Abstract
Accumulating evidence suggests that the pineal hormone melatonin displays protective effects against renal fibrosis, but the mechanisms remain poorly understood. Here, we investigate the effect of the pineal hormone on transdifferentiation of renal fibroblasts to myofibroblasts invoked by transforming growth factor-β1 (TGF-β1). Increased proliferation and activation of renal interstitial fibroblasts after TGF-β1 treatment were attenuated by melatonin pretreatment. Mechanistically, melatonin suppressed Smad2/3 phosphorylation and nuclear co-localization of their phosphorylated forms and Smad4 after TGF-β1 stimulation. In addition, increased phosphorylations of Akt, extracellular signal-regulated kinase 1/2, and p38 after TGF-β1 treatment were also suppressed by the hormone. These effects of melatonin were not affected by pharmacological and genetic inhibition of its membrane receptors. Furthermore, melatonin significantly reversed an increase of intracellular reactive oxygen species (ROS) and malondialdehyde levels, and a decrease of the reduced glutathione/oxidized glutathione ratio after TGF-β1 treatment. Finally, TGF-β1-induced proliferation and activation were also suppressed by N-acetylcysteine. Altogether, these findings suggest that the pineal hormone melatonin prevents TGF-β1-induced transdifferentiation of renal interstitial fibroblasts to myofibroblasts via inhibition of Smad and non-Smad signaling cadcades by inhibiting ROS-mediated mechanisms in its receptor-independent manner.
1. Introduction
Renal fibrosis is critically involved in the pathogenesis of chronic kidney disease (CKD) and attributed to excessive deposition of extracellular matrix (ECM). Its underlying mechanisms are complex and involve various cellular pathways [1]. Among them, fibroblast-myofibroblast transdifferentiation is one of the critical steps in the fibrotic process. Myofibroblasts synthesize and secrete large amount of ECM into the renal interstitial region. Thus, blocking fibroblast-myofibroblast transdifferentiation would be a promising preventive or therapeutic approach against renal fibrosis.
Transforming growth factor-β1 (TGF-β1) is a crucial mediator in the pathophysiology of fibrotic diseases such as renal fibrosis [2,3]. Its downstream signaling involves Smad and non-Smad signaling cascades that regulate gene expression required for fibrotic processes including fibroblast-myofibroblast transdifferentiation. It has been shown that Smad proteins are overactivated in the kidneys of patients and animals with CKD [4,5,6,7]. TGF-β1 induces phosphorylation of Smad proteins including Smad2 and Smad3, which form complexes with Smad4 [2]. Then, the complexes are translocated into the nucleus to modulate expression of fibrosis-related genes. In addition, the cytokine can also activate various non-Smad signaling pathways such as Akt and mitogen-activated protein kinase (MAPK) pathways [8].
The pineal hormone melatonin plays an essential role in regulating the sleep–wake cycle [9]. Besides, the hormone has been reported to exert multiple biological actions such as anti-inflammatory and anti-oxidant effects [10,11]. Accumulating evidence suggests that fibrotic processes in a variety of organs were ameliorated by the hormone [12]. Indeed, melatonin was shown to have protective effects against renal fibrosis in several animal models [13,14,15,16]. However, mechanisms for the beneficial effects of the hormone against renal fibrosis remain poorly understood.
The present study aimed to explore the effects of melatonin on TGF-β1-stimulated fibroblast-myofibroblast transdifferentiation and investigate its underlying mechanisms. We noted that melatonin prevents TGF-β1-induced proliferation and activation of renal fibroblasts through suppressing Smad and non-Smad signaling cascades. These effects of melatonin were mediated by inhibiting reactive oxygen species (ROS)-mediated mechanisms in its receptor-independent manner. These findings provide a novel mechanistic insight into the preventive effects of melatonin against renal fibrosis.
2. Materials and Methods
2.1. Cell Culture and Treatments
The rat kidney interstitial fibroblast cell line NRK-49F cells were purchased from the American Type Culture Collection (Rockville, MD, USA). Cells were grown in Dulbecco’s Modified Eagle’s Medium containing 10% fetal bovine serum at 37 °C under 5% CO₂ and 95% air. To explore the effect of melatonin on TGF-β1-stimulated activation of fibroblasts, the cells were incubated with TGF-β1 (5 ng/mL; R&D Systems, Minneapolis, MN, USA) for 24 h after pretreatment with melatonin (0.1 mM or 1 mM) for 30 min in the presence or absence of luzindole (20 μM or 100 μM). In addition, the cells were treated with TGF-β1 (5 ng/mL) for 24 h after preincubation with N-acetylcysteine (NAC, 10 mM) or 1 mM melatonin for 30 min. Melatonin, lunzindole, and NAC were purchased from Sigma-Aldrich (St. Louis, MO, USA). Melatonin and luzindole were dissolved in dimethyl sulfoxide (DMSO). The solvent was added to the control cells in the experiments with these compounds. The final concentrations of DMSO in each well did not exceed 0.5% (v/v), which by itself did not affect the cell viability.
2.2. Cell Viability Assay
To evaluate the effect of melatonin on TGF-β1-stimulated proliferation of fibroblasts, NRK-49F cells were treated with TGF-β1 (5 ng/mL) after preincubation with melatonin (0.01 mM, 0.1 mM, or 1 mM) for 30 min in the presence or absence of luzindole (100 μM). In another experiment, cells were treated with TGF-β1 (5 ng/mL) after preincubation with NAC (10 mM) or melatonin (1 mM) for 30 min. Cell viability was analyzed using the Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan) assay at 0, 24, and 48 h after TGF-β1 stimulation according to the manufacturer’s instructions. The absorbance at 450 nm was assessed using a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA).
2.3. Western Blot Analysis
Western blotting was performed as described previously [17]. Briefly, protein samples were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred from the gels onto nitrocellulose membranes. The membranes were proved with primary antibodies overnight at 4 °C, followed by incubation with a horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. The following primary antibodies were used in this study: anti-collagen Ⅰ (1:1000; ab34710; Abcam, Cambridge, MA, USA), anti-fibronectin (1:1000; ab2413; Abcam), anti-α-smooth muscle actin (α-SMA; 1:1000; A2547; Sigma-Aldrich), anti-p-Smad2/3 (1:1000; #8828; Cell Signaling, Danvers, MA, USA), anti-Smad2/3 (1:1000; #3102; Cell Signaling), anti-p-Akt (1:1000; #9271; Cell Signaling), anti-Akt (1:1000; #9272; Cell Signaling), anti-p-extracellular signal-regulated kinase 1/2 (p-ERK1/2; 1:1000; #4370; Cell Signaling), anti-ERK1/2 (1:1000; #9102; Cell Signaling), anti-p-p38 (1:1000; #9215; Cell Signaling), anti-p38 (1:1000; #8690; Cell Signaling), anti-melatonin receptor type 1A (MT1; 1:1000; orb11085; Biorbyt, San Francisco, CA, USA), anti-melatonin receptor type 1B (MT2; 1:1000; NLS932; Novus Biologicals, Littleton, CO, USA), and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:3000; #2118; Cell Signaling) antibody. The protein expression levels were normalized with GAPDH. Quantitative analysis of protein levels was performed using NIH ImageJ software (National Institutes of Health, Bethesda, MD, USA).
2.4. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from cells with TRIzol (Invitrogen, Carlsbad, CA, USA) and cDNA was synthesized from 2 ug of total RNA using RNA to cDNA EcoDry Premix (TaKaRa, Tokyo, Japan) according to the manufacturer’s instructions. The cDNA was amplified by PCR with the following primers: MT1 (NM_008639.3), 5′-TGTCAGCGAGCTGCTCAATG-3′ and 5′-GGTACACAGACAGGATGACCA-3′; MT2 (NM_145712.2), 5′-GAACAGCTCAATCCCTAACTGC-3′ and 5′-ACGACTACTGTAGATAGCATGGG-3′, GAPDH (NM_008084.2), 5′-GACAACTTTGGCATCGTGGA-3′ and 5′–ATGCAGGGATGATGTTCTGG-3′.
2.5. Knockdown of MT1 and MT2
NRK-49F cells were seeded onto a 60-mm culture dish and transfected with rat MT1/MT2 small interfering RNA (siRNA) (#1330001; Thermo Fisher Scientific) or control siRNA (AM4611; Invitrogen) using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. At 24 h after transfection, the cells were pretreated with melatonin (1 mM) for 30 min and then incubated with TGF-β1 (5 ng/mL).
2.6. Evaluation of Iintracellular ROS and Redox Status
Amounts of intracellular ROS and malondialdehyde (MDA) were measured using the 2’,7’-dichlorofluorescin diacetate (DCFDA)-Cellular ROS Assay Kit (ab113851; Abcam) and the Lipid Peroxidation (MDA) Assay Kit (MAK085; Sigma-Aldrich), respectively, according to the manufacturer’s instructions. The reduced glutathione/oxidized glutathione ratio (GSH/GSSG) were measured using the Glutathione (GSSG/GSH) Detection Kit (ADI-900-160; Enzo Life Sciences, Farmingdale, NY, USA) according to the manufacturer’s instructions.
2.7. Immunofluorescence Analysis
NRK-49F cells were fixed for 20 min at room temperature with 4% paraformaldehyde in phosphate-buffered saline. After permeabilization and blocking, the cells were probed with primary antibodies against p-Smad2/3 (1:200; #8828; Cell Signaling), Smad4 (1:200; sc7966; Santa Cruz Biotechnology, Santa Cruz, CA, USA), or α-SMA (1:200; A2547; Sigma-Aldrich), followed by incubation with secondary antibodies directed against the primary antibody. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). The stained cells were visualized using a confocal microscope (Nikon, Tokyo, Japan).
2.8. Statistical Analysis
Data are presented as the mean ± standard error of the mean (SEM). Comparisons between groups were assessed using one-way ANOVA with Bonferroni’s post-hoc tests. All experiments were performed at least 2 times. A p value less than 0.05 was considered statistically significant.
3. Results
3.1. Melatonin Inhibits TGF-β1-Induced Proliferation and Activation in NRK-49F Cells
Given that proliferation and activation of fibroblasts are key processes for their transdifferentiation to myiofibroblasts, we first investigated the effects of melatonin on TGF-β1-stimulated proliferation of renal interstitial fibroblasts. NRK-49F cells were preincubated with melatonin (1 mM) and then treated with TGF-β1 (5 ng/mL). Cell viability was evaluated using CCK-8 assay at 0, 24, and 48 h. Pretreatment with melatonin significantly suppressed TGF-β1-stimulated proliferation, while melatonin alone did not affect cell proliferation (Figure 1A).
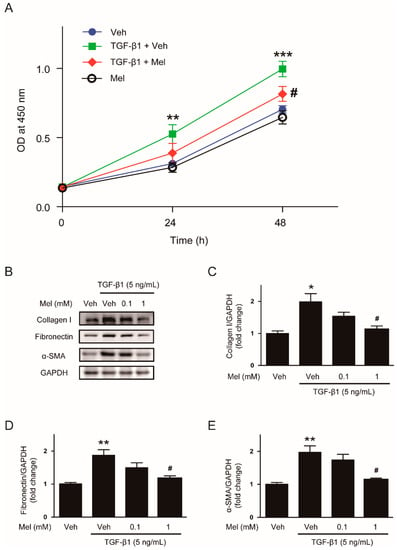
Figure 1.
Effects of melatonin on transforming growth factor-β1 (TGF-β1)-stimulated proliferation and activation in renal interstitial fibroblasts. (A) NRK-49F cells were treated with TGF-β1 (5 ng/mL) after preincubation with vehicle (Veh) or melatonin (Mel; 1 mM) for 30 min. Cell viability was analyzed using the Cell Counting Kit-8 (CCK-8) assay at 0, 24, and 48 h. The optical density (OD) was measured at 450 nm. (B) Western blot analysis for collagen Ⅰ, fibronectin, and α-smooth muscle actin (α-SMA). Cells were treated with TGF-β1 (5 ng/mL) for 24 h after preincubation with Veh or Mel (0.1 mM or 1 mM) for 30 min. The graphs show the results of quantitative analysis of collagen Ⅰ (C), fibronectin (D), and α-SMA (E). * p < 0.05, ** p < 0.01, and *** p < 0.001 vs. Veh-treated cells. # p < 0.05 vs. TGF-β1-treated cells.
We next examined the effects of the hormone on fibroblast activation invoked by TGF-β1. Treatment with TGF-β1 (5 ng/mL) significantly increased expression of ECM proteins including collagen Ⅰ and fibronectin, and α-SMA when compared with the control (Figure 1B–E). These changes were significantly suppressed by melatonin (1 mM).
3.2. Melatonin Suppresses TGF-β1-Induced Smad and Non-Smad Signaling Cascades
In order to explore mechanisms for the inhibitory effects of the hormone on fibroblast-myofibroblast transdifferentiation, we first investigated its effects on TGF-β1/Smad signaling pathway. TGF-β1 induces phosphorylation of Smad2 and Smad3, which form a heteromeric complex with Smad4 [2]. Then, the complex is translocated into the nucleus to regulate expression of fibrosis-related genes. We found that pretreatment with melatonin (1 mM) suppressed TGF-β1-induced phosphorylation of Smad2/3 (Figure 2A,B). Immunofluorescent staining revealed that increased nuclear co-localization of their phosphorylated forms and Smad4 after TGF-β1 treatment was decreased by melatonin (Figure 2C).
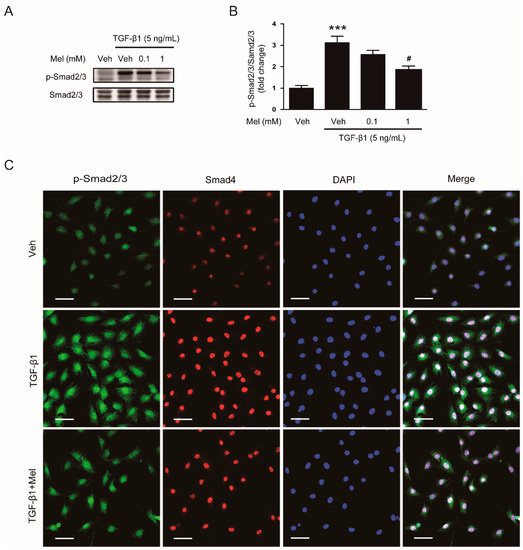
Figure 2.
Effects of melatonin on TGF-β1-stimulated activation of Smad signaling pathway in renal interstitial fibroblasts. NRK-49F cells were treated with TGF-β1 (5 ng/mL) for 24 h after preincubation with vehicle (Veh) or melatonin (Mel; 0.1 mM, or 1 mM) for 30 min. (A) Western blot analysis for p-Smad2/3 and Smad2/3. (B) The graph shows the result of quantitative analysis of p-Smad2/3 (C) Representative immunofluorescence staining of p-Smad2/3 (green) and Smad4 (red) in cells treated with Veh, cells treated with TGF-β1 (5 ng/mL), or cells treated with TGF-β1 (5 ng/mL) plus Mel (1mM). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bar: 50 μm. *** p < 0.001 vs. Veh-treated cells. # p < 0.05 vs. TGF-β1-treated cells.
In addition, the cytokine can also induce activation of non-Smad signaling pathways such as Akt or MAPK cascades [8]. We observed that phosphorylations of Akt, ERK1/2, and p38 after TGF-β1 treatment were also significantly inhibited by the hormone (1 mM) (Figure 3A–D). Collectively, these findings indicate that melatonin suppresses Smad and non-Smad signaling pathways stimulated by TGF-β1.
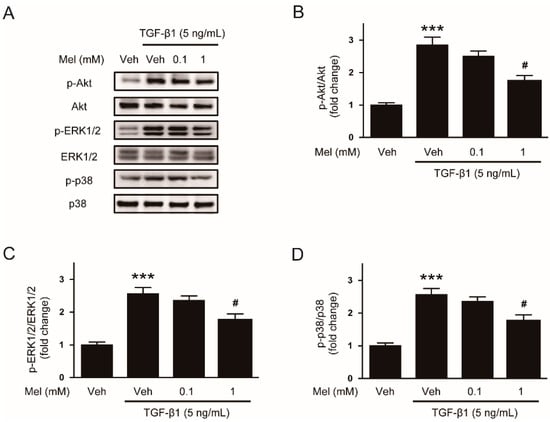
Figure 3.
Effects of melatonin on TGF-β1-induced activation of non-Smad signaling pathway in renal interstitial fibroblasts. NRK-49F cells were treated with TGF-β1 (5 ng/mL) for 30 min after preincubation with vehicle (Veh) or melatonin (Mel; 0.1 mM, or 1 mM) for 30 min. (A) Western blot analysis for p-Akt, Akt, p-extracellular signal-regulated kinase 1/2 (ERK1/2), ERK1/2, p-p38, and p38. The graphs show the results of quantitative analysis of p-Akt (B), p-ERK1/2 (C), and p-p38 (D). *** p < 0.001 vs. Veh-treated cells. # p < 0.05 vs. TGF-β1-treated cells.
3.3. Inhibitory Effects of Melatonin on TGF-β1-Induced Proliferation and Activation Is Independent of Its Membrane Receptors
It has been shown that melatonin displays multiple actions through its membrane receptor-dependent and -independent mechanisms [10]. To date, two subtypes of melatonin membrane receptors, MT1 and MT2, have been identified. To evaluate whether the inhibitory effects of the hormone on proliferation and activation invoked by TGF-β1 is dependent on its receptors, we evaluated the effects of luzindole, an antagonist of melatonin receptors, on the action of melatonin. We first confirmed the presence of MT1 and MT2 in NRK-49F cells using RT-PCR (Figure 4A). Treatment with luzindole at a routinely used concentration (100 μM) [18,19] did not affect the inhibitory effects of melatonin on TGF-β1-stimulated proliferation (Figure 4B). Additionally, increased levels of α-SMA and phosphorylated Smad2/3 after TGF-β1 treatment were not significantly modified by the compound (Figure 4C–E), suggesting that the receptors are dispensable for the suppressive effects of the hormone on TGF-β1-stimulated fibroblast-myofibroblast transdifferentiation.
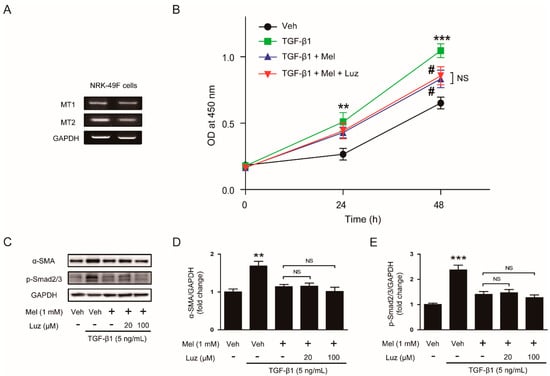
Figure 4.
Effects of luzindole on inhibitory action of melatonin on TGF-β1-stimulated proliferation and activation in renal interstitial fibroblasts. (A) Reverse transcription-polymerase chain reaction analysis of melatonin receptor type 1A (MT1) and type 1B (MT2) in NRK-49F cells. (B) NRK-49F cells were treated with with TGF-β1 (5 ng/mL) after preincubation with vehicle (Veh) or melatonin (Mel; 1mM) in the presence or absence of luzindole (Luz; 100 μM) for 30 min. Cell viability was assessed using CCK-8 assay at 0, 24, and 48 h. The OD was measured at 450 nm. (C) Western blot analysis for α-SMA and p-Smad2/3. Cells were treated with TGF-β1 (5 ng/mL) for 24 h after preincubation with Veh or Mel (1 mM) in the presence or absence of luzindole (Luz; 20 μM or 100 μM) for 30 min. The graphs show the results of quantitative analysis of α-SMA (D) and p-Smad2/3 (E). ** p < 0.01 and *** p < 0.001 vs. Veh-treated cells. # p < 0.05 vs. TGF-β1-treated cells. NS: not significant.
To more clearly demonstrate that the inhibitory effects of melatonin on fibroblast-myofibroblast transdifferentiation is independent of its receptors, we next examined the effects of genetic inhibition of MT1 and MT2 using siRNA on the action of the hormone. We found that knockdown of melatonin receptors (MT1 and MT2) using siRNA did not significantly affect the inhibitory effects of the hormone on TGF-β1-induced proliferation (Figure 5A) and expression of fibronectin and α-SMA (Figure 5B–D). Altogether, these results suggest that the inhibitory action of melatonin on TGF-β1-stimulated proliferation and activation is independent of its receptors.
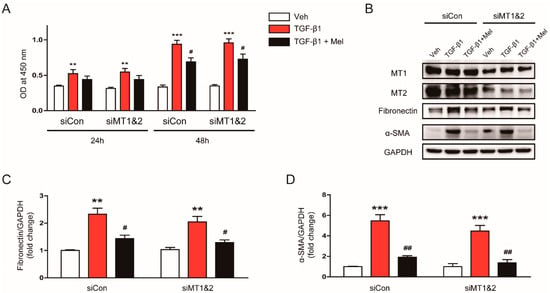
Figure 5.
Effects of siRNA-mediated knockdown of melatonin receptors on inhibitory effects of melatonin on TGF-β1-induced proliferation and activation in renal interstitial fibroblasts. NRK-49F cells were treated with negative control siRNA (siCon) or siRNAs targeting MT1 and MT2 (siMT1&B). After 24 h, the cells were pretreated with vehicle (Veh) or melatonin (Mel; 1 mM) and then incubated with TGF-β1 (5 ng/mL) for 24 or 48 h. (A) Cell viability was assessed using CCK-8 assay at 24 and 48 h. The OD was measured at 450 nm. (B) Western blot analysis for MT1, MT2, fibronectin, and α-SMA. Cells were incubated with TGF-β1 (5 ng/mL) for 24 h after pretreatment with Veh or Mel (1 mM). The graphs show the results of quantitative analysis of fibronectin (C) and α-SMA (D). ** p < 0.01 and *** p < 0.001 vs. Veh-treated cells. # p < 0.05 and ## p < 0.01 vs. TGF-β1-treated cells.
3.4. Inhibitory Effects of Melatonin on TGF-β1-Induced Proliferation and Activation Is attributed to the Inhibition of ROS-Mediated Mechanisms
Melatonin can act as a direct scavenger of ROS. Accumulating evidence suggests that the antioxidant property of melatonin is closely associated with its membrane receptor-independent mechanisms [10]. Thus, we next aimed to explore whether inhibitory action of the hormone on TGF-β1-stimulated fibroblast-myofibroblast transdifferentiation is attributed to the deactivation of ROS-mediated mechanisms. We first investigated the effects of melatonin on ROS generation in NRK-49F cells stimulated by TGF-β1. As expected, treatment with TGF-β1 elevated intracellular levels of ROS and MDA (Figure 6A,B). These effects were significantly attenuated by pretreatment with melatonin or NAC. Decreased GSH/GSSG ratio after TGF-β1 treatment was also significantly reversed by the hormone or NAC (Figure 6C). Altogether, these results suggest that melatonin effectively reduces ROS generation and lipid peroxidation, and changes intracellular redox status in TGF-β1-treated renal interstitial fibroblasts.
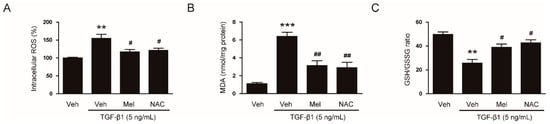
Figure 6.
Effects of melatonin or N-acetylcysteine (NAC) on TGF-β1-stimulated generation of reactive oxygen species (ROS) and alterations in redox status. NRK-49F cells were pretreated with vehicle (Veh), melatonin (Mel; 1 mM), or NAC (10 mM) and then incubated with TGF-β1 (5 ng/mL) for 24 h. (A) Intracellular ROS levels. (B) Intracellular malondialdehyde (MDA) levels. (C) The reduced glutathione/oxidized glutathione ratio (GSH/GSSG). ** p < 0.01 and *** p < 0.001 vs. Veh-treated cells. # p < 0.05 and ## p < 0.01 vs. TGF-β1-treated cells.
We next examined whether NAC can also exert inhibitory effects on proliferation and activation in renal interstitial fibroblasts stimulated with TGF-β1. We observed that TGF-β1-stimulated proliferation was significantly inhibited by NAC or melatonin (Figure 7A). Pretreatment with NAC or melatonin also decreased protein expression of collagen Ⅰ and α-SMA after TGF-β1 treatment (Figure 7B–D). Furthermore, immunofluorescent staining revealed that an elevation in α-SMA expression (Figure 7E) after TGF-β1 treatment was reversed by NAC as well as melatonin.
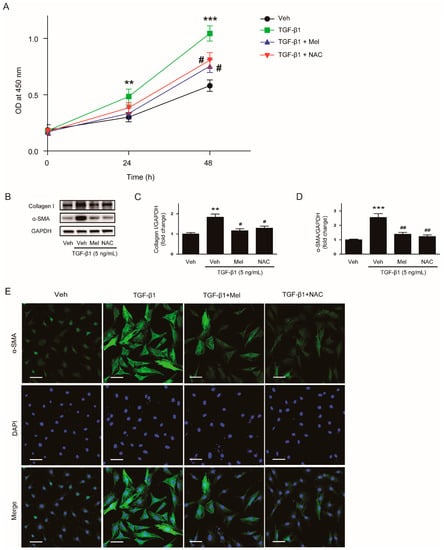
Figure 7.
Effects of melatonin or NAC on TGF-β1-stimulated proliferation and activation in renal interstitial fibroblasts. (A) NRK-49F cells were treated with TGF-β1 (5 ng/mL) after preincubation with vehicle (Veh), melatonin (Mel; 1mM), or NAC (10 mM) for 30 min. Cell viability was assessed using CCK-8 assay at 0, 24, and 48 h. The OD was measured at 450 nm. (B) Western blot analysis for collagen Ⅰ and α-SMA. Cells were pretreated with Veh, Mel (1 mM), or NAC (10 mM) and then incubated with TGF-β1 (5 ng/mL) for 24 h. The graphs show the results of quantitative analysis of collagen Ⅰ (C) and α-SMA (D). (E) Representative immunofluorescence staining of α-SMA (green) in cells treated with Veh, TGF-β1 (5 ng/mL), TGF-β1 (5 ng/mL) plus Mel (1mM), or TGF-β1 (5 ng/mL) plus NAC (10 mM). Nuclei were counterstained with DAPI (blue). Scale bar: 50 μm. ** p < 0.01, and *** p < 0.001 vs. Veh-treated cells. # p < 0.05 and ## p < 0.01 vs. TGF-β1-treated cells.
These molecules also suppressed nuclear localization of p-Smad2/3 (Figure 8A) and reversed increased expression of p-Akt, p-ERK1/2, and p-p38 (Figure 8B–E) invoked by TGF-β1. Taken together, these findings indicate that the suppressive effects of melatonin on fibroblast-myofibroblast transdifferentiation invoked by TGF-β1 is presumably attributed to the suppression of ROS-dependent mechanisms.
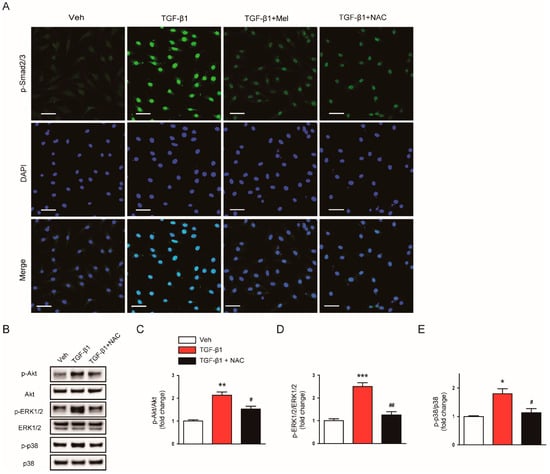
Figure 8.
Effects of melatonin or NAC on TGF-β1-stimulated nuclear localization of p-Smad2/3 in renal interstitial fibroblasts. NRK-49F cells were pretreated with vehicle (Veh), melatonin (Mel; 1mM), or NAC (10 mM) and then incubated with TGF-β1 (5 ng/mL) for 24 h. (A) The images show representative immunofluorescence staining of p-Smad2/3 (green). Nuclei were counterstained with DAPI (blue). Scale bar: 50 μm. (B) Western blot analysis for p-Akt, Akt, p-ERk1/2, ERK1/2, p-p38, and p38. The graphs show the results of quantitative analysis of p-Akt (C), p-ERK1/2 (D), and p-p38 (E). * p < 0.05, ** p < 0.01, and *** p < 0.001 vs. Veh-treated cells. # p < 0.05 and ## p < 0.01 vs. TGF-β1-treated cells.
4. Discussion
In this study, we demonstrated that melatonin inhibited TGF-β1-stimulated transdifferentiation of renal interstitial fibroblasts to myofibroblasts. Pretreatment with melatonin effectively suppressed TGF-β1-stimulated proliferation. Increased levels of activation-related markers after TGF-β1 treatment was also inhibited by the hormone. These effects of the hormone were accompanied by suppression of Smad and non-Smad signaling pathways (Akt, ERK1/2, and p38). Additionally, pharmacological ad genetic inhibition of melatonin receptors (MT1 and MT2) did not modify the action of melatonin, indicating that the receptors are not required for the suppressive effects of the hormone on TGF-β1-induced proliferation and activation. Furthermore, we found that the suppressive effects of melatonin on TGF-β1-induced transdifferentiation of fibroblasts to myofibroblasts are presumably attributed to the suppression of ROS-dependent mechanisms. These results provide a novel mechanistic insight into the preventive effects of the pineal hormone on renal fibrosis (Figure 9).
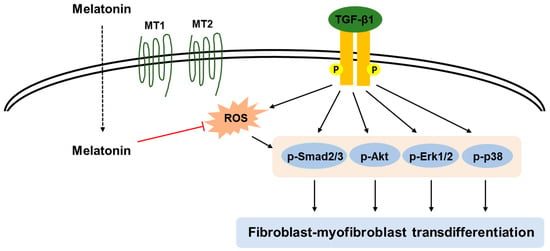
Figure 9.
A graphical representation of the mechanism for the inhibitory action of melatonin on TGF-β1-induced transdifferentiation of fibroblasts to myofibroblasts. Melatonin prevents TGF-β1-stimulated proliferation and activation by suppressing Smad and non-Smad signaling cascades (Akt, ERK1/2, and p38) in renal interstitial fibroblasts. These effects of melatonin were attributed to the deactivation of ROS-dependent mechanisms in its membrane receptors (MT1 and MT2)-independent manner.
The pineal hormone melatonin plays an essential role in modulating the sleep–wake cycle [9]. Besides, the hormone has been shown to exert preventive and/or therapeutic effects against various diseases [10,11]. The favorable actions of melatonin were primarily attributed to its anti-inflammatory and anti-oxidant activities. Emerging evidence also reveals that the hormone exerts strong anti-fibrotic activities in various organs [12]. It has been shown that melatonin ameliorated cyclosporine A [13]- or carbon tetrachloride [14]-induced renal fibrosis. Additionally, melatonin was found to suppress the fibrotic process invoked by unilateral ureteral obstruction (UUO) in a rodent model of CKD [15]. A recent study also showed that melatonin ameliorated renal fibrosis in animals with diabetic kidney disease [16]. However, molecular mechanisms for the favorable effects of the hormone against renal fibrosis remain unclear. In the present study, we demonstrated that melatonin prevents transdifferentiation of renal interstitial fibroblasts to myofibroblasts invoked by TGF-β1. During the development and progression of fibrosis, myofibroblasts synthesize and secret ECM components including collagen and fibronectin. Given that fibroblast-myofibroblast transdifferentiation is a critical process in the pathophysiology of renal fibrosis [1], these findings provide a novel mechanistic insight into the preventive effects of melatonin against renal fibrosis. Consistent with our findings, a previous study showed that melatonin inhibits TGF-β1-induced epithelial-mesenchymal transition in lung alveolar epithelial cells through suppressing Smad and Wnt/β-catenin signaling pathways [20]. The hormone was also found to suppress fibrotic process in rat kidneys and human renal proximal tubular epithelial cells by inhibiting Smad and MAPK signaling pathways [21]. Additionally, it was recently reported that the anti-fibrotic effects of melatonin against liver fibrosis induced by carbon tetrachloride was associated with its inhibitory action on TGF-β1/Smad signaling cascade [22].
Canonical TGF-β/Smad signaling cascade plays a key role in the regulation of fibroblast-myofibroblast transdifferentiation [2,3]. TGF-β1 induces phosphorylation of Smad2 and Smad3, which form a heteromeric complex with Smad4. Then, the complex is translocated into the nucleus to regulate expression of fibrosis-related genes. In this study, we noted that melatonin suppressed TGF-β1-stimulated phosphorylation of Smad2/3. Furthermore, immunofluorescent staining showed that the hormone significantly attenuated TGF-β1-induced nuclear co-localization of their phosphorylated forms and Smad4. In addition, the cytokine can activate various non-Smad signaling pathways [8]. We also observed that melatonin significantly suppressed an increase in the phosphorylations of Akt, ERK1/2, and p38 after TGF-β1 treatment. Previous studies showed that Akt and ERK1/2 in ligated kidneys were activated in the UUO model [23]. Pharmacological inhibition of Akt or ERK1/2 induces a reduction in levels of myofibroblast markers in the kidneys. In addition, phosphorylation of p38 was also shown to be increased in the UUO model [24,25] and a genetic model of CKD [26]. Treatment with a specific inhibitor of p38 significantly reduced accumulation of interstitial myofibroblasts and renal fibrosis in both models. Collectively, our findings suggest that melatonin dampens TGF-β1-induced fibroblast-myofibroblast transdifferentiation through inhibition of Smad and non-Smad signaling pathways.
Melatonin displays various biological actions through both its receptor-dependent and -independent mechanisms [10]. To date, two subtypes of mammalian membrane receptors, MT1 and MT2, have been identified. Membrane receptor-dependent action of melatonin includes regulation of circadian rhythm and anti-cancer effect [10]. In addition, a number of studies have reported the receptor-independent action of melatonin on various cellular functions despite existence of melatonin receptors [27,28,29,30]. In the present study, luzindole, an antagonist of melatonin receptors, was used to evaluate whether the suppressive effects of the hormone on TGF-β1-stimulated fibroblast-myofibroblast transdifferentiation are dependent on its receptors. We found that the compound did not modify the suppressive effects of melatonin on TGF-β1-induced proliferation and activation of fibroblasts. Furthermore, the effects of melatonin were not significantly affected by siRNA-mediated knockdown of MT1 and MT2, indicating that the receptors are not required for the suppressive effects of the hormone. The membrane receptor-independent actions of melatonin have been known to be related to its ROS scavenging property [10]. Because of its high lipophilicity, melatonin can easily enter into the cytosol through passing the plasma membrane. In cytosol, the hormone can directly scavenge ROS. In the present study, we noted an elevation of intracellular ROS levels after TGF-β1 treatment. This observation is consistent with the results of previous reports [31,32,33]. As expected, melatonin significantly attenuated ROS production and changed intracellular redox status in renal interstitial fibroblasts stimulated by TGF-β1, as evidenced by decreased intracellular levels of ROS and MDA and increased GSH/GSSG ratio. We also found that the antioxidant NAC also exerted inhibitory effects on TGF-β1-induced proliferation and activation. Immunofluorescent staining clearly revealed that an elevation in α-SMA expression and nuclear localization of p-Smad2/3 after TGF-β1 treatment were reversed by NAC as well as melatonin. Pretreatment with NAS also suppressed phosphorylations of Akt, ERK1/2, and p38 after TGF-β1 treatment. Taken together, these findings indicate that the suppressive action of melatonin on fibroblast-myofibroblast transdifferentiation invoked by TGF-β1 is presumably attributed to the suppression of ROS-mediated mechanisms in its receptor-independent manner.
ROS can act as an intracellular second messenger during cell differentiation [34]. Indeed, accumulating evidence suggests that nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-generated ROS is critically involved in fibroblast-myofibroblast transdifferentiation and progression of kidney fibrosis [31,35]. TGF-β1-stimulated conversion of fibroblasts into myofibroblasts in other organs including heart [36], lung [37], intestine [38], and skin [39] was also mediated by ROS derived from NADPH. Thus, targeting of NADPH oxidase is considered as a potential preventive and therapeutic strategy against fibrotic diseases. Altogether, our findings suggest that the ROS scavenging property of melatonin mainly contributes to its suppressive effects on TGF-β1-stimulated fibroblast-myofibroblast transdifferentiation.
5. Conclusions
These results demonstrate that the pineal hormone melatonin prevents TGF-β1-stimulated transdifferentiation of fibroblasts to myofibroblasts by suppressing Smad and non-Smad signaling cascades. These effects of melatonin were attributed to the deactivation of ROS-mediated mechanisms in its receptor-independent manner. These results strengthen the idea that melatonin may be a promising preventive option against fibrotic diseases.
Author Contributions
Conceptualization, J.-Y.K., J.-H.P. and J.L.; Data curation, J.-Y.K.; Formal analysis, J.-Y.K., J.-H.P., and E.J.J.; Funding acquisition, E.J.J., J.L.; Investigation, J.-Y.K.; Methodology, J.-Y.K., E.J.J., and K.-K.P.; Project administration, J.-H.P. and J.L.; Supervision, E.J.J. and J.L.; Writing—original draft, J.-Y.K., J.-H.P., and J.L.; Writing—review and editing, J.-Y.K., J.-H.P., E.J.J., J.L., and K.-K.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT & Future Planning (MSIP) (NRF-2017R1E1A2A02023467 and NRF-2017R1C1B5076755) and the Ministry of Education (NRF-2017R1D1A1B03035278).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Farris, A.B.; Colvin, R.B. Renal interstitial fibrosis: Mechanisms and evaluation. Curr. Opin. Nephrol. Hypertens. 2012, 21, 289–300. [Google Scholar] [CrossRef]
- Meng, X.M.; Tang, P.M.; Li, J.; Lan, H.Y. TGF-β/Smad signaling in renal fibrosis. Front. Physiol. 2015, 6, 82. [Google Scholar] [CrossRef]
- Isaka, Y. Targeting TGF-β signaling in kidney fibrosis. Int. J. Mol. Sci. 2018, 19, 2532. [Google Scholar] [CrossRef]
- Kim, M.K.; Maeng, Y.I.; Sung, W.J.; Oh, H.K.; Park, J.B.; Yoon, G.S.; Cho, C.H.; Park, K.K. The differential expression of TGF-β1, ILK and wnt signaling inducing epithelial to mesenchymal transition in human renal fibrogenesis: An immunohistochemical study. Int. J. Clin. Exp. Pathol. 2013, 6, 1747–1758. [Google Scholar] [PubMed]
- Thomsen, L.H.; Fog-Tonnesen, M.; Nielsen Fink, L.; Norlin, J.; García de Vinuesa, A.; Hansen, T.K.; de Heer, E.; Ten Dijke, P.; Rosendahl, A. Disparate phospho-Smad2 levels in advanced type 2 diabetes patients with diabetic nephropathy and early experimental db/db mouse model. Ren. Fail. 2017, 39, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Jang, K.M.; An, H.J.; Kim, J.Y.; Gwon, M.G.; Gu, H.; Park, B.; Park, K.K. Pomolic Acid Ameliorates Fibroblast Activation and Renal Interstitial Fibrosis through Inhibition of SMAD-STAT Signaling Pathways. Molecules 2018, 23, 2236. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.J.; Kim, K.H.; Kim, Y.J.; Chang, Y.C.; Lee, I.H.; Park, K.K. Antifibrotic effect of synthetic Smad/Sp1 chimeric decoy oligodeoxynucleotide through the regulation of epithelial mesenchymal transition in unilateral ureteral obstruction model of mice. Exp. Mol. Pathol. 2013, 95, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Zisapel, N. New perspectives on the role of melatonin in human sleep, circadian rhythms and their regulation. Br. J. Pharmacol. 2018, 175, 3190–3199. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Galano, A. Melatonin: Exceeding expectations. Physiology 2014, 29, 325–333. [Google Scholar] [CrossRef]
- Opie, L.H.; Lecour, S. Melatonin has multiorgan effects. Eur. Heart J. Cardiovasc. Pharmacother. 2016, 2, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Ma, Z.; Jiang, S.; Fan, C.; Deng, C.; Yan, X.; Di, S.; Lv, J.; Reiter, R.J.; Yang, Y. Melatonin: The dawning of a treatment for fibrosis? J. Pineal Res. 2016, 60, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Eşrefoğlu, M.; Kuruş, M.; Sahna, E. The beneficial effect of melatonin on chronic cyclosporin A nephrotoxicity in rats. J. Int. Med. Res. 2003, 31, 42–44. [Google Scholar] [CrossRef] [PubMed]
- Ogeturk, M.; Kus, I.; Kavakli, A.; Oner, J.; Kukner, A.; Sarsilmaz, M. Reduction of carbon tetrachloride-induced nephropathy by melatonin administration. Cell Biochem. Funct. 2005, 23, 85–92. [Google Scholar] [CrossRef]
- Ozbek, E.; Ilbey, Y.O.; Ozbek, M.; Simsek, A.; Cekmen, M.; Somay, A. Melatonin attenuates unilateral ureteral obstruction-induced renal injury by reducing oxidative stress, iNOS, MAPK, and NF-kB expression. J. Endourol. 2009, 23, 1165–1173. [Google Scholar] [CrossRef]
- Li, J.; Li, N.; Yan, S.; Lu, Y.; Miao, X.; Gu, Z.; Shao, Y. Melatonin attenuates renal fibrosis in diabetic mice by activating the AMPK/PGC1α signaling pathway and rescuing mitochondrial function. Mol. Med. Rep. 2019, 19, 1318–1330. [Google Scholar] [CrossRef]
- Kim, J.Y.; Jo, J.; Kim, K.; An, H.J.; Gwon, M.G.; Gu, H.; Kim, H.J.; Yang, A.Y.; Kim, S.W.; Jeon, E.J.; et al. Pharmacological Activation of Sirt1 Ameliorates Cisplatin-Induced Acute Kidney Injury by Suppressing Apoptosis, Oxidative Stress, and Inflammation in Mice. Antioxidants 2019, 8, 322. [Google Scholar] [CrossRef]
- Lin, J.J.; Lin, Y.; Zhao, T.Z.; Zhang, C.K.; Zhang, T.; Chen, X.L.; Ding, J.Q.; Chang, T.; Zhang, Z.; Sun, C.; et al. Melatonin suppresses neuropathic pain via MT2-dependent and -independent pathways in dorsal root ganglia neurons of mice. Theranostics 2017, 7, 2015–2032. [Google Scholar] [CrossRef]
- Suofu, Y.; Li, W.; Jean-Alphonse, F.G.; Jia, J.; Khattar, N.K.; Li, J.; Baranov, S.V.; Leronni, D.; Mihalik, A.C.; He, Y.; et al. Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome c release. Proc. Natl. Acad. Sci. USA 2017, 114, E7997–E8006. [Google Scholar] [CrossRef]
- Yu, N.; Sun, Y.T.; Su, X.M.; He, M.; Dai, B.; Kang, J. Melatonin attenuates TGFβ1-induced epithelial-mesenchymal transition in lung alveolar epithelial cells. Mol. Med. Rep. 2016, 14, 5567–5572. [Google Scholar] [CrossRef]
- Chen, D.Q.; Cao, G.; Zhao, H.; Chen, L.; Yang, T.; Wang, M.; Vaziri, N.D.; Guo, Y.; Zhao, Y.Y. Combined melatonin and poricoic acid A inhibits renal fibrosis through modulating the interaction of Smad3 and β-catenin pathway in AKI-to-CKD continuum. Ther. Adv. Chronic Dis. 2019, 10, 2040622319869116. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.R.; Hong, R.T.; Xie, Y.Y.; Xu, J.M. Melatonin Ameliorates Liver Fibrosis Induced by Carbon Tetrachloride in Rats via Inhibiting TGF-β1/Smad Signaling Pathway. Curr. Med. Sci. 2018, 38, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Peña, A.B.; Grande, M.T.; Eleno, N.; Arévalo, M.; Guerrero, C.; Santos, E.; López-Novoa, J.M. Activation of Erk1/2 and Akt following unilateral ureteral obstruction. Kidney Int. 2008, 74, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Stambe, C.; Atkins, R.C.; Tesch, G.H.; Masaki, T.; Schreiner, G.F.; Nikolic-Paterson, D.J. The role of p38alpha mitogen-activated protein kinase activation in renal fibrosis. J. Am. Soc. Nephrol. 2004, 15, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Okumura, Y.; Sato, H.; Hamaoka, K. Delayed inhibition of p38 mitogen-activated protein kinase ameliorates renal fibrosis in obstructive nephropathy. Nephrol. Dial. Transplant. 2008, 23, 2520–2524. [Google Scholar] [CrossRef]
- Sugiyama, N.; Kohno, M.; Yokoyama, T. Inhibition of the p38 MAPK pathway ameliorates renal fibrosis in an NPHP2 mouse model. Nephrol. Dial. Transplant. 2012, 27, 1351–1358. [Google Scholar] [CrossRef]
- Xu, X.; Wang, G.; Ai, L.; Shi, J.; Zhang, J.; Chen, Y.X. Melatonin suppresses TLR9-triggered proinflammatory cytokine production in macrophages by inhibiting ERK1/2 and AKT activation. Sci. Rep. 2018, 8, 15579. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, H.J.; Bae, M.K.; Kim, Y.D. Suppression of osteoclastogenesis by melatonin: A melatonin receptor-independent action. Int. J. Mol. Sci. 2017, 18, 1142. [Google Scholar] [CrossRef]
- Zielińska, M.; Jarmuż, A.; Sałaga, M.; Kordek, R.; Laudon, M.; Storr, M.; Fichna, J. Melatonin, but not melatonin receptor agonists Neu-P11 and Neu-P67, attenuates TNBS-induced colitis in mice. Naunyn Schmiedebergs Arch. Pharmacol. 2016, 389, 511–519. [Google Scholar] [CrossRef]
- Cheng, X.P.; Sun, H.; Ye, Z.Y.; Zhou, J.N. Melatonin modulates the GABAergic response in cultured rat hippocampal neurons. J. Pharmacol. Sci. 2012, 119, 177–185. [Google Scholar] [CrossRef]
- Bondi, C.D.; Manickam, N.; Lee, D.Y.; Block, K.; Gorin, Y.; Abboud, H.E.; Barnes, J.L. NAD(P)H oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J. Am. Soc. Nephrol. 2010, 21, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Manickam, N.; Patel, M.; Griendling, K.K.; Gorin, Y.; Barnes, J.L. RhoA/Rho kinase mediates TGF-β1-induced kidney myofibroblast activation through Poldip2/Nox4-derived reactive oxygen species. Am. J. Physiol.-Ren. Physiol. 2014, 307, F159–F171. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.I.; Ma, S.K.; Bae, E.H.; Lee, J.; Kim, S.W. Peroxiredoxin 5 Protects TGF-β Induced Fibrosis by Inhibiting Stat3 Activation in Rat Kidney Interstitial Fibroblast Cells. PLoS ONE 2016, 11, e0149266. [Google Scholar] [CrossRef] [PubMed]
- Sauer, H.; Wartenberg, M.; Hescheler, J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell. Physiol. Biochem. 2004, 101, 2259–2264. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.L.; Gorin, Y. Myofibroblast differentiation during fibrosis: Role of NAD(P)H oxidases. Kidney Int. 2011, 79, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef]
- Hotta, Y.; Uchiyama, K.; Takagi, T.; Kashiwagi, S.; Nakano, T.; Mukai, R.; Toyokawa, Y.; Yasuda, T.; Ueda, T.; Suyama, Y.; et al. Transforming growth factor β1-induced collagen production in myofibroblasts is mediated by reactive oxygen species derived from NADPH oxidase 4. Biochem. Biophys. Res. Commun. 2018, 506, 557–562. [Google Scholar] [CrossRef]
- Dosoki, H.; Stegemann, A.; Taha, M.; Schnittler, H.; Luger, T.A.; Schröder, K.; Distler, J.H.; Kerkhoff, C.; Böhm, M. Targeting of NADPH oxidase in vitro and in vivo suppresses fibroblast activation and experimental skin fibrosis. Exp. Dermatol. 2017, 26, 73–81. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).