The Protective Role of Bioactive Quinones in Stress-induced Senescence Phenotype of Endothelial Cells Exposed to Cigarette Smoke Extract

 , ,
, , 

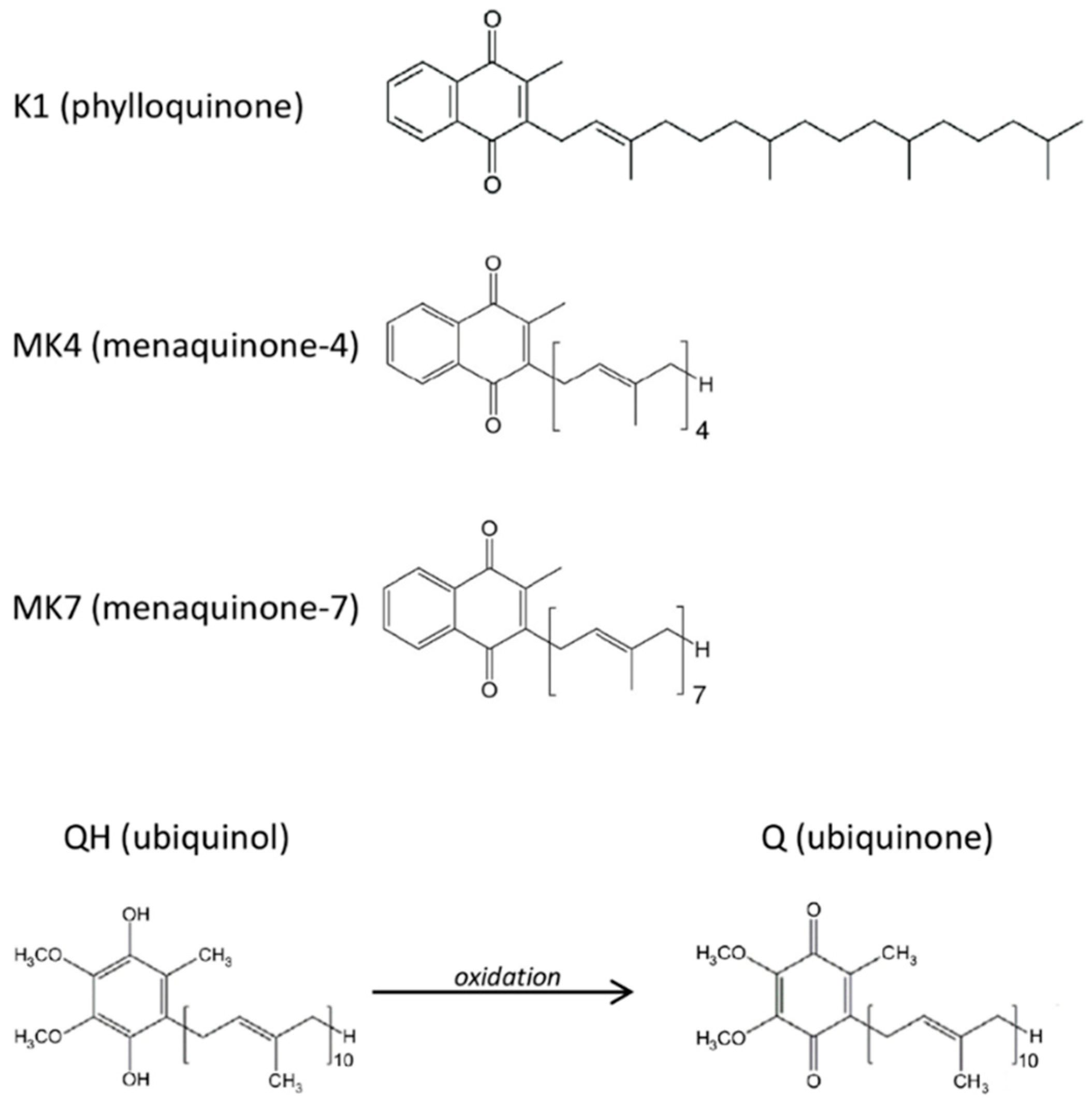{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cellular Model
2.2. Cigarette Smoke Extract Preparation
2.3. Quinones Bioavailability
2.4. Viability, Cellular and Mitochondrial ROS Content and mPTP Assay
2.5. Activated Caspase-1 Detection
2.6. Senescence-associated β-galactosidase Staining
2.7. Data Analysis
3. Results
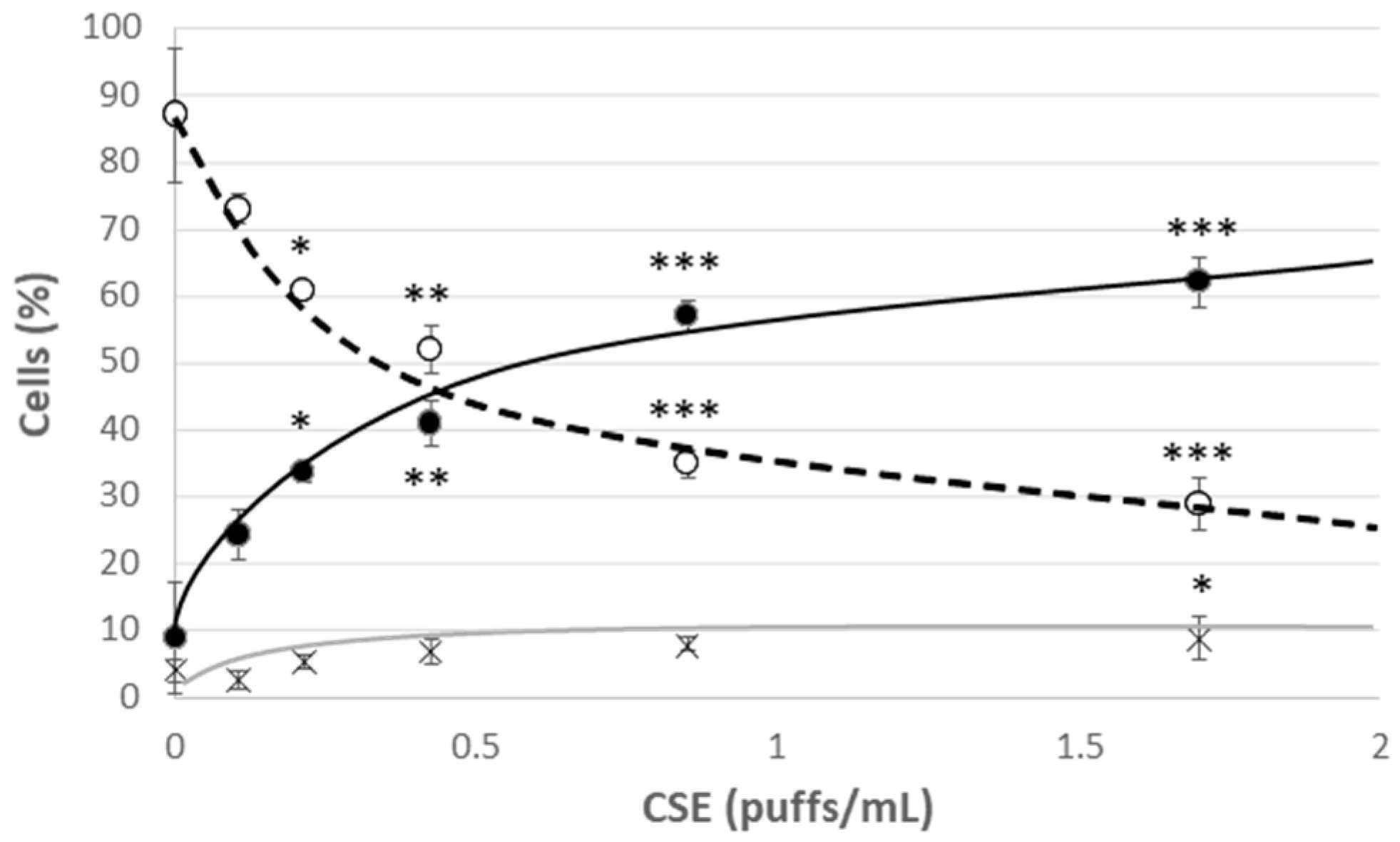
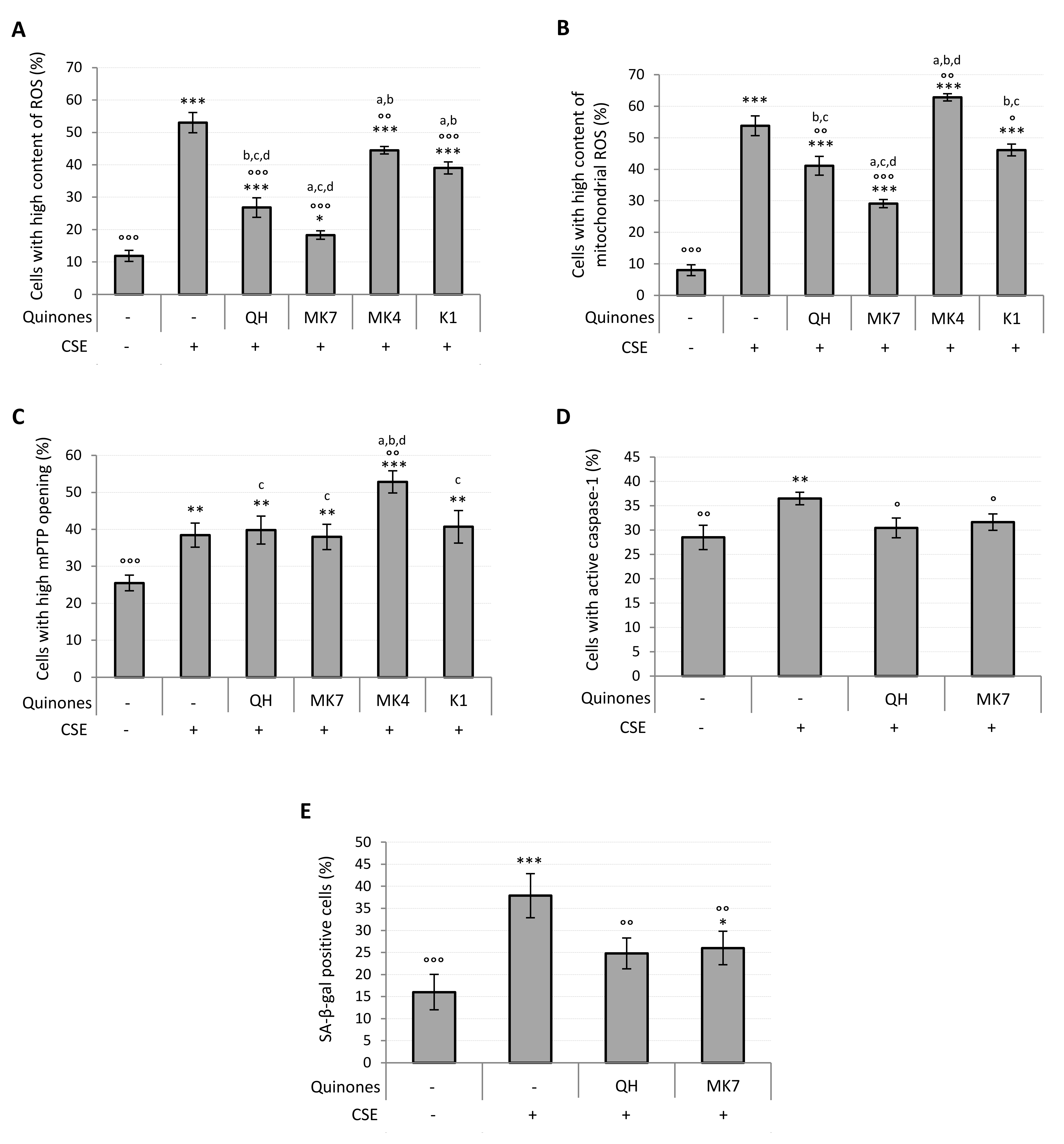
3.1. CSE Altered Oxidative Status, Mitochondrial Health, Pro-Inflammatory Caspase-1 Activity and Promoted Cellular Senescence
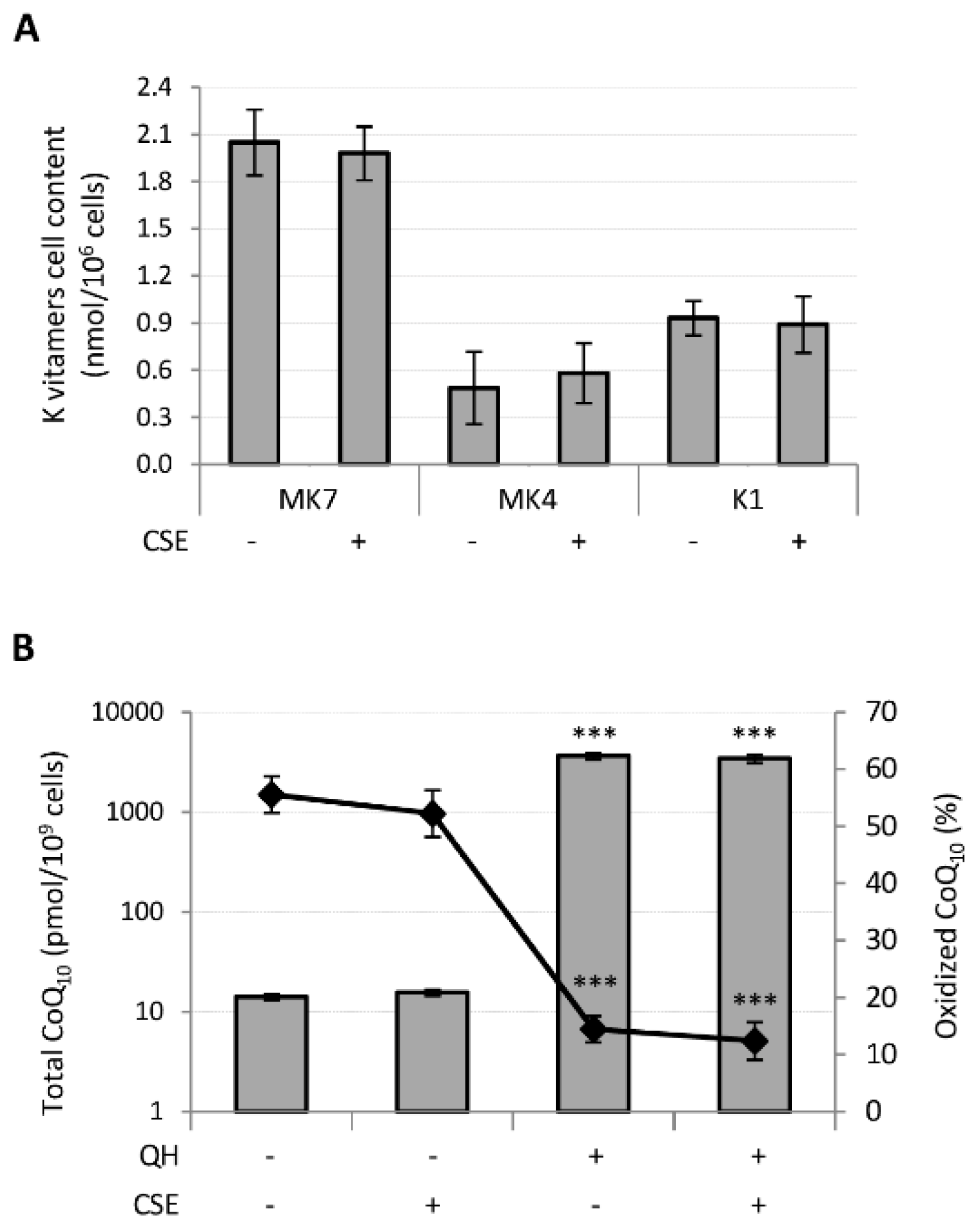
3.2. CSE Did not Affect Exogenous Quinones Bioavailability and CoQ10 Oxidative Status
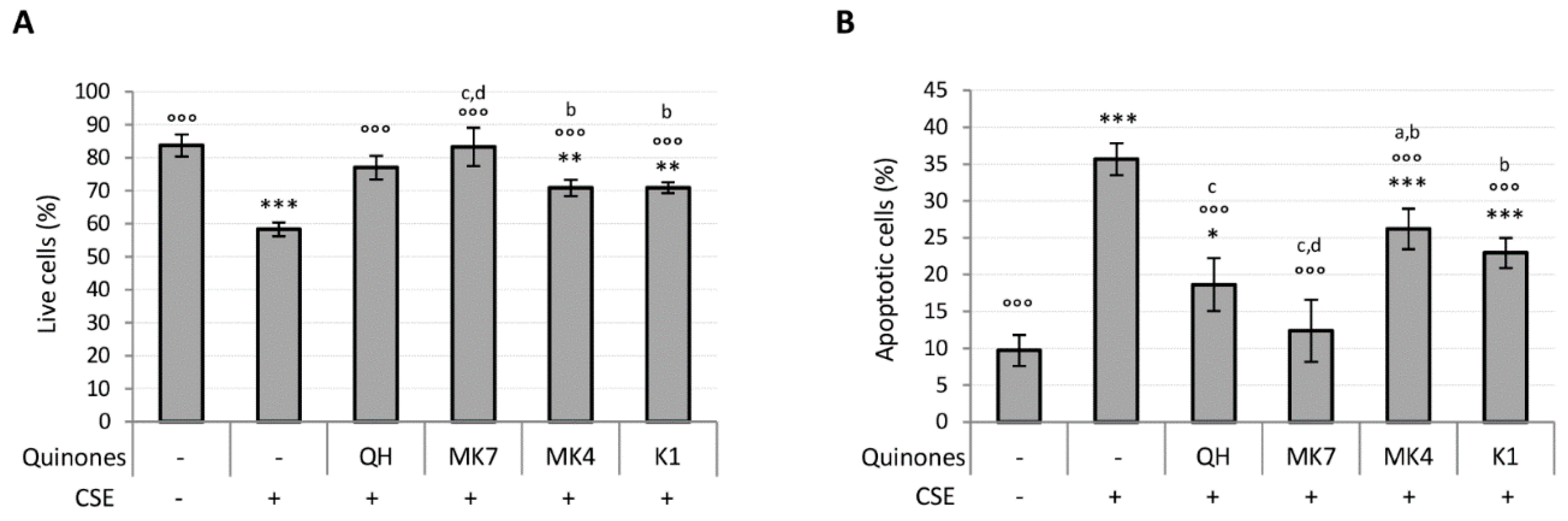
3.3. Quinones Differentially Protect against CSE-Induced Cytotoxicity, Oxidative Stress, Mitochondrial Dysfunction, Pro-Inflammatory Caspase-1 Activation and Cellular Senesence
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Ezzati, M.; Obermeyer, Z.; Tzoulaki, I.; Mayosi, B.M.; Elliott, P.; Leon, D.A. Contributions of risk factor trends and medical care to cardiovascular mortality trends. Nat. Rev. Cardiol. 2015, 12, 508–530. [Google Scholar] [CrossRef] [PubMed]
- Rodgman, A.; Perfetti, T.A. The Chemical Components of Tobacco and Tobacco Smoke, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2013; pp. 1221–1297. [Google Scholar]
- Farhat, N.; Thorin-Trescases, N.; Voghel, G.; Villeneuve, L.; Mamarbachi, M.; Perrault, L.P.; Carrier, M.; Thorin, E. Stress-induced senescence predominates in endothelial cells isolated from atherosclerotic chronic smokers. Can. J. Physiol. Pharmacol. 2008, 86, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Mehta, J.L. Intracellular signaling of LOX-1 in endothelial cell apoptosis. Circ. Res. 2009, 104, 566–568. [Google Scholar] [CrossRef] [PubMed]
- Frey, R.S.; Ushio-Fukai, M.; Malik, A.B. NADPH oxidase-dependent signaling in endothelial cells: Role in physiology and pathology. Antioxid. Redox Signal. 2009, 11, 791–810. [Google Scholar] [CrossRef]
- Orosz, Z.; Csiszar, A.; Labinskyy, N.; Smith, K.; Kaminski, P.M.; Ferdinandy, P.; Wolin, M.S.; Rivera, A.; Ungvari, Z. Cigarette smoke-induced proinflammatory alterations in the endothelial phenotype: Role of NAD(P)H oxidase activation. Am. J. Physiol. Circ. Physiol. 2007, 292, H130–H139. [Google Scholar] [CrossRef]
- Yang, G.Y.; Zhang, C.L.; Liu, X.C.; Qian, G.; Deng, D.Q. Effects of cigarette smoke extracts on the growth and senescence of skin fibroblasts in vitro. Int. J. Biol. Sci. 2013, 9, 613–623. [Google Scholar] [CrossRef]
- Wu, Q.; Jiang, D.; Matsuda, J.L.; Ternyak, K.; Zhang, B.; Chu, H.W. Cigarette smoke induces human airway epithelial senescence via growth differentiation factor 15 production. Am. J. Respir. Cell Mol. Biol. 2016, 55, 429–438. [Google Scholar] [CrossRef]
- Liu, A.; Wu, J.; Li, A.; Bi, W.; Liu, T.; Cao, L.; Liu, Y.; Dong, L. The inhibitory mechanism of Cordyceps sinensis on cigarette smoke extract-induced senescence in human bronchial epithelial cells. Int. J. Chronic Obstr. Pulm. Dis. 2016, 11, 1721–1731. [Google Scholar] [CrossRef]
- He, Z.; Chen, Y.; Hou, C.; He, W.; Chen, P. Cigarette smoke extract changes expression of endothelial nitric oxide synthase (eNOS) and p16(INK4a) and is related to endothelial progenitor cell dysfunction. Med. Sci. Monit. 2017, 23, 3224–3231. [Google Scholar] [CrossRef][Green Version]
- Bentinger, M.; Brismar, K.; Dallner, G. The antioxidant role of coenzyme Q. Mitochondrion 2007, 7, S41–S50. [Google Scholar] [CrossRef] [PubMed]
- Failla, M.L.; Chitchumroonchokchai, C.; Aoki, F. Increased bioavailability of ubiquinol compared to that of ubiquinone is due to more efficient micellarization during digestion and greater GSH-dependent uptake and basolateral secretion by Caco-2 cells. J. Agric. Food Chem. 2014, 62, 7174–7182. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.L.; Huang, Y.H.; Kao, C.L.; Yang, D.M.; Lee, H.C.; Chou, H.Y.; Chen, Y.C.; Chiou, G.Y.; Chen, L.H.; Yang, Y.P.; et al. A novel mechanism of coenzyme Q10 protects against human endothelial cells from oxidative stress-induced injury by modulating NO-related pathways. J. Nutr. Biochem. 2012, 23, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Schmelzer, C.; Lorenz, G.; Rimbach, G.; Doring, F. In vitro effects of the reduced form of coenzyme Q (10) on secretion levels of TNF-α and chemokines in response to LPS in the human monocytic cell line THP-1. J. Clin. Biochem. Nutr. 2009, 44, 62–66. [Google Scholar] [CrossRef]
- Lee, B.J.; Tseng, Y.F.; Yen, C.H.; Lin, P.T. Effects of coenzyme Q10 supplementation (300 mg/day) on antioxidation and anti-inflammation in coronary artery disease patients during statins therapy: A randomized, placebo-controlled trial. Nutr. J. 2013, 12, 142. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Lazzarini, R.; Babini, L.; Prattichizzo, F.; Rippo, M.R.; Tiano, L.; Di Nuzzo, S.; Graciotti, L.; Festa, R.; Bruge, F.; et al. Anti-inflammatory effect of ubiquinol-10 on young and senescent endothelial cells via miR-146a modulation. Free Radic. Biol. Med. 2013, 63, 410–520. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.; Xu, Z.; Hosoe, K.; Kubo, H.; Miyahara, H.; Dai, J.; Mori, M.; Sawashita, J.; Higuchi, K. Coenzyme Q10 prevents senescence and dysfunction caused by oxidative stress in vascular endothelial cells. Oxidative Med. Cell. Longev. 2018, 2018, 3181759. [Google Scholar] [CrossRef]
- Niklowitz, P.; Onur, S.; Fischer, A.; Laudes, M.; Palussen, M.; Menke, T.; Doring, F. Coenzyme Q10 serum concentration and redox status in European adults: Influence of age, sex, and lipoprotein concentration. J. Clin. Biochem. Nutr. 2016, 58, 240–245. [Google Scholar] [CrossRef]
- Schottlaender, L.V.; Bettencourt, C.; Kiely, A.P.; Chalasani, A.; Neergheen, V.; Holton, J.L.; Hargreaves, I.; Houlden, H. Coenzyme Q10 levels are decreased in the cerebellum of multiple-system atrophy patients. PLoS ONE 2016, 11, e0149557. [Google Scholar] [CrossRef]
- Shearer, M.J.; Newman, P. Recent trends in the metabolism and cell biology of vitamin K with special reference to vitamin K cycling and MK-4 biosynthesis. J. Lipid Res. 2014, 55, 345–362. [Google Scholar] [CrossRef]
- Ohsaki, Y.; Shirakawa, H.; Miura, A.; Giriwono, P.E.; Sato, S.; Ohashi, A.; Iribe, M.; Goto, T.; Komai, M. Vitamin K suppresses the lipopolysaccharide-induced expression of inflammatory cytokines in cultured macrophage-like cells via the inhibition of the activation of nuclear factor kB through the repression of IKKα/β phosphorylation. J. Nutr. Biochem. 2010, 21, 1120–1126. [Google Scholar] [CrossRef]
- Lee, H.; Park, J.R.; Kim, E.J.; Kim, W.J.; Hong, S.H.; Park, S.M.; Yang, S.R. Cigarette smoke-mediated oxidative stress induces apoptosis via the MAPKs/STAT1 pathway in mouse lung fibroblasts. Toxicol. Lett. 2016, 240, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Carp, H.; Janoff, A. Possible mechanisms of emphysema in smokers. In vitro suppression of serum elastase-inhibitory capacity by fresh cigarette smoke and its prevention by antioxidants. Am. Rev. Respir. Dis. 1978, 118, 617–621. [Google Scholar] [CrossRef]
- Giannubilo, S.R.; Orlando, P.; Silvestri, S.; Cirilli, I.; Marcheggiani, F.; Ciavattini, A.; Tiano, L. CoQ10 supplementation in patients undergoing IVF-ET: The relationship with follicular fluid content and oocyte Maturity. Antioxidants 2018, 7, 141. [Google Scholar] [CrossRef]
- Damiani, E.; Brugè, F.; Cirilli, I.; Marcheggiani, F.; Olivieri, F.; Armeni, T.; Cianfruglia, L.; Giuliani, A.; Orlando, P.; Tiano, L. Modulation of oxidative status by normoxia and hypoxia on cultures of human dermal fibroblasts: How does it affect cell aging? Oxidative Med. Cell. Longev. 2018, 2018, 5469159. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, A.; Cirilli, I.; Prattichizzo, F.; Mensà, E.; Fulgenzi, G.; Sabbatinelli, J.; Graciotti, L.; Olivieri, F.; Procopio, A.D.; Tiano, L.; et al. The mitomiR/Bcl-2 axis affects mitochondrial function and autophagic vacuole formation in senescent endothelial cells. Aging 2018, 10, 2855–2873. [Google Scholar] [CrossRef] [PubMed]
- Carnevali, S.; Petruzzelli, S.; Longoni, B.; Vanacore, R.; Barale, R.; Cipollini, M.; Scatena, F.; Paggiaro, P.; Celi, A.; Giuntini, C. Cigarette smoke extract induces oxidative stress and apoptosis in human lung fibroblasts. Am. J. Physiol. Cell. Mol. Physiol. 2003, 284, L955–L963. [Google Scholar] [CrossRef]
- Csiszar, A.; Labinskyy, N.; Podlutsky, A.; Kaminski, P.M.; Wolin, M.S.; Zhang, C.; Mukhopadhyay, P.; Pacher, P.; Hu, F.; de Cabo, R.; et al. Vasoprotective effects of resveratrol and SIRT1: Attenuation of cigarette smoke-induced oxidative stress and proinflammatory phenotypic alterations. Am. J. Physiol. Circ. Physiol. 2008, 294, H2721–H2735. [Google Scholar] [CrossRef]
- Csordas, A.; Kreutmayer, S.; Ploner, C.; Braun, P.R.; Karlas, A.; Backovic, A.; Wick, G.; Bernhard, D. Cigarette smoke extract induces prolonged endoplasmic reticulum stress and autophagic cell death in human umbilical vein endothelial cells. Cardiovasc. Res. 2011, 92, 141–148. [Google Scholar] [CrossRef]
- Messner, B.; Frotschnig, S.; Steinacher-Nigisch, A.; Winter, B.; Eichmair, E.; Gebetsberger, J.; Schwaiger, S.; Ploner, C.; Laufer, G.; Bernhard, D. Apoptosis and necrosis: Two different outcomes of cigarette smoke condensate-induced endothelial cell death. Cell Death Dis. 2012, 3, e424. [Google Scholar] [CrossRef]
- Faux, S.P.; Tai, T.; Thorne, D.; Xu, Y.; Breheny, D.; Gaca, M. The role of oxidative stress in the biological responses of lung epithelial cells to cigarette smoke. Biomarkers 2009, 14 (Suppl. 1), 90–96. [Google Scholar] [CrossRef]
- Slebos, D.J.; Ryter, S.W.; van der Toorn, M.; Liu, F.; Guo, F.; Baty, C.J.; Karlsson, J.M.; Watkins, S.C.; Kim, H.P.; Wang, X.; et al. Mitochondrial localization and function of heme oxygenase-1 in cigarette smoke-induced cell death. Am. J. Respir. Cell Mol. Biol. 2007, 36, 409–417. [Google Scholar] [CrossRef]
- Csiszar, A.; Podlutsky, A.; Wolin, M.S.; Losonczy, G.; Pacher, P.; Ungvari, Z. Oxidative stress and accelerated vascular aging: Implications for cigarette smoking. Front. Biosci. 2009, 14, 3128–3144. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed]
- Kurz, D.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D. Senescence-associated (β)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 2000, 113, 3613–3622. [Google Scholar] [PubMed]
- Hossain, M.; Mazzone, P.; Tierney, W.; Cucullo, L. In vitro assessment of tobacco smoke toxicity at the BBB: Do antioxidant supplements have a protective role? BMC Neurosci. 2011, 12, 92. [Google Scholar] [CrossRef]
- Kaisar, M.A.; Prasad, S.; Cucullo, L. Protecting the BBB endothelium against cigarette smoke-induced oxidative stress using popular antioxidants: Are they really beneficial? Brain Res. 2015, 1627, 90–100. [Google Scholar] [CrossRef]
- Willems, B.A.; Vermeer, C.; Reutelingsperger, C.P.; Schurgers, L.J. The realm of vitamin K dependent proteins: Shifting from coagulation toward calcification. Mol. Nutr. Food Res. 2014, 58, 1620–1635. [Google Scholar] [CrossRef]
- Van der Meer, J.H.; van der Poll, T.; Van’t Veer, C. TAM receptors, Gas6, and protein S: Roles in inflammation and hemostasis. Blood 2014, 123, 2460–2469. [Google Scholar] [CrossRef]
- Gairola, C.G.; Howatt, D.A.; Daugherty, A. Dietary coenzyme Q10 does not protect against cigarette smoke-augmented atherosclerosis in apoE-deficient mice. Free Radic. Biol. Med. 2010, 48, 1535–1539. [Google Scholar] [CrossRef]
- Tian, G.; Sawashita, J.; Kubo, H.; Nishio, S.Y.; Hashimoto, S.; Suzuki, N.; Yoshimura, H.; Tsuruoka, M.; Wang, Y.; Liu, Y.; et al. Ubiquinol-10 supplementation activates mitochondria functions to decelerate senescence in senescence-accelerated mice. Antioxid. Redox Signal. 2014, 20, 2606–2620. [Google Scholar] [CrossRef] [PubMed]
- Nowicka, B.; Kruk, J. Occurrence, biosynthesis and function of isoprenoid quinones. Biochim. Biophys. Acta BBA Bioenerg. 2010, 9, 1587–1605. [Google Scholar] [CrossRef]
- Schwalfenberg, G.K. Vitamins K1 and K2: The emerging group of vitamins required for human health. J. Nutr. Metab. 2017, 2017, 6254836. [Google Scholar] [CrossRef]
- Ivanova, D.; Zhelev, Z.; Getso, P.; Nikolova, B.; Aoki, I.; Higashi, T.; Bakalova, R. Vitamin K: Redox-modulation, prevention of mitochondrial dysfunction and anticancer effect. Redox Biol. 2018, 352–358. [Google Scholar] [CrossRef]
- Vos, M.; Esposito, G.; Edirisinghe, J.N.; Vilain, S.; Haddad, D.M.; Slabbaert, J.R.; Van Meensel, S.; Schaap, O.; De Strooper, B.; Meganathan, R.; et al. Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science 2012, 336, 1306–1310. [Google Scholar] [CrossRef]
- Yang, Y.M.; Liu, G.T. Damaging effect of cigarette smoke extract on primary cultured human umbilical vein endothelial cells and its mechanism. Biomed. Environ. Sci. 2004, 17, 121–134. [Google Scholar]
- Shamas-Din, A.; Satsoura, D.; Khan, O.; Zhu, W.; Leber, B.; Fradin, C.; Andrews, D.W. Multiple partners can kiss-and-run: Bax transfers between multiple membranes and permeabilizes those primed by tBid. Cell Death Dis. 2014, 5, e1277. [Google Scholar] [CrossRef]
- Wu, K.; Luan, G.; Xu, Y.; Shen, S.; Qian, S.; Zhu, Z.; Zhang, X.; Yin, S.; Ye, J. Cigarette smoke extract increases mitochondrial membrane permeability through activation of adenine nucleotide translocator (ANT) in lung epithelial cells. Biochem. Biophys. Res. Commun. 2020, 525, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Syed, F.M.; Hahn, H.S.; Odley, A.; Guo, Y.; Vallejo, J.G.; Lynch, R.A.; Mann, D.L.; Bolli, R.; Dorn, G.W., II. Proapoptotic effects of caspase-1/interleukin-converting enzyme dominate in myocardial ischemia. Circ. Res. 2005, 96, 1103–1109. [Google Scholar] [CrossRef]
- Müller, T.; Hengstermann, A. Nrf2: Friend and foe in preventing cigarette smoking-dependent lung disease. Chem. Res. Toxicol. 2012, 25, 1805–1824. [Google Scholar] [CrossRef] [PubMed]
- Giebe, S.; Cockcroft, N.; Hewitt, K.; Brux, M.; Hofmann, A.; Morawietz, H.; Brunssen, C. Cigarette smoke extract counteracts atheroprotective effects of high laminar flow on endothelial function. Redox Biol. 2017, 12, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Fratta Pasini, A.; Albiero, A.; Stranieri, C.; Cominacini, M.; Pasini, A.; Mozzini, C.; Vallerio, P.; Cominacini, L.; Garbin, U. Serum oxidative stress-induced repression of Nrf2 and GSH depletion: A mechanism potentially involved in endothelial dysfunction of young smokers. PLoS ONE 2012, 7, e30291. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Schurgers, L.J.; Uenishi, K. Comparison of menaquinone-4 and menaquinone-7 bioavailability in healthy women. Nutr. J. 2012, 11, 93. [Google Scholar] [CrossRef] [PubMed]
- Shea, M.K.; Holden, R.M. Vitamin K status and vascular calcification: Evidence from observational and clinical studies. Adv. Nutr. 2012, 3, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, E.; Aoki, M.; Watanabe, F.; Kamimura, A. Low-dose menaquinone-4 improves γ-carboxylation of osteocalcin in young males: A non-placebo-controlled dose-response study. Nutr. J. 2014, 13, 1–4. [Google Scholar] [CrossRef][Green Version]
- Knapen, M.H.; Drummen, N.E.; Smit, E.; Vermeer, C.; Theuwissen, E. Three-year low-dose menaquinone-7 supplementation helps decrease bone loss in healthy postmenopausal women. Osteoporos. Int. 2013, 24, 2499–2507. [Google Scholar] [CrossRef]
- Halder, M.; Petsophonsakul, P.; Akbulut, A.C.; Pavlic, A.; Bohan, F.; Anderson, E.; Maresz, K.; Kramann, R.; Schurgers, L. Vitamin K: Double bonds beyond coagulation insights into differences between Vitamin K1 and K2 in health and disease. Int. J. Mol. Sci. 2019, 20, 896. [Google Scholar] [CrossRef]
- Roche, Y.; Peretti, P.; Bernard, S. Influence of the chain length of ubiquinones on their interaction with DPPC in mixed monolayers. Biochim. Biophys. Acta BBA Biomembr. 2006, 1758, 468–478. [Google Scholar] [CrossRef][Green Version]
- Okada, K.; Kainou, T.; Matsuda, H.; Kawamukai, M. Biological significance of the side chain length of ubiquinone in Saccharomyces cerevisiae. FEBS Lett. 1998, 431, 241–244. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cirilli, I.; Orlando, P.; Marcheggiani, F.; Dludla, P.V.; Silvestri, S.; Damiani, E.; Tiano, L. The Protective Role of Bioactive Quinones in Stress-induced Senescence Phenotype of Endothelial Cells Exposed to Cigarette Smoke Extract. Antioxidants 2020, 9, 1008. https://doi.org/10.3390/antiox9101008
Cirilli I, Orlando P, Marcheggiani F, Dludla PV, Silvestri S, Damiani E, Tiano L. The Protective Role of Bioactive Quinones in Stress-induced Senescence Phenotype of Endothelial Cells Exposed to Cigarette Smoke Extract. Antioxidants. 2020; 9(10):1008. https://doi.org/10.3390/antiox9101008
Chicago/Turabian StyleCirilli, Ilenia, Patrick Orlando, Fabio Marcheggiani, Phiwayinkosi V. Dludla, Sonia Silvestri, Elisabetta Damiani, and Luca Tiano. 2020. "The Protective Role of Bioactive Quinones in Stress-induced Senescence Phenotype of Endothelial Cells Exposed to Cigarette Smoke Extract" Antioxidants 9, no. 10: 1008. https://doi.org/10.3390/antiox9101008
APA StyleCirilli, I., Orlando, P., Marcheggiani, F., Dludla, P. V., Silvestri, S., Damiani, E., & Tiano, L. (2020). The Protective Role of Bioactive Quinones in Stress-induced Senescence Phenotype of Endothelial Cells Exposed to Cigarette Smoke Extract. Antioxidants, 9(10), 1008. https://doi.org/10.3390/antiox9101008