Protein Redox State Monitoring Studies of Thiol Reactivity

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Protein Expression and Purification
2.2. Cell Culture
2.3. Protein Redox State Monitoring
2.4. Statistical Analysis
3. Results
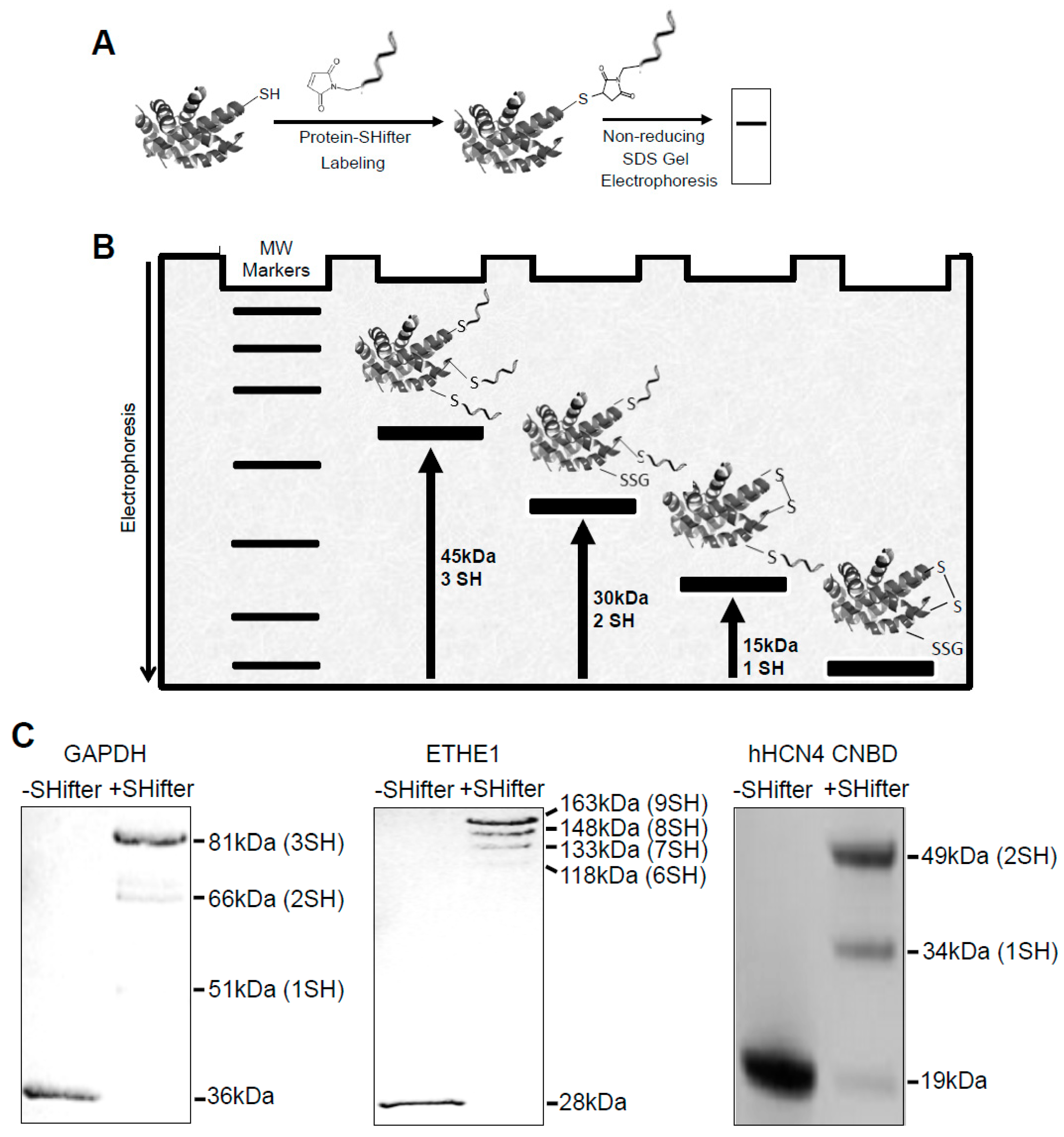
3.1. -SulfoBiotics- Protein Redox State Monitoring Kit to Assess Thiol States of Purified Proteins
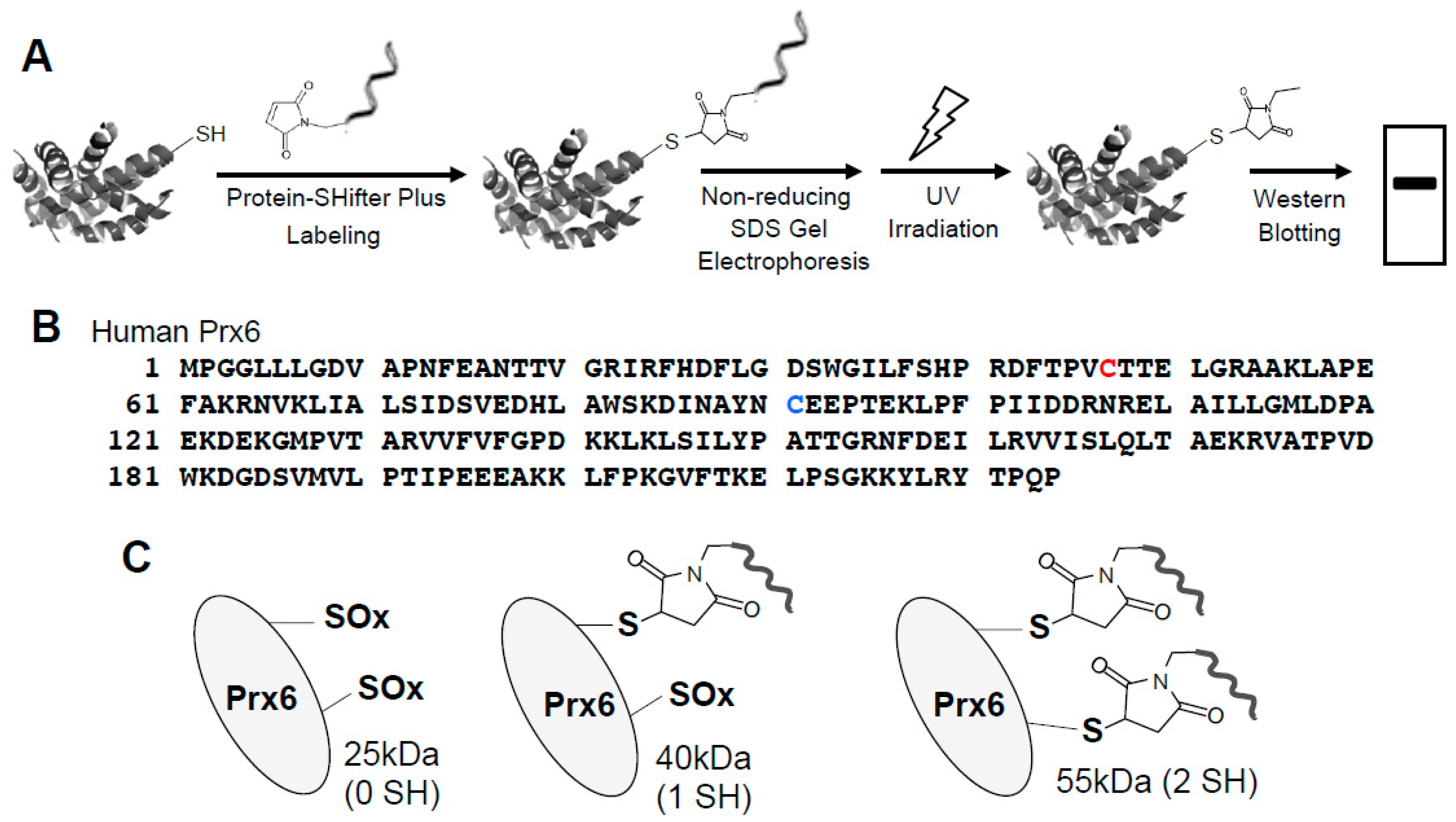
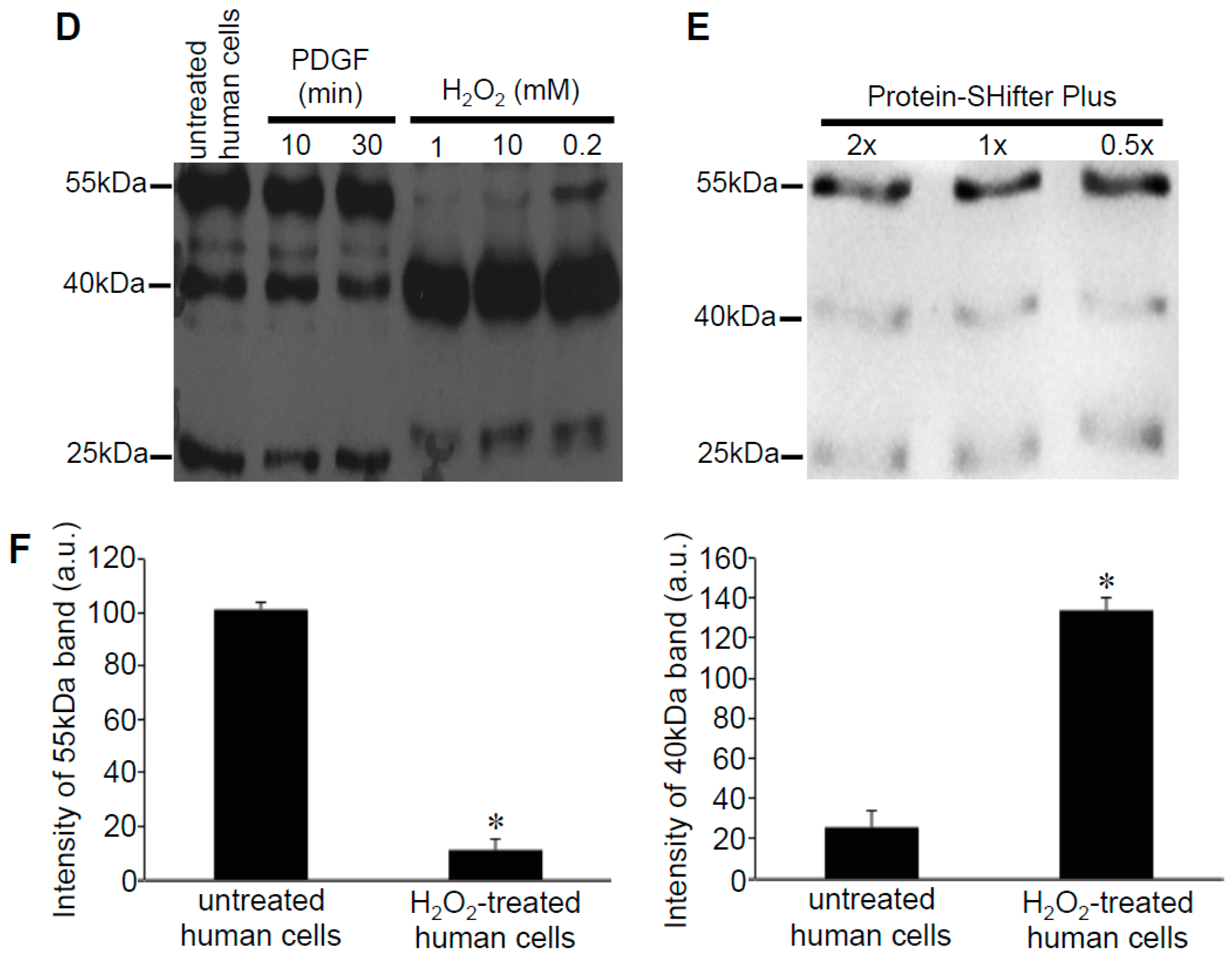
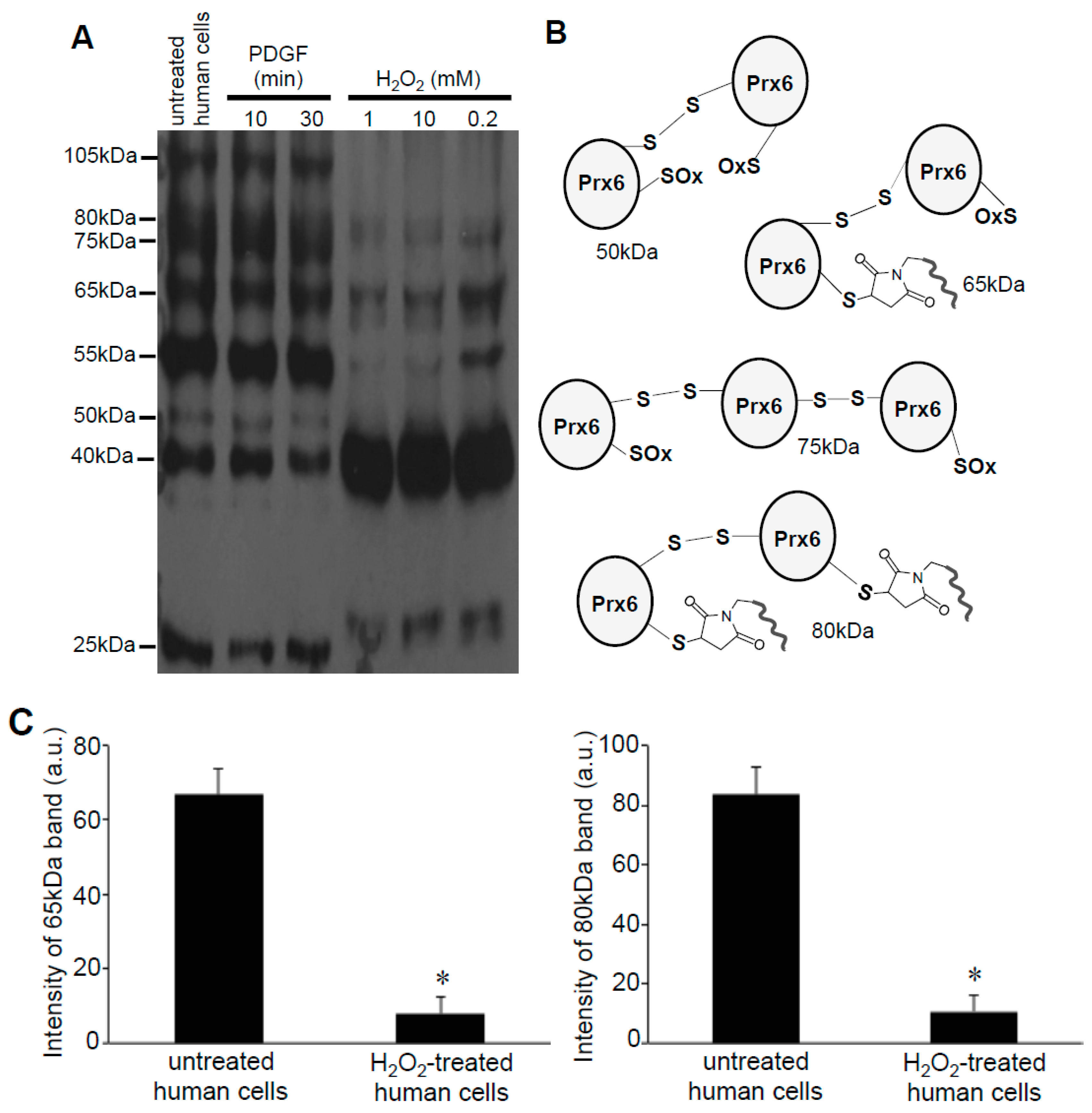
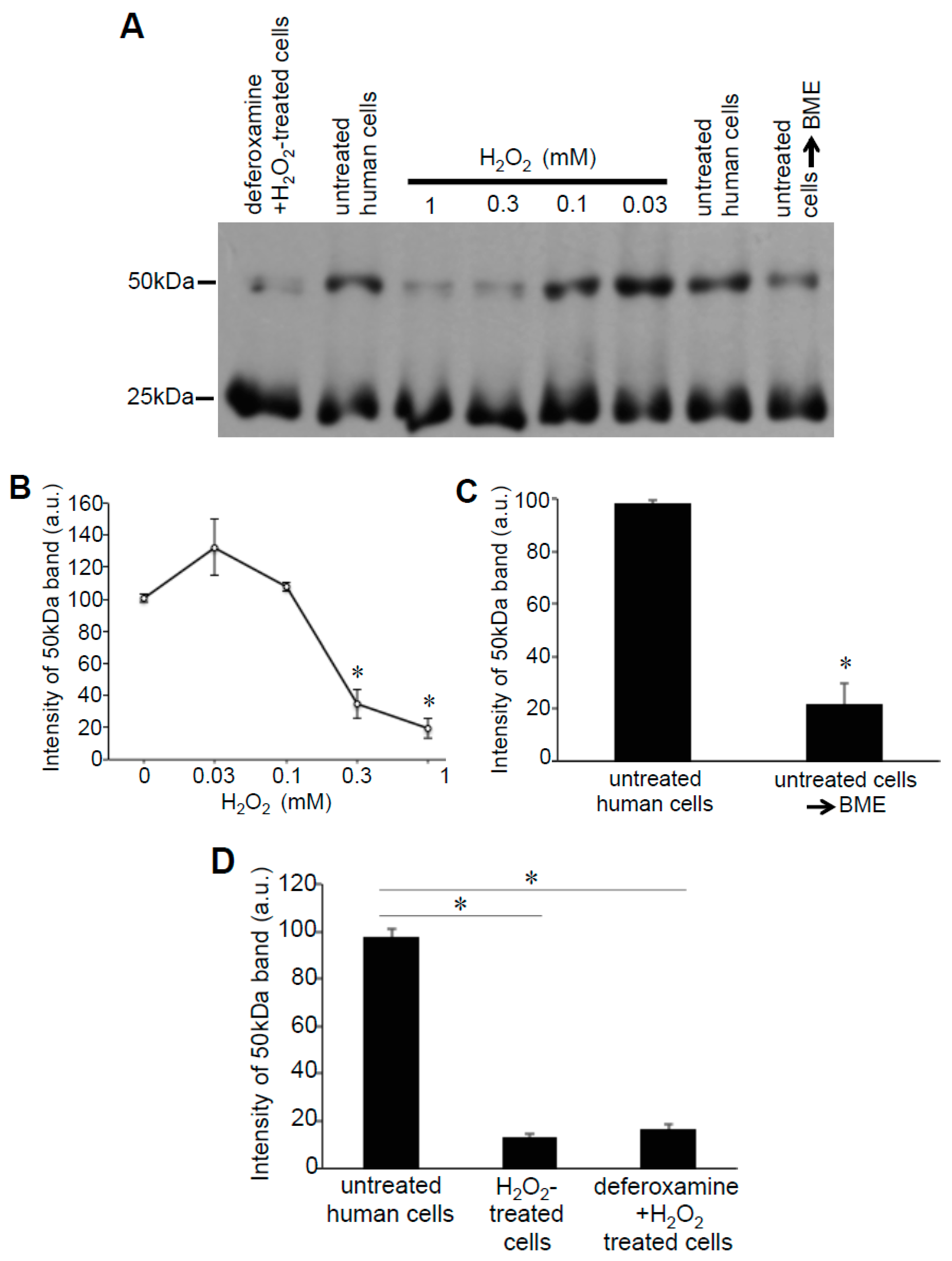
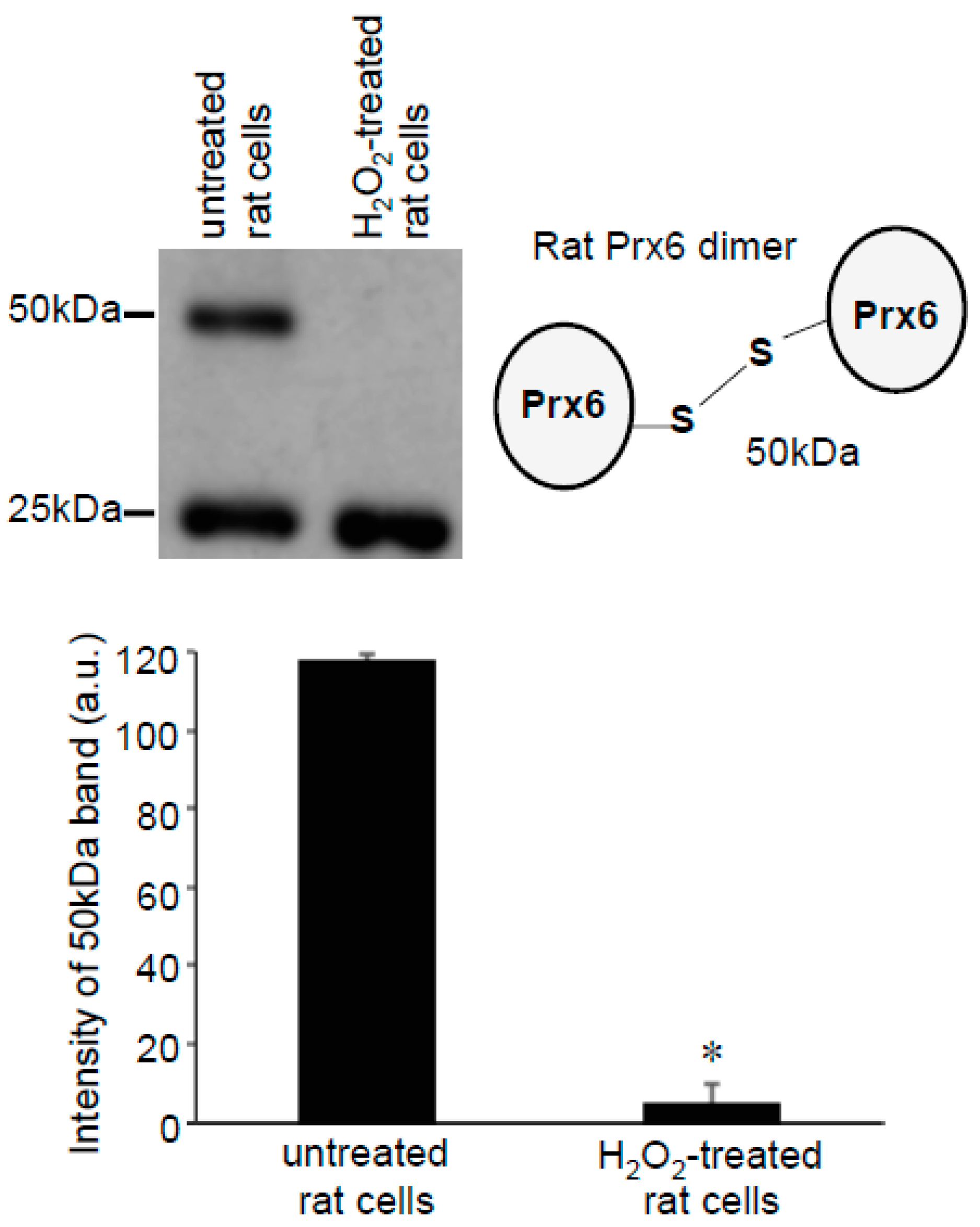
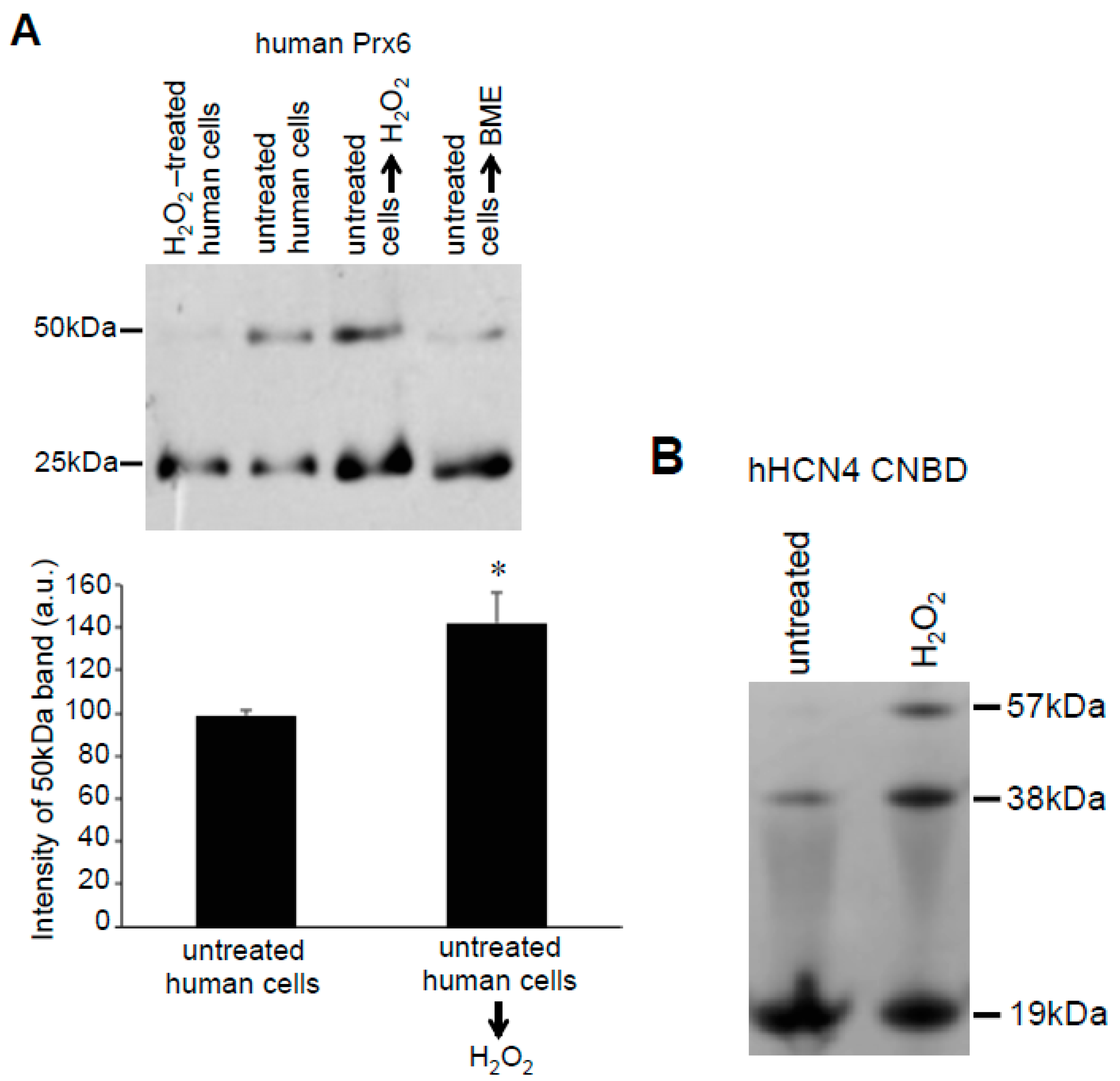
3.2. Redox Studies of Prx6 in Cultured Cells Using -Sulfobiotics- Protein Redox State Monitoring Kit Plus
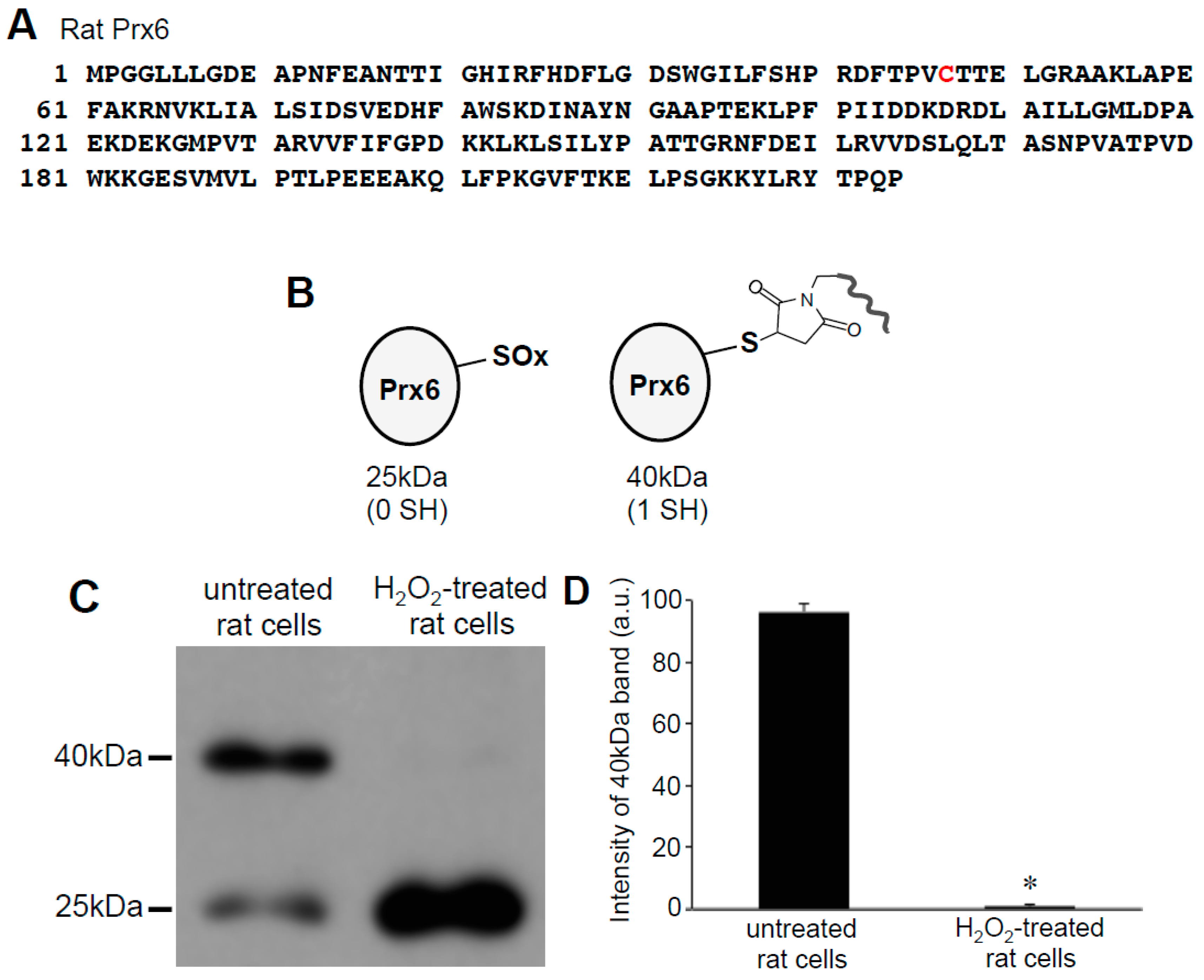
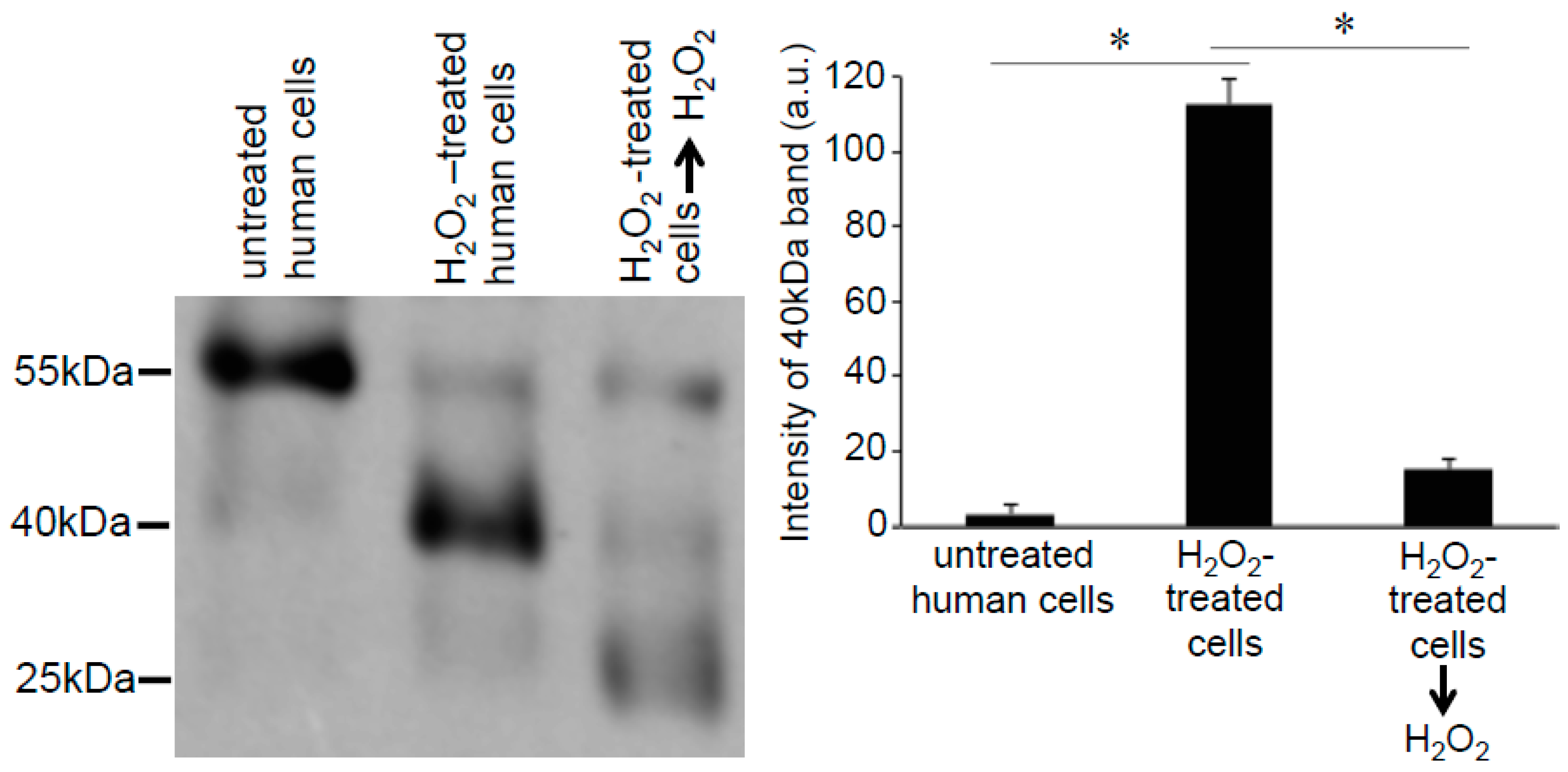
3.3. H2O2 Can Decrease Disulfide Bonds in the Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boronat, S.; Domènech, A.; Hidalgo, E. Proteomic characterization of reversible thiol oxidations in proteomes and proteins. Antioxid. Redox Signal. 2017, 26, 329–344. [Google Scholar] [CrossRef]
- Kasamatsu, S.; Nishimura, A.; Morita, M.; Matsunaga, T.; Abdul Hamid, H.; Akaike, T. Redox signaling regulated by cysteine persulfide and protein polysulfidation. Molecules 2016, 21, 1721. [Google Scholar] [CrossRef]
- Chung, H.S.; Wang, S.B.; Venkatraman, V.; Murray, C.I.; Van Eyk, J.E. Cysteine oxidative posttranslational modifications: Emerging regulation in the cardiovascular system. Circ. Res. 2013, 112, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Mieyal, J.J.; Chock, P.B. Posttranslational modification of cysteine in redox signaling and oxidative stress: Focus on s-glutathionylation. Antioxid. Redox Signal. 2012, 16, 471–475. [Google Scholar] [CrossRef]
- Brandes, N.; Schmitt, S.; Jakob, U. Thiol-based redox switches in eukaryotic proteins. Antioxid. Redox Signal. 2009, 11, 997–1014. [Google Scholar] [CrossRef]
- Sen, C.K. Cellular thiols and redox-regulated signal transduction. Curr. Top. Cell. Regul. 2000, 36, 1–30. [Google Scholar] [PubMed]
- Suzuki, Y.J.; Forman, H.J.; Sevanian, A. Oxidants as stimulators of signal transduction. Free Radic. Biol. Med. 1997, 22, 269–285. [Google Scholar] [CrossRef]
- Rudyk, O.; Eaton, P. Biochemical methods for monitoring protein thiol redox states in biological systems. Redox Biol. 2014, 2, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Kil, I.S. Multiple Functions and Regulation of Mammalian Peroxiredoxins. Annu. Rev. Biochem. 2017, 86, 749–775. [Google Scholar] [CrossRef]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar] [CrossRef]
- Kim, K.; Kim, I.H.; Lee, K.Y.; Rhee, S.G.; Stadtman, E.R. The isolation and purification of a specific ‘protector’ protein which inhibits enzyme inactivation by a thiol/Fe(III)/O2 mixed-function oxidation system. J. Biol. Chem. 1988, 263, 4704–4711. [Google Scholar]
- Seo, M.S.; Kang, S.W.; Kim, K.; Baines, I.C.; Lee, T.H.; Rhee, S.G. Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J. Biol. Chem. 2000, 275, 20346–20354. [Google Scholar] [CrossRef]
- Fisher, A.B. Peroxiredoxin 6 in the repair of peroxidized cell membranes and cell signaling. Arch. Biochem. Biophys. 2017, 617, 68–83. [Google Scholar] [CrossRef]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. Antioxid. Redox Signal. 2011, 15, 831–844. [Google Scholar] [CrossRef]
- Wahl-Schott, C.; Biel, M. HCN channels: Structure, cellular regulation and physiological function. Cell. Mol. Life Sci. 2009, 66, 470–494. [Google Scholar] [CrossRef] [PubMed]
- James, Z.M.; Zagotta, W.N. Structural insights into the mechanisms of CNBD channel function. J. Gen. Physiol. 2018, 150, 225–244. [Google Scholar] [CrossRef] [PubMed]
- Hayoz, S.; Tiwari, P.B.; Piszczek, G.; Uren, A.; Brelidze, T.I. Investigating cyclic nucleotide and cyclic dinucleotide binding to HCN channels by surface plasmon resonance. PLoS ONE 2017, 12, e0185359. [Google Scholar] [CrossRef] [PubMed]
- Hara, S.; Nojima, T.; Seio, K.; Yoshida, M.; Hisabori, T. DNA-maleimide: An improved maleimide compound for electrophoresis-based titration of reactive thiols in a specific protein. Biochim. Biophys. Acta 2013, 1830, 3077–3081. [Google Scholar] [CrossRef]
- Hara, S.; Tatenaka, Y.; Ohuchi, Y.; Hisabori, T. Direct determination of the redox status of cysteine residues in proteins in vivo. Biochem. Biophys. Res. Commun. 2015, 456, 339–343. [Google Scholar] [CrossRef]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef]
- Thamsen, M.; Jakob, U. The redoxome: Proteomic analysis of cellular redox networks. Curr. Opin. Chem. Biol. 2011, 15, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Carroll, K.S.; Liebler, D.C. The Expanding Landscape of the Thiol Redox Proteome. Mol. Cell. Proteom. 2016, 15, 1–11. [Google Scholar] [CrossRef]
- Couvertier, S.M.; Zhou, Y.; Weerapana, E. Chemical-proteomic strategies to investigate cysteine posttranslational modifications. Biochim. Biophys. Acta 2014, 1844, 2315–2330. [Google Scholar] [CrossRef]
- Leichert, L.I.; Gehrke, F.; Gudiseva, H.V.; Blackwell, T.; Ilbert, M.; Walker, A.K.; Strahler, J.R.; Andrews, P.C.; Jakob, U. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 8197–8202. [Google Scholar] [CrossRef]
- Paoli, P.; Giannoni, E.; Pescitelli, R.; Camici, G.; Manao, G.; Ramponi, G. Hydrogen peroxide triggers the formation of a disulfide dimer of muscle acylphosphatase and modifies some functional properties of the enzyme. J. Biol. Chem. 2001, 276, 41862–41869. [Google Scholar] [CrossRef] [PubMed]
- Van Der Wijk, T.; Overvoorde, J.; den Hertog, J. H2O2-induced intermolecular disulfide bond formation between receptor protein-tyrosine phosphatases. J. Biol. Chem. 2004, 279, 44355–44361. [Google Scholar] [CrossRef] [PubMed]
- Bindoli, A.; Fukuto, J.M.; Forman, H.J. Thiol chemistry in peroxidase catalysis and redox signaling. Antioxid. Redox Signal. 2008, 10, 1549–1564. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki, Y.J.; Marcocci, L.; Shimomura, T.; Tatenaka, Y.; Ohuchi, Y.; Brelidze, T.I. Protein Redox State Monitoring Studies of Thiol Reactivity. Antioxidants 2019, 8, 143. https://doi.org/10.3390/antiox8050143
Suzuki YJ, Marcocci L, Shimomura T, Tatenaka Y, Ohuchi Y, Brelidze TI. Protein Redox State Monitoring Studies of Thiol Reactivity. Antioxidants. 2019; 8(5):143. https://doi.org/10.3390/antiox8050143
Chicago/Turabian StyleSuzuki, Yuichiro J., Lucia Marcocci, Takashi Shimomura, Yuki Tatenaka, Yuya Ohuchi, and Tinatin I. Brelidze. 2019. "Protein Redox State Monitoring Studies of Thiol Reactivity" Antioxidants 8, no. 5: 143. https://doi.org/10.3390/antiox8050143
APA StyleSuzuki, Y. J., Marcocci, L., Shimomura, T., Tatenaka, Y., Ohuchi, Y., & Brelidze, T. I. (2019). Protein Redox State Monitoring Studies of Thiol Reactivity. Antioxidants, 8(5), 143. https://doi.org/10.3390/antiox8050143