenase, and aspartate oxidase. J. Biol. Chem. 2002, 277, 42563–42571. [Google Scholar] [CrossRef]
- Grivennikova, V.G.; Kozlovsky, V.S.; Vinogradov, A.D. Respiratory complex II: ROS production and the kinetics of ubiquinone reduction. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 109–117. [Google Scholar] [CrossRef]
- Manhas, N.; Duong, Q.V.; Lee, P.; Richardson, J.D.; Robertson, J.D.; Moxley, M.A.; Bazil, J.N. Computationally modeling mammalian succinate dehydrogenase kinetics identifies the origins and primary determinants of ROS production. J. Biol. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. The protonmotive Q cycle: A general formulation. FEBS Lett. 1975, 59, 137–139. [Google Scholar] [CrossRef]
- Crofts, A.R. The Cytochromebc1Complex: Function in the Context of Structure. Ann. Rev. Physiol. 2004, 66, 689–733. [Google Scholar] [CrossRef] [PubMed]
- Boveris, A.; Cadenas, E.; Stoppani, A.O.M. Role of ubiquinone in the mitochondrial generation of hydrogen peroxide. Biochem. J. 1976, 156, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Boveris, A.; Ragan, C.; Stoppani, A.O. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch. Biochem. Biophys. 1977, 180, 248–257. [Google Scholar] [CrossRef]
- Bleier, L.; Dröse, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta Bioenerg. 2013, 1827, 1320–1331. [Google Scholar] [CrossRef]
- Muller, F.L. The nature and mechanism of superoxide production by the electron transport chain: Its relevance to aging. J. Am. Aging Assoc. 2000, 23, 227–253. [Google Scholar] [CrossRef]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef]
- De Grey, A.D. HO2: The forgotten radical. DNA Cell Biol. 2002, 21, 251–257. [Google Scholar] [CrossRef]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef]
- Świerczek, M.; Cieluch, E.; Sarewicz, M.; Borek, A.; Moser, C.C.; Dutton, P.L.; Osyczka, A. An electronic bus bar lies in the core of cytochrome bc1. Science 2010, 329, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Drose, S.; Brandt, U. The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex. J. Biol. Chem. 2008, 283, 21649–21654. [Google Scholar] [CrossRef] [PubMed]
- Bielski, B.H.J.; Arudi, R.; Sutherland, M. A study of the reactivity of HO2/O2-with unsaturated fatty acids. J. Biol. Chem. 1983, 258, 4759–4761. [Google Scholar] [PubMed]
- Bielski, B.H.J. Reactivity of HO2/O2-radicals in aqueous solution. J. Phys. Chem. 1985, 14, 1041–1091. [Google Scholar]
- Quinlan, C.L.; Gerencser, A.A.; Treberg, J.R.; Brand, M.D. The Mechanism of Superoxide Production by the Antimycin-inhibited Mitochondrial Q-cycle. J. Biol. Chem. 2011, 286, 31361–31372. [Google Scholar] [CrossRef]
- Cadenas, E.; Boveris, A. Enhancement of hydrogen peroxide formation by protophores and ionophores in antimycin-supplemented mitochondria. Biochem. J. 1980, 188, 31–37. [Google Scholar] [CrossRef]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef]
- Guillaud, F.; Drose, S.; Kowald, A.; Brandt, U.; Klipp, E. Superoxide production by cytochrome bc1 complex: A mathematical model. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 1643–1652. [Google Scholar] [CrossRef][Green Version]
- Mailloux, R.J. Protein S-glutathionylation reactions as a global inhibitor of cell metabolism for the desensitization of hydrogen peroxide signals. Redox Biol. 2020, 32, 101472. [Google Scholar] [CrossRef]
- Orr, A.L.; Quinlan, C.L.; Perevoshchikova, I.V.; Brand, M.D. A refined analysis of superoxide production by mitochondrialsn-glycerol 3-phosphate dehydrogenase. J. Biol. Chem. 2012, 287, 42921–42935. [Google Scholar] [CrossRef]
- Hey-Mogensen, M.; Gonçalves, R.L.; Orr, A.L.; Brand, M.D. Production of superoxide/H2O2 by dihydroorotate dehydrogenase in rat skeletal muscle mitochondria. Free. Radic. Biol. Med. 2014, 72, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Perevoshchikova, I.V.; Quinlan, C.L.; Orr, A.L.; Gerencser, A.A.; Brand, M.D. Sites of superoxide and hydrogen peroxide production during fatty acid oxidation in rat skeletal muscle mitochondria. Free. Radic. Biol. Med. 2013, 61, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, R.L.; Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Brand, M.D. Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessedex vivounder conditions mimicking rest and exercise. J. Biol. Chem. 2015, 290, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Sidlauskaite, E.; Gibson, J.W.; Megson, I.L.; Whitfield, P.D.; Tovmasyan, A.; Batinic-Haberle, I.; Murphy, M.P.; Moult, P.R.; Cobley, J.N. Mitochondrial ROS cause motor deficits induced by synaptic inactivity: Implications for synapse pruning. Redox Biol. 2018, 16, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N. Synapse pruning: Mitochondrial ROS with their hands on the shears. BioEssays 2018, 40, e1800031. [Google Scholar] [CrossRef]
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide activates mitochondrial uncoupling proteins. Nature 2002, 415, 96–99. [Google Scholar] [CrossRef]
- Berry, B.J.; Trewin, A.J.; Amitrano, A.M.; Kim, M.; Wojtovich, A.P. Use the protonmotive force: Mitochondrial uncoupling and reactive oxygen species. J. Mol. Biol. 2018, 430, 3873–3891. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Affourtit, C.; Esteves, T.C.; Green, K.; Lambert, A.J.; Miwa, S.; Pakay, J.L.; Parker, N. Mitochondrial superoxide: Production, biological effects, and activation of uncoupling proteins. Free. Radic. Biol. Med. 2004, 37, 755–767. [Google Scholar] [CrossRef]
- Blaza, J.N.; Vinothkumar, K.R.; Hirst, J. Structure of the deactive state of mammalian respiratory complex I. Structure 2018, 26, 312–319. [Google Scholar] [CrossRef]
- Galkin, A.; Meyer, B.; Wittig, I.; Karas, M.; Schägger, H.; Vinogradov, A.; Brandt, U. Identification of the mitochondrial ND3 subunit as a structural component involved in the active/deactive enzyme transition of respiratory complex I. J. Biol. Chem. 2008, 283, 20907–20913. [Google Scholar] [CrossRef] [PubMed]
- Babot, M.; Birch, A.; Labarbuta, P.; Galkin, A. Characterisation of the active/de-active transition of mitochondrial complex I. Biochim. Biophys. Acta-Bioenerg. 2014, 1837, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Dröse, S.; Stepanova, A.; Galkin, A. Ischemic A/D transition of mitochondrial complex I and its role in ROS generation. Biochim. Biophys. Acta-Bioenerg. 2016, 1857, 946–957. [Google Scholar] [CrossRef] [PubMed]
- Pikó, L.; Taylor, K.D. Amounts of mitochondrial DNA and abundance of some mitochondrial gene transcripts in early mouse embryos. Dev. Biol. 1987, 123, 364–374. [Google Scholar] [CrossRef]
- Chronopoulou, E.; Harper, J.C. IVF culture media: Past, present and future. Hum. Reprod. Update 2014, 21, 39–55. [Google Scholar] [CrossRef]
- Place, T.L.; Domann, F.E.; Case, A.J. Limitations of oxygen delivery to cells in culture: An underappreciated problem in basic and translational research. Free. Radic. Biol. Med. 2017, 113, 311–322. [Google Scholar] [CrossRef]
- Nastri, C.O.; Nóbrega, B.N.; Teixeira, D.M.; Amorim, J.; Diniz, L.M.; Barbosa, M.W.; Giorgi, V.S.I.; Pileggi, V.N.; Martins, W.P. Low versus atmospheric oxygen tension for embryo culture in assisted reproduction: A systematic review and meta-analysis. Fertil. Steril. 2016, 106, 95–104. [Google Scholar] [CrossRef]
- Nasr-Esfahani, M.H.; Winston, N.J.; Johnson, M.H. Effects of glucose, glutamine, ethylenediaminetetraacetic acid and oxygen tension on the concentration of reactive oxygen species and on development of the mouse preimplantation embryo in vitro. J. Reprod. Fertil. 1992, 96, 219–231. [Google Scholar] [CrossRef]
- Dumollard, R.; Carroll, J.; Duchen, M.R.; Campbell, K.; Swann, K. Mitochondrial function and redox state in mammalian embryos. Semin. Cell Dev. Biol. 2009, 20, 346–353. [Google Scholar] [CrossRef]
- Jain, I.H.; Zazzeron, L.; Goli, R.; Alexa, K.; Schatzman-Bone, S.; Dhillon, H.; Goldberger, O.; Peng, J.; Shalem, O.; Sanjana, N.E.; et al. Hypoxia as a therapy for mitochondrial disease. Science 2016, 352, 54–61. [Google Scholar] [CrossRef]
- Kelley, R.L.; Gardner, D. Individual culture and atmospheric oxygen during culture affect mouse preimplantation embryo metabolism and post-implantation development. Reprod. Biomed. Online 2019, 39, 3–18. [Google Scholar] [CrossRef]
- Di Mascio, P.; Martinez, G.R.; Miyamoto, S.; Ronsein, G.E.; Medeiros, M.; Cadet, J. Singlet molecular oxygen reactions with nucleic acids, lipids, and proteins. Chem. Rev. 2019, 119, 2043–2086. [Google Scholar] [CrossRef]
- Takenaka, M.; Horiuchi, T.; Yanagimachi, R. Effects of light on development of mammalian zygotes. Proc. Natl. Acad. Sci. USA 2007, 104, 14289–14293. [Google Scholar] [CrossRef]
- Hockberger, P.E.; Skimina, T.A.; Centonze, V.E.; Lavin, C.; Chu, S.; Dadras, S.; Reddy, J.K.; White, J.G. Activation of flavin-containing oxidases underlies light-induced production of H2O2 in mammalian cells. Proc. Natl. Acad. Sci. USA 1999, 96, 6255–6260. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.M.; Janes, M.S.; Pehar, M.; Monette, J.S.; Ross, M.F.; Hagen, T.M.; Murphy, M.P.; Beckman, J.S. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc. Natl. Acad. Sci. USA 2006, 103, 15038–15043. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B.; Darley-Usmar, V.M.; Davies, K.P.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J.; Ischiropoulos, H. Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free. Radic. Biol. Med. 2011, 52, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Nasr-Esfahani, M.M.; Johnson, M.H. The origin of reactive oxygen species in mouse embryos cultured in vitro. Development 1991, 113, 551–560. [Google Scholar] [PubMed]
- Bonini, M.G.; Rota, C.; Tomasi, A.; Mason, R.P. The oxidation of 2′,7′-dichlorofluorescin to reactive oxygen species: A self-fulfilling prophesy? Free. Radic. Biol. Med. 2006, 40, 968–975. [Google Scholar] [CrossRef]
- Wardman, P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: Progress, pitfalls, and prospects. Free. Radic. Biol. Med. 2007, 43, 995–1022. [Google Scholar] [CrossRef]
- Zielonka, J.; Kalyanaraman, B. Small-molecule luminescent probes for the detection of cellular oxidizing and nitrating species. Free. Radic. Biol. Med. 2018, 128, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Van Blerkom, J. Mitochondrial function in the human oocyte and embryo and their role in developmental competence. Mitochondrion 2011, 11, 797–813. [Google Scholar] [CrossRef] [PubMed]
- Van Blerkom, J.; Davis, P.; Mathwig, V.; Alexander, S. Domains of high-polarized and low-polarized mitochondria may occur in mouse and human oocytes and early embryos. Hum. Reprod. 2002, 17, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Wilding, M.; Dale, B.; Marino, M.; Di Matteo, L.; Alviggi, C.; Pisaturo, M.L.; Lombardi, L.; De Placido, G. Mitochondrial aggregation patterns and activity in human oocytes and preimplantation embryos. Hum. Reprod. 2001, 16, 909–917. [Google Scholar] [CrossRef]
- Van Blerkom, J.; Davis, P. High-polarized (ΔΨmHIGH) mitochondria are spatially polarized in human oocytes and early embryos in stable subplasmalemmal domains: Developmental significance and the concept of vanguard mitochondria. Reprod. Biomed. Online 2006, 13, 246–254. [Google Scholar] [CrossRef]
- Van Blerkom, J.; Davis, P. Mitochondrial signaling and fertilization. Mol. Hum. Reprod. 2007, 13, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Dumollard, R.; Duchen, M.R.; Carroll, J. The role of mitochondrial function in the oocyte and embryo. Curr. Top. Dev. Biol. 2007, 77, 21–49. [Google Scholar] [CrossRef]
- Barshop, B.A.; Gangoiti, J.A. Analysis of coenzyme Q in human blood and tissues. Mitochondrion 2007, 7, S89–S93. [Google Scholar] [CrossRef]
- Dumollard, R.; Marangos, P.; Fitzharris, G.; Swann, K.; Duchen, M.R.; Carroll, J. Sperm-triggered [Ca2+] oscillations and Ca2+ homeostasis in the mouse egg have an absolute requirement for mitochondrial ATP production. Development 2004, 131, 3057–3067. [Google Scholar] [CrossRef]
- Clementi, E.; Brown, G.C.; Feelisch, M.; Moncada, S. Persistent inhibition of cell respiration by nitric oxide: Crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc. Natl. Acad. Sci. USA 1998, 95, 7631–7636. [Google Scholar] [CrossRef]
- Toledo, J.O.; Augusto, O. Connecting the chemical and biological properties of nitric oxide. Chem. Res. Toxicol. 2012, 25, 975–989. [Google Scholar] [CrossRef] [PubMed]
- Van Blerkom, J.; Davis, P.; Thalhammer, V. Regulation of mitochondrial polarity in mouse and human oocytes: The influence of cumulus derived nitric oxide. Mol. Hum. Reprod. 2008, 14, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Dröse, S. Differential effects of complex II on mitochondrial ROS production and their relation to cardioprotective pre- and postconditioning. Biochim. Biophys. Acta-Bioenerg. 2013, 1827, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.L.; Vargas, L.; Turk, C.N.; Baaten, J.E.; Matzen, J.T.; Dardov, V.J.; Attle, S.J.; Li, J.; Quackenbush, D.C.; Goncalves, R.L.S.; et al. Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 2015, 11, 834–836. [Google Scholar] [CrossRef]
- Brand, M.D.; Goncalves, R.L.; Orr, A.L.; Vargas, L.; Gerencser, A.A.; Jensen, M.B.; Wang, Y.T.; Melov, S.; Turk, C.N.; Matzen, J.T.; et al. Suppressors of superoxide-H2O2 production at site I Q of mitochondrial complex i protect against stem cell hyperplasia and ischemia-reperfusion injury. Cell Metab. 2016, 24, 582–592. [Google Scholar] [CrossRef]
- Wong, H.-S.; Monternier, P.-A.; Brand, M.D. S1QELs suppress mitochondrial superoxide/hydrogen peroxide production from site IQ without inhibiting reverse electron flow through Complex I. Free. Radic. Biol. Med. 2019, 143, 545–559. [Google Scholar] [CrossRef]
- Goncalves, R.L.; Watson, M.A.; Wong, H.-S.; Orr, A.L.; Brand, M.D. The use of site-specific suppressors to measure the relative contributions of different mitochondrial sites to skeletal muscle superoxide and hydrogen peroxide production. Redox Biol. 2020, 28, 101341. [Google Scholar] [CrossRef]
- Banba, A.; Tsuji, A.; Kimura, H.; Murai, M.; Miyoshi, H. Defining the mechanism of action of S1QELs, specific suppressors of superoxide production in the quinone-reaction site in mitochondrial complex I. J. Biol. Chem. 2019, 294, 6550–6561. [Google Scholar] [CrossRef]
- Margaritelis, N.; Cobley, J.; Paschalis, V.; Veskoukis, A.; Theodorou, A.; Kyparos, A.; Nikolaidis, M.G. Going retro: Oxidative stress biomarkers in modern redox biology. Free. Radic. Biol. Med. 2016, 98, 2–12. [Google Scholar] [CrossRef]
- Margaritelis, N.V.; Cobley, J.N.; Paschalis, V.; Veskoukis, A.S.; Theodorou, A.A.; Kyparos, A.; Nikolaidis, M.G. Principles for integrating reactive species into in vivo biological processes: Examples from exercise physiology. Cell. Sign. 2016, 28, 256–271. [Google Scholar] [CrossRef]
- James, A.M.; Cochemé, H.M.; Smith, R.A.J.; Murphy, M.P. Interactions of Mitochondria-targeted and Untargeted Ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. J. Biol. Chem. 2005, 280, 21295–21312. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. Antioxidants as therapies: Can we improve on nature? Free. Radic. Biol. Med. 2014, 66, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Finichiu, P.G.; Larsen, D.S.; Evans, C.; Larsen, L.; Bright, T.P.; Robb, E.L.; Trnka, J.; Prime, T.A.; James, A.M.; Smith, R.A.; et al. A mitochondria-targeted derivative of ascorbate: MitoC. Free. Radic. Biol. Med. 2015, 89, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Murphy, M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci. USA 2010, 1201, 96–103. [Google Scholar] [CrossRef]
- Smith, R.A.J.; Hartley, R.C.; Murphy, M.P. Mitochondria-targeted small molecule therapeutics and probes. Antioxid. Redox Sign. 2011, 15, 3021–3038. [Google Scholar] [CrossRef]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.J.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef]
- Smith, R.A.J.; Porteous, C.M.; Coulter, C.V.; Murphy, M.P. Selective targeting of an antioxidant to mitochondria. Eur. J. Chem. 1999, 263, 709–716. [Google Scholar] [CrossRef]
- Batinic-Haberle, I.; Rebouças, J.S.; Spasojevic, I. superoxide dismutase mimics: Chemistry, pharmacology, and therapeutic potential. Antioxid. Redox Sign. 2010, 13, 877–918. [Google Scholar] [CrossRef]
- Batinic-Haberle, I.; Tovmasyan, A.; Roberts, E.R.H.; Vujaskovic, Z.; Leong, K.W.; Spasojevic, I. SOD therapeutics: Latest insights into their structure-activity relationships and impact on the cellular redox-based signaling pathways. Antioxid. Redox Sign. 2014, 20, 2372–2415. [Google Scholar] [CrossRef]
- Batinic-Haberle, I.; Tovmasyan, A.; Spasojevic, I. An educational overview of the chemistry, biochemistry and therapeutic aspects of Mn porphyrins–From superoxide dismutation to H2O2-driven pathways. Redox Biol. 2015, 5, 43–65. [Google Scholar] [CrossRef]
- Gutteridge, J.M.; Halliwell, B. Antioxidants: Molecules, medicines, and myths. Biochem. Biophys. Res. Commun. 2010, 393, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Niki, E. Do antioxidants impair signaling by reactive oxygen species and lipid oxidation products? FEBS Lett. 2012, 586, 3767–3770. [Google Scholar] [CrossRef] [PubMed]
- Niki, E. Role of vitamin E as a lipid-soluble peroxyl radical scavenger: In vitro and in vivo evidence. Free. Radic. Biol. Med. 2014, 66, 3–12. [Google Scholar] [CrossRef]
- Meyer, A.J.; Dick, T.P. Fluorescent Protein-Based Redox Probes. Antioxid. Redox Sign. 2010, 13, 621–650. [Google Scholar] [CrossRef] [PubMed]
- Cochemé, H.M.; Quin, C.; McQuaker, S.J.; Cabreiro, F.G.; Logan, A.; Prime, T.A.; Abakumova, I.; Patel, J.V.; Fearnley, I.M.; James, A.M.; et al. Measurement of H2O2 within living drosophila during aging using a ratiometric mass spectrometry probe targeted to the mitochondrial matrix. Cell Metab. 2011, 13, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Logan, A.; Cochemé, H.M.; Pun, P.B.L.; Apostolova, N.; Smith, R.A.; Larsen, L.; Larsen, D.S.; James, A.M.; Fearnley, I.M.; Rogatti, S.; et al. Using exomarkers to assess mitochondrial reactive species in vivo. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2014, 1840, 923–930. [Google Scholar] [CrossRef]
- Shchepinova, M.M.; Cairns, A.G.; Prime, T.A.; Logan, A.; James, A.M.; Hall, A.R.; Vidoni, S.; Arndt, S.; Caldwell, S.T.; Prag, H.A.; et al. MitoNeoD: A mitochondria-targeted superoxide probe. Cell Chem. Biol. 2017, 24, 1285–1298. [Google Scholar] [CrossRef]
- Zielonka, J.; Kalyanaraman, B. Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: Another inconvenient truth. Free. Radic. Biol. Med. 2010, 48, 983–1001. [Google Scholar] [CrossRef]
- Held, J.M. Redox Systems Biology: Harnessing the Sentinels of the Cysteine Redoxome. Antioxid. Redox Sign. 2020, 32, 659–676. [Google Scholar] [CrossRef]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A Quantitative tissue-specific landscape of protein redox regulation during aging. Cell 2020, 180, 968–983.e24. [Google Scholar] [CrossRef]
- Cobley, J.N.; Sakellariou, G.K.; Husi, H.; McDonagh, B. Proteomic strategies to unravel age-related redox signalling defects in skeletal muscle. Free. Radic. Biol. Med. 2019, 132, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Carroll, K.S.; Liebler, D.C. The expanding landscape of the thiol redox proteome. Mol. Cell. Proteom. 2015, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gould, N.S.; Evans, P.; Martínez-Acedo, P.; Marino, S.M.; Gladyshev, V.N.; Carroll, K.S.; Ischiropoulos, H. Site-specific proteomic mapping identifies selectively modified regulatory cysteine residues in functionally distinct protein networks. Chem. Biol. 2015, 22, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, J.R.; Oviosu, O.; Eaton, P. The PEG-switch assay: A fast semi-quantitative method to determine protein reversible cysteine oxidation. J. Pharmacol. Toxicol. Methods 2013, 68, 297–301. [Google Scholar] [CrossRef]
- Van Leeuwen, L.A.; Hinchy, E.C.; Murphy, M.P.; Robb, E.L.; Cochemé, H.M. Click-PEGylation - A mobility shift approach to assess the redox state of cysteines in candidate proteins. Free. Radic. Biol. Med. 2017, 108, 374–382. [Google Scholar] [CrossRef]
- Cobley, J.N.; Noble, A.; Fernández, E.J.; Moya, M.-T.V.; Guille, M.; Husi, H. Catalyst-free Click PEGylation reveals substantial mitochondrial ATP synthase sub-unit alpha oxidation before and after fertilisation. Redox Biol. 2019, 26, 101258. [Google Scholar] [CrossRef]
- Cobley, J.N.; Husi, H. Immunological techniques to assess protein thiol redox state: Opportunities, challenges and solutions. Antioxidants 2020, 9, 315. [Google Scholar] [CrossRef]
- Logan, A.; Pell, V.R.; Shaffer, K.J.; Evans, C.; Stanley, N.J.; Robb, E.L.; Prime, T.A.; Chouchani, E.T.; Cochemé, H.M.; Fearnley, I.M.; et al. Assessing the mitochondrial membrane potential in cells and in vivo using targeted click chemistry and mass spectrometry. Cell Metab. 2016, 23, 379–385. [Google Scholar] [CrossRef]
- Burger, N.; Logan, A.; Prime, T.A.; Mottahedin, A.; Caldwell, S.T.; Krieg, T.; Hartley, R.C.; James, A.M.; Murphy, M.P. A sensitive mass spectrometric assay for mitochondrial CoQ pool redox state in vivo. Free. Radic. Biol. Med. 2020, 147, 37–47. [Google Scholar] [CrossRef]
- Robb, E.L.; Gawel, J.M.; Aksentijević, D.; Cochemé, H.M.; Stewart, T.; Shchepinova, M.M.; Qiang, H.; Prime, T.A.; Bright, T.P.; James, A.M.; et al. Selective superoxide generation within mitochondria by the targeted redox cycler MitoParaquat. Free. Radic. Biol. Med. 2015, 89, 883–894. [Google Scholar] [CrossRef]
- Booty, L.M.; Gawel, J.M.; Cvetko, F.; Caldwell, S.T.; Hall, A.R.; Mulvey, J.F.; James, A.M.; Hinchy, E.C.; Prime, T.A.; Arndt, S.; et al. Selective disruption of mitochondrial thiol redox state in cells and in vivo. Cell Chem. Biol. 2019, 26, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Dalle-Donne, I.; Milzani, A.D.G.; Fanti, P.; Rossi, R. Analysis of GSH and GSSG after derivatization with N-ethylmaleimide. Nat. Protoc. 2013, 8, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Weydert, C.J.; Cullen, J.J. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat. Protoc. 2010, 5, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Dumollard, R.; Duchen, M.R.; Sardet, C. Calcium signals and mitochondria at fertilisation. Semin. Cell Dev. Biol. 2006, 17, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Muller, B.; Lewis, N.; Adeniyi, T.; Leese, H.J.; Brison, D.R.; Sturmey, R.G. Application of extracellular flux analysis for determining mitochondrial function in mammalian oocytes and early embryos. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Van Blerkom, J.; Davis, P.W.; Lee, J. Fertilization and early embryolgoy: ATP content of human oocytes and developmental potential and outcome after in-vitro fertilization and embryo transfer. Hum. Reprod. 1995, 10, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Allen, J. Separate Sexes and the Mitochondrial Theory of Ageing. J. Theor. Biol. 1996, 180, 135–140. [Google Scholar] [CrossRef]
- Hernansanz-Agustín, P.; Choya-Foces, C.; Carregal-Romero, S.; Ramos, E.; Oliva, T.; Villa-Piña, T.; Moreno, L.; Izquierdo-Álvarez, A.; Cabrera-García, J.D.; Cortés, A.; et al. Na+ controls hypoxic signalling by the mitochondrial respiratory chain. Nature 2020. [Google Scholar] [CrossRef]
- Biggers, J.D.; Whittingham, D.G.; Donahue, R.P. The pattern of energy metabolism in the mouse oocyte and zygote. Proc. Nat. Acad. Sci. USA 1967, 58, 560–567. [Google Scholar] [CrossRef]
- Houghton, F.D.; Christopher, G.T.; Leese, H.J. Oxygen consumption and energy metabolism of the early mouse embryo. Mol. Reprod. Dev. 1996, 485, 476–485. [Google Scholar] [CrossRef]
- O’Brien, M.; Chalker, J.; Slade, L.; Gardiner, D.; Mailloux, R.J. Protein S-glutathionylation alters superoxide/hydrogen peroxide emission from pyruvate dehydrogenase complex. Free. Radic. Biol. Med. 2017, 106, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Gill, R.M.; O’Brien, M.; Young, A.; Gardiner, D.; Mailloux, R.J. Protein S-glutathionylation lowers superoxide/hydrogen peroxide release from skeletal muscle mitochondria through modification of complex I and inhibition of pyruvate uptake. PLoS ONE 2018, 13, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lane, M.; Gardner, D. Mitochondrial malate-aspartate shuttle regulates mouse embryo nutrient consumption. J. Biol. Chem. 2005, 280, 18361–18367. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, R.; Sharpley, M.S.; Chi, F.; Braas, D.; Zhou, Y.; Kim, R.; Clark, A.T.; Banerjee, U. Nuclear localization of mitochondrial TCA cycle enzymes as a critical step in mammalian zygotic genome activation. Cell 2017, 168, 210–223.e11. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, J.; Dahan, P.; Lu, V.; Zhang, C.; Li, H.; Teitell, M.A. Metabolism in pluripotent stem cells and early mammalian development. Cell Metab. 2018, 27, 332–338. [Google Scholar] [CrossRef]
- Sturmey, R.; Leese, H.; Sturmey, R.G. Energy metabolism in pig oocytes and early embryos. Reproduction 2003, 126, 197–204. [Google Scholar] [CrossRef]
- Motta, P.M.; Nottola, S.A.; Makabe, S.; Heyn, R. Mitochondrial morphology in human fetal and adult female germ cells. Hum. Reprod. 2000, 15 (Suppl. S2), 129–147. [Google Scholar] [CrossRef]
- Petrova, B.; Liu, K.; Tian, C.; Kitaoka, M.; Freinkman, E.; Yang, J.; Orr-Weaver, T.L. Dynamic redox balance directs the oocyte-to-embryo transition via developmentally controlled reactive cysteine changes. Proc. Natl. Acad. Sci. USA 2018, 115, E7978–E7986. [Google Scholar] [CrossRef]
- Smith, D.G.; Sturmey, R.G. Parallels between embryo and cancer cell metabolism. Biochem. Soc. Trans. 2013, 41, 664–669. [Google Scholar] [CrossRef]
- Muramoto, K.; Ohta, K.; Shinzawa-Itoh, K.; Kanda, K.; Taniguchi, M.; Nabekura, H.; Yamashita, E.; Tsukihara, T.; Yoshikawa, S. Bovine cytochromecoxidase structures enable O2 reduction with minimization of reactive oxygens and provide a proton-pumping gate. Proc. Natl. Acad. Sci. USA 2010, 107, 7740–7745. [Google Scholar] [CrossRef]
- Barja, G. Oxygen radicals, a failure or sucess of evolution. Free Radic. Res. 1993, 18, 63–70. [Google Scholar]
- Buettner, G.R. Superoxide dismutase in redox biology: The roles of superoxide and hydrogen peroxide. Anti-Cancer Agents Med. Chem. 2011, 11, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Lane, N. The Vital Question: Energy, Evolution, and the Origins of Complex Life; Profile Books Limited: London, UK, 2015. [Google Scholar]
- Finkel, T. Signal transduction by mitochondrial oxidants. J. Biol. Chem. 2011, 287, 4434–4440. [Google Scholar] [CrossRef] [PubMed]
- Allen, J. Why chloroplasts and mitochondria retain their own genomes and genetic systems: Colocation for redox regulation of gene expression. Proc. Natl. Acad. Sci. USA 2015, 112, 10231–10238. [Google Scholar] [CrossRef] [PubMed]
- Vandaele, L.; Thys, M.; Bijttebier, J.; Van Langendonckt, A.; Donnay, I.; Maes, D.; Meyer, E.; Van Soom, A. Short-term exposure to hydrogen peroxide during oocyte maturation improves bovine embryo development. Reproduction 2010, 139, 505–511. [Google Scholar] [CrossRef]
- Buettner, G.R.; Ng, C.F.; Wang, M.; Rodgers, V.G.; Schafer, F.Q. A new paradigm: Manganese superoxide dismutase influences the production of H2O2 in cells and thereby their biological state. Free. Radic. Biol. Med. 2006, 41, 1338–1350. [Google Scholar] [CrossRef]
- Brown, G.C. Nitric oxide and mitochondrial respiration. Biochim. Biophys. Acta (BBA)-Bioenerg. 1999, 1411, 351–369. [Google Scholar] [CrossRef]
- Vásquez-Vivar, J.; Kalyanaraman, B.; Kennedy, M.C. Mitochondrial Aconitase Is a Source of Hydroxyl Radical. J. Biol. Chem. 2000, 275, 14064–14069. [Google Scholar] [CrossRef]
- Gardner, P.R.; Fridovich, I. Superoxide sensitivity of the Escherichia coli aconitase. J. Biol. Chem. 1991, 266, 19328–19333. [Google Scholar]
- Abreu, I.A.; Cabelli, D.E. Superoxide dismutases—A review of the metal-associated mechanistic variations. Biochim. Biophys. Acta-Proteins Proteom. 2010, 1804, 263–274. [Google Scholar] [CrossRef]
- Sheng, Y.; Abreu, I.A.; Cabelli, D.E.; Maroney, M.J.; Miller, A.-F.; Teixeira, M.; Valentine, J.S. Superoxide Dismutases and Superoxide Reductases. Chem. Rev. 2014, 114, 3854–3918. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Biological chemistry of superoxide radicals. ChemTexts 2020, 6, 1–13. [Google Scholar] [CrossRef]
- Imlay, J.A. The molecular mechanisms and physiological consequences of oxidative stress: Lessons from a model bacterium. Nat. Rev. Microbiol. 2013, 11, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Ganini, D.; Santos, J.H.; Bonini, M.G.; Mason, R.P. Switch of mitochondrial superoxide dismutase into a prooxidant peroxidase in manganese-deficient cells and mice. Cell Chem. Biol. 2018, 25, 413–425. [Google Scholar] [CrossRef]
- Palma, F.R.; He, C.; Danes, M.J.M.; Paviani, V.; Coelho, D.R.; Gantner, B.N.; Bonini, M.G.; Sampaio, V.P. Mitochondrial superoxide dismutase: What the established, the intriguing, and the novel reveal about a key cellular redox switch. Antioxid. Redox Sign. 2020, 32, 701–714. [Google Scholar] [CrossRef]
- Lustgarten, M.S.; Bhattacharya, A.; Muller, F.L.; Jang, Y.C.; Shimizu, T.; Shirasawa, T.; Richardson, A.; Van Remmen, H. Complex I generated, mitochondrial matrix-directed superoxide is released from the mitochondria through voltage dependent anion channels. Biochem. Biophys. Res. Commun. 2012, 422, 515–521. [Google Scholar] [CrossRef]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef]
- Radi, R. Peroxynitrite, a stealthy biological oxidant. J. Biol. Chem. 2013, 288, 26464–26472. [Google Scholar] [CrossRef]
- Carballal, S.; Bartesaghi, S.; Radi, R. Kinetic and mechanistic considerations to assess the biological fate of peroxynitrite. Biochim. Biophys. Acta-Gen. Subj. 2014, 1840, 768–780. [Google Scholar] [CrossRef]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 2010, 425, 313–325. [Google Scholar] [CrossRef]
- Murphy, M.P. Mitochondrial thiols in antioxidant protection and redox signaling: Distinct roles for glutathionylation and other thiol modifications. Antioxid. Redox Sign. 2012, 16, 476–495. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.D.; Schmidt, C.A.; Lin, C.-T.; Fisher-Wellman, K.H.; Neufer, P. Flux through mitochondrial redox circuits linked to nicotinamide nucleotide transhydrogenase generates counterbalance changes in energy expenditure. J. Biol. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sobotta, M.C.; Liou, W.; Stöcker, S.; Talwar, D.; Oehler, M.; Ruppert, T.; Scharf, A.N.D.; Dick, T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015, 11, 64–70. [Google Scholar] [CrossRef]
- Pak, V.V.; Ezeriņa, D.; Lyublinskaya, O.G.; Pedre, B.; Tyurin-Kuzmin, P.A.; Mishina, N.M.; Thauvin, M.; Young, D.; Wahni, K.; Gache, S.A.M.; et al. Ultrasensitive genetically encoded indicator for hydrogen peroxide identifies roles for the oxidant in cell migration and mitochondrial function. Cell Metab. 2020, 31, 642–653.e6. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Biological Production, Detection, and Fate of Hydrogen Peroxide. Antioxid. Redox Sign. 2018, 20, 541–551. [Google Scholar] [CrossRef]
- Sies, H. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007, 35, 1147–1151. [Google Scholar] [CrossRef]
- Zhang, Y.; Marcillat, O.; Giulivi, C.; Ernster, L.; Davies, K.J. The oxidative inactivation of mitochondrial electron transport chain components and ATPase. J. Biol. Chem. 1990, 265, 16330–16336. [Google Scholar]
- Shoubridge, E.A.; Wai, T. Mitochondrial DNA and the mammalian oocyte. Curr. Top. Dev. Biol. 2007, 77, 87–111. [Google Scholar] [CrossRef]
- Cobley, J.N.; Sakellariou, G.K.; Murray, S.; Waldron, S.; Gregson, W.; Burniston, J.G.; Morton, J.P.; Iwanejko, L.A.; Close, G.L. Lifelong endurance training attenuates age-related genotoxic stress in human skeletal muscle. Longev. Health 2013, 2, 11. [Google Scholar] [CrossRef]
- Cobley, J.N.; Margaritelis, N.V.; Morton, J.P.; Close, G.L.; Nikolaidis, M.G.; Malone, J.K. The basic chemistry of exercise-induced DNA oxidation: Oxidative damage, redox signaling, and their interplay. Front. Physiol. 2015, 6, 1–8. [Google Scholar] [CrossRef]
- Williamson, J.; Hughes, C.M.; Cobley, J.N.; Davison, G.W. The mitochondria-targeted antioxidant MitoQ, attenuates exercise-induced mitochondrial DNA damage. Redox Biol. 2020, 36, 101673. [Google Scholar] [CrossRef] [PubMed]
- Lane, N. Mitonuclear match: Optimizing fitness and fertility over generations drives ageing within generations. BioEssays 2011, 33, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Latorre-Pellicer, A.; Moreno-Loshuertos, R.; Lechuga-Vieco, A.V.; Sánchez-Cabo, F.; Torroja, C.; Acín-Pérez, R.; Calvo, E.; Aix, E.; González-Guerra, A.; Logan, A.; et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 2016, 535, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Carroll, K.S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Parvez, S.; Long, M.J.C.; Poganik, J.R.; Aye, Y. Redox signaling by reactive electrophiles and oxidants. Chem. Rev. 2018, 118, 8798–8888. [Google Scholar] [CrossRef]
- Janssen-Heininger, Y.M.; Mossman, B.T.; Heintz, N.H.; Forman, H.J.; Kalyanaraman, B.; Finkel, T.; Stamler, J.S.; Rhee, S.G.; Van Der Vliet, A. Redox-based regulation of signal transduction: Principles, pitfalls, and promises. Free. Radic. Biol. Med. 2008, 45, 1–17. [Google Scholar] [CrossRef]
- Cobley, J.N.; McHardy, H.; Morton, J.P.; Nikolaidis, M.G.; Close, G.L. Influence of vitamin C and vitamin E on redox signaling: Implications for exercise adaptations. Free. Radic. Biol. Med. 2015, 84, 65–76. [Google Scholar] [CrossRef]
- Bersweiler, A.; D’Autréaux, B.; Mazon, H.; Kriznik, A.; Belli, G.; Delaunay-Moisan, A.; Toledano, M.B.; Rahuel-Clermont, S. A scaffold protein that chaperones a cysteine-sulfenic acid in H2O2 signaling. Nat. Chem. Rev. Biol. 2017, 13, 909–915. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Go, Y.-M.; Chandler, J.D.; Jones, D.P. The cysteine proteome. Free. Radic. Biol. Med. 2015, 84, 227–245. [Google Scholar] [CrossRef]
- Collins, Y.; Chouchani, E.T.; Menger, K.E.; Murphy, M.P.; Cochemé, H.M.; James, A.M. Mitochondrial redox signalling at a glance. J. Cell Sci. 2012, 125, 1837. [Google Scholar] [CrossRef]
- Requejo, R.; Hurd, T.R.; Costa, N.J.; Murphy, M.P. Cysteine residues exposed on protein surfaces are the dominant intramitochondrial thiol and may protect against oxidative damage. FEBS J. 2010, 277, 1465–1480. [Google Scholar] [CrossRef] [PubMed]
- Dröse, S.; Brandt, U.; Wittig, I. Mitochondrial respiratory chain complexes as sources and targets of thiol-based redox-regulation. Biochim. Biophys. Acta-Proteins Proteom. 2014, 1844, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Hurd, T.R.; Requejo, R.; Filipovska, A.; Brown, S.; Prime, T.A.; Robinson, A.J.; Fearnley, I.M.; Murphy, M.P. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit. J. Biol. Chem. 2008, 283, 24801–24815. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-B.; Foster, D.B.; Rucker, J.; O’Rourke, B.; Kass, D.A.; Van Eyk, J.E. Redox regulation of mitochondrial ATP synthase: Implications for cardiac resynchronization therapy. Circ. Res. 2011, 109, 750–757. [Google Scholar] [CrossRef]
- Wang, S.-B.; Murray, C.I.; Chung, H.S.; Van Eyk, J. Redox regulation of mitochondrial ATP synthase. Trends Cardiovasc. Med. 2013, 23, 18. [Google Scholar] [CrossRef]
- Cobley, J.N.; Noble, A.; Bessell, R.; Guille, M.; Husi, H. Reversible thiol oxidation inhibits the mitochondrial ATP Synthase in Xenopus laevis Oocytes. Antioxidants 2020, 9, 215. [Google Scholar] [CrossRef]
- Ramalho-Santos, J.; Varum, S.; Amaral, S.; Mota, P.C.; Sousa, A.P.M.; Amaral, A. Mitochondrial functionality in reproduction: From gonads and gametes to embryos and embryonic stem cells. Hum. Reprod. Update 2009, 15, 553–572. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free. Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Flohé, L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Sign. 2011, 15, 2335–2381. [Google Scholar] [CrossRef]
- Stöcker, S.; Van Laer, K.; Mijuskovic, A.; Dick, T.P. The conundrum of hydrogen peroxide signaling and the emerging role of peroxiredoxins as redox relay hubs. Antioxid. Redox Sign. 2018, 28, 558–573. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.-Y.; Rhee, S.G. Inactivation of peroxiredoxin i by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Reczek, C.R.; Chandel, N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; Close, G.L.; Bailey, D.M.; Davison, G.W. Exercise redox biochemistry: Conceptual, methodological and technical recommendations. Redox Biol. 2017, 12, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Hydrogen peroxide reactivity and specificity in thiol-based cell signalling. Biochem. Soc. Trans. 2020, 48, 1–10. [Google Scholar] [CrossRef]
- Dagnell, M.; Cheng, Q.; Rizvi, S.H.M.; Pace, P.E.; Boivin, B.; Winterbourn, C.C.; Arnér, E.S.J. Bicarbonate is essential for protein-tyrosine phosphatase 1B (PTP1B) oxidation and cellular signaling through EGF-triggered phosphorylation cascades. J. Biol. Chem. 2019, 294, 12330–12338. [Google Scholar] [CrossRef] [PubMed]
- Augusto, O.; Bonini, M.G.; Amanso, A.M.; Linares, E.; Santos, C.C.; De Menezes, S.L. Nitrogen dioxide and carbonate radical anion: Two emerging radicals in biology. Free. Radic. Biol. Med. 2002, 32, 841–859. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Revisiting the reactions of superoxide with glutathione and other thiols. Arch. Biochem. Biophys. 2016, 595, 68–71. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Superoxide as an intracellular radical sink. Free. Radic. Biol. Med. 1993, 14, 85–90. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Are free radicals involved in thiol-based redox signaling? Free. Radic. Biol. Med. 2015, 80, 164–170. [Google Scholar] [CrossRef]
- Kang, P.T.; Zhang, L.; Chen, C.-L.; Chen, J.; Green, K.B.; Chen, Y.-R. Protein thiyl radical mediates S-glutathionylation of complex I. Free. Radic. Biol. Med. 2012, 53, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, J.R.; Liu, L.; Porterfield, D.M.; Smith, P.J.; Keefe, D.L. Oxidative phosphorylation-dependent and -independent oxygen consumption by individual preimplantation mouse embryos1. Biol. Reprod. 2000, 62, 1866–1874. [Google Scholar] [CrossRef] [PubMed]
- Nasr-Esfahani, M.H.; Aitken, J.R.; Johnson, M.H. Hydrogen peroxide levels in mouse oocytes and early cleavage stage embryos developed in vitro or in vivo. Development 1990, 109, 501–507. [Google Scholar] [PubMed]
- Foerder, C.A.; Klebanoff, S.J.; Shapiro, B.M. Hydrogen peroxide production, chemiluminescence, and the respiratory burst of fertilization: Interrelated events in early sea urchin development. Proc. Natl. Acad. Sci. USA 1978, 75, 3183–3187. [Google Scholar] [CrossRef]
- Heinecke, J.W.; Shapiro, B.M. Respiratory burst oxidase of fertilization. Proc. Natl. Acad. Sci. USA 1989, 86, 1259–1263. [Google Scholar] [CrossRef]
- Wong, J.L.; Creton, R.; Wessel, G. The oxidative burst at fertilization is dependent upon activation of the dual oxidase Udx1. Dev. Cell 2004, 7, 801–814. [Google Scholar] [CrossRef]
- Han, Y.; Ishibashi, S.; Iglesias-Gonzalez, J.; Chen, Y.; Love, N.R.; Amaya, E. Ca 2+ Induced mitochondrial ros regulate the early embryonic cell cycle. Cell Rep. 2018, 22, 218–231. [Google Scholar] [CrossRef]
- Natsuyama, S.; Noda, Y.; Narimoto, K.; Umaoka, Y.; Mori, T. Release of two-cell block by reduction of protein disulfide with thioredoxin from Escherichia coli in mice. J. Reprod. Fertil. 1992, 95, 649–656. [Google Scholar] [CrossRef]
- Natsuyama, S.; Noda, Y.; Yamashita, M.; Nagahama, Y.; Mori, T. Superoxide dismutase and thioredoxin restore defective p34cdc2 kinase activation in mouse two-cell block. BBA-Mol. Cell Res. 1993, 1176, 90–94. [Google Scholar] [CrossRef]
- Ufer, C.; Wang, C.C.; Borchert, A.; Heydeck, D.; Kuhn, H. Redox control in mammalian embryo development. Antioxid. Redox Sign. 2010, 13, 833–875. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic. Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.G.; Murphy, M.P.; Frezza, C.; Prag, H.A.; Chouchani, E.T.; O’Neill, L.A.; Mills, E.L. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat. Metab. 2018, 1, 16–33. [Google Scholar] [CrossRef]
- Harvey, A.J. Mitochondria in early development: Linking the microenvironment, metabolism and the epigenome. Reproduction 2019, 157, R159–R179. [Google Scholar] [CrossRef] [PubMed]
- Hitchler, M.J.; Domann, F.E. An epigenetic perspective on the free radical theory of development. Free Radic. Biol. Med. 2007, 43, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Bazopoulou, D.; Knoefler, D.; Zheng, Y.; Ulrich, K.; Oleson, B.J.; Xie, L.; Kim, M.; Kaufmann, A.; Lee, Y.-T.; Dou, Y.; et al. Developmental ROS individualizes organismal stress resistance and lifespan. Nature 2019, 576, 301–305. [Google Scholar] [CrossRef]
- Knoefler, D.; Thamsen, M.; Koniczek, M.; Niemuth, N.J.; Diederich, A.-K.; Jakob, U. Quantitative in vivo redox sensors uncover oxidative stress as an early event in life. Mol. Cell 2012, 47, 767–776. [Google Scholar] [CrossRef]
- Lozoya, O.A.; Xu, F.; Grenet, D.; Wang, T.; Grimm, S.A.; Godfrey, V.; Waidyanatha, S.; Woychik, R.P.; Santos, J.H. Single nucleotide resolution analysis reveals pervasive, long-lasting DNA methylation changes by developmental exposure to a mitochondrial toxicant. Cell Rep. 2020, 32. [Google Scholar] [CrossRef]
- Chowdhury, A.R.; Zielonka, J.; Kalyanaraman, B.; Hartley, R.C.; Murphy, M.P.; Avadhani, N.G. Mitochondria-targeted paraquat and metformin mediate ROS production to induce multiple pathways of retrograde signaling: A dose-dependent phenomenon. Redox Biol. 2020, 36, 101606. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Substrate | Group | Source/Topology | Comments |
|---|---|---|---|---|
| Alpha keto glutarate dehydrogenase | Alpha keto glutarate. | NAD+/NADH | FAD of E3 dihydrolipoamide dehydrogenase. Matrix. | ROS production is inhibited by high ATP and aspartate [69] and regulated by reversible thiol oxidation [105]. Favored by high NAD+/NADH ratio. May also produce H2O2. Requires substrate. Ca2+ sensitive. |
| Pyruvate dehydrogenase | Pyruvate. | NAD+/NADH | FAD of E3 dihydrolipoamide dehydrogenase. Matrix. | Regulated by reversible thiol oxidation [105] and phosphorylation. ROS production is favored by high NAD+/ NADH ratio. May also produce H2O2. Requires substrate. |
| Branched-chain 2-oxoacid dehydrogenase complex | Branched chain 2-oxoacids (e.g., 3-methyl-2-oxopentanoate). | NAD+/NADH | FAD of E3 dihydrolipoamide dehydrogenase. Matrix | Favored by high NAD+/NADH ratio. Requires substrate. |
| Aminoadipate dehydrogenase complex | 2-oxoadipate. | NAD+/NADH | FAD of E3 dihydrolipoamide dehydrogenase. Matrix | ROS production is favored by high NADH/NAD+ ratio. Requires substrate. |
| sn-glycerol-3-phosphate dehydrogenase | Glycerol 3-phosphate | UQ/UQH2 | UQ binding site but may also involve a flavin. Intermembrane space and matrix. | Ca2+ sensitive—can enhance ROS production at low substrate levels [106]. Requires substrate. Much emanates from complex II. |
| Dihydroorotate dehydrogenase | Dihydroorotate | UQ/UQH2 | UQ binding site. Matrix. | Relatively low superoxide producing capacity but can drive other sites to high rates by reducing the Q pool [107]. |
| Electron transferring-flavoprotein: ubiquinone oxidoreductase | Electron transferring flavoprotein (involved in lipid + amino acid metabolism). | UQ/UQH2 | May emanate from the flavin but origin is unclear [69]. | ROS production is quite low even when other sites are inhibited [108], suggesting main contribution under native conditions is to reduce the Q pool. |
| Mode | OXPHOS | Respiration | Δp | NADH | QH2 | Dominant ETC Site(s) |
|---|---|---|---|---|---|---|
| 1 | High | High | Mod | Low to mod | Low | Complex I (forward) |
| 2 | Low-mod | Low-mod | X | High | Intermediate | Complex I (forward), complex II |
| 3 | Low-mod | Low-mod | Mod | X | Intermediate | Complex III, complex II |
| 4 | Negligible | Some * | High | X | High | Complex I (reverse) |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cobley, J.N. Mechanisms of Mitochondrial ROS Production in Assisted Reproduction: The Known, the Unknown, and the Intriguing. Antioxidants 2020, 9, 933. https://doi.org/10.3390/antiox9100933
Cobley JN. Mechanisms of Mitochondrial ROS Production in Assisted Reproduction: The Known, the Unknown, and the Intriguing. Antioxidants. 2020; 9(10):933. https://doi.org/10.3390/antiox9100933
Chicago/Turabian StyleCobley, James N. 2020. "Mechanisms of Mitochondrial ROS Production in Assisted Reproduction: The Known, the Unknown, and the Intriguing" Antioxidants 9, no. 10: 933. https://doi.org/10.3390/antiox9100933
APA StyleCobley, J. N. (2020). Mechanisms of Mitochondrial ROS Production in Assisted Reproduction: The Known, the Unknown, and the Intriguing. Antioxidants, 9(10), 933. https://doi.org/10.3390/antiox9100933