Therapeutic Hypothermia Improves Hind Limb Motor Outcome and Attenuates Oxidative Stress and Neuronal Damage in the Lumbar Spinal Cord Following Cardiac Arrest

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Groups
2.2. Induction of ACA and CPR
2.3. Hypothermic Therapy
2.4. Evaluation of Motor Function
2.5. Tissue Preparation for Histological Analyses
2.6. Cresyl Violet (CV) and Fluoro-Jade B (FJB) Histofluorescence Staining
2.7. Immunohistochemistry
2.8. Dihydroethidium (DHE) Fluorescence Staining
2.9. Data Analyses
2.10. Statistical Analysis
3. Results
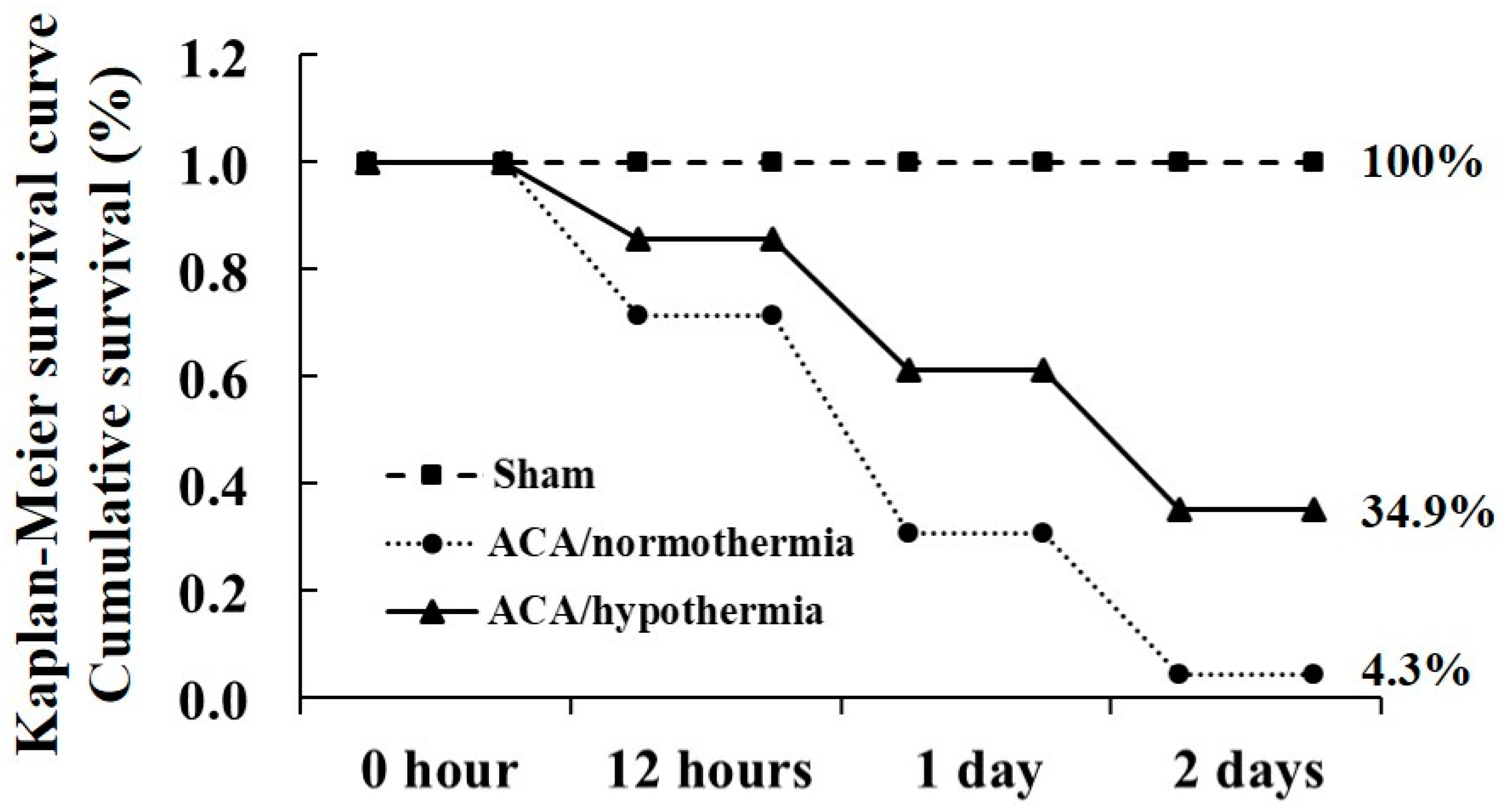
3.1. Survival Rate by Hypothermia
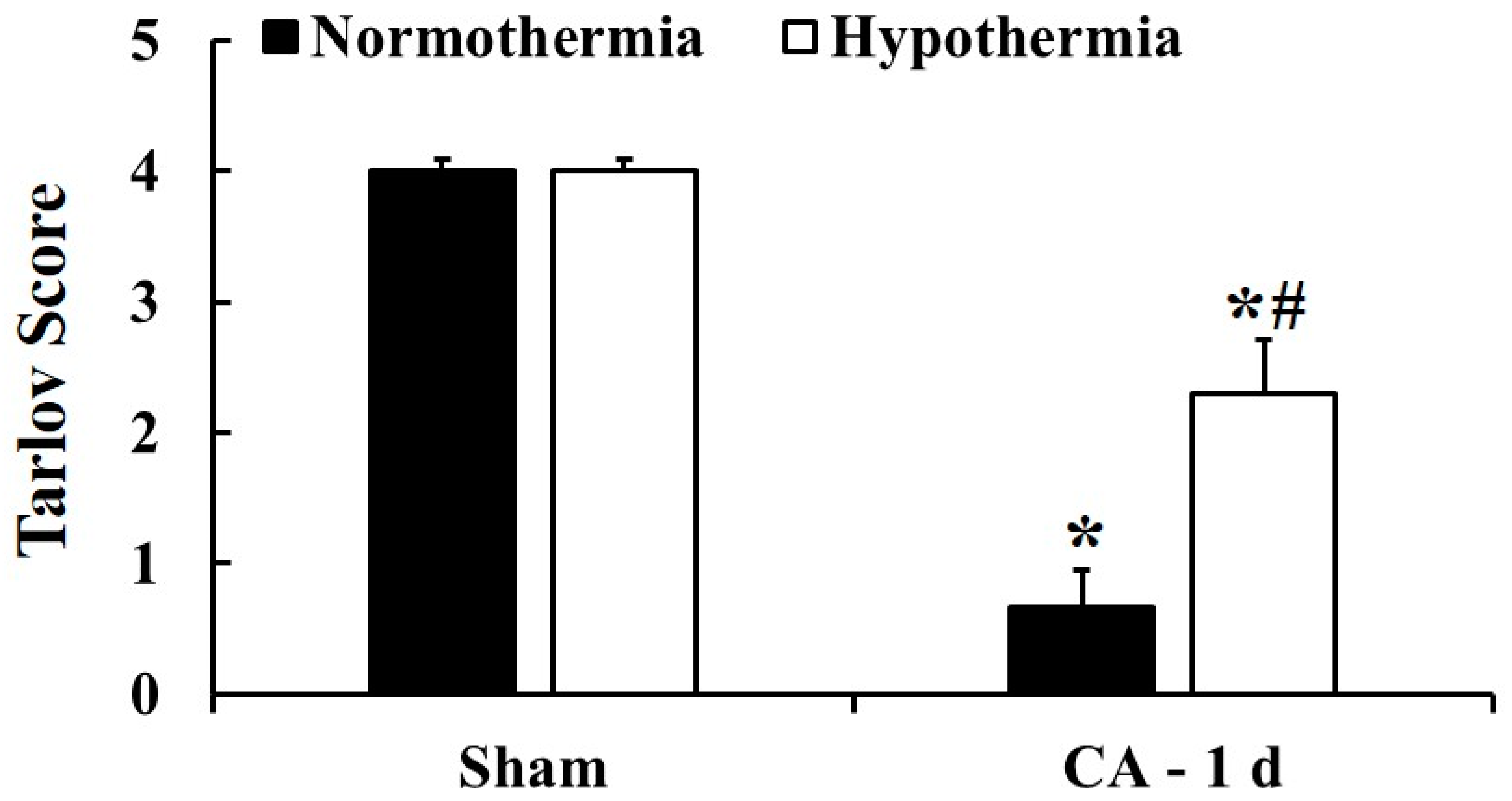
3.2. Motor Deficit Score by Hypothermia
3.3. Neuroprotection by Hypothermia
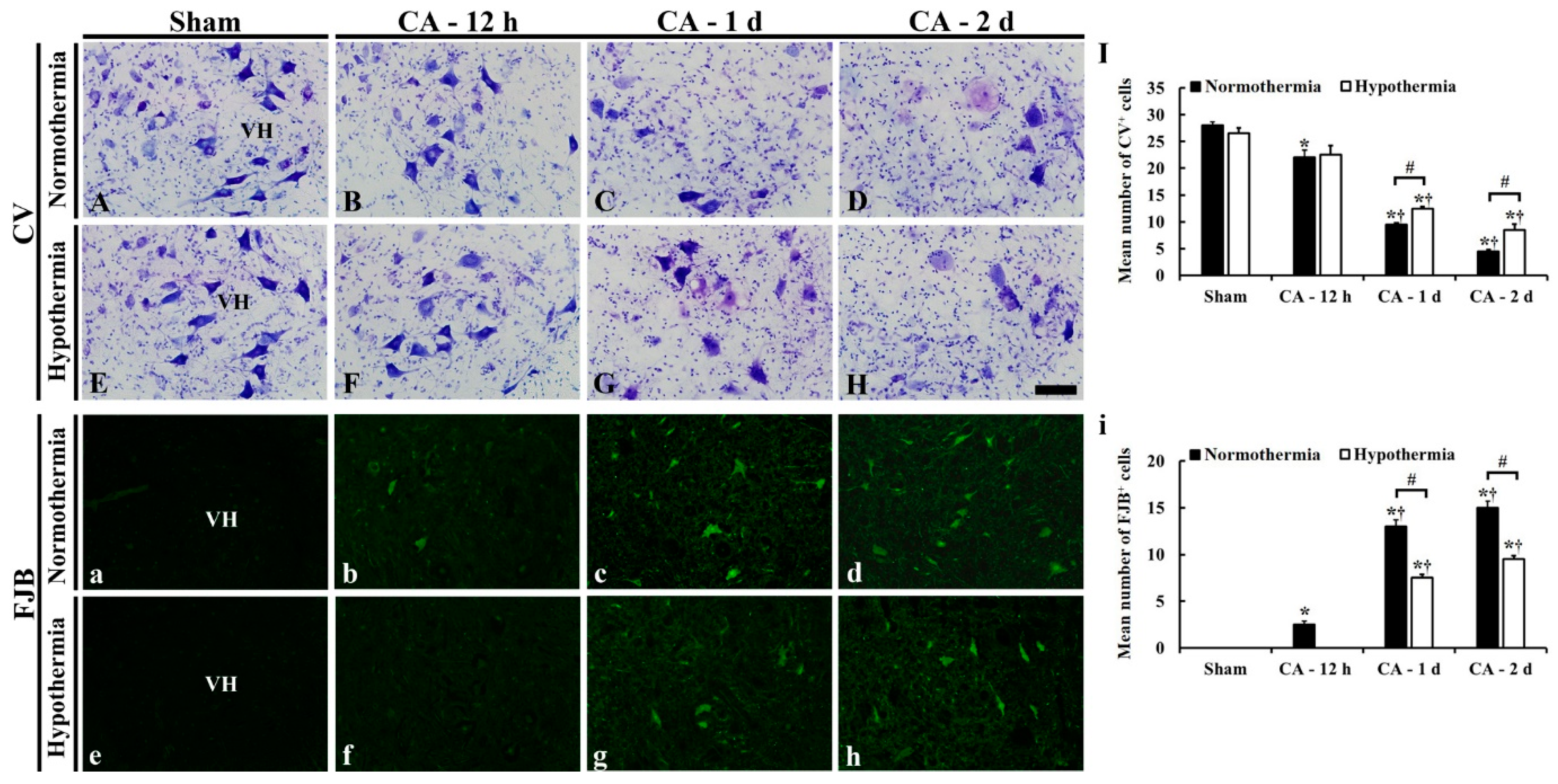
3.3.1. CV- and FJB-Positive Cells
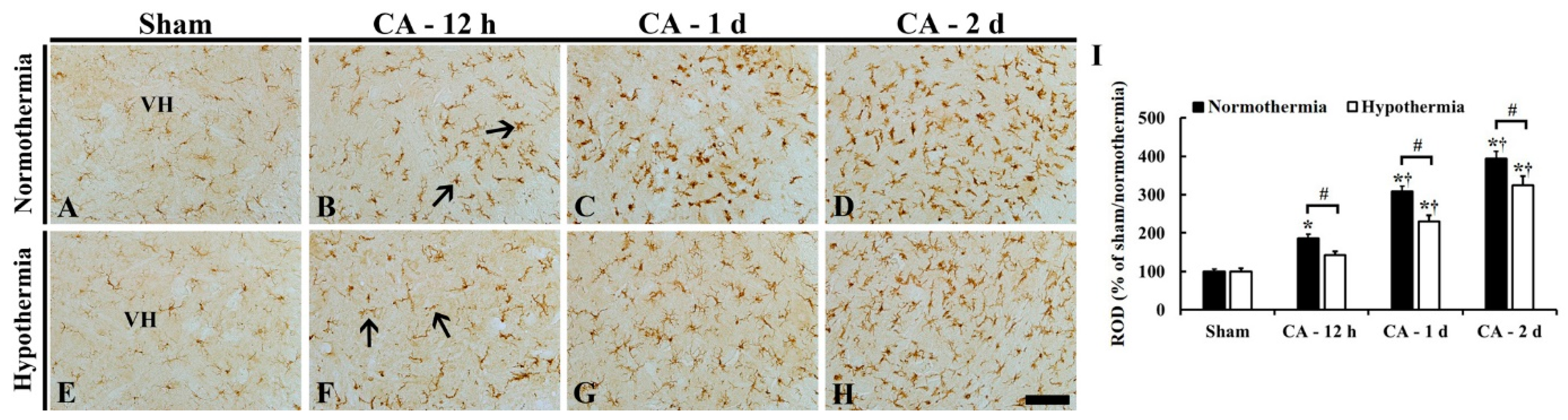
3.3.2. Reduced Microglia Activation by Hypothermia
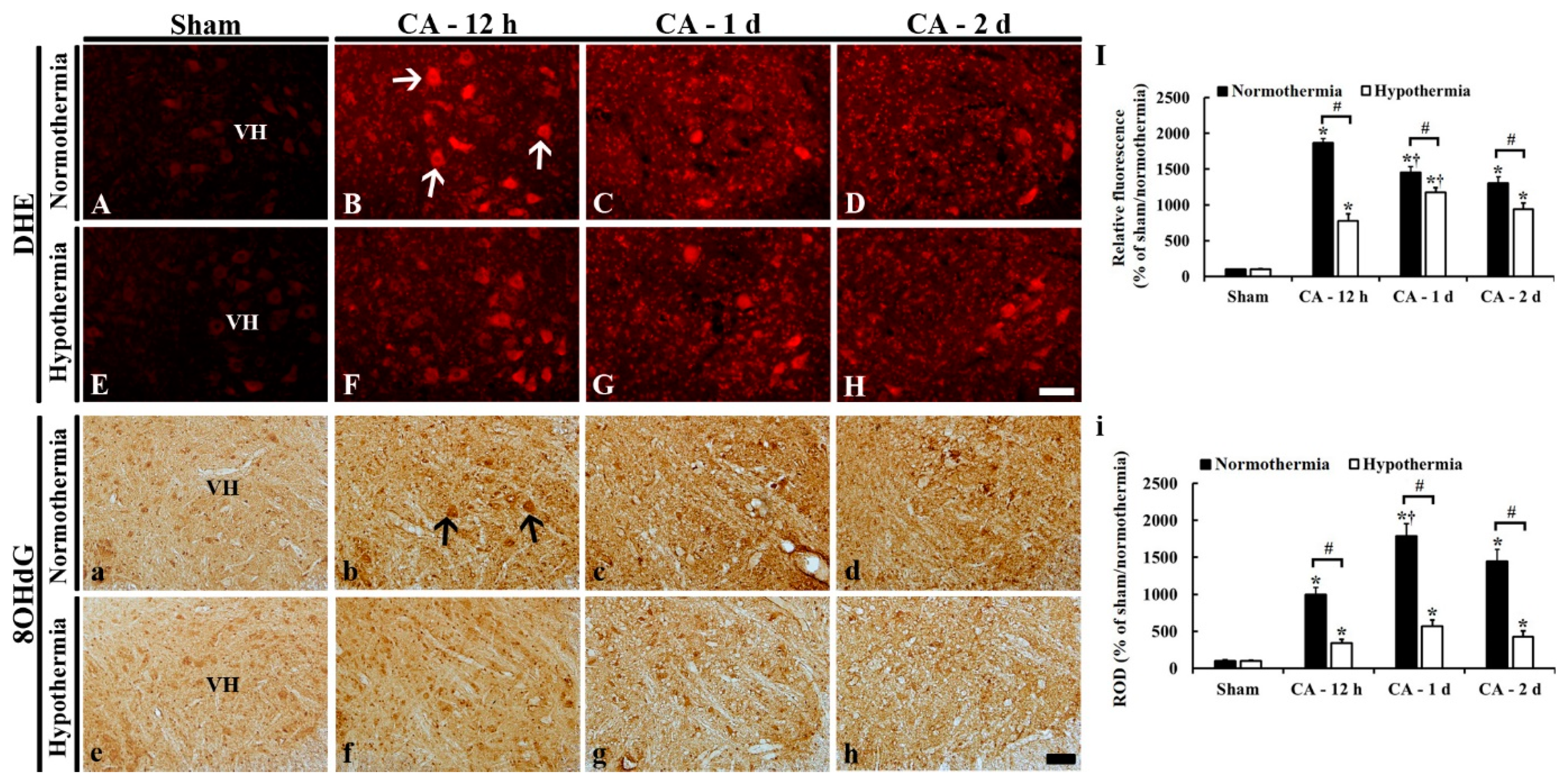
3.4. Reduced Oxidative Stress by Hypothermia
3.4.1. DHE Fluorescence
3.4.2. 8OHdG Immunoreactivity
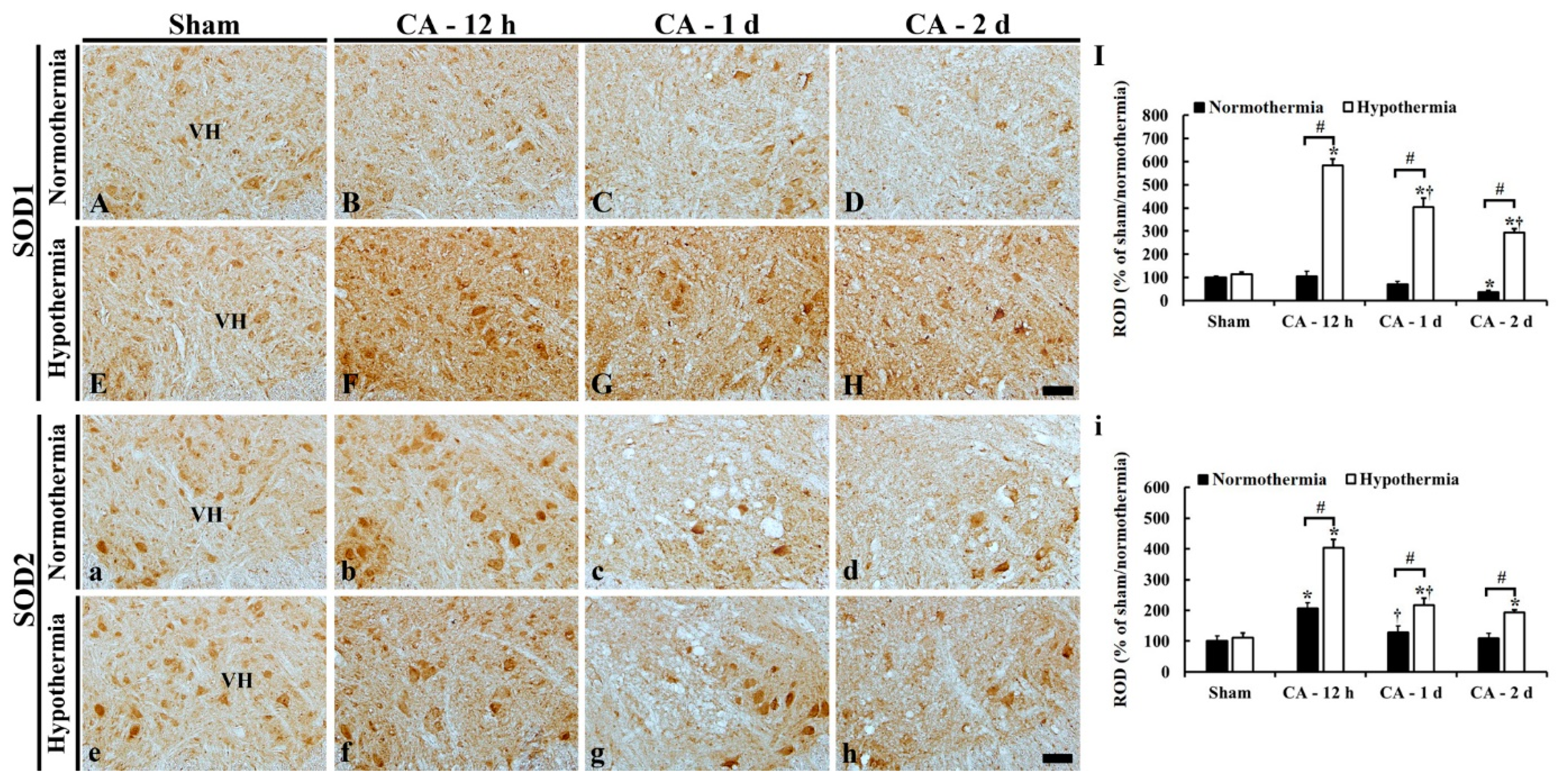
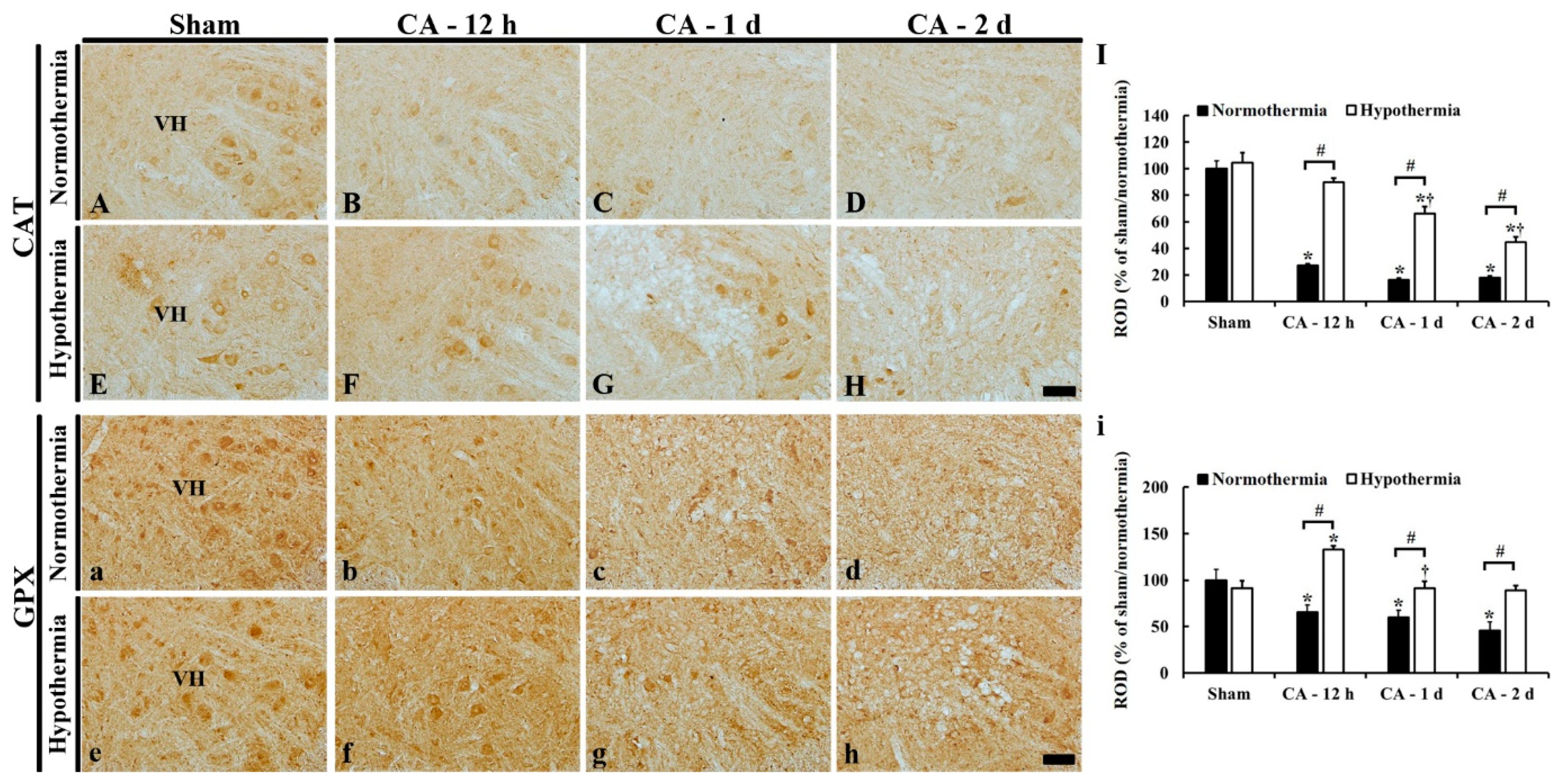
3.5. Increased Anti-Oxidant Enzymes by Hypothermia
3.5.1. SOD1 Immunoreactivity
3.5.2. SOD2 Immunoreactivity
3.5.3. CAT Immunoreactivity
3.5.4. GPX Immunoreactivity
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Bianchetti, E.; Mladinic, M.; Nistri, A. Mechanisms underlying cell death in ischemia-like damage to the rat spinal cord in vitro. Cell Death Dis. 2013, 4, e707. [Google Scholar] [CrossRef][Green Version]
- Imaizumi, H.; Ujike, Y.; Asai, Y.; Kaneko, M.; Chiba, S. Spinal cord ischemia after cardiac arrest. J. Emerg. Med. 1994, 12, 789–793. [Google Scholar] [CrossRef]
- Kudo, Y.; Ohtaki, H.; Dohi, K.; Yin, L.; Nakamachi, T.; Endo, S.; Yofu, S.; Hiraizumi, Y.; Miyaoka, H.; Shioda, S. Neuronal damage in rat brain and spinal cord after cardiac arrest and massive hemorrhagic shock. Crit. Care Med. 2006, 34, 2820–2826. [Google Scholar] [CrossRef]
- Duggal, N.; Lach, B. Selective vulnerability of the lumbosacral spinal cord after cardiac arrest and hypotension. Stroke 2002, 33, 116–121. [Google Scholar] [CrossRef]
- Wang, J.; Pearse, D.D. Therapeutic hypothermia in spinal cord injury: The status of its use and open questions. Int. J. Mol. Sci. 2015, 16, 16848–16879. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Ischemia/Reperfusion. Compr. Physiol. 2016, 7, 113–170. [Google Scholar]
- Darwazeh, R.; Yan, Y. Mild hypothermia as a treatment for central nervous system injuries: Positive or negative effects. Neural Regen. Res. 2013, 8, 2677–2686. [Google Scholar]
- Gong, P.; Li, C.S.; Hua, R.; Zhao, H.; Tang, Z.R.; Mei, X.; Zhang, M.Y.; Cui, J. Mild hypothermia attenuates mitochondrial oxidative stress by protecting respiratory enzymes and upregulating MnSOD in a pig model of cardiac arrest. PLoS ONE 2012, 7, e35313. [Google Scholar] [CrossRef]
- Zhao, H.; Li, C.S.; Gong, P.; Tang, Z.R.; Hua, R.; Mei, X.; Zhang, M.Y.; Cui, J. Molecular mechanisms of therapeutic hypothermia on neurological function in a swine model of cardiopulmonary resuscitation. Resuscitation 2012, 83, 913–920. [Google Scholar] [CrossRef]
- Gong, P.; Zhao, H.; Hua, R.; Zhang, M.; Tang, Z.; Mei, X.; Cui, J.; Li, C. Mild hypothermia inhibits systemic and cerebral complement activation in a swine model of cardiac arrest. J. Cereb. Blood Flow Metab. 2015, 35, 1289–1295. [Google Scholar] [CrossRef]
- Lee, J.C.; Tae, H.J.; Cho, J.H.; Kim, I.S.; Lee, T.K.; Park, C.W.; Park, Y.E.; Ahn, J.H.; Park, J.H.; Yan, B.C.; et al. Therapeutic hypothermia attenuates paraplegia and neuronal damage in the lumbar spinal cord in a rat model of asphyxial cardiac arrest. J. Biol. 2019, 83, 1–7. [Google Scholar] [CrossRef]
- Tae, H.J.; Kang, I.J.; Lee, T.K.; Cho, J.H.; Lee, J.C.; Shin, M.C.; Kim, Y.S.; Kim, J.D.; Ahn, J.H.; Park, J.H.; et al. Neuronal injury and tumor necrosis factor-alpha immunoreactivity in the rat hippocampus in the early period of asphyxia-induced cardiac arrest under normothermia. Neural Regen. Res. 2017, 12, 2007–2013. [Google Scholar]
- Lee, J.H.; Lee, T.K.; Kim, I.H.; Lee, J.C.; Won, M.H.; Park, J.H.; Ahn, J.H.; Shin, M.C.; Ohk, T.G.; Moon, J.B.; et al. Changes in histopathology and tumor necrosis factor-alpha levels in the hearts of rats following asphyxial cardiac arrest. Clin. Exp. Emerg. Med. 2017, 4, 160–167. [Google Scholar] [CrossRef]
- Drabek, T.; Janata, A.; Wilson, C.D.; Stezoski, J.; Janesko-Feldman, K.; Tisherman, S.A.; Foley, L.M.; Verrier, J.D.; Kochanek, P.M. Minocycline attenuates brain tissue levels of TNF-alpha produced by neurons after prolonged hypothermic cardiac arrest in rats. Resuscitation 2014, 85, 284–291. [Google Scholar] [CrossRef]
- Han, F.; Boller, M.; Guo, W.; Merchant, R.M.; Lampe, J.W.; Smith, T.M.; Becker, L.B. A rodent model of emergency cardiopulmonary bypass resuscitation with different temperatures after asphyxial cardiac arrest. Resuscitation 2010, 81, 93–99. [Google Scholar] [CrossRef]
- Che, D.; Li, L.; Kopil, C.M.; Liu, Z.; Guo, W.; Neumar, R.W. Impact of therapeutic hypothermia onset and duration on survival, neurologic function, and neurodegeneration after cardiac arrest. Crit. Care Med. 2011, 39, 1423–1430. [Google Scholar] [CrossRef]
- Idris, A.H.; Becker, L.B.; Ornato, J.P.; Hedges, J.R.; Bircher, N.G.; Chandra, N.C.; Cummins, R.O.; Dick, W.; Ebmeyer, U.; Halperin, H.R.; et al. Utstein-style guidelines for uniform reporting of laboratory CPR research. A statement for healthcare professionals from a Task Force of the American Heart Association, the American College of Emergency Physicians, the American College of Cardiology, the European Resuscitation Council, the Heart and Stroke Foundation of Canada, the Institute of Critical Care Medicine, the Safar Center for Resuscitation Research, and the Society for Academic Emergency Medicine. Resuscitation 1996, 33, 69–84. [Google Scholar]
- Liachenko, S.; Tang, P.; Hamilton, R.L.; Xu, Y. A reproducible model of circulatory arrest and remote resuscitation in rats for NMR investigation. Stroke 1998, 29, 1229–1238. [Google Scholar] [CrossRef][Green Version]
- Jia, X.; Koenig, M.A.; Shin, H.C.; Zhen, G.; Pardo, C.A.; Hanley, D.F.; Thakor, N.V.; Geocadin, R.G. Improving neurological outcomes post-cardiac arrest in a rat model: Immediate hypothermia and quantitative EEG monitoring. Resuscitation 2008, 76, 431–442. [Google Scholar] [CrossRef]
- Tarlov, I.M. Acute spinal cord compression paralysis. J. Neurosurg. 1972, 36, 10–20. [Google Scholar] [CrossRef]
- Lee, C.H.; Hwang, I.K.; Choi, J.H.; Yoo, K.Y.; Han, T.H.; Park, O.K.; Lee, S.Y.; Ryu, P.D.; Won, M.H. Calcium binding proteins immunoreactivity in the rat basolateral amygdala following myocardial infarction. Cell Mol. Neurobiol. 2010, 30, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.K.; Park, J.H.; Ahn, J.H.; Shin, M.C.; Cho, J.H.; Bae, E.J.; Kim, Y.M.; Won, M.H.; Lee, C.H. Pretreated duloxetine protects hippocampal CA1 pyramidal neurons from ischemia-reperfusion injury through decreases of glial activation and oxidative stress. J. Neurol. Sci. 2016, 370, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Ahn, J.H.; Song, M.; Kim, D.W.; Lee, T.K.; Lee, J.C.; Kim, Y.M.; Kim, J.D.; Cho, J.H.; Hwang, I.K.; et al. Pretreated fucoidan confers neuroprotection against transient global cerebral ischemic injury in the gerbil hippocampal CA1 area via reducing of glial cell activation and oxidative stress. Biomed. Pharm. 2019, 109, 1718–1727. [Google Scholar] [CrossRef]
- Lee, J.C.; Park, J.H.; Kim, I.H.; Cho, G.S.; Ahn, J.H.; Tae, H.J.; Choi, S.Y.; Cho, J.H.; Kim, D.W.; Kwon, Y.G.; et al. Neuroprotection of ischemic preconditioning is mediated by thioredoxin 2 in the hippocampal CA1 region following a subsequent transient cerebral ischemia. Brain Pathol. 2017, 27, 276–291. [Google Scholar] [CrossRef]
- Song, M.; Ahn, J.H.; Kim, H.; Kim, D.W.; Lee, T.K.; Lee, J.C.; Kim, Y.M.; Lee, C.H.; Hwang, I.K.; Yan, B.C.; et al. Chronic high-fat diet-induced obesity in gerbils increases pro-inflammatory cytokines and mTOR activation, and elicits neuronal death in the striatum following brief transient ischemia. Neurochem. Int. 2018, 121, 75–85. [Google Scholar] [CrossRef]
- Turkoz, A.; Gulcan, Ö.; Kizilkilic, O.; Kocum, A.; Turkoz, R. Spinal cord ischemia caused by cardiac arrest secondary to pericardial effusion. J. Cardiothorac. Vasc. Anesth. 2007, 21, 91–92. [Google Scholar] [CrossRef]
- Lu, K.; Liang, C.L.; Chen, H.J.; Chen, S.D.; Hsu, H.C.; Liliang, P.C.; Lin, T.K.; Cho, C.L. Injury severity and cell death mechanisms: Effects of concomitant hypovolemic hypotension on spinal cord ischemia-reperfusion in rats. Exp. Neurol. 2004, 185, 120–132. [Google Scholar] [CrossRef]
- Ye, S.; Weng, Y.; Sun, S.; Chen, W.; Wu, X.; Li, Z.; Weil, M.H.; Tang, W. Comparison of the durations of mild therapeutic hypothermia on outcome after cardiopulmonary resuscitation in the rat. Circulation 2012, 125, 123–129. [Google Scholar] [CrossRef]
- Gong, P.; Zhao, S.; Wang, J.; Yang, Z.; Qian, J.; Wu, X.; Cahoon, J.; Tang, W. Mild hypothermia preserves cerebral cortex microcirculation after resuscitation in a rat model of cardiac arrest. Resuscitation 2015, 97, 109–114. [Google Scholar] [CrossRef]
- Yu, C.G.; Jimenez, O.; Marcillo, A.E.; Weider, B.; Bangerter, K.; Dietrich, W.D.; Castro, S.; Yezierski, R.P. Beneficial effects of modest systemic hypothermia on locomotor function and histopathological damage following contusion-induced spinal cord injury in rats. J. Neurosurg. Spine 2000, 93, 85–93. [Google Scholar] [CrossRef]
- Lo, T.P., Jr.; Cho, K.S.; Garg, M.S.; Lynch, M.P.; Marcillo, A.E.; Koivisto, D.L.; Stagg, M.; Abril, R.M.; Patel, S.; Dietrich, W.D. Systemic hypothermia improves histological and functional outcome after cervical spinal cord contusion in rats. J. Comp. Neurol. 2009, 514, 433–448. [Google Scholar]
- Seo, J.-Y.; Kim, Y.-H.; Kim, J.-W.; Kim, S.-I.; Ha, K.-Y. Effects of therapeutic hypothermia on apoptosis and autophagy after spinal cord injury in rats. Spine 2015, 40, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Neumar, R.W.; Nolan, J.P.; Adrie, C.; Aibiki, M.; Berg, R.A.; Bottiger, B.W.; Callaway, C.; Clark, R.S.; Geocadin, R.G.; Jauch, E.C.; et al. Post-cardiac arrest syndrome: Epidemiology, pathophysiology, treatment, and prognostication. A consensus statement from the International Liaison Committee on Resuscitation (American Heart Association, Australian and New Zealand Council on Resuscitation, European Resuscitation Council, Heart and Stroke Foundation of Canada, InterAmerican Heart Foundation, Resuscitation Council of Asia, and the Resuscitation Council of Southern Africa); the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; and the Stroke Council. Circulation 2008, 118, 2452–2483. [Google Scholar] [PubMed]
- Topjian, A.A.; de Caen, A.; Wainwright, M.S.; Abella, B.S.; Abend, N.S.; Atkins, D.L.; Bembea, M.M.; Fink, E.L.; Guerguerian, A.M.; Haskell, S.E.; et al. Pediatric Post-Cardiac Arrest Care: A Scientific Statement from the American Heart Association. Circulation 2019, 140, e194–e233. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, N.; Zhu, L.; Lin, Y.; Cheng, H. The microglial activation profile and associated factors after experimental spinal cord injury in rats. Neuropsychiatr. Dis. Treat. 2018, 14, 2401–2413. [Google Scholar] [CrossRef]
- Abdanipour, A.; Tiraihi, T.; Taheri, T.; Kazemi, H. Microglial activation in rat experimental spinal cord injury model. Iran. Biomed. J. 2013, 17, 214–220. [Google Scholar]
- Dietrich, W.D.; Atkins, C.M.; Bramlett, H.M. Protection in animal models of brain and spinal cord injury with mild to moderate hypothermia. J. Neurotrauma 2009, 26, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Huang, X.; Li, J.; Sun, A.; Yu, J.; Xie, N.; Xi, Y.; Ye, X. Methane Suppresses Microglial Activation Related to Oxidative, Inflammatory, and Apoptotic Injury during Spinal Cord Injury in Rats. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Morino, T.; Ogata, T.; Takeba, J.; Yamamoto, H. Microglia inhibition is a target of mild hypothermic treatment after the spinal cord injury. Spinal Cord 2008, 46, 425–431. [Google Scholar] [CrossRef]
- Ok, J.H.; Kim, Y.H.; Ha, K.Y. Neuroprotective effects of hypothermia after spinal cord injury in rats: Comparative study between epidural hypothermia and systemic hypothermia. Spine 2012, 37, E1551–E1559. [Google Scholar] [CrossRef]
- Becker, L.B. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc. Res. 2004, 61, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Dohi, K.; Miyamoto, K.; Fukuda, K.; Nakamura, S.; Hayashi, M.; Ohtaki, H.; Shioda, S.; Aruga, T. Status of systemic oxidative stress during therapeutic hypothermia in patients with post-cardiac arrest syndrome. Oxid. Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Bao, F.; Dekaban, G.A.; Weaver, L.C. Anti-CD11d antibody treatment reduces free radical formation and cell death in the injured spinal cord of rats. J. Neurochem. 2005, 94, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, M.; Nagata, T.; Abe, K.; Horinouchi, T.; Itoyama, Y.; Tabayashi, K. Oxidative damage and reduction of redox factor-1 expression after transient spinal cord ischemia in rabbits. J. Vasc. Surg. 2003, 37, 446–452. [Google Scholar] [CrossRef]
- Warita, H.; Hayashi, T.; Murakami, T.; Manabe, Y.; Abe, K. Oxidative damage to mitochondrial DNA in spinal motoneurons of transgenic ALS mice. Mol. Brain Res. 2001, 89, 147–152. [Google Scholar] [CrossRef]
- Fagová, Z.; Domoráková, I.; Danková, M.; Mechírová, E.; Kunová, A.; Stebnický, M. Ubiquitin and endogenous antioxidant enzymes participate in neuroprotection of the rabbit spinal cord after ischemia and bradykinin postconditioning. Acta Histochem. 2019, 121, 732–741. [Google Scholar] [CrossRef]
- Villa, J.V.; Villamar, D.M.P.; Zapien, J.A.T.; Espinoza, L.B.; García, J.H.; García, R.S. Current Developments in Antioxidant Therapies for Spinal Cord Injury. In Spinal Cord Injury Therapy; IntechOpen: Hamilton, NJ, USA, 2019. [Google Scholar]
- Alkabie, S.; Boileau, A.J. The Role of Therapeutic Hypothermia after Traumatic Spinal Cord Injury—A Systematic Review. World Neurosurg. 2016, 86, 432–449. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, J.H.; Lee, T.-K.; Kim, B.; Lee, J.-C.; Tae, H.-J.; Cho, J.H.; Park, Y.; Shin, M.C.; Ohk, T.G.; Park, C.W.; et al. Therapeutic Hypothermia Improves Hind Limb Motor Outcome and Attenuates Oxidative Stress and Neuronal Damage in the Lumbar Spinal Cord Following Cardiac Arrest. Antioxidants 2020, 9, 38. https://doi.org/10.3390/antiox9010038
Ahn JH, Lee T-K, Kim B, Lee J-C, Tae H-J, Cho JH, Park Y, Shin MC, Ohk TG, Park CW, et al. Therapeutic Hypothermia Improves Hind Limb Motor Outcome and Attenuates Oxidative Stress and Neuronal Damage in the Lumbar Spinal Cord Following Cardiac Arrest. Antioxidants. 2020; 9(1):38. https://doi.org/10.3390/antiox9010038
Chicago/Turabian StyleAhn, Ji Hyeon, Tae-Kyeong Lee, Bora Kim, Jae-Chul Lee, Hyun-Jin Tae, Jeong Hwi Cho, Yoonsoo Park, Myoung Cheol Shin, Taek Geun Ohk, Chan Woo Park, and et al. 2020. "Therapeutic Hypothermia Improves Hind Limb Motor Outcome and Attenuates Oxidative Stress and Neuronal Damage in the Lumbar Spinal Cord Following Cardiac Arrest" Antioxidants 9, no. 1: 38. https://doi.org/10.3390/antiox9010038
APA StyleAhn, J. H., Lee, T.-K., Kim, B., Lee, J.-C., Tae, H.-J., Cho, J. H., Park, Y., Shin, M. C., Ohk, T. G., Park, C. W., Cho, J. H., Hong, S., Park, J. H., Choi, S. Y., & Won, M.-H. (2020). Therapeutic Hypothermia Improves Hind Limb Motor Outcome and Attenuates Oxidative Stress and Neuronal Damage in the Lumbar Spinal Cord Following Cardiac Arrest. Antioxidants, 9(1), 38. https://doi.org/10.3390/antiox9010038