Diabetic Retinopathy: The Role of Mitochondria in the Neural Retina and Microvascular Disease




Abstract
1. Introduction
2. Normal Retinal Structure
3. Pathophysiology of DR
4. Retinal Bioenergetics and Mitochondrial Substrate Selection
4.1. ATP-Consuming Processes in the Retina
4.2. ATP-Generating Processes in the Retina and the Heterogeneity of Retinal Bioenergetics
4.3. Substrate Selection and Energy Production
5. Alterations in Mitochondrial Oxidative Metabolism in the Neural Retina
5.1. Localization of Mitochondria in the Retina
5.2. Diabetes Alters Mitochondrial Function in the Retina
6. Mitochondria-Derived Oxidative Stress in the Diabetic Retina
7. NAD Pool and the NADH/NAD+ Redox Ratio in the Diabetic Retina
7.1. NAD Pool
7.2. NADH/NAD+ Redox Ratio
8. Alterations in Mitochondrial Turnover
8.1. Mitochondrial Biogenesis and Mitophagy
8.2. Fusion–Fission Dynamics in the Retina
9. Consequences of Increased Oxidative Stress in the Neural Retina
9.1. Diabetic Milieu Alters Ion Channel Homeostasis in the Retina
9.2. Apoptosis
10. Therapeutic Implications
10.1. Therapies Focused on Maintaining the Integrity of Retinal Mitochondria
10.2. Clinical Trials and the Role of Antioxidants in the Management of DR
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed]
- Yau, J.W.Y.; Rogers, S.L.; Kawasaki, R.; Lamoureux, E.L.; Kowalski, J.W.; Bek, T.; Chen, S.J.; Dekker, J.M.; Fletcher, A.; Grauslund, J.; et al. Global Prevalence and Major Risk Factors of Diabetic Retinopathy. Diabetes Care 2012, 35, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Knudtson, M.D.; Lee, K.E.; Gangnon, R.; Klein, B.E.K. The Wisconsin Epidemiologic Study of Diabetic Retinopathy: XXII the twenty-five-year progression of retinopathy in persons with type 1 diabetes. Ophthalmology 2008, 115, 1859–1868. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, I.B.; Brownlee, M. Beyond Hemoglobin A1c—Need for Additional Markers of Risk for Diabetic Microvascular Complications. JAMA 2010, 303, 2291–2292. [Google Scholar] [CrossRef] [PubMed]
- Royle, P.; Mistry, H.; Auguste, P.; Shyangdan, D.; Freeman, K.; Lois, N.; Waugh, N. Pan-retinal photocoagulation and other forms of laser treatment and drug therapies for non-proliferative diabetic retinopathy: Systematic review and economic evaluation. Health Technol. Assess. 2015, 19, 1–247. [Google Scholar] [CrossRef]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef]
- Antonetti, D.A.; Barber, A.J.; Khin, S.; Lieth, E.; Tarbell, J.M.; Gardner, T.W. Vascular Permeability in Experimental Diabetes Is Associated With Reduced Endothelial Occludin Content: Vascular endothelial growth factor decreases occludin in retinal endothelial cells. Penn State Retina Research Group. Diabetes 1998, 47, 1953–1959. [Google Scholar] [CrossRef]
- Roy, S.; Maiello, M.; Lorenzi, M. Increased expression of basement membrane collagen in human diabetic retinopathy. J. Clin. Investig. 1994, 93, 438–442. [Google Scholar] [CrossRef]
- Biallosterski, C.; Van Velthoven, M.E.J.; Michels, R.P.J.; Schlingemann, R.O.; Devries, J.H.; Verbraak, F.D. Decreased optical coherence tomography-measured pericentral retinal thickness in patients with diabetes mellitus type 1 with minimal diabetic retinopathy. Br. J. Ophthalmol. 2007, 91, 1135–1138. [Google Scholar] [CrossRef]
- Van Dijk, H.W.; Kok, P.H.B.; Garvin, M.; Sonka, M.; Devries, J.H.; Michels, R.P.J.; Van Velthoven, M.E.J.; Schlingemann, R.O.; Verbraak, F.D.; AbraMoff, M.D. Selective Loss of Inner Retinal Layer Thickness in Type 1 Diabetic Patients with Minimal Diabetic Retinopathy. Investig. Opthalmol. Vis. Sci. 2009, 50, 340–3409. [Google Scholar] [CrossRef]
- Simo, R.; Hernandez, C.; European Consortium for the Early Treatment of Diabetic Retinopathy (EUROCONDOR). Neurodegeneration is an early event in diabetic retinopathy: Therapeutic implications. Br. J. Ophthalmol. 2012, 96, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Vujosevic, S.; Midena, E. Retinal Layers Changes in Human Preclinical and Early Clinical Diabetic Retinopathy Support Early Retinal Neuronal and Müller Cells Alterations. J. Diabetes Res. 2013, 905058. [Google Scholar] [CrossRef]
- Yang, J.H.; Kwak, H.W.; Kim, T.G.; Han, J.; Moon, S.W.; Yu, S.Y. Retinal Neurodegeneration in Type II Diabetic Otsuka Long-Evans Tokushima Fatty Rats. Investig. Opthalmol. Vis. Sci. 2013, 54, 3844–3851. [Google Scholar] [CrossRef] [PubMed]
- Wolter, J.R. Diabetic Retinopathy. Am. J. Ophthalmol. 1961, 51, 1123/1251–1141/1269. [Google Scholar] [CrossRef]
- Barber, A.J.; Lieth, E.; Khin, S.A.; Antonetti, D.A.; Buchanan, A.G.; Gardner, T.W. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J. Clin. Investig. 1998, 102, 783–791. [Google Scholar] [CrossRef]
- Sokol, S.; Moskowitz, A.; Skarf, B.; Evans, R.; Molitch, M.; Senior, B. Contrast Sensitivity in Diabetes With and without Background Retinopathy. Arch. Ophthalmol. 1985, 103, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.S.; Gunkel, R.D.; Podgor, M.J. Color Vision Defects in Early Diabetic Retinopathy. Arch. Ophthalmol. 1986, 104, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.-C. Selective Loss of S-Cones in Diabetic Retinopathy. Arch. Ophthalmol. 2000, 118, 1393–1400. [Google Scholar] [CrossRef] [PubMed]
- Wachtmeister, L. Oscillatory potentials in the retina: What do they reveal. Prog. Retin Eye Res. 1998, 17, 485–521. [Google Scholar] [CrossRef]
- Bresnick, G.H.; Korth, K.; Groo, A.; Palta, M. Electroretinographic oscillatory potentials predict progression of diabetic retinopathy. Preliminary report. Arch. Ophthalmol. 1984, 102, 1307–1311. [Google Scholar] [CrossRef] [PubMed]
- Coupland, S.G. A comparison of oscillatory potential and pattern electroretinogram measures in diabetic retinopathy. Doc. Ophthalmol. 1987, 66, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Metea, M.R.; Newman, E.A. Signalling within the neurovascular unit in the mammalian retina. Exp. Physiol. 2007, 92, 635–640. [Google Scholar] [CrossRef]
- Bilimoria, P.M.; Stevens, B. Microglia function during brain development: New insights from animal models. Brain Res. 2015, 1617, 7–17. [Google Scholar] [CrossRef]
- Koehler, R.C.; Gebremedhin, D.; Harder, D.R. Role of astrocytes in cerebrovascular regulation. J. Appl. Physiol. 2006, 100, 307–317. [Google Scholar] [CrossRef][Green Version]
- Kurihara, T. Development and pathological changes of neurovascular unit regulated by hypoxia response in the retina. Prog. Brain Res. 2016, 225, 201–211. [Google Scholar] [CrossRef]
- Gardner, T.W.; Davila, J.R. The neurovascular unit and the pathophysiologic basis of diabetic retinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 2017, 255, 1–6. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Chan, P.S. Oxidative stress and diabetic retinopathy. Exp. Diabetes Res. 2007, 2007, 43603. [Google Scholar] [CrossRef]
- Hammes, H.P. Diabetic retinopathy: Hyperglycaemia, oxidative stress and beyond. Diabetologia 2018, 61, 29–38. [Google Scholar] [CrossRef]
- Kim, J.A.; Wei, Y.; Sowers, J.R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef]
- Du, X.L.; Edelstein, D.; Rossetti, L.; Fantus, I.G.; Goldberg, H.; Ziyadeh, F.; Wu, J.; Brownlee, M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12222–12226. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.-I.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.-P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Veenstra, A.; Palczewski, K.; Kern, T.S. Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc. Natl. Acad. Sci. USA 2013, 110, 16586–16591. [Google Scholar] [CrossRef] [PubMed]
- Tonade, D.; Liu, H.; Kern, T.S. Photoreceptor Cells Produce Inflammatory Mediators That Contribute to Endothelial Cell Death in Diabetes. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4264–4271. [Google Scholar] [CrossRef]
- Arden, G.B. The absence of diabetic retinopathy in patients with retinitis pigmentosa: Implications for pathophysiology and possible treatment. Br. J. Ophthalmol. 2001, 85, 366–370. [Google Scholar] [CrossRef] [PubMed]
- De Gooyer, T.E.; Stevenson, K.A.; Humphries, P.; Simpson, D.A.C.; Gardiner, T.A.; Stitt, A.W. Retinopathy Is Reduced during Experimental Diabetes in a Mouse Model of Outer Retinal Degeneration. Investig. Opthalmol. Vis. Sci. 2006, 47, 5561–5568. [Google Scholar] [CrossRef] [PubMed]
- Jezek, P.; Holendova, B.; Plecita-Hlavata, L. Redox Signaling from Mitochondria: Signal Propagation and Its Targets. Biomolecules 2020, 10, 93. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Newman, N.J. Inherited eye-related disorders due to mitochondrial dysfunction. Hum. Mol. Genet. 2017, 26, R12–R20. [Google Scholar] [CrossRef]
- Hoegger, M.J.; Lieven, C.J.; Levin, L.A. Differential production of superoxide by neuronal mitochondria. BMC Neurosci. 2008, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Remor, A.P.; de Matos, F.J.; Ghisoni, K.; da Silva, T.L.; Eidt, G.; Burigo, M.; de Bem, A.F.; Silveira, P.C.; de Leon, A.; Sanchez, M.C.; et al. Differential effects of insulin on peripheral diabetes-related changes in mitochondrial bioenergetics: Involvement of advanced glycosylated end products. Biochim. Biophys. Acta 2011, 1812, 1460–1471. [Google Scholar] [CrossRef]
- Cogan, D.G.; Kuwabara, T. Capillary Shunts in the Pathogenesis of Diabetic Retinopathy. Diabetes 1963, 12, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, M.; Kern, T.S.; Lorenzi, M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J. Clin. Investig. 1996, 97, 2883–2890. [Google Scholar] [CrossRef] [PubMed]
- Franco, C.A.; Jones, M.L.; Bernabeu, M.O.; Geudens, I.; Mathivet, T.; Rosa, A.; Lopes, F.M.; Lima, A.P.; Ragab, A.; Collins, R.T.; et al. Dynamic Endothelial Cell Rearrangements Drive Developmental Vessel Regression. PLoS Biol. 2015, 13, e1002125. [Google Scholar] [CrossRef]
- Lenard, A.; Daetwyler, S.; Betz, C.; Ellertsdottir, E.; Belting, H.-G.; Huisken, J.; Affolter, M. Endothelial Cell Self-fusion during Vascular Pruning. PLoS Biol. 2015, 13, e1002126. [Google Scholar] [CrossRef]
- Cogan, D.G.; Kuwabara, T. The Mural Cell in Perspective. Arch. Ophthalmol. 1967, 78, 133–139. [Google Scholar] [CrossRef]
- Pfister, F.; Feng, Y.; vom Hagen, F.; Hoffmann, S.; Molema, G.; Hillebrands, J.L.; Shani, M.; Deutsch, U.; Hammes, H.P. Pericyte migration: A novel mechanism of pericyte loss in experimental diabetic retinopathy. Diabetes 2008, 57, 2495–2502. [Google Scholar] [CrossRef]
- Warmke, N.; Griffin, K.J.; Cubbon, R.M. Pericytes in diabetes-associated vascular disease. J. Diabetes Complicat. 2016, 30, 1643–1650. [Google Scholar] [CrossRef]
- Valdez, C.N.; Arboleda-Velasquez, J.F.; Amarnani, D.S.; Kim, L.A.; D’Amore, P.A. Retinal Microangiopathy in a Mouse Model of Inducible Mural Cell Loss. Am. J. Pathol. 2014, 184, 2618–2626. [Google Scholar] [CrossRef]
- Hammes, H.-P.; Feng, Y.; Pfister, F.; Brownlee, M. Diabetic Retinopathy: Targeting Vasoregression. Diabetes 2011, 60, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Santos, G.S.P.; Prazeres, P.; Mintz, A.; Birbrair, A. Role of pericytes in the retina. Eye 2018, 32, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; von Wendt, G.; Wanger, P.; Martin, L. Early detection of macular changes in patients with diabetes using Rarebit Fovea Test and optical coherence tomography. Br. J. Ophthalmol. 2007, 91, 1596–1598. [Google Scholar] [CrossRef] [PubMed]
- Valverde, A.M.; Miranda, S.; García-Ramírez, M.; González-Rodriguez, Á.; Hernández, C.; Simó, R. Proapoptotic and survival signaling in the neuroretina at early stages of diabetic retinopathy. Mol. Vis. 2013, 19, 47–53. [Google Scholar]
- Trudeau, K.; Molina, A.J.A.; Guo, W.; Roy, S. High Glucose Disrupts Mitochondrial Morphology in Retinal Endothelial Cells. Am. J. Pathol. 2010, 177, 447–455. [Google Scholar] [CrossRef]
- Kern, T.S.; Barber, A.J. Retinal ganglion cells in diabetes. J. Physiol. 2008, 586, 4401–4408. [Google Scholar] [CrossRef] [PubMed]
- Gastinger, M.J.; Singh, R.S.J.; Barber, A.J. Loss of Cholinergic and Dopaminergic Amacrine Cells in Streptozotocin-Diabetic Rat and Ins2Akita-Diabetic Mouse Retinas. Investig. Opthalmol. Vis. Sci. 2006, 47, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Szabo, K.; Enzsoly, A.; Dekany, B.; Szabo, A.; Hajdu, R.I.; Radovits, T.; Matyas, C.; Olah, A.; Laurik, L.K.; Somfai, G.M.; et al. Histological Evaluation of Diabetic Neurodegeneration in the Retina of Zucker Diabetic Fatty (ZDF) Rats. Sci. Rep. 2017, 7, 8891. [Google Scholar] [CrossRef]
- Rodrigues, E.B.; Urias, M.G.; Penha, F.M.; Badaro, E.; Novais, E.; Meirelles, R.; Farah, M.E. Diabetes induces changes in neuroretina before retinal vessels: A spectral-domain optical coherence tomography study. Int. J. Retina Vitreous 2015, 1, 4. [Google Scholar] [CrossRef]
- Barber, A.J.; Gardner, T.W.; Abcouwer, S.F. The Significance of Vascular and Neural Apoptosis to the Pathology of Diabetic Retinopathy. Investig. Opthalmol. Vis. Sci. 2011, 52, 1156–1163. [Google Scholar] [CrossRef]
- Enzsoly, A.; Szabo, A.; Kantor, O.; David, C.; Szalay, P.; Szabo, K.; Szel, A.; Nemeth, J.; Lukats, A. Pathologic alterations of the outer retina in streptozotocin-induced diabetes. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3686–3699. [Google Scholar] [CrossRef] [PubMed]
- Hombrebueno, J.R.; Chen, M.; Penalva, R.G.; Xu, H. Loss of synaptic connectivity, particularly in second order neurons is a key feature of diabetic retinal neuropathy in the Ins2Akita mouse. PLoS ONE 2014, 9, e97970. [Google Scholar] [CrossRef] [PubMed]
- Zarebska, A.; Czerny, K.; Bakiera, K.; Cichacz-Kwiatkowska, B.; Lis-Sochocka, M.; Kis, G.; Wojtowicz, Z. Histological changes in the retina in experimental alloxan-induced diabetes in rabbits. Ann. Univ. Mariae Curie Sklodowska Med. 2001, 56, 81–84. [Google Scholar] [PubMed]
- Alvarez, Y.; Chen, K.; Reynolds, A.L.; Waghorne, N.; O’Connor, J.J.; Kennedy, B.N. Predominant cone photoreceptor dysfunction in a hyperglycaemic model of non-proliferative diabetic retinopathy. Dis. Model. Mech. 2010, 3, 236–245. [Google Scholar] [CrossRef]
- Shin, H.J.; Chung, H.; Kim, H.C. Association between integrity of foveal photoreceptor layer and visual outcome in retinal vein occlusion. Acta Ophthalmol. 2011, 89, e35–e40. [Google Scholar] [CrossRef]
- Forooghian, F.; Stetson, P.F.; Meyer, S.A.; Chew, E.Y.; Wong, W.T.; Cukras, C.; Meyerle, C.B.; Ferris, F.L., 3rd. Relationship between photoreceptor outer segment length and visual acuity in diabetic macular edema. Retina 2010, 30, 63–70. [Google Scholar] [CrossRef]
- Ito, S.; Miyamoto, N.; Ishida, K.; Kurimoto, Y. Association between external limiting membrane status and visual acuity in diabetic macular oedema. Br. J. Ophthalmol. 2013, 97, 228–232. [Google Scholar] [CrossRef]
- Park, S.H.; Park, J.W.; Park, S.J.; Kim, K.Y.; Chung, J.W.; Chun, M.H.; Oh, S.J. Apoptotic death of photoreceptors in the streptozotocin-induced diabetic rat retina. Diabetologia 2003, 46, 1260–1268. [Google Scholar] [CrossRef]
- Ottlecz, A.; Bensaoula, T. Captopril ameliorates the decreased Na+,K(+)-ATPase activity in the retina of streptozotocin-induced diabetic rats. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1633–1641. [Google Scholar]
- Ottlecz, A.; Garcia, C.A.; Eichberg, J.; Fox, D.A. Alterations in retinal Na+, K(+)-ATPase in diabetes: Streptozotocin-induced and Zucker diabetic fatty rats. Curr. Eye Res. 1993, 12, 1111–1121. [Google Scholar] [CrossRef]
- Phipps, J.A.; Fletcher, E.L.; Vingrys, A.J. Paired-flash identification of rod and cone dysfunction in the diabetic rat. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4592–4600. [Google Scholar] [CrossRef]
- Kern, T.S.; Berkowitz, B.A. Photoreceptors in diabetic retinopathy. J. Diabetes Investig. 2015, 6, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Country, M.W. Retinal metabolism: A comparative look at energetics in the retina. Brain Res. 2017, 1672, 50–57. [Google Scholar] [CrossRef]
- Tang, P.H.; Kono, M.; Koutalos, Y.; Ablonczy, Z.; Crouch, R.K. New insights into retinoid metabolism and cycling within the retina. Prog. Retin Eye Res. 2013, 32, 48–63. [Google Scholar] [CrossRef]
- Kolesnikov, A.V.; Ala-Laurila, P.; Shukolyukov, S.A.; Crouch, R.K.; Wiggert, B.; Estevez, M.E.; Govardovskii, V.I.; Cornwall, M.C. Visual cycle and its metabolic support in gecko photoreceptors. Vision Res. 2007, 47, 363–374. [Google Scholar] [CrossRef] [PubMed][Green Version]
- LaVail, M.M. Rod outer segment disk shedding in rat retina: Relationship to cyclic lighting. Science 1976, 194, 1071–1074. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Iannaccone, A.; Jablonski, M.M. Contribution of Muller cells toward the regulation of photoreceptor outer segment assembly. Neuron Glia Biol. 2005, 1, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, M.M.; Iannaccone, A. Targeted disruption of Muller cell metabolism induces photoreceptor dysmorphogenesis. Glia 2000, 32, 192–204. [Google Scholar] [CrossRef]
- Hurley, J.B. Warburg’s vision. Elife 2017, 6, e29217. [Google Scholar] [CrossRef]
- Rueda, E.M.; Johnson, J.E., Jr.; Giddabasappa, A.; Swaroop, A.; Brooks, M.J.; Sigel, I.; Chaney, S.Y.; Fox, D.A. The cellular and compartmental profile of mouse retinal glycolysis, tricarboxylic acid cycle, oxidative phosphorylation, and ~P transferring kinases. Mol. Vis. 2016, 22, 847–885. [Google Scholar]
- Mitchell, P. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. 1966. Biochim. Biophys. Acta 2011, 1807, 1507–1538. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Rountree, A.; Cleghorn, W.M.; Contreras, L.; Lindsay, K.J.; Sadilek, M.; Gu, H.; Djukovic, D.; Raftery, D.; Satrústegui, J.; et al. Phototransduction Influences Metabolic Flux and Nucleotide Metabolism in Mouse Retina. J. Biol. Chem. 2016, 291, 4698–4710. [Google Scholar] [CrossRef] [PubMed]
- Linsenmeier, R.A. Effects of light and darkness on oxygen distribution and consumption in the cat retina. J. Gen. Physiol. 1986, 88, 521–542. [Google Scholar] [CrossRef] [PubMed]
- Ames, A., 3rd; Li, Y.Y.; Heher, E.C.; Kimble, C.R. Energy metabolism of rabbit retina as related to function: High cost of Na+ transport. J. Neurosci. 1992, 12, 840–853. [Google Scholar] [CrossRef]
- Perezleon, J.A.; Osorio-Paz, I.; Francois, L.; Salceda, R. Immunohistochemical localization of glycogen synthase and GSK3beta: Control of glycogen content in retina. Neurochem. Res. 2013, 38, 1063–1069. [Google Scholar] [CrossRef]
- Winkler, B.S.; Arnold, M.J.; Brassell, M.A.; Puro, D.G. Energy metabolism in human retinal Muller cells. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3183–3190. [Google Scholar]
- Toft-Kehler, A.K.; Skytt, D.M.; Svare, A.; Lefevere, E.; Van Hove, I.; Moons, L.; Waagepetersen, H.S.; Kolko, M. Mitochondrial function in Müller cells—Does it matter? Mitochondrion 2017, 36, 43–51. [Google Scholar] [CrossRef]
- Tsacopoulos, M.; Poitry-Yamate, C.L.; MacLeish, P.R.; Poitry, S. Trafficking of molecules and metabolic signals in the retina. Prog. Retin Eye Res. 1998, 17, 429–442. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Bayeva, M.; Sawicki, K.T.; Ardehali, H. Taking diabetes to heart--deregulation of myocardial lipid metabolism in diabetic cardiomyopathy. J. Am. Heart Assoc. 2013, 2, e000433. [Google Scholar] [CrossRef]
- Muoio, D.M. Metabolic inflexibility: When mitochondrial indecision leads to metabolic gridlock. Cell 2014, 159, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Joyal, J.S.; Sun, Y.; Gantner, M.L.; Shao, Z.; Evans, L.P.; Saba, N.; Fredrick, T.; Burnim, S.; Kim, J.S.; Patel, G.; et al. Retinal lipid and glucose metabolism dictates angiogenesis through the lipid sensor Ffar1. Nat. Med. 2016, 22, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Tyni, T.; Johnson, M.; Eaton, S.; Pourfarzam, M.; Andrews, R.; Turnbull, D.M. Mitochondrial fatty acid beta-oxidation in the retinal pigment epithelium. Pediatr. Res. 2002, 52, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Atsuzawa, K.; Nakazawa, A.; Mizutani, K.; Fukasawa, M.; Yamamoto, N.; Hashimoto, T.; Usuda, N. Immunohistochemical localization of mitochondrial fatty acid beta-oxidation enzymes in Muller cells of the retina. Histochem. Cell. Biol. 2010, 134, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.L.; Pennesi, M.E.; Harding, C.O.; Weleber, R.G.; Gillingham, M.B. Observations regarding retinopathy in mitochondrial trifunctional protein deficiencies. Mol. Genet. Metab. 2012, 106, 18–24. [Google Scholar] [CrossRef]
- Pearsall, E.A.; Cheng, R.; Matsuzaki, S.; Zhou, K.; Ding, L.; Ahn, B.; Kinter, M.; Humphries, K.M.; Quiambao, A.B.; Farjo, R.A.; et al. Neuroprotective effects of PPARalpha in retinopathy of type 1 diabetes. PLoS ONE 2019, 14, e0208399. [Google Scholar] [CrossRef]
- Group, A.S.; Group, A.E.S.; Chew, E.Y.; Ambrosius, W.T.; Davis, M.D.; Danis, R.P.; Gangaputra, S.; Greven, C.M.; Hubbard, L.; Esser, B.A.; et al. Effects of medical therapies on retinopathy progression in type 2 diabetes. N. Engl. J. Med. 2010, 363, 233–244. [Google Scholar] [CrossRef]
- Keech, A.C.; Mitchell, P.; Summanen, P.A.; O’Day, J.; Davis, T.M.; Moffitt, M.S.; Taskinen, M.R.; Simes, R.J.; Tse, D.; Williamson, E.; et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): A randomised controlled trial. Lancet 2007, 370, 1687–1697. [Google Scholar] [CrossRef]
- Schoors, S.; Bruning, U.; Missiaen, R.; Queiroz, K.C.; Borgers, G.; Elia, I.; Zecchin, A.; Cantelmo, A.R.; Christen, S.; Goveia, J.; et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015, 520, 192–197. [Google Scholar] [CrossRef]
- Masser, D.R.; Otalora, L.; Clark, N.W.; Kinter, M.T.; Elliott, M.H.; Freeman, W.M. Functional changes in the neural retina occur in the absence of mitochondrial dysfunction in a rodent model of diabetic retinopathy. J. Neurochem. 2017, 143, 595–608. [Google Scholar] [CrossRef]
- Lopaschuk, G.D. Fatty Acid Oxidation and Its Relation with Insulin Resistance and Associated Disorders. Ann. Nutr. Metab. 2016, 68 (Suppl. 3), 15–20. [Google Scholar] [CrossRef]
- Swarup, A.; Samuels, I.S.; Bell, B.A.; Han, J.Y.S.; Du, J.; Massenzio, E.; Abel, E.D.; Boesze-Battaglia, K.; Peachey, N.S.; Philp, N.J. Modulating GLUT1 expression in retinal pigment epithelium decreases glucose levels in the retina: Impact on photoreceptors and Muller glial cells. Am. J. Physiol. Cell Physiol. 2019, 316, C121–C133. [Google Scholar] [CrossRef]
- Sanchez-Chavez, G.; Pena-Rangel, M.T.; Riesgo-Escovar, J.R.; Martinez-Martinez, A.; Salceda, R. Insulin stimulated-glucose transporter Glut 4 is expressed in the retina. PLoS ONE 2012, 7, e52959. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H.; et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 2003, 422, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Falomir-Lockhart, L.J.; Cavazzutti, G.F.; Gimenez, E.; Toscani, A.M. Fatty Acid Signaling Mechanisms in Neural Cells: Fatty Acid Receptors. Front. Cell Neurosci. 2019, 13, 162. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Mansfield, K.D.; Bertozzi, C.C.; Rudenko, V.; Chan, D.A.; Giaccia, A.J.; Simon, M.C. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol. Cell Biol. 2007, 27, 912–925. [Google Scholar] [CrossRef] [PubMed]
- Germer, A.; Biedermann, B.; Wolburg, H.; Schuck, J.; Grosche, J.; Kuhrt, H.; Reichelt, W.; Schousboe, A.; Paasche, G.; Mack, A.F.; et al. Distribution of mitochondria within Muller cells--I. Correlation with retinal vascularization in different mammalian species. J. Neurocytol. 1998, 27, 329–345. [Google Scholar] [CrossRef]
- Stone, J.; van Driel, D.; Valter, K.; Rees, S.; Provis, J. The locations of mitochondria in mammalian photoreceptors: Relation to retinal vasculature. Brain Res. 2008, 1189, 58–69. [Google Scholar] [CrossRef]
- Osorio-Paz, I.; Uribe-Carvajal, S.; Salceda, R. In the Early Stages of Diabetes, Rat Retinal Mitochondria Undergo Mild Uncoupling due to UCP2 Activity. PLoS ONE 2015, 10, e0122727. [Google Scholar] [CrossRef]
- Han, W.H.; Gotzmann, J.; Kuny, S.; Huang, H.; Chan, C.B.; Lemieux, H.; Sauve, Y. Modifications in Retinal Mitochondrial Respiration Precede Type 2 Diabetes and Protracted Microvascular Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3826–3839. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Mishra, M.; Kowluru, A.; Kumar, B. Hyperlipidemia and the development of diabetic retinopathy: Comparison between type 1 and type 2 animal models. Metabolism 2016, 65, 1570–1581. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, M.; Chan, P.S.; Kern, T.S.; Kowluru, R.A. Oxidative damage in the retinal mitochondria of diabetic mice: Possible protection by superoxide dismutase. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3805–3811. [Google Scholar] [CrossRef] [PubMed]
- Madsen-Bouterse, S.A.; Mohammad, G.; Kanwar, M.; Kowluru, R.A. Role of mitochondrial DNA damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxid. Redox Signal. 2010, 13, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; Tewari, S.; Kowluru, R.A. A compensatory mechanism protects retinal mitochondria from initial insult in diabetic retinopathy. Free Radic. Biol. Med. 2012, 53, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zheng, Z.; Gong, Y.; Zhu, B.; Xu, X. U83836E inhibits retinal neurodegeneration in early-stage streptozotocin-induced diabetic rats. Ophthalmic Res. 2011, 46, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Lai, J.; Yuan, Y.; Wang, L.; Wang, Q.; Yuan, F. Taurine Protects Retinal Cells and Improves Synaptic Connections in Early Diabetic Rats. Curr. Eye Res. 2020, 45, 52–63. [Google Scholar] [CrossRef]
- Li, X.; Zhang, M.; Zhou, H. The morphological features and mitochondrial oxidative stress mechanism of the retinal neurons apoptosis in early diabetic rats. J. Diabetes Res. 2014, 2014, 678123. [Google Scholar] [CrossRef]
- Chang, J.Y.; Yu, F.; Shi, L.; Ko, M.L.; Ko, G.Y. Melatonin Affects Mitochondrial Fission/Fusion Dynamics in the Diabetic Retina. J. Diabetes Res. 2019, 2019, 8463125. [Google Scholar] [CrossRef]
- Devi, T.S.; Lee, I.; Huttemann, M.; Kumar, A.; Nantwi, K.D.; Singh, L.P. TXNIP links innate host defense mechanisms to oxidative stress and inflammation in retinal Muller glia under chronic hyperglycemia: Implications for diabetic retinopathy. Exp. Diabetes Res. 2012, 2012, 438238. [Google Scholar] [CrossRef]
- Devi, T.S.; Somayajulu, M.; Kowluru, R.A.; Singh, L.P. TXNIP regulates mitophagy in retinal Muller cells under high-glucose conditions: Implications for diabetic retinopathy. Cell Death Dis. 2017, 8, e2777. [Google Scholar] [CrossRef]
- Hombrebueno, J.R.; Cairns, L.; Dutton, L.R.; Lyons, T.J.; Brazil, D.P.; Moynagh, P.; Curtis, T.M.; Xu, H. Uncoupled turnover disrupts mitochondrial quality control in diabetic retinopathy. JCI Insight 2019, 4, e129760. [Google Scholar] [CrossRef] [PubMed]
- Krugel, K.; Wurm, A.; Pannicke, T.; Hollborn, M.; Karl, A.; Wiedemann, P.; Reichenbach, A.; Kohen, L.; Bringmann, A. Involvement of oxidative stress and mitochondrial dysfunction in the osmotic swelling of retinal glial cells from diabetic rats. Exp. Eye Res. 2011, 92, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Tien, T.; Zhang, J.; Muto, T.; Kim, D.; Sarthy, V.P.; Roy, S. High Glucose Induces Mitochondrial Dysfunction in Retinal Muller Cells: Implications for Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2915–2921. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Xie, W.; Meng, X.; Zhai, Y.; Dong, X.; Zhang, X.; Sun, G.; Sun, X. Notoginsenoside R1 Ameliorates Diabetic Retinopathy through PINK1-Dependent Activation of Mitophagy. Cells 2019, 8, 213. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Kowluru, R.A. DNA Methylation-a Potential Source of Mitochondria DNA Base Mismatch in the Development of Diabetic Retinopathy. Mol. Neurobiol. 2019, 56, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.; Duraisamy, A.J.; Kowluru, A.; Kowluru, R.A. Functional Regulation of an Oxidative Stress Mediator, Rac1, in Diabetic Retinopathy. Mol. Neurobiol. 2019, 56, 8643–8655. [Google Scholar] [CrossRef]
- Santos, J.M.; Kowluru, R.A. Role of mitochondria biogenesis in the metabolic memory associated with the continued progression of diabetic retinopathy and its regulation by lipoic acid. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8791–8798. [Google Scholar] [CrossRef]
- Santos, J.M.; Kowluru, R.A. Impaired transport of mitochondrial transcription factor A (TFAM) and the metabolic memory phenomenon associated with the progression of diabetic retinopathy. Diabetes Metab. Res. Rev. 2013, 29, 204–213. [Google Scholar] [CrossRef]
- Santos, J.M.; Tewari, S.; Goldberg, A.F.; Kowluru, R.A. Mitochondrial biogenesis and the development of diabetic retinopathy. Free Radic. Biol. Med. 2011, 51, 1849–1860. [Google Scholar] [CrossRef]
- Schulze, P.C.; Yoshioka, J.; Takahashi, T.; He, Z.; King, G.L.; Lee, R.T. Hyperglycemia promotes oxidative stress through inhibition of thioredoxin function by thioredoxin-interacting protein. J. Biol. Chem. 2004, 279, 30369–30374. [Google Scholar] [CrossRef]
- Zou, Y.L.; Luo, W.B.; Xie, L.; Mao, X.B.; Wu, C.; You, Z.P. Targeting human 8-oxoguanine DNA glycosylase to mitochondria protects cells from high glucose-induced apoptosis. Endocrine 2018, 60, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, W.; Zhao, Y.; Shu, X.; Wang, W.; Wang, D.; Yang, Y.; He, Z.; Wang, X.; Ying, Y. GSK3beta-mediated tau hyperphosphorylation triggers diabetic retinal neurodegeneration by disrupting synaptic and mitochondrial functions. Mol. Neurodegener. 2018, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.Y.; Liou, S.S.; Hong, T.Y.; Liu, I.M. Protective Effects of Hesperidin (Citrus Flavonone) on High Glucose Induced Oxidative Stress and Apoptosis in a Cellular Model for Diabetic Retinopathy. Nutrients 2017, 9, 1312. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mishra, M.; Kumar, B. Diabetic retinopathy and transcriptional regulation of a small molecular weight G-Protein, Rac1. Exp. Eye Res. 2016, 147, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Devi, T.S.; Hosoya, K.; Terasaki, T.; Singh, L.P. Critical role of TXNIP in oxidative stress, DNA damage and retinal pericyte apoptosis under high glucose: Implications for diabetic retinopathy. Exp. Cell Res. 2013, 319, 1001–1012. [Google Scholar] [CrossRef]
- Duraisamy, A.J.; Mohammad, G.; Kowluru, R.A. Mitochondrial fusion and maintenance of mitochondrial homeostasis in diabetic retinopathy. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1617–1626. [Google Scholar] [CrossRef]
- Leal, E.C.; Aveleira, C.A.; Castilho, A.F.; Serra, A.M.; Baptista, F.I.; Hosoya, K.; Forrester, J.V.; Ambrosio, A.F. High glucose and oxidative/nitrosative stress conditions induce apoptosis in retinal endothelial cells by a caspase-independent pathway. Exp. Eye Res. 2009, 88, 983–991. [Google Scholar] [CrossRef]
- Madsen-Bouterse, S.A.; Zhong, Q.; Mohammad, G.; Ho, Y.S.; Kowluru, R.A. Oxidative damage of mitochondrial DNA in diabetes and its protection by manganese superoxide dismutase. Free Radic. Res. 2010, 44, 313–321. [Google Scholar] [CrossRef]
- Xie, L.; Zhu, X.; Hu, Y.; Li, T.; Gao, Y.; Shi, Y.; Tang, S. Mitochondrial DNA oxidative damage triggering mitochondrial dysfunction and apoptosis in high glucose-induced HRECs. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4203–4209. [Google Scholar] [CrossRef]
- Van Meerloo, J.; Kaspers, G.J.; Cloos, J. Cell sensitivity assays: The MTT assay. Methods Mol. Biol. 2011, 731, 237–245. [Google Scholar] [CrossRef]
- Betteridge, D.J. What is oxidative stress? Metabolism 2000, 49, 3–8. [Google Scholar] [CrossRef]
- Kowluru, R.A. Diabetic retinopathy, metabolic memory and epigenetic modifications. Vision Res. 2017, 139, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Tang, J.; Kern, T.S. Abnormalities of retinal metabolism in diabetes and experimental galactosemia. VII. Effect of long-term administration of antioxidants on the development of retinopathy. Diabetes 2001, 50, 1938–1942. [Google Scholar] [CrossRef]
- Kowluru, R.; Kern, T.S.; Engerman, R.L. Abnormalities of retinal metabolism in diabetes or galactosemia. II. Comparison of gamma-glutamyl transpeptidase in retina and cerebral cortex, and effects of antioxidant therapy. Curr. Eye Res. 1994, 13, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, B.A.; Gradianu, M.; Bissig, D.; Kern, T.S.; Roberts, R. Retinal ion regulation in a mouse model of diabetic retinopathy: Natural history and the effect of Cu/Zn superoxide dismutase overexpression. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2351–2358. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Abbas, S.N. Diabetes-induced mitochondrial dysfunction in the retina. Investig. Ophthalmol. Vis. Sci. 2003, 44, 5327–5334. [Google Scholar] [CrossRef]
- Madsen-Bouterse, S.; Mohammad, G.; Kowluru, R.A. Glyceraldehyde-3-phosphate dehydrogenase in retinal microvasculature: Implications for the development and progression of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1765–1772. [Google Scholar] [CrossRef]
- Lott, M.E.; Slocomb, J.E.; Shivkumar, V.; Smith, B.; Gabbay, R.A.; Quillen, D.; Gardner, T.W.; Bettermann, K. Comparison of retinal vasodilator and constrictor responses in type 2 diabetes. Acta Ophthalmol. 2012, 90, e434–e441. [Google Scholar] [CrossRef]
- Du, Y.; Miller, C.M.; Kern, T.S. Hyperglycemia increases mitochondrial superoxide in retina and retinal cells. Free Radic. Biol. Med. 2003, 35, 1491–1499. [Google Scholar] [CrossRef]
- Adam-Vizi, V. Production of reactive oxygen species in brain mitochondria: Contribution by electron transport chain and non-electron transport chain sources. Antioxid. Redox. Signal. 2005, 7, 1140–1149. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bek, T. Mitochondrial dysfunction and diabetic retinopathy. Mitochondrion 2017, 36, 4–6. [Google Scholar] [CrossRef]
- Mekala, N.K.; Kurdys, J.; Depuydt, M.M.; Vazquez, E.J.; Rosca, M.G. Apoptosis inducing factor deficiency causes retinal photoreceptor degeneration. The protective role of the redox compound methylene blue. Redox. Biol. 2019, 20, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.; Klimcakova, E.; St-Pierre, J. Impact of PGC-1alpha on the topology and rate of superoxide production by the mitochondrial electron transport chain. Free Radic. Biol. Med. 2011, 51, 2243–2248. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Vazquez, E.J.; Chen, Q.; Kerner, J.; Kern, T.S.; Hoppel, C.L. Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes 2012, 61, 2074–2083. [Google Scholar] [CrossRef]
- Ji, L.L.; Gomez-Cabrera, M.C.; Vina, J. Exercise and hormesis: Activation of cellular antioxidant signaling pathway. Ann. N. Y. Acad. Sci. 2006, 1067, 425–435. [Google Scholar] [CrossRef]
- Roy, S.; Kern, T.S.; Song, B.; Stuebe, C. Mechanistic Insights into Pathological Changes in the Diabetic Retina: Implications for Targeting Diabetic Retinopathy. Am. J. Pathol. 2017, 187, 9–19. [Google Scholar] [CrossRef]
- Tang, J.; Kern, T.S. Inflammation in diabetic retinopathy. Prog. Retin Eye Res. 2011, 30, 343–358. [Google Scholar] [CrossRef]
- Tewari, S.; Santos, J.M.; Kowluru, R.A. Damaged mitochondrial DNA replication system and the development of diabetic retinopathy. Antioxid. Redox. Signal. 2012, 17, 492–504. [Google Scholar] [CrossRef]
- Tewari, S.; Zhong, Q.; Santos, J.M.; Kowluru, R.A. Mitochondria DNA replication and DNA methylation in the metabolic memory associated with continued progression of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4881–4888. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Brachmann, C.B.; Celic, I.; Kenna, M.A.; Muhammad, S.; Starai, V.J.; Avalos, J.L.; Escalante-Semerena, J.C.; Grubmeyer, C.; Wolberger, C.; et al. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc. Natl. Acad. Sci. USA 2000, 97, 6658–6663. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Sauve, A.A.; Bai, P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol. Aspects Med. 2013, 34, 1168–1201. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.B.; Kubota, S.; Ban, N.; Yoshida, M.; Santeford, A.; Sene, A.; Nakamura, R.; Zapata, N.; Kubota, M.; Tsubota, K.; et al. NAMPT-Mediated NAD(+) Biosynthesis Is Essential for Vision In Mice. Cell Rep. 2016, 17, 69–85. [Google Scholar] [CrossRef]
- Ido, Y.; Nyengaard, J.R.; Chang, K.; Tilton, R.G.; Kilo, C.; Mylari, B.L.; Oates, P.J.; Williamson, J.R. Early neural and vascular dysfunctions in diabetic rats are largely sequelae of increased sorbitol oxidation. Antioxid. Redox. Signal. 2010, 12, 39–51. [Google Scholar] [CrossRef]
- Williamson, J.R.; Chang, K.; Frangos, M.; Hasan, K.S.; Ido, Y.; Kawamura, T.; Nyengaard, J.R.; van den Enden, M.; Kilo, C.; Tilton, R.G. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 1993, 42, 801–813. [Google Scholar] [CrossRef]
- Lorenzi, M. The polyol pathway as a mechanism for diabetic retinopathy: Attractive, elusive, and resilient. Exp. Diabetes Res. 2007, 2007, 61038. [Google Scholar] [CrossRef]
- Li, Q.; Hwang, Y.C.; Ananthakrishnan, R.; Oates, P.J.; Guberski, D.; Ramasamy, R. Polyol pathway and modulation of ischemia-reperfusion injury in Type 2 diabetic BBZ rat hearts. Cardiovasc. Diabetol. 2008, 7, 33. [Google Scholar] [CrossRef]
- Obrosova, I.G.; Drel, V.R.; Kumagai, A.K.; Szabo, C.; Pacher, P.; Stevens, M.J. Early diabetes-induced biochemical changes in the retina: Comparison of rat and mouse models. Diabetologia 2006, 49, 2525–2533. [Google Scholar] [CrossRef]
- Tang, J.; Du, Y.; Petrash, J.M.; Sheibani, N.; Kern, T.S. Deletion of aldose reductase from mice inhibits diabetes-induced retinal capillary degeneration and superoxide generation. PLoS ONE 2013, 8, e62081. [Google Scholar] [CrossRef]
- Berthiaume, J.M.; Kurdys, J.G.; Muntean, D.M.; Rosca, M.G. Mitochondrial NAD(+)/NADH Redox State and Diabetic Cardiomyopathy. Antioxid. Redox Signal. 2019, 30, 375–398. [Google Scholar] [CrossRef]
- Diederen, R.M.H.; Starnes, C.A.; Berkowitz, B.A.; Winkler, B.S. Reexamining the Hyperglycemic Pseudohypoxia Hypothesis of Diabetic Oculopathy. Investig. Ophtal. Vis. Sci. 2006, 47, 2726. [Google Scholar] [CrossRef] [PubMed]
- Karamanlidis, G.; Lee, C.F.; Garcia-Menendez, L.; Kolwicz, S.C., Jr.; Suthammarak, W.; Gong, G.; Sedensky, M.M.; Morgan, P.G.; Wang, W.; Tian, R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013, 18, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.J.; Ma, W.; Wang, P.Y.; Hynes, J.; O’Riordan, T.C.; Combs, C.A.; McCoy, J.P., Jr.; Bunz, F.; Kang, J.G.; Hwang, P.M. Mitochondrial respiration protects against oxygen-associated DNA damage. Nat. Commun. 2010, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Akie, T.E.; Liu, L.; Nam, M.; Lei, S.; Cooper, M.P. OXPHOS-Mediated Induction of NAD+ Promotes Complete Oxidation of Fatty Acids and Interdicts Non-Alcoholic Fatty Liver Disease. PLoS ONE 2015, 10, e0125617. [Google Scholar] [CrossRef]
- Masutani, M.; Suzuki, H.; Kamada, N.; Watanabe, M.; Ueda, O.; Nozaki, T.; Jishage, K.; Watanabe, T.; Sugimoto, T.; Nakagama, H.; et al. Poly(ADP-ribose) polymerase gene disruption conferred mice resistant to streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. USA 1999, 96, 2301–2304. [Google Scholar] [CrossRef]
- Bai, P.; Canto, C.; Oudart, H.; Brunyanszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef]
- Mishra, M.; Duraisamy, A.J.; Kowluru, R.A. Sirt1- A Guardian of the Development of Diabetic Retinopathy. Diabetes 2018, 67, 745–754. [Google Scholar] [CrossRef]
- Rydstrom, J. Mitochondrial NADPH, transhydrogenase and disease. Biochim. Biophys. Acta 2006, 1757, 721–726. [Google Scholar] [CrossRef]
- Nickel, A.G.; von Hardenberg, A.; Hohl, M.; Loffler, J.R.; Kohlhaas, M.; Becker, J.; Reil, J.C.; Kazakov, A.; Bonnekoh, J.; Stadelmaier, M.; et al. Reversal of Mitochondrial Transhydrogenase Causes Oxidative Stress in Heart Failure. Cell Metab. 2015, 22, 472–484. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.H.; Lin, C.T.; Ryan, T.E.; Reese, L.R.; Gilliam, L.A.; Cathey, B.L.; Lark, D.S.; Smith, C.D.; Muoio, D.M.; Neufer, P.D. Pyruvate dehydrogenase complex and nicotinamide nucleotide transhydrogenase constitute an energy-consuming redox circuit. Biochem. J. 2015, 467, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, J.A.; Francisco, A.; Passos, L.A.; Figueira, T.R.; Castilho, R.F. The Contribution of Nicotinamide Nucleotide Transhydrogenase to Peroxide Detoxification Is Dependent on the Respiratory State and Counterbalanced by Other Sources of NADPH in Liver Mitochondria. J. Biol. Chem. 2016, 291, 20173–20187. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of PGC-1alpha. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; Mishra, M.; Kowluru, R.A. Posttranslational modification of mitochondrial transcription factor A in impaired mitochondria biogenesis: Implications in diabetic retinopathy and metabolic memory phenomenon. Exp. Eye Res. 2014, 121, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Devi, T.S.; Yumnamcha, T.; Yao, F.; Somayajulu, M.; Kowluru, R.A.; Singh, L.P. TXNIP mediates high glucose-induced mitophagic flux and lysosome enlargement in human retinal pigment epithelial cells. Biol. Open 2019, 8, bio038521. [Google Scholar] [CrossRef]
- Santos, J.M.; Mohammad, G.; Zhong, Q.; Kowluru, R.A. Diabetic retinopathy, superoxide damage and antioxidants. Curr. Pharm. Biotechnol. 2011, 12, 352–361. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Zhong, Q.; Kowluru, R.A. Diabetic retinopathy and damage to mitochondrial structure and transport machinery. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8739–8746. [Google Scholar] [CrossRef]
- Berkowitz, B.A.; Roberts, R.; Luan, H.; Bissig, D.; Bui, B.V.; Gradianu, M.; Calkins, D.J.; Vingrys, A.J. Manganese-enhanced MRI studies of alterations of intraretinal ion demand in models of ocular injury. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3796–3804. [Google Scholar] [CrossRef]
- Bangi, B.B.; Ginjupally, U.; Nadendla, L.K.; Mekala, M.R.; Lakshmi B, J.; Kakumani, A. Evaluation of Gustatory Function in Oral Submucous Fibrosis Patients and Gutka Chewers. Asian Pac. J. Cancer Prev. 2019, 20, 569–573. [Google Scholar] [CrossRef]
- Haider, S.Z.; Sadanandan, N.P.; Joshi, P.G.; Mehta, B. Early Diabetes Induces Changes in Mitochondrial Physiology of Inner Retinal Neurons. Neuroscience 2019, 406, 140–149. [Google Scholar] [CrossRef]
- Kern, T.S.; Kowluru, R.A.; Engerman, R.L. Abnormalities of retinal metabolism in diabetes or galactosemia: ATPases and glutathione. Investig. Ophthalmol. Vis. Sci. 1994, 35, 2962–2967. [Google Scholar]
- Berkowitz, B.A.; Roberts, R.; Stemmler, A.; Luan, H.; Gradianu, M. Impaired apparent ion demand in experimental diabetic retinopathy: Correction by lipoic Acid. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4753–4758. [Google Scholar] [CrossRef][Green Version]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Chen, C.; Peng, S.; Chen, F.; Liu, L.; Li, Z.; Zeng, G.; Huang, Q. Protective effects of pioglitazone on vascular endothelial cell dysfunction induced by high glucose via inhibition of IKKalpha/beta-NFkappaB signaling mediated by PPARgamma in vitro. Can. J. Physiol. Pharmacol. 2017, 95, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol. 2017, 5, 90. [Google Scholar] [CrossRef]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef]
- Szeto, H.H.; Liu, S.; Soong, Y.; Birk, A.V. Improving mitochondrial bioenergetics under ischemic conditions increases warm ischemia tolerance in the kidney. Am. J. Physiol. Renal Physiol. 2015, 308, F11–F21. [Google Scholar] [CrossRef]
- Alam, N.M.; Mills, W.C.t.; Wong, A.A.; Douglas, R.M.; Szeto, H.H.; Prusky, G.T. A mitochondrial therapeutic reverses visual decline in mouse models of diabetes. Dis. Model. Mech. 2015, 8, 701–710. [Google Scholar] [CrossRef]
- Huang, J.; Li, X.; Li, M.; Li, J.; Xiao, W.; Ma, W.; Chen, X.; Liang, X.; Tang, S.; Luo, Y. Mitochondria-targeted antioxidant peptide SS31 protects the retinas of diabetic rats. Curr. Mol. Med. 2013, 13, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Lopez, I.; Diaz-Morales, N.; Iannantuoni, F.; Lopez-Domenech, S.; de Maranon, A.M.; Abad-Jimenez, Z.; Banuls, C.; Rovira-Llopis, S.; Herance, J.R.; Rocha, M.; et al. The mitochondrial antioxidant SS-31 increases SIRT1 levels and ameliorates inflammation, oxidative stress and leukocyte-endothelium interactions in type 2 diabetes. Sci. Rep. 2018, 8, 15862. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Esteves, T.C. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005, 2, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef]
- Arsenijevic, D.; Onuma, H.; Pecqueur, C.; Raimbault, S.; Manning, B.S.; Miroux, B.; Couplan, E.; Alves-Guerra, M.C.; Goubern, M.; Surwit, R.; et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 2000, 26, 435–439. [Google Scholar] [CrossRef]
- He, Y.; Luan, Z.; Fu, X.; Xu, X. Overexpression of uncoupling protein 2 inhibits the high glucose-induced apoptosis of human umbilical vein endothelial cells. Int. J. Mol. Med. 2016, 37, 631–638. [Google Scholar] [CrossRef]
- Cui, Y.; Xu, X.; Bi, H.; Zhu, Q.; Wu, J.; Xia, X.; Qiushi, R.; Ho, P.C. Expression modification of uncoupling proteins and MnSOD in retinal endothelial cells and pericytes induced by high glucose: The role of reactive oxygen species in diabetic retinopathy. Exp. Eye Res. 2006, 83, 807–816. [Google Scholar] [CrossRef]
- Hinder, L.M.; Sas, K.M.; O’Brien, P.D.; Backus, C.; Kayampilly, P.; Hayes, J.M.; Lin, C.M.; Zhang, H.; Shanmugam, S.; Rumora, A.E.; et al. Mitochondrial uncoupling has no effect on microvascular complications in type 2 diabetes. Sci. Rep. 2019, 9, 881. [Google Scholar] [CrossRef]
- Wen, Y.; Li, W.; Poteet, E.C.; Xie, L.; Tan, C.; Yan, L.J.; Ju, X.; Liu, R.; Qian, H.; Marvin, M.A.; et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J. Biol. Chem. 2011, 286, 16504–16515. [Google Scholar] [CrossRef]
- Haefeli, R.H.; Erb, M.; Gemperli, A.C.; Robay, D.; Courdier Fruh, I.; Anklin, C.; Dallmann, R.; Gueven, N. NQO1-dependent redox cycling of idebenone: Effects on cellular redox potential and energy levels. PLoS ONE 2011, 6, e17963. [Google Scholar] [CrossRef]
- Mordente, A.; Martorana, G.E.; Minotti, G.; Giardina, B. Antioxidant properties of 2,3-dimethoxy-5-methyl-6-(10-hydroxydecyl)-1,4-benzoquinone (idebenone). Chem. Res. Toxicol. 1998, 11, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Heitz, F.D.; Erb, M.; Anklin, C.; Robay, D.; Pernet, V.; Gueven, N. Idebenone protects against retinal damage and loss of vision in a mouse model of Leber’s hereditary optic neuropathy. PLoS ONE 2012, 7, e45182. [Google Scholar] [CrossRef] [PubMed]
- Erb, M.; Hoffmann-Enger, B.; Deppe, H.; Soeberdt, M.; Haefeli, R.H.; Rummey, C.; Feurer, A.; Gueven, N. Features of idebenone and related short-chain quinones that rescue ATP levels under conditions of impaired mitochondrial complex I. PLoS ONE 2012, 7, e36153. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Soiferman, D.; Moore, D.G.; Burte, F.; Saada, A. Evaluating the therapeutic potential of idebenone and related quinone analogues in Leber hereditary optic neuropathy. Mitochondrion 2017, 36, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Dombi, E.; Diot, A.; Morten, K.; Carver, J.; Lodge, T.; Fratter, C.; Ng, Y.S.; Liao, C.; Muir, R.; Blakely, E.L.; et al. The m.13051G>A mitochondrial DNA mutation results in variable neurology and activated mitophagy. Neurology 2016, 86, 1921–1923. [Google Scholar] [CrossRef]
- Klopstock, T.; Yu-Wai-Man, P.; Dimitriadis, K.; Rouleau, J.; Heck, S.; Bailie, M.; Atawan, A.; Chattopadhyay, S.; Schubert, M.; Garip, A.; et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain 2011, 134, 2677–2686. [Google Scholar] [CrossRef]
- Pemp, B.; Kircher, K.; Reitner, A. Visual function in chronic Leber’s hereditary optic neuropathy during idebenone treatment initiated 5 to 50 years after onset. Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 2751–2757. [Google Scholar] [CrossRef]
- Garcia-Medina, J.J.; Rubio-Velazquez, E.; Foulquie-Moreno, E.; Casaroli-Marano, R.P.; Pinazo-Duran, M.D.; Zanon-Moreno, V.; Del-Rio-Vellosillo, M. Update on the Effects of Antioxidants on Diabetic Retinopathy: In Vitro Experiments, Animal Studies and Clinical Trials. Antioxidants 2020, 9, 561. [Google Scholar] [CrossRef]
- Chous, A.P.; Richer, S.P.; Gerson, J.D.; Kowluru, R.A. The Diabetes Visual Function Supplement Study (DiVFuSS). Br. J. Ophthalmol. 2016, 100, 227–234. [Google Scholar] [CrossRef]
- Hu, B.J.; Hu, Y.N.; Lin, S.; Ma, W.J.; Li, X.R. Application of Lutein and Zeaxanthin in nonproliferative diabetic retinopathy. Int. J. Ophthalmol. 2011, 4, 303–306. [Google Scholar] [CrossRef]
- Garcia-Medina, J.J.; Pinazo-Duran, M.D.; Garcia-Medina, M.; Zanon-Moreno, V.; Pons-Vazquez, S. A 5-year follow-up of antioxidant supplementation in type 2 diabetic retinopathy. Eur. J. Ophthalmol. 2011, 21, 637–643. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
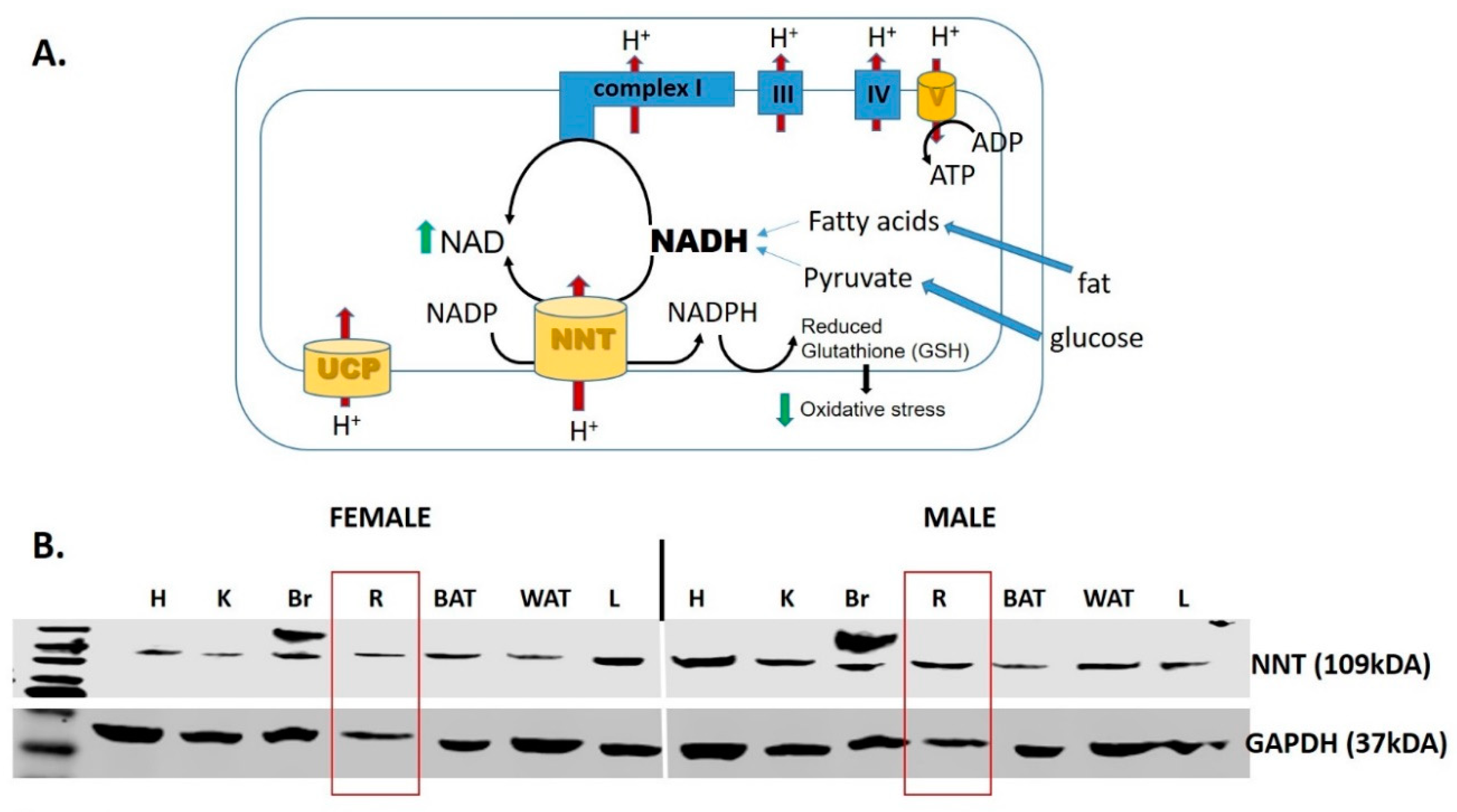{kind=link}
| Cell Type | Oxidative Stress | Mitochondrial Morphology | Mitochondrial Turnover | References |
|---|---|---|---|---|
| RGCs | ✔ | ✔ | Unknown | [115,116,117] |
| Bipolar cells | ✔ | ✔ | Unknown | [117] |
| Müller cells | ✔ | ✔ | ✔ | [118,119,120,121,122,123,124] |
| Photoreceptors | ✔ | ✔ | ✔ | [121] |
| Endothelial cells | ✔ | ✔ | ✔ | [113,114,125,126,127,128,129,130,131] |
| Cell Type | Complex I | Complex II † | Complex III | Complex IV | Complex V (ATP Synthase) | References |
|---|---|---|---|---|---|---|
| RGCs | ↓ | ↓ | Unknown | ↓ | Unknown | [132,133] |
| Bipolar cells | Unknown | Unknown | Unknown | Unknown | Unknown | |
| Müller cells | Unknown | ↔ | Unknown | ↔ | Unknown | [119] |
| Photoreceptors | Unknown | Unknown | Unknown | Varies | Unknown | [121] |
| Retinal homogenate | Biphasic ‡ | Biphasic | Biphasic | Biphasic | Varies | [100,109,110,111,112,121,134] |
| Pericytes | Unknown | ↓ | Unknown | Unknown | Unknown | [135] |
| Endothelial cells | ↓ | ↓ | ↓ | ↓ | ↓ | [129,136,137,138,139] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, D.J.; Cascio, M.A.; Rosca, M.G. Diabetic Retinopathy: The Role of Mitochondria in the Neural Retina and Microvascular Disease. Antioxidants 2020, 9, 905. https://doi.org/10.3390/antiox9100905
Miller DJ, Cascio MA, Rosca MG. Diabetic Retinopathy: The Role of Mitochondria in the Neural Retina and Microvascular Disease. Antioxidants. 2020; 9(10):905. https://doi.org/10.3390/antiox9100905
Chicago/Turabian StyleMiller, David J., M. Ariel Cascio, and Mariana G. Rosca. 2020. "Diabetic Retinopathy: The Role of Mitochondria in the Neural Retina and Microvascular Disease" Antioxidants 9, no. 10: 905. https://doi.org/10.3390/antiox9100905
APA StyleMiller, D. J., Cascio, M. A., & Rosca, M. G. (2020). Diabetic Retinopathy: The Role of Mitochondria in the Neural Retina and Microvascular Disease. Antioxidants, 9(10), 905. https://doi.org/10.3390/antiox9100905