Xanthohumol, a Prenylated Flavonoid from Hops, Induces Caspase-Dependent Degradation of Oncoprotein BCR-ABL in K562 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Drug
2.2. Cell Lines and Cell Culture
2.3. Cell Viability Assessment
2.4. Cell Cycle Analysis
2.5. Drug Combination and Calculation of Synergism
2.6. Westerrn Blotting Assay
2.7. Cychloheximide (CHX) Chase Assay
2.8. Cell Apoptosis Analysis
2.9. Statistical Analysis
3. Results
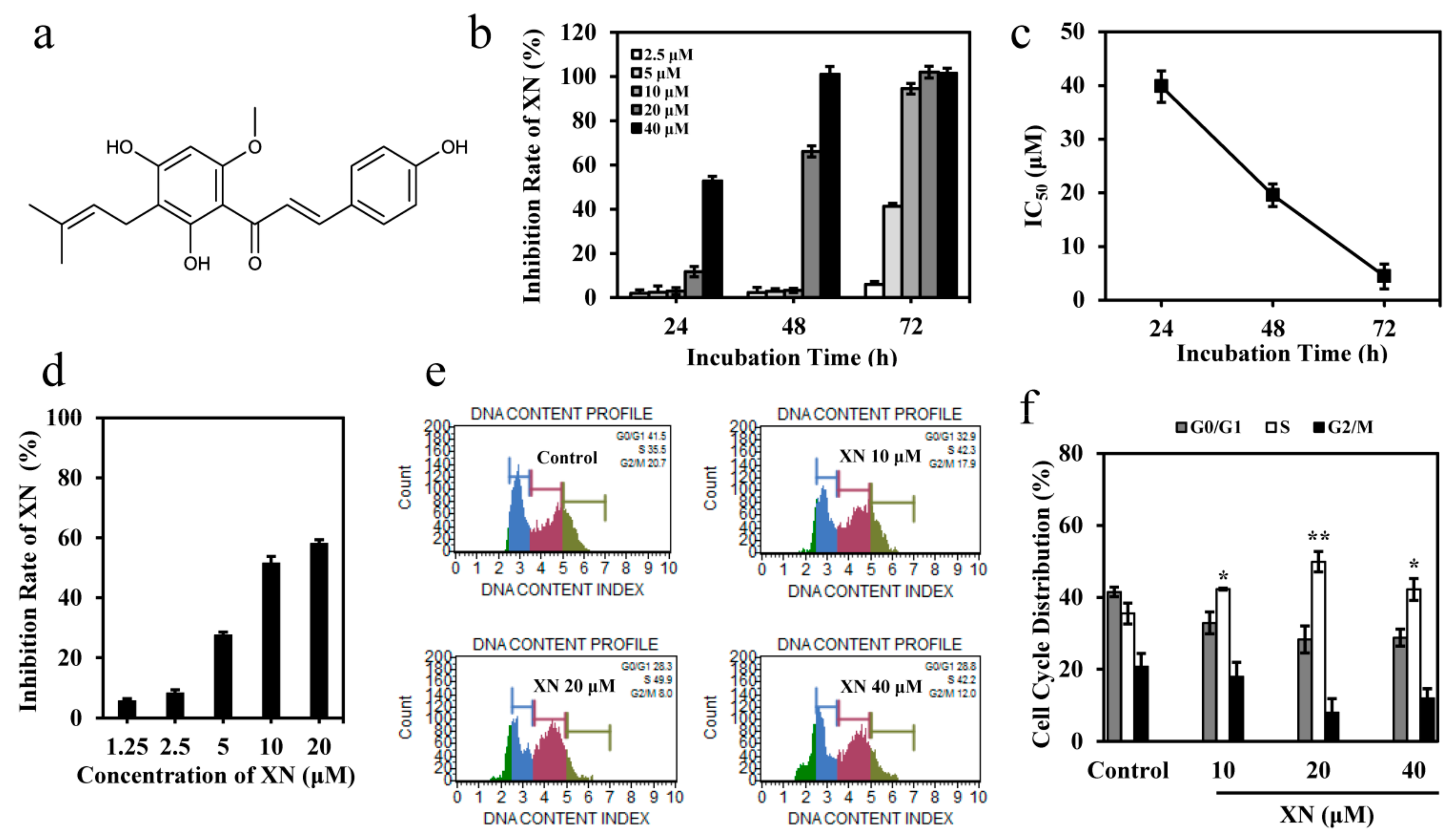
3.1. XN Inhibits the Viability of K562 Cells
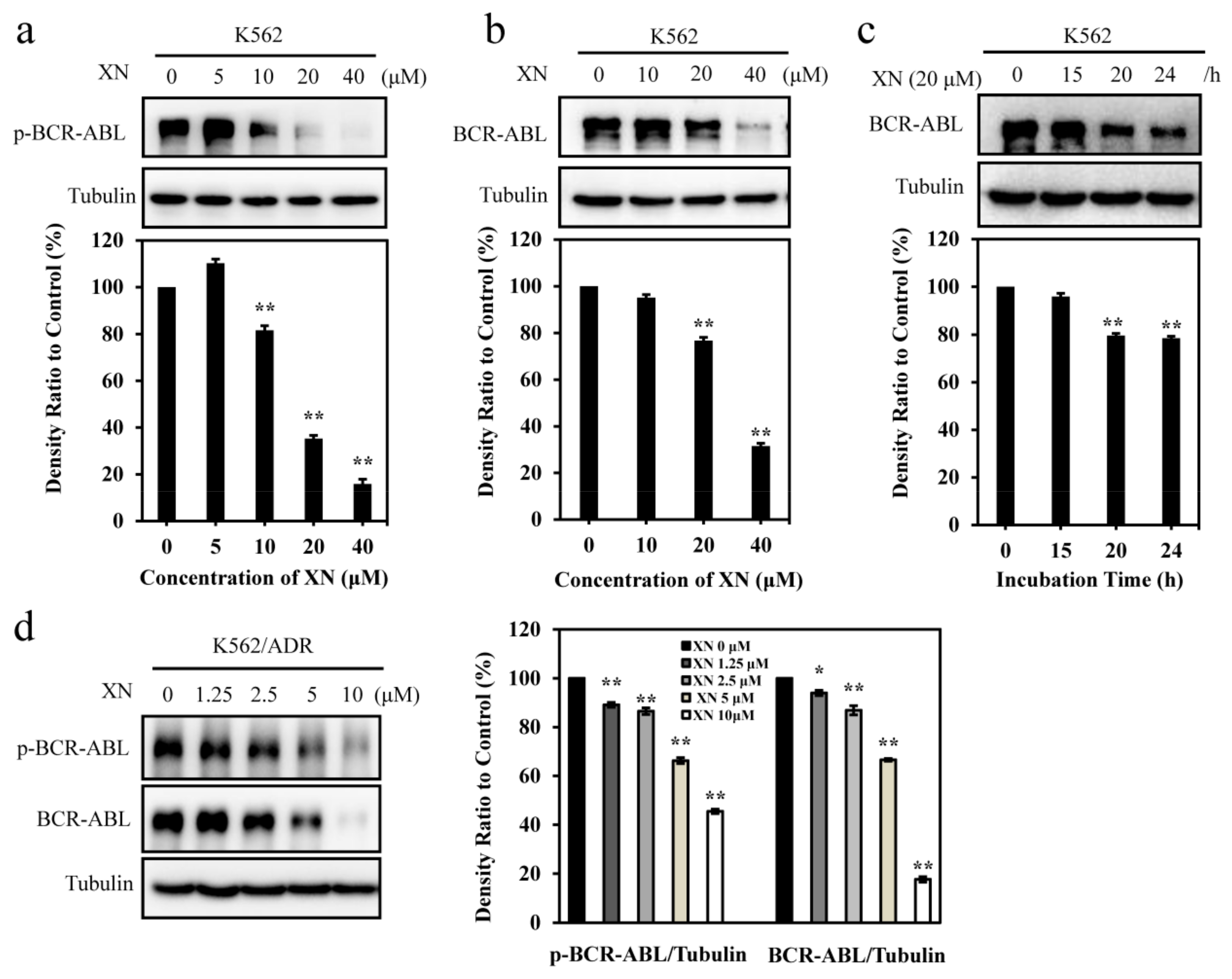
3.2. XN Inhibits Oncoprotein BCR-ABL Expression
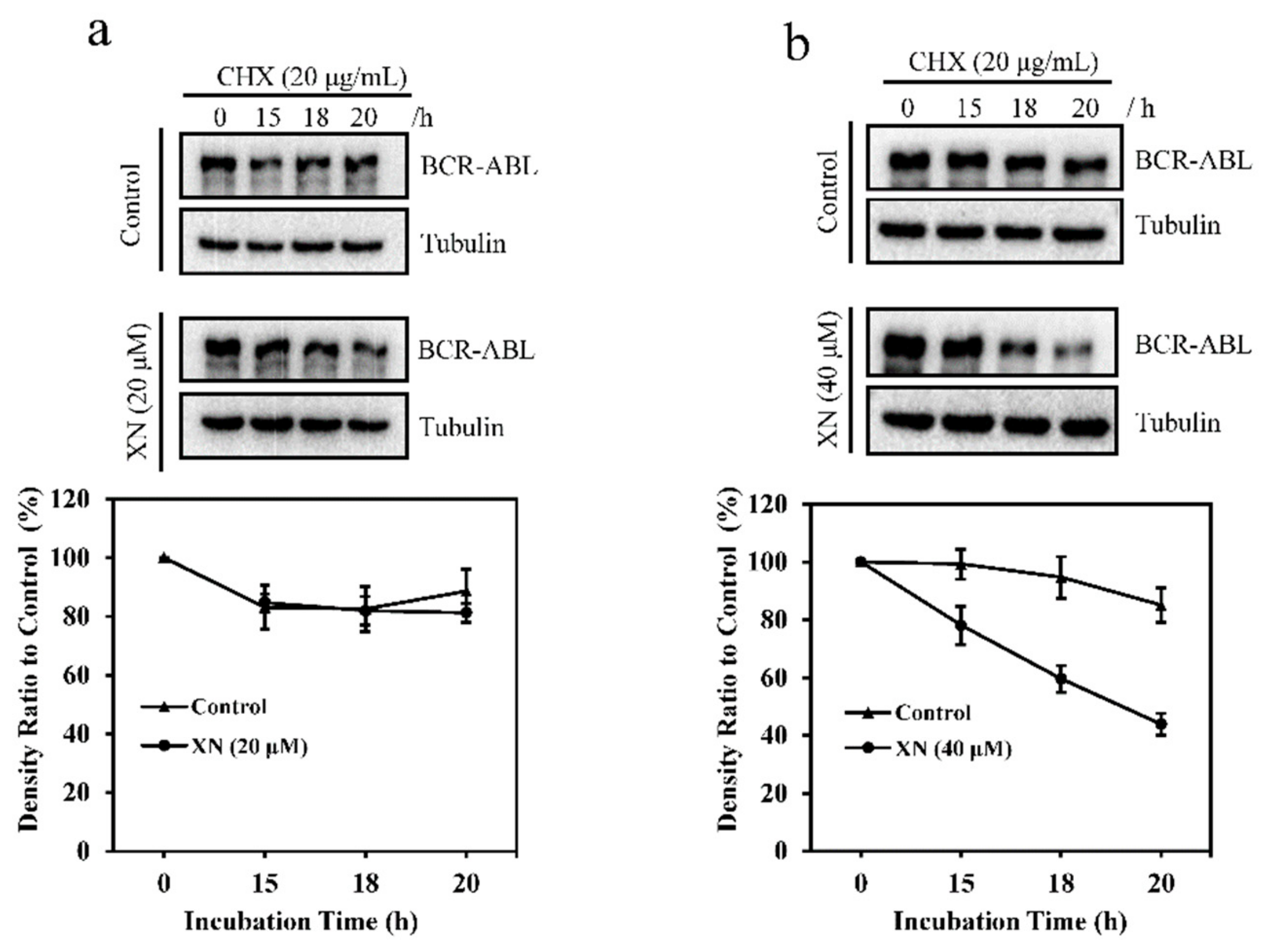
3.3. XN Promotes the Degradation of BCR-ABL Oncoprotein
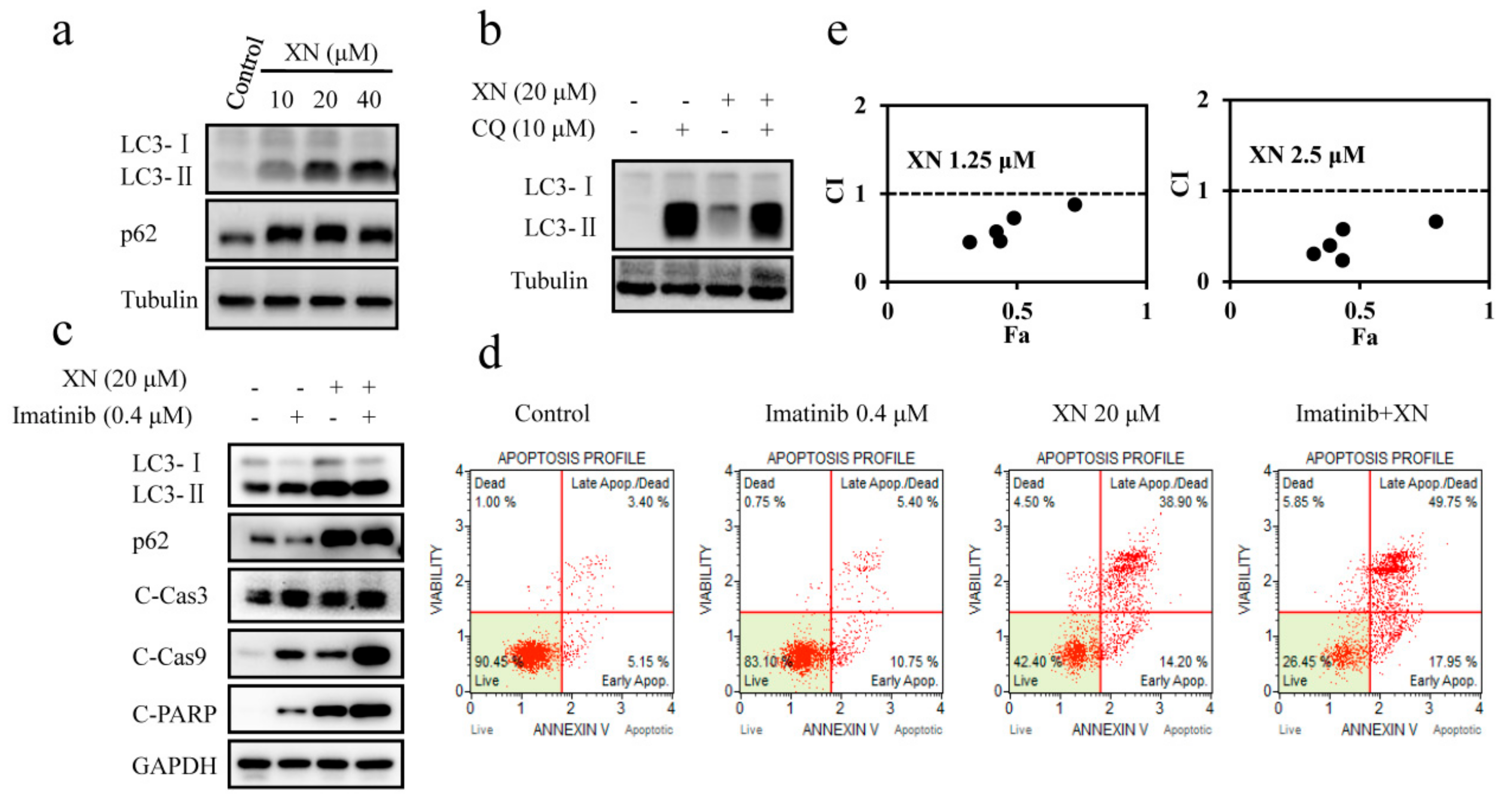
3.4. XN Inhibits Autophagosome Maturation and Autophagy is not Involved in BCR-ABL Degradation
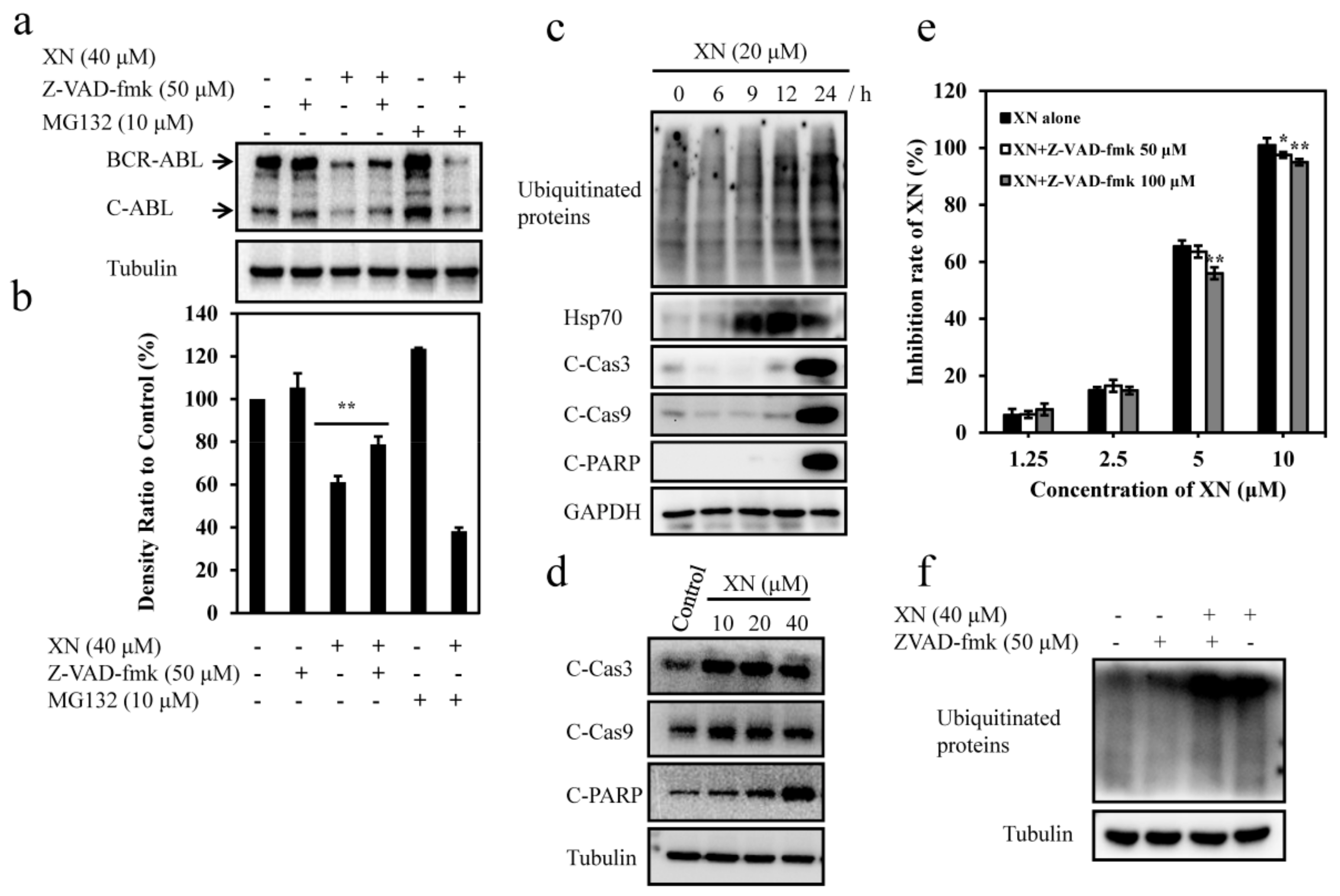
3.5. XN-Induced BCR-ABL Degradation is Caspase-Dependent
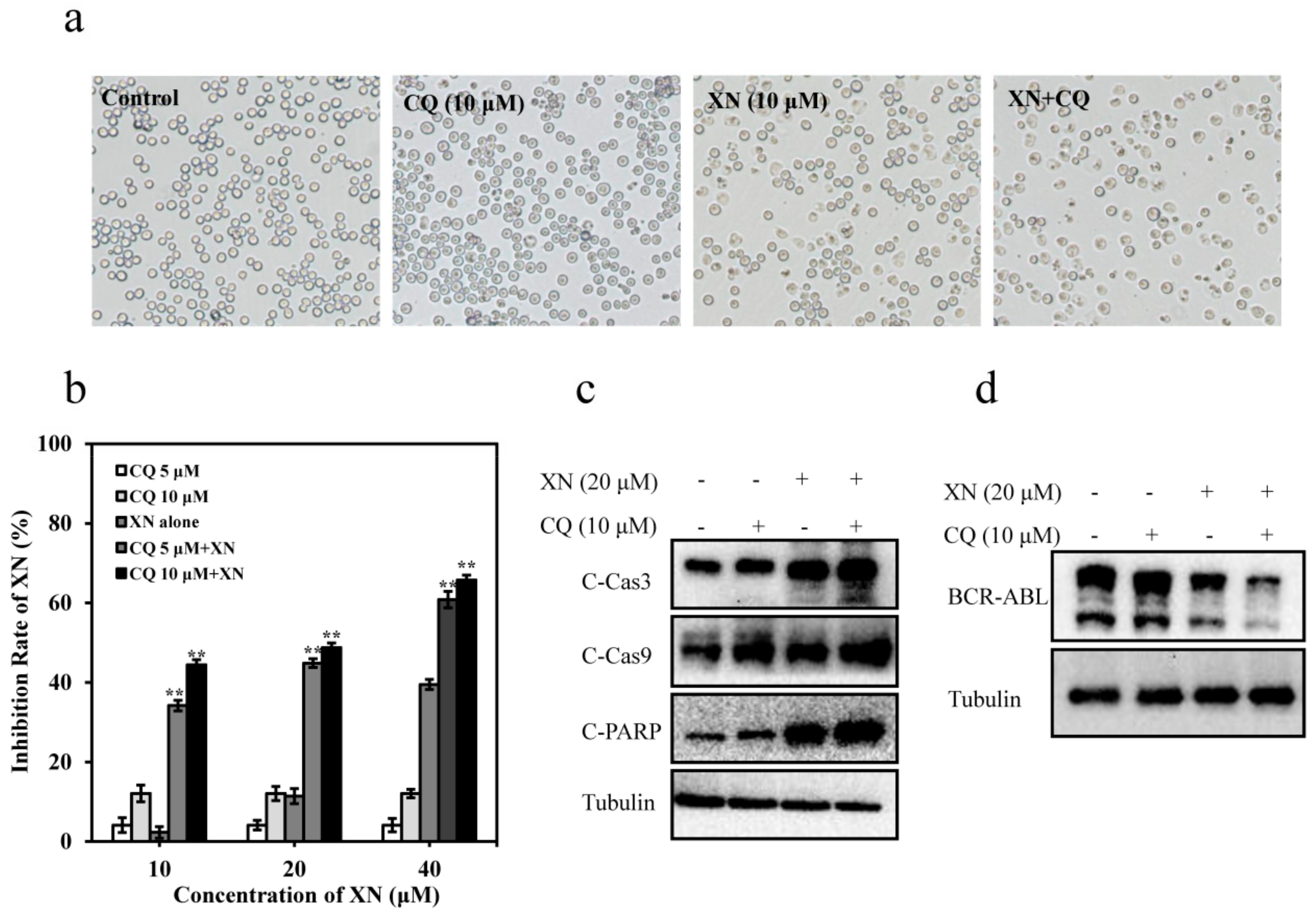
3.6. XN-Induced Caspase Activation and BCR-ABL Degradation were Enhanced by Autophagy Inhibitor CQ
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Deininger, M.W.N.; Goldman, J.M.; Melo, J.V. The molecular biology of chronic myeloid leukemia. Blood 2000, 96, 3343–3356. [Google Scholar] [PubMed]
- Quintás-Cardama, A.; Cortes, J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood 2009, 113, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.N.; Rothman, P. JAK-STAT signaling activated by Abl oncogenes. Oncogene 2000, 19, 2523–2531. [Google Scholar] [CrossRef] [PubMed]
- Lugo, T.G.; Pendergast, A.M.; Muller, A.J.; Witte, O.N. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science 1990, 247, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Quintás-Cardama, A.; Kantarjian, H.; Cortes, J. Imatinib and beyond—Exploring the full potential of targeted therapy for CML. Nat. Rev. Clin. Oncol. 2009, 6, 535. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.P.; Tran, C.; Lee, F.Y.; Chen, P.; Norris, D.; Sawyers, C.L. Overriding Imatinib Resistance with a Novel ABL Kinase Inhibitor. Science 2004, 305, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Jones, D.; Kantarjian, H.M.; O’Brien, S.; Tam, C.; Koller, C.; Burger, J.A.; Borthakur, G.; Wierda, W.G.; Cortes, J. Long-term outcome of patients with chronic myeloid leukemia treated with second-generation tyrosine kinase inhibitors after imatinib failure is predicted by the in vitro sensitivity of BCR-ABL kinase domain mutations. Blood 2009, 114, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Monteghirfo, S.; Tosetti, F.; Ambrosini, C.; Stigliani, S.; Pozzi, S.; Frassoni, F.; Fassina, G.; Soverini, S.; Albini, A.; Ferrari, N. Antileukemia effects of xanthohumol in Bcr/Abl-transformed cells involve nuclear factor-κB and p53 modulation. Mol. Cancer Ther. 2008, 7, 2692. [Google Scholar] [CrossRef] [PubMed]
- Ablain, J.; Nasr, R.; Bazarbachi, A.; de Thé, H. The Drug-Induced Degradation of Oncoproteins: An Unexpected Achilles’ Heel of Cancer Cells? Cancer Discov. 2011, 1, 117. [Google Scholar] [CrossRef]
- Ray, D.; Cuneo, K.C.; Rehemtulla, A.; Lawrence, T.S.; Nyati, M.K. Inducing Oncoprotein Degradation to Improve Targeted Cancer Therapy. Neoplasia 2015, 17, 697–703. [Google Scholar] [CrossRef]
- Huang, H.; Weng, H.; Dong, B.; Zhao, P.; Zhou, H.; Qu, L. Oridonin Triggers Chaperon-mediated Proteasomal Degradation of BCR-ABL in Leukemia. Sci. Rep. 2017, 7, 41525. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.-G.; Estrov, Z.; Wang, Y.; O’Brien, S.; Faderl, S.; Harris, D.M.; Van Pham, Q.; Hazan-Halevy, I.; Liu, Z.; Koch, P.; et al. The synthetic heat shock protein 90 (Hsp90) inhibitor EC141 induces degradation of Bcr-Abl p190 protein and apoptosis of Ph-positive acute lymphoblastic leukemia cells. Investig. New Drugs 2011, 29, 1206–1212. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Goussetis, D.J.; Gounaris, E.; Wu, E.J.; Vakana, E.; Sharma, B.; Bogyo, M.; Altman, J.K.; Platanias, L.C. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood 2012, 120, 3555–3562. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Chen, X.; Li, X.; Lan, X.; Zhao, C.; Liu, S.; Huang, H.; Liu, N.; Liao, S.; Song, W.; et al. Gambogic acid induces apoptosis in imatinib-resistant chronic myeloid leukemia cells via inducing proteasome inhibition and caspase-dependent Bcr-Abl downregulation. Clin. Cancer Res. 2014, 20, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Zhao, C.; Chen, X.; Zhang, P.; Zang, D.; Wu, J.; Chen, J.; Long, H.; Yang, L.; Huang, H.; et al. Platinum pyrithione induces apoptosis in chronic myeloid leukemia cells resistant to imatinib via DUB inhibition-dependent caspase activation and Bcr-Abl downregulation. Cell Death Dis. 2017, 8, e2913. [Google Scholar] [CrossRef]
- Chen, Q.-H.; Fu, M.-L.; Chen, M.-M.; Liu, J.; Liu, X.-J.; He, G.-Q.; Pu, S.-C. Preparative isolation and purification of xanthohumol from hops (Humulus lupulus L.) by high-speed counter-current chromatography. Food Chem. 2012, 132, 619–623. [Google Scholar] [CrossRef]
- Jiang, C.-H.; Sun, T.-L.; Xiang, D.-X.; Wei, S.-S.; Li, W.-Q. Anticancer Activity and Mechanism of Xanthohumol: A Prenylated Flavonoid From Hops (Humulus lupulus L.). Front. Pharmacol. 2018, 9, 530. [Google Scholar] [CrossRef]
- Dorn, C.; Bataille, F.; Gaebele, E.; Heilmann, J.; Hellerbrand, C. Xanthohumol feeding does not impair organ function and homoeostasis in mice. Food Chem. Toxicol. 2010, 48, 1890–1897. [Google Scholar] [CrossRef]
- Liu, M.; Hansen, P.E.; Wang, G.; Qiu, L.; Dong, J.; Yin, H.; Qian, Z.; Yang, M.; Miao, J. Pharmacological profile of xanthohumol, a prenylated flavonoid from hops (Humulus lupulus). Molecules 2015, 20, 754–779. [Google Scholar] [CrossRef]
- Carvalho, D.O.; Freitas, J.; Nogueira, P.; Henriques, S.N.; Carmo, A.M.; Castro, M.A.; Guido, L.F. Xanthohumol inhibits cell proliferation and induces apoptosis in human thyroid cells. Food Chem. Toxicol. 2018, 121, 450–457. [Google Scholar] [CrossRef]
- Tronina, T.; Bartmańska, A.; Filip-Psurska, B.; Wietrzyk, J.; Popłoński, J.; Huszcza, E. Fungal metabolites of xanthohumol with potent antiproliferative activity on human cancer cell lines in vitro. Bioorg. Med. Chem. 2013, 21, 2001–2006. [Google Scholar] [CrossRef]
- Miranda, C.L.; Stevens, J.F.; Helmrich, A.; Henderson, M.C.; Rodriguez, R.J.; Yang, Y.H.; Deinzer, M.L.; Barnes, D.W.; Buhler, D.R. Antiproliferative and cytotoxic effects of prenylated flavonoids from hops (Humulus lupulus) in human cancer cell lines. Food Chem. Toxicol. 1999, 37, 271–285. [Google Scholar] [CrossRef]
- Harikumar, K.B.; Kunnumakkara, A.B.; Ahn, K.S.; Anand, P.; Krishnan, S.; Guha, S.; Aggarwal, B.B. Modification of the cysteine residues in IkappaBalpha kinase and NF-kappaB (p65) by xanthohumol leads to suppression of NF-kappaB-regulated gene products and potentiation of apoptosis in leukemia cells. Blood 2009, 113, 2003–2013. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Ross, T.S.; Mgbemena, V.E. Re-evaluating the role of BCR/ABL in chronic myelogenous leukemia. Mol. Cell. Oncol. 2014, 1, e963450. [Google Scholar] [CrossRef]
- Auberger, P. BCR-ABL/p62/SQSTM1: A cannibal embrace. Blood 2012, 120, 3389. [Google Scholar] [CrossRef][Green Version]
- Tong, Y.; Liu, Y.-Y.; You, L.-S.; Qian, W.-B. Perifosine induces protective autophagy and upregulation of ATG5 in human chronic myelogenous leukemia cells in vitro. Acta Pharmacol. Sin. 2012, 33, 542–550. [Google Scholar] [CrossRef]
- Bellodi, C.; Lidonnici, M.R.; Hamilton, A.; Helgason, G.V.; Soliera, A.R.; Ronchetti, M.; Galavotti, S.; Young, K.W.; Selmi, T.; Yacobi, R.; et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J. Clin. Investig. 2009, 119, 1109–1123. [Google Scholar] [CrossRef]
- Tsukahara, F.; Maru, Y. Bag1 directly routes immature BCR-ABL for proteasomal degradation. Blood 2010, 116, 3582. [Google Scholar] [CrossRef]
- Geng, Y.; Kohli, L.; Klocke, B.J.; Roth, K.A. Chloroquine-induced autophagic vacuole accumulation and cell death in glioma cells is p53 independent. Neuro Oncol. 2010, 12, 473–481. [Google Scholar] [CrossRef][Green Version]
- Ben-Neriah, Y.; Daley, G.Q.; Mes-Masson, A.M.; Witte, O.N.; Baltimore, D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science 1986, 233, 212. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discov. Today 2019, 31, 15–27. [Google Scholar] [CrossRef]
- Huang, H.-L.; Weng, H.-Y.; Wang, L.-Q.; Yu, C.-H.; Huang, Q.-J.; Zhao, P.-P.; Wen, J.-Z.; Zhou, H.; Qu, L.-H. Triggering Fbw7-Mediated Proteasomal Degradation of c-Myc by Oridonin Induces Cell Growth Inhibition and Apoptosis. Mol. Cancer Ther. 2012, 11, 1155–1165. [Google Scholar] [CrossRef]
- Monteiro, R.; Calhau, C.; Silva, A.O.; Pinheiro-Silva, S.; Guerreiro, S.; Gärtner, F.; Azevedo, I.; Soares, R. Xanthohumol inhibits inflammatory factor production and angiogenesis in breast cancer xenografts. J. Cell. Biochem. 2008, 104, 1699–1707. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Jiang, K.; Liang, B.; Huang, X. Anticancer effect of xanthohumol induces growth inhibition and apoptosis of human liver cancer through NF-κB/p53-apoptosis signaling pathway. Oncol. Rep. 2016, 35, 669–675. [Google Scholar] [CrossRef]
- Guo, D.; Zhang, B.; Liu, S.; Jin, M. Xanthohumol induces apoptosis via caspase activation, regulation of Bcl-2, and inhibition of PI3K/Akt/mTOR-kinase in human gastric cancer cells. Biomed. Pharmacother. 2018, 106, 1300–1306. [Google Scholar] [CrossRef] [PubMed]
- Colgate, E.C.; Miranda, C.L.; Stevens, J.F.; Bray, T.M.; Ho, E. Xanthohumol, a prenylflavonoid derived from hops induces apoptosis and inhibits NF-kappaB activation in prostate epithelial cells. Cancer Lett. 2007, 246, 201–209. [Google Scholar] [CrossRef]
- Sun, Z.; Zhou, C.; Liu, F.; Zhang, W.; Chen, J.; Pan, Y.; Ma, L.; Liu, Q.; Du, Y.; Yang, J.; et al. Inhibition of breast cancer cell survival by Xanthohumol via modulation of the Notch signaling pathway in vivo and in vitro. Oncol. Lett. 2018, 15, 908–916. [Google Scholar] [CrossRef]
- Albini, A.; Dell’Eva, R.; Vené, R.; Ferrari, N.; Buhler, D.R.; Noonan, D.M.; Fassina, G. Mechanisms of the antiangiogenic activity by the hop flavonoid xanthohumol: NF-κB and Akt as targets. FASEB J. 2005, 20, 527–529. [Google Scholar] [CrossRef]
- Inoue, J.; Miyata, S.; Shimizu, M.; Sato, R. Isoxanthohumol stimulates ubiquitin-proteasome-dependent degradation of precursor forms of sterol regulatory element-binding proteins. Biosci. Biotechnol. Biochem. 2018, 82, 1591–1598. [Google Scholar] [CrossRef]
- Puissant, A.; Colosetti, P.; Robert, G.; Cassuto, J.P.; Raynaud, S.; Auberger, P. Cathepsin B release after imatinib-mediated lysosomal membrane permeabilization triggers BCR–ABL cleavage and elimination of chronic myelogenous leukemia cells. Leukemia 2009, 24, 115. [Google Scholar] [CrossRef] [PubMed]
- Sasazawa, Y.; Kanagaki, S.; Tashiro, E.; Nogawa, T.; Muroi, M.; Kondoh, Y.; Osada, H.; Imoto, M. Xanthohumol Impairs Autophagosome Maturation through Direct Inhibition of Valosin-Containing Protein. ACS Chem. Biol. 2012, 7, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Mi, X.; Wang, C.; Sun, C.; Chen, X.; Huo, X.; Zhang, Y.; Li, G.; Xu, B.; Zhang, J.; Xie, J.; et al. Xanthohumol induces paraptosis of leukemia cells through p38 mitogen activated protein kinase signaling pathway. Oncotarget 2017, 8, 31297–31304. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-Y.; Zupkó, I.; Chang, F.-R.; Hunyadi, A.; Wu, C.-C.; Weng, T.-S.; Wang, H.-C. Dietary flavonoid derivatives enhance chemotherapeutic effect by inhibiting the DNA damage response pathway. Toxicol. Appl. Pharmacol. 2016, 311, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-C.; Lee, A.Y.-L.; Chou, W.-C.; Wu, C.-C.; Tseng, C.-N.; Liu, K.Y.-T.; Lin, W.-L.; Chang, F.-R.; Chuang, D.-W.; Hunyadi, A.; et al. Inhibition of ATR-Dependent Signaling by Protoapigenone and Its Derivative Sensitizes Cancer Cells to Interstrand Cross-link–Generating Agents In Vitro and In Vivo. Mol. Cancer Ther. 2012, 11, 1443. [Google Scholar] [CrossRef] [PubMed]
- Albero, M.P.; Vaquer, J.M.; Andreu, E.J.; Villanueva, J.J.; Franch, L.; Ivorra, C.; Poch, E.; Agirre, X.; Prosper, F.; Pérez-Roger, I. Bortezomib decreases Rb phosphorylation and induces caspase-dependent apoptosis in Imatinib-sensitive and -resistant Bcr-Abl1-expressing cells. Oncogene 2010, 29, 3276. [Google Scholar] [CrossRef] [PubMed]
- Logan, I.E.; Miranda, C.L.; Lowry, M.B.; Maier, C.S.; Stevens, J.F.; Gombart, A.F. Antiproliferative and Cytotoxic Activity of Xanthohumol and Its Non-Estrogenic Derivatives in Colon and Hepatocellular Carcinoma Cell Lines. Int. J. Mol. Sci. 2019, 20, 1203. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, X.; Geng, J.; Zhang, J.; Miao, J.; Liu, M. Xanthohumol, a Prenylated Flavonoid from Hops, Induces Caspase-Dependent Degradation of Oncoprotein BCR-ABL in K562 Cells. Antioxidants 2019, 8, 402. https://doi.org/10.3390/antiox8090402
Lu X, Geng J, Zhang J, Miao J, Liu M. Xanthohumol, a Prenylated Flavonoid from Hops, Induces Caspase-Dependent Degradation of Oncoprotein BCR-ABL in K562 Cells. Antioxidants. 2019; 8(9):402. https://doi.org/10.3390/antiox8090402
Chicago/Turabian StyleLu, Xuxiu, Jiajia Geng, Jinman Zhang, Jinlai Miao, and Ming Liu. 2019. "Xanthohumol, a Prenylated Flavonoid from Hops, Induces Caspase-Dependent Degradation of Oncoprotein BCR-ABL in K562 Cells" Antioxidants 8, no. 9: 402. https://doi.org/10.3390/antiox8090402
APA StyleLu, X., Geng, J., Zhang, J., Miao, J., & Liu, M. (2019). Xanthohumol, a Prenylated Flavonoid from Hops, Induces Caspase-Dependent Degradation of Oncoprotein BCR-ABL in K562 Cells. Antioxidants, 8(9), 402. https://doi.org/10.3390/antiox8090402