Cytoprotective Effects of Delphinidin for Human Chondrocytes against Oxidative Stress through Activation of Autophagy

 ,
,  and
and 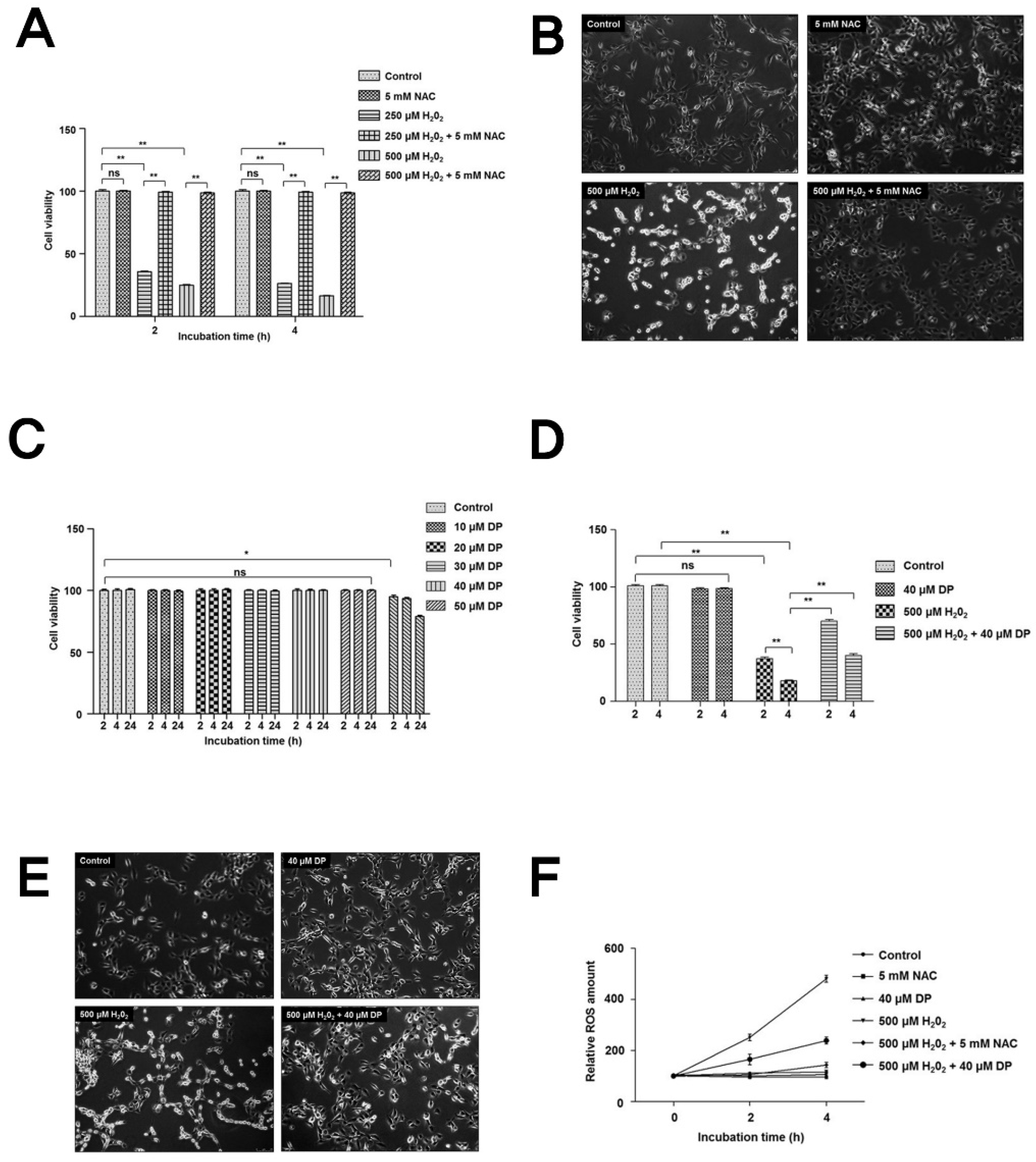{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Reagents
2.2. Cell Viability
2.3. Determination of Intracellular ROS
2.4. TUNEL Assay
2.5. Western Blot Analysis
2.6. Immunofluorescence Staining
2.7. Acridine Orange Dye Staining
2.8. Monodansylcadaverine (MDC) Staining for Autophagic Vacuoles
2.9. Statistical Analyses
3. Results
3.1. Delphinidin Has Cytoprotective Effects in Chondrocytes under Oxidative Stress
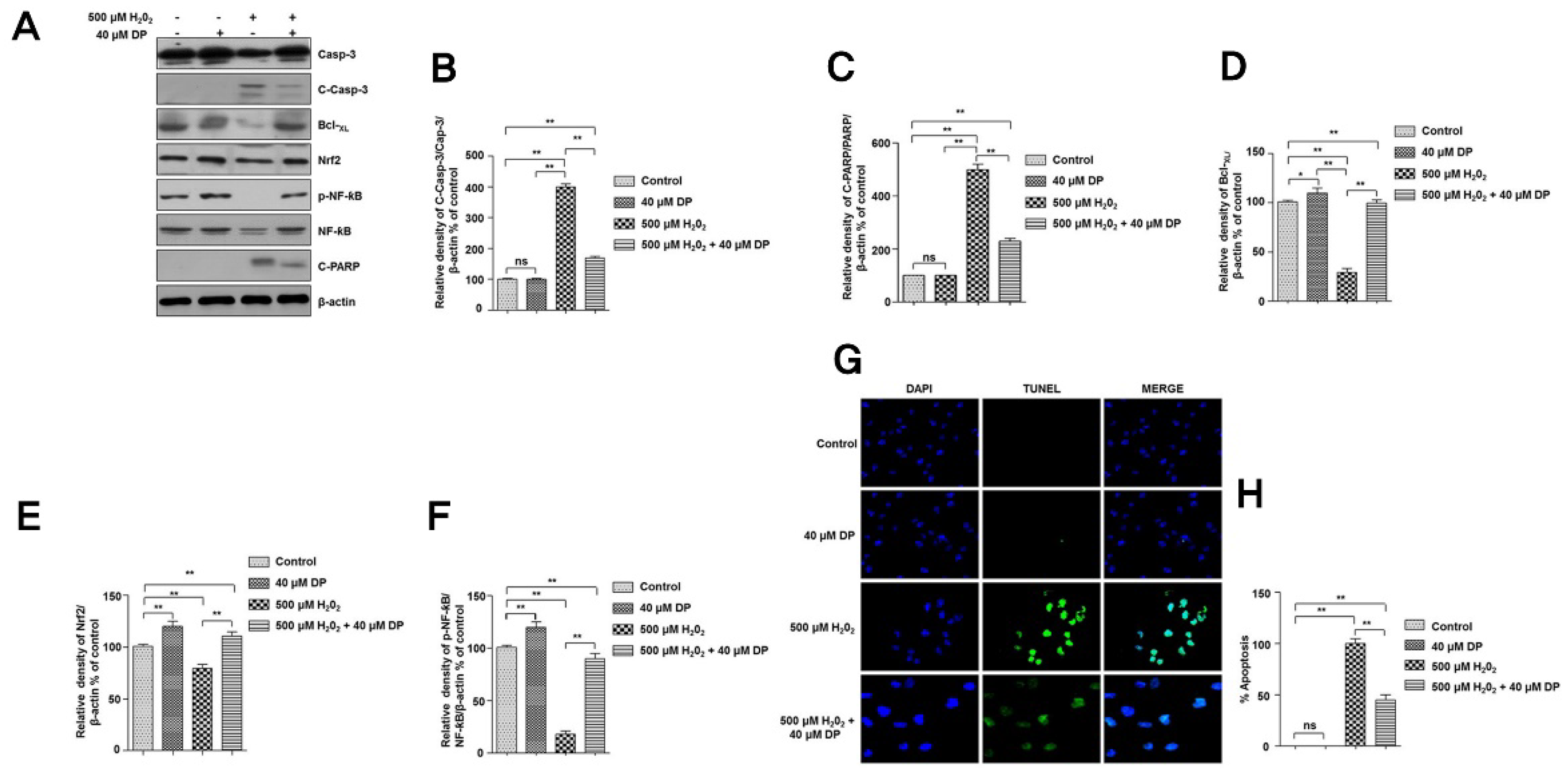
3.2. Delphinidin Inhibits Apoptotic Cell Death in Chondrocytes during Oxidative Stress
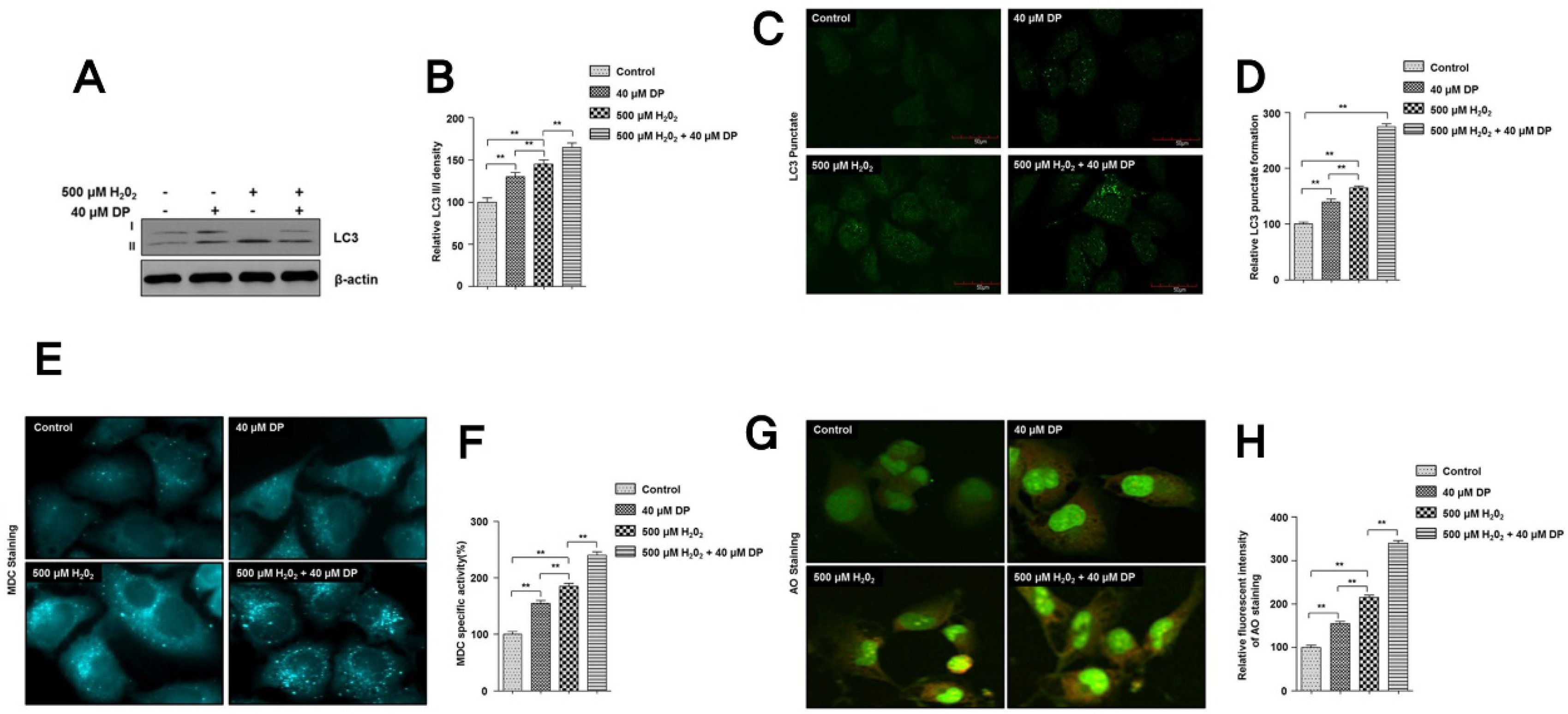
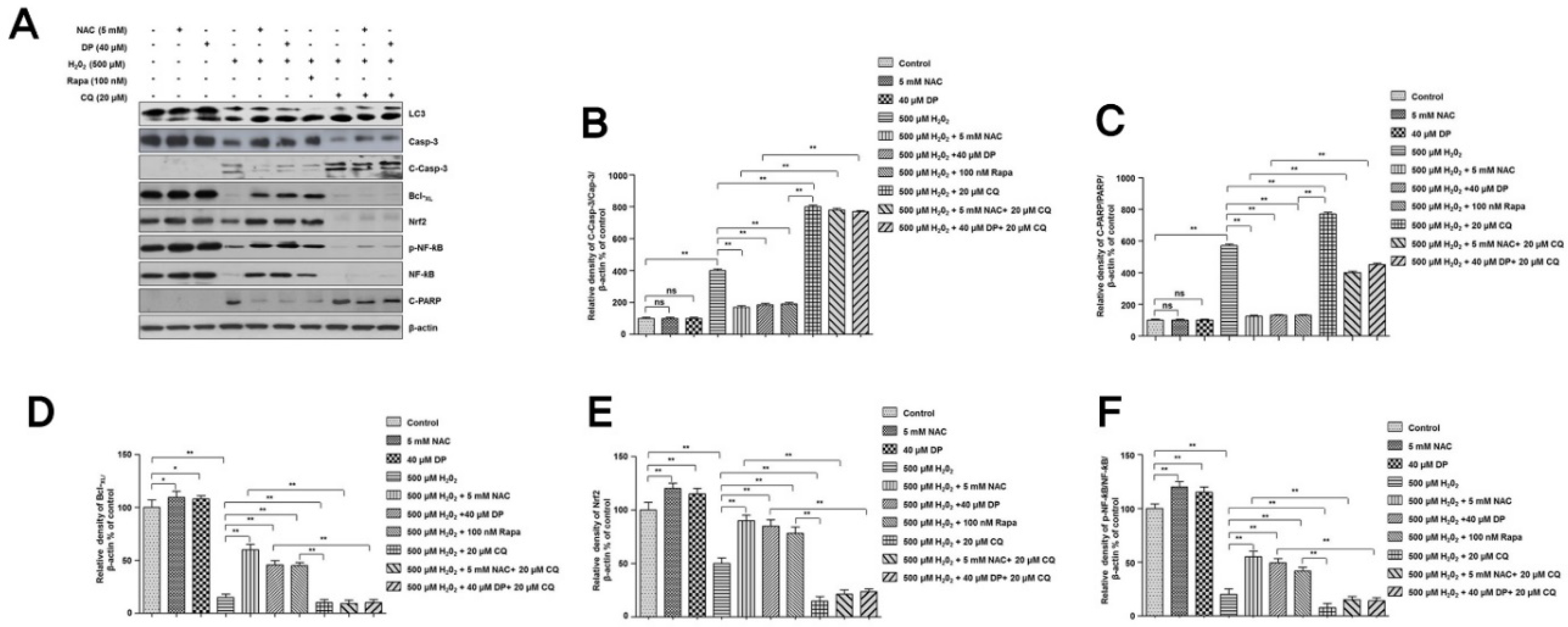
3.3. Autophagy Is Involved in the Protection of Delphinidin against H2O2-Induced Oxidative Stress
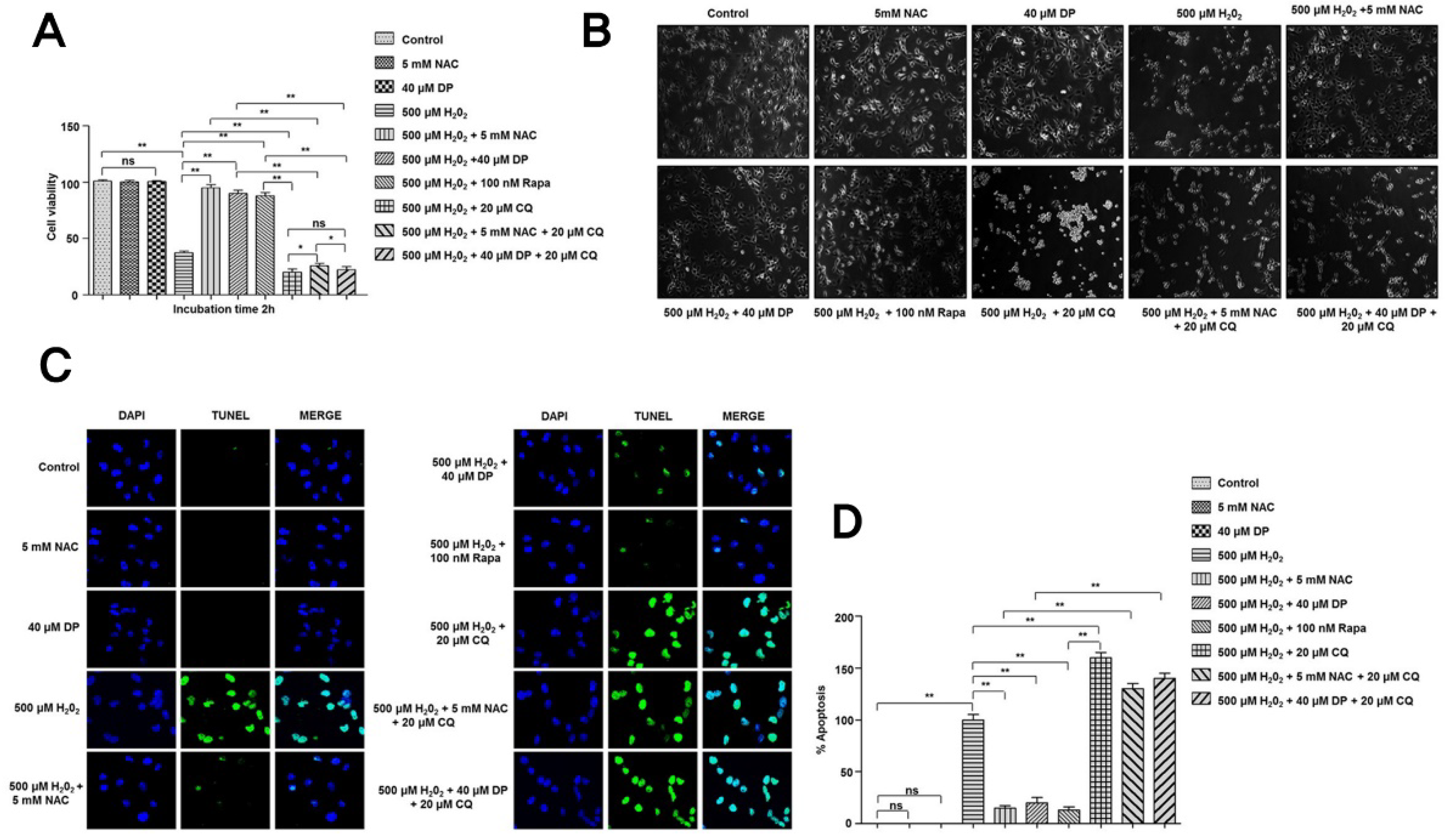
3.4. Cytoprotective Role of Delphinidin Is Modulated by Activation of Autophagy in Oxidative Stress
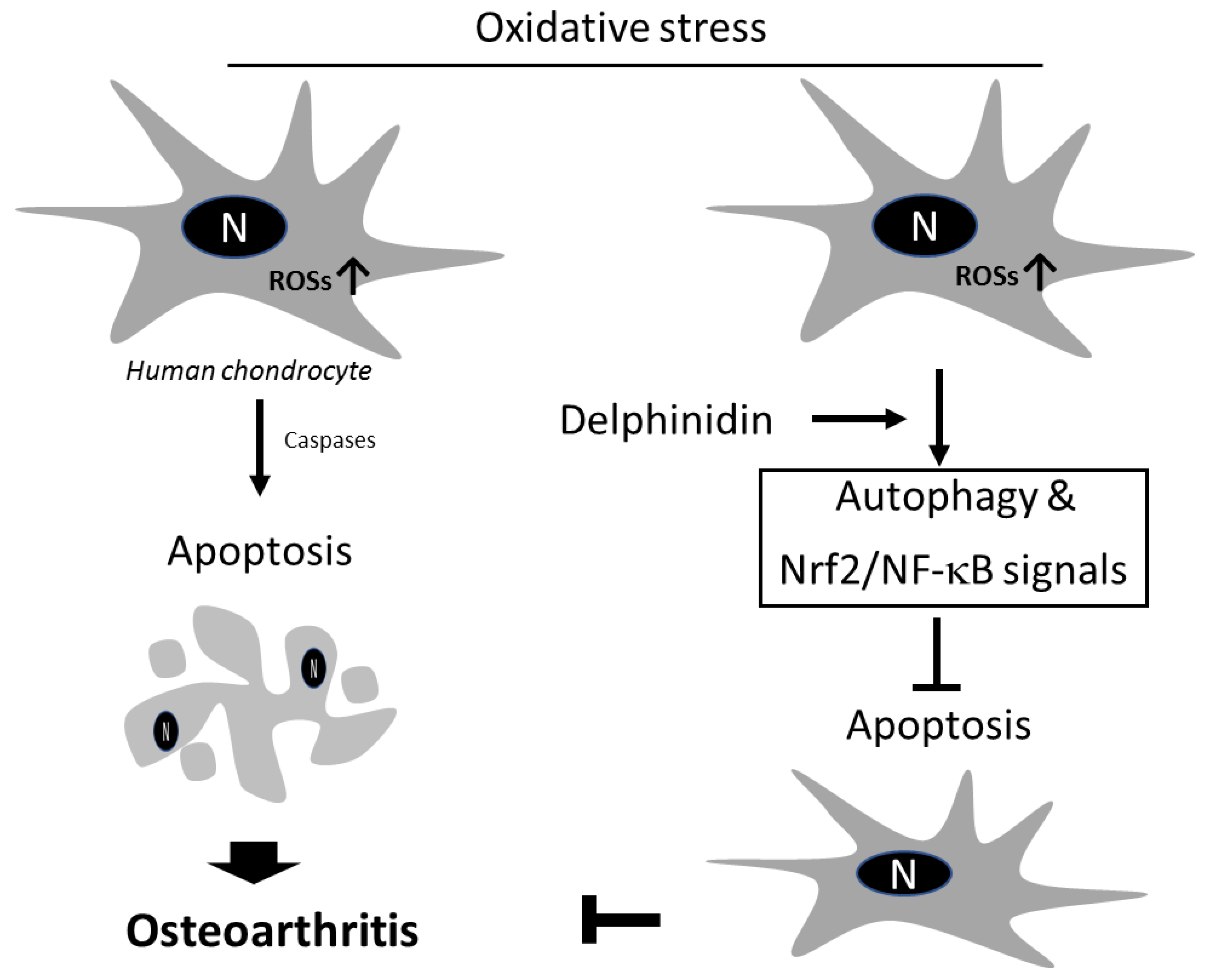
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hunter, D.J.; Felson, D.T. Osteoarthritis. BMJ 2006, 332, 639–642. [Google Scholar] [CrossRef]
- Vos, T.; Flaxman, A.D.; Naghavi, M.; Lozano, R.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; Aboyans, V.; et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2163–2196. [Google Scholar] [CrossRef]
- Cross, M.; Smith, E.; Hoy, D.; Nolte, S.; Ackerman, I.; Fransen, M.; Bridgett, L.; Williams, S.; Guillemin, F.; Hill, C.L.; et al. The global burden of hip and knee osteoarthritis: Estimates from the global burden of disease 2010 study. Ann. Rheum. Dis. 2014, 73, 1323–1330. [Google Scholar] [CrossRef]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Chopra, A. The COPCORD world of musculoskeletal pain and arthritis. Rheumatology 2013, 52, 1925–1928. [Google Scholar] [CrossRef] [PubMed]
- Maetzel, A.; Li, L.C.; Pencharz, J.; Tomlinson, G.; Bombardier, C.; Community, H.; Arthritis Project Study Team. The economic burden associated with osteoarthritis, rheumatoid arthritis, and hypertension: A comparative study. Ann. Rheum. Dis. 2004, 63, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Gupte, C.; Akhtar, K.; Smith, P.; Cobb, J. The Global Economic Cost of Osteoarthritis: How the UK Compares. Arthritis 2012, 2012, 1–6. [Google Scholar] [CrossRef]
- Ruiz, D., Jr.; Koenig, L.; Dall, T.M.; Gallo, P.; Narzikul, A.; Parvizi, J.; Tongue, J. The direct and indirect costs to society of treatment for end-stage knee osteoarthritis. J. Bone Jt. Surg. Am. 2013, 95, 1473–1480. [Google Scholar] [CrossRef]
- Yelin, E.; Weinstein, S.; King, T. An update on the burden of musculoskeletal diseases in the U.S. Semin. Arthritis Rheum. 2019, 49, 1–2. [Google Scholar] [CrossRef]
- Kye, S.Y.; Park, K. Suicidal ideation and suicidal attempts among adults with chronic diseases: A cross-sectional study. Compr. Psychiatry 2017, 73, 160–167. [Google Scholar] [CrossRef]
- Carlo, M.D., Jr.; Loeser, R.F. Increased oxidative stress with aging reduces chondrocyte survival: Correlation with intracellular glutathione levels. Arthritis Rheum. 2003, 48, 3419–3430. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Rego, I.; Ruiz-Romero, C. The role of mitochondria in osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Kuszel, L.; Trzeciak, T.; Richter, M.; Czarny-Ratajczak, M. Osteoarthritis and telomere shortening. J. Appl. Genet. 2015, 56, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Collins, J.A.; Diekman, B.O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 412–420. [Google Scholar] [CrossRef]
- Gloire, G.; Legrand-Poels, S.; Piette, J. NF-kappaB activation by reactive oxygen species: Fifteen years later. Biochem. Pharmacol. 2006, 72, 1493–1505. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Onodera, Y.; Teramura, T.; Takehara, T.; Fukuda, K. Hyaluronic acid regulates a key redox control factor Nrf2 via phosphorylation of Akt in bovine articular chondrocytes. FEBS Open Bio 2015, 5, 476–484. [Google Scholar] [CrossRef]
- Cai, D.; Feng, W.; Liu, J.; Jiang, L.; Chen, S.; Yuan, T.; Yu, C.; Xie, H.; Geng, D.; Qin, J. 7,8-Dihydroxyflavone activates Nrf2/HO-1 signaling pathways and protects against osteoarthritis. Exp. Ther. Med. 2019, 18, 1677–1684. [Google Scholar] [CrossRef]
- Li, B.; Jiang, T.; Liu, H.; Miao, Z.; Fang, D.; Zheng, L.; Zhao, J. Andrographolide protects chondrocytes from oxidative stress injury by activation of the Keap1-Nrf2-Are signaling pathway. J. Cell Physiol. 2018, 234, 561–571. [Google Scholar] [CrossRef]
- Roman-Blas, J.A.; Jimenez, S.A. NF-kappaB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthr. Cartil. 2006, 14, 839–848. [Google Scholar] [CrossRef]
- Ye, Z.W.; Zhang, J.; Townsend, D.M.; Tew, K.D. Oxidative stress, redox regulation and diseases of cellular differentiation. Biochim. Biophys. Acta 2015, 1850, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Jallali, N.; Ridha, H.; Thrasivoulou, C.; Underwood, C.; Butler, P.E.; Cowen, T. Vulnerability to ROS-induced cell death in ageing articular cartilage: The role of antioxidant enzyme activity. Osteoarthr. Cartil. 2005, 13, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.R. Can serum biomarker assays measure the progression of cartilage degeneration in osteoarthritis? Arthritis Rheum. 2002, 46, 2549–2552. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.M.; Chia, L.S.; Goh, N.K.; Chia, T.F.; Brouillard, R. Analysis and biological activities of anthocyanins. Phytochemistry 2003, 64, 923–933. [Google Scholar] [CrossRef]
- Dai, T.; Shi, K.; Chen, G.; Shen, Y.; Pan, T. Malvidin attenuates pain and inflammation in rats with osteoarthritis by suppressing NF-kappaB signaling pathway. Inflamm. Res. 2017, 66, 1075–1084. [Google Scholar] [CrossRef]
- Wongwichai, T.; Teeyakasem, P.; Pruksakorn, D.; Kongtawelert, P.; Pothacharoen, P. Anthocyanins and metabolites from purple rice inhibit IL-1beta-induced matrix metalloproteinases expression in human articular chondrocytes through the NF-kappaB and ERK/MAPK pathway. Biomed. Pharmacother. 2019, 112, 108610. [Google Scholar] [CrossRef]
- Yoon, C.H.; Chung, S.J.; Lee, S.W.; Park, Y.B.; Lee, S.K.; Park, M.C. Gallic acid, a natural polyphenolic acid, induces apoptosis and inhibits proinflammatory gene expressions in rheumatoid arthritis fibroblast-like synoviocytes. Jt. Bone Spine 2013, 80, 274–279. [Google Scholar] [CrossRef]
- Haseeb, A.; Chen, D.; Haqqi, T.M. Delphinidin inhibits IL-1beta-induced activation of NF-kappaB by modulating the phosphorylation of IRAK-1(Ser376) in human articular chondrocytes. Rheumatology 2013, 52, 998–1008. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef]
- Almonte-Becerril, M.; Navarro-Garcia, F.; Gonzalez-Robles, A.; Vega-Lopez, M.A.; Lavalle, C.; Kouri, J.B. Cell death of chondrocytes is a combination between apoptosis and autophagy during the pathogenesis of Osteoarthritis within an experimental model. Apoptosis 2010, 15, 631–638. [Google Scholar] [CrossRef]
- Hwang, H.S.; Yang, C.M.; Park, S.J.; Kim, H.A. Monosodium Urate Crystal-Induced Chondrocyte Death via Autophagic Process. Int. J. Mol. Sci. 2015, 16, 29265–29277. [Google Scholar] [CrossRef]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.; Botbol, Y.; Macian, F.; Cuervo, A.M. Autophagy and disease: Always two sides to a problem. J. Pathol. 2012, 226, 255–273. [Google Scholar] [CrossRef] [PubMed]
- Cinque, L.; Forrester, A.; Bartolomeo, R.; Svelto, M.; Venditti, R.; Montefusco, S.; Polishchuk, E.; Nusco, E.; Rossi, A.; Medina, D.L.; et al. FGF signalling regulates bone growth through autophagy. Nature 2015, 528, 272–275. [Google Scholar] [CrossRef]
- Barranco, C. Osteoarthritis: Activate autophagy to prevent cartilage degeneration? Nat. Rev. Rheumatol. 2015, 11, 127. [Google Scholar] [CrossRef] [PubMed]
- Carames, B.; Taniguchi, N.; Otsuki, S.; Blanco, F.J.; Lotz, M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010, 62, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Lotz, M.; Carames, B. Autophagy: A new therapeutic target in cartilage injury and osteoarthritis. J. Am. Acad. Orthop. Surg. 2012, 20, 261–262. [Google Scholar] [CrossRef] [PubMed]
- Lotz, M.K.; Carames, B. Autophagy and cartilage homeostasis mechanisms in joint health, aging and OA. Nat. Rev. Rheumatol. 2011, 7, 579–587. [Google Scholar] [CrossRef]
- Vuppalapati, K.K.; Bouderlique, T.; Newton, P.T.; Kaminskyy, V.O.; Wehtje, H.; Ohlsson, C.; Zhivotovsky, B.; Chagin, A.S. Targeted Deletion of Autophagy Genes Atg5 or Atg7 in the Chondrocytes Promotes Caspase-Dependent Cell Death and Leads to Mild Growth Retardation. J. Bone Miner. Res. 2015, 30, 2249–2261. [Google Scholar] [CrossRef]
- Kang, X.; Yang, W.; Feng, D.; Jin, X.; Ma, Z.; Qian, Z.; Xie, T.; Li, H.; Liu, J.; Wang, R.; et al. Cartilage-Specific Autophagy Deficiency Promotes ER Stress and Impairs Chondrogenesis in PERK-ATF4-CHOP-Dependent Manner. J. Bone Miner. Res. 2017, 32, 2128–2141. [Google Scholar] [CrossRef] [PubMed]
- Bouderlique, T.; Vuppalapati, K.K.; Newton, P.T.; Li, L.; Barenius, B.; Chagin, A.S. Targeted deletion of Atg5 in chondrocytes promotes age-related osteoarthritis. Ann. Rheum. Dis. 2016, 75, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Horigome, Y.; Ida-Yonemochi, H.; Waguri, S.; Shibata, S.; Endo, N.; Komatsu, M. Loss of autophagy in chondrocytes causes severe growth retardation. Autophagy 2019, 1–11. [Google Scholar] [CrossRef]
- Henrotin, Y.; Kurz, B.; Aigner, T. Oxygen and reactive oxygen species in cartilage degradation: Friends or foes? Osteoarthr. Cartil. 2005, 13, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xie, G.; Wang, W. Reactive oxygen species: The 2-edged sword of osteoarthritis. Am. J. Med. Sci. 2012, 344, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Henrotin, Y.E.; Bruckner, P.; Pujol, J.P. The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthr. Cartil. 2003, 11, 747–755. [Google Scholar] [CrossRef]
- Henrotin, Y.; Deby-Dupont, G.; Deby, C.; De Bruyn, M.; Lamy, M.; Franchimont, P. Production of active oxygen species by isolated human chondrocytes. Br. J. Rheumatol. 1993, 32, 562–567. [Google Scholar] [CrossRef]
- Rathakrishnan, C.; Tiku, K.; Raghavan, A.; Tiku, M.L. Release of oxygen radicals by articular chondrocytes: A study of luminol-dependent chemiluminescence and hydrogen peroxide secretion. J. Bone Miner. Res. 1992, 7, 1139–1148. [Google Scholar] [CrossRef]
- Tiku, M.L.; Liesch, J.B.; Robertson, F.M. Production of hydrogen peroxide by rabbit articular chondrocytes. Enhancement by cytokines. J. Immunol. 1990, 145, 690–696. [Google Scholar]
- Altay, M.A.; Erturk, C.; Bilge, A.; Yapti, M.; Levent, A.; Aksoy, N. Evaluation of prolidase activity and oxidative status in patients with knee osteoarthritis: Relationships with radiographic severity and clinical parameters. Rheumatol. Int. 2015, 35, 1725–1731. [Google Scholar] [CrossRef]
- Erturk, C.; Altay, M.A.; Selek, S.; Kocyigit, A. Paraoxonase-1 activity and oxidative status in patients with knee osteoarthritis and their relationship with radiological and clinical parameters. Scand. J. Clin. Lab. Investig. 2012, 72, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Altindag, O.; Erel, O.; Aksoy, N.; Selek, S.; Celik, H.; Karaoglanoglu, M. Increased oxidative stress and its relation with collagen metabolism in knee osteoarthritis. Rheumatol. Int. 2007, 27, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Lepetsos, P.; Papavassiliou, A.G. ROS/oxidative stress signaling in osteoarthritis. Biochim. Biophys. Acta 2016, 1862, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ouyang, H.; Dass, C.R.; Xu, J. Current research on pharmacologic and regenerative therapies for osteoarthritis. Bone Res. 2016, 4, 15040. [Google Scholar] [CrossRef]
- Hwang, H.S.; Kim, H.A. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 26035–26054. [Google Scholar] [CrossRef]
- Blanco, F.J.; Guitian, R.; Vazquez-Martul, E.; de Toro, F.J.; Galdo, F. Osteoarthritis chondrocytes die by apoptosis. A possible pathway for osteoarthritis pathology. Arthritis Rheum. 1998, 41, 284–289. [Google Scholar] [CrossRef]
- Aigner, T.; Hemmel, M.; Neureiter, D.; Gebhard, P.M.; Zeiler, G.; Kirchner, T.; McKenna, L. Apoptotic cell death is not a widespread phenomenon in normal aging and osteoarthritis human articular knee cartilage: A study of proliferation, programmed cell death (apoptosis), and viability of chondrocytes in normal and osteoarthritic human knee cartilage. Arthritis Rheum. 2001, 44, 1304–1312. [Google Scholar]
- Kim, H.A.; Lee, Y.J.; Seong, S.C.; Choe, K.W.; Song, Y.W. Apoptotic chondrocyte death in human osteoarthritis. J. Rheumatol. 2000, 27, 455–462. [Google Scholar]
- Kouri, J.B.; Aguilera, J.M.; Reyes, J.; Lozoya, K.A.; Gonzalez, S. Apoptotic chondrocytes from osteoarthrotic human articular cartilage and abnormal calcification of subchondral bone. J. Rheumatol. 2000, 27, 1005–1019. [Google Scholar]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Sha, W.C.; Bronson, R.T.; Ghosh, S.; Baltimore, D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature 1995, 376, 167–170. [Google Scholar] [CrossRef]
- Li, Q.; Van Antwerp, D.; Mercurio, F.; Lee, K.F.; Verma, I.M. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science 1999, 284, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Marchev, A.S.; Dimitrova, P.A.; Burns, A.J.; Kostov, R.V.; Dinkova-Kostova, A.T.; Georgiev, M.I. Oxidative stress and chronic inflammation in osteoarthritis: Can NRF2 counteract these partners in crime? Ann. N. Y. Acad. Sci. 2017, 1401, 114–135. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Jia, J.; Jin, X.; Tong, W.; Tian, H. Resveratrol ameliorates inflammatory damage and protects against osteoarthritis in a rat model of osteoarthritis. Mol. Med. Rep. 2018, 17, 1493–1498. [Google Scholar] [CrossRef] [PubMed]
- Jia, T.; Qiao, J.; Guan, D.; Chen, T. Anti-Inflammatory Effects of Licochalcone A on IL-1beta-Stimulated Human Osteoarthritis Chondrocytes. Inflammation 2017, 40, 1894–1902. [Google Scholar] [CrossRef] [PubMed]
- Hosseinzadeh, A.; Jafari, D.; Kamarul, T.; Bagheri, A.; Sharifi, A.M. Evaluating the Protective Effects and Mechanisms of Diallyl Disulfide on Interlukin-1beta-Induced Oxidative Stress and Mitochondrial Apoptotic Signaling Pathways in Cultured Chondrocytes. J. Cell. Biochem. 2017, 118, 1879–1888. [Google Scholar] [CrossRef]
- Xue, E.X.; Lin, J.P.; Zhang, Y.; Sheng, S.R.; Liu, H.X.; Zhou, Y.L.; Xu, H. Pterostilbene inhibits inflammation and ROS production in chondrocytes by activating Nrf2 pathway. Oncotarget 2017, 8, 41988–42000. [Google Scholar] [CrossRef]
- Khan, N.M.; Haseeb, A.; Ansari, M.Y.; Devarapalli, P.; Haynie, S.; Haqqi, T.M. Wogonin, a plant derived small molecule, exerts potent anti-inflammatory and chondroprotective effects through the activation of ROS/ERK/Nrf2 signaling pathways in human Osteoarthritis chondrocytes. Free Radic. Biol. Med. 2017, 106, 288–301. [Google Scholar] [CrossRef]
- Abusarah, J.; Benabdoune, H.; Shi, Q.; Lussier, B.; Martel-Pelletier, J.; Malo, M.; Fernandes, J.C.; de Souza, F.P.; Fahmi, H.; Benderdour, M. Elucidating the Role of Protandim and 6-Gingerol in Protection Against Osteoarthritis. J. Cell. Biochem. 2017, 118, 1003–1013. [Google Scholar] [CrossRef]
- Bai, Y.; Gong, X.; Dou, C.; Cao, Z.; Dong, S. Redox control of chondrocyte differentiation and chondrogenesis. Free Radic. Biol. Med. 2019, 132, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.M.; Guilak, F.; Weinberg, J.B.; Fermor, B. Reactive nitrogen and oxygen species in interleukin-1-mediated DNA damage associated with osteoarthritis. Osteoarthr. Cartil. 2008, 16, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Schafer, A.; Chovanova, Z.; Muchova, J.; Sumegova, K.; Liptakova, A.; Durackova, Z.; Hogger, P. Inhibition of COX-1 and COX-2 activity by plasma of human volunteers after ingestion of French maritime pine bark extract (Pycnogenol). Biomed. Pharmacother. 2005, 60, 5–9. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.-Y.; Park, Y.-J.; Song, M.-G.; Kim, D.R.; Zada, S.; Kim, D.-H. Cytoprotective Effects of Delphinidin for Human Chondrocytes against Oxidative Stress through Activation of Autophagy. Antioxidants 2020, 9, 83. https://doi.org/10.3390/antiox9010083
Lee D-Y, Park Y-J, Song M-G, Kim DR, Zada S, Kim D-H. Cytoprotective Effects of Delphinidin for Human Chondrocytes against Oxidative Stress through Activation of Autophagy. Antioxidants. 2020; 9(1):83. https://doi.org/10.3390/antiox9010083
Chicago/Turabian StyleLee, Dong-Yeong, Young-Jin Park, Myung-Geun Song, Deok Ryong Kim, Sahib Zada, and Dong-Hee Kim. 2020. "Cytoprotective Effects of Delphinidin for Human Chondrocytes against Oxidative Stress through Activation of Autophagy" Antioxidants 9, no. 1: 83. https://doi.org/10.3390/antiox9010083
APA StyleLee, D.-Y., Park, Y.-J., Song, M.-G., Kim, D. R., Zada, S., & Kim, D.-H. (2020). Cytoprotective Effects of Delphinidin for Human Chondrocytes against Oxidative Stress through Activation of Autophagy. Antioxidants, 9(1), 83. https://doi.org/10.3390/antiox9010083