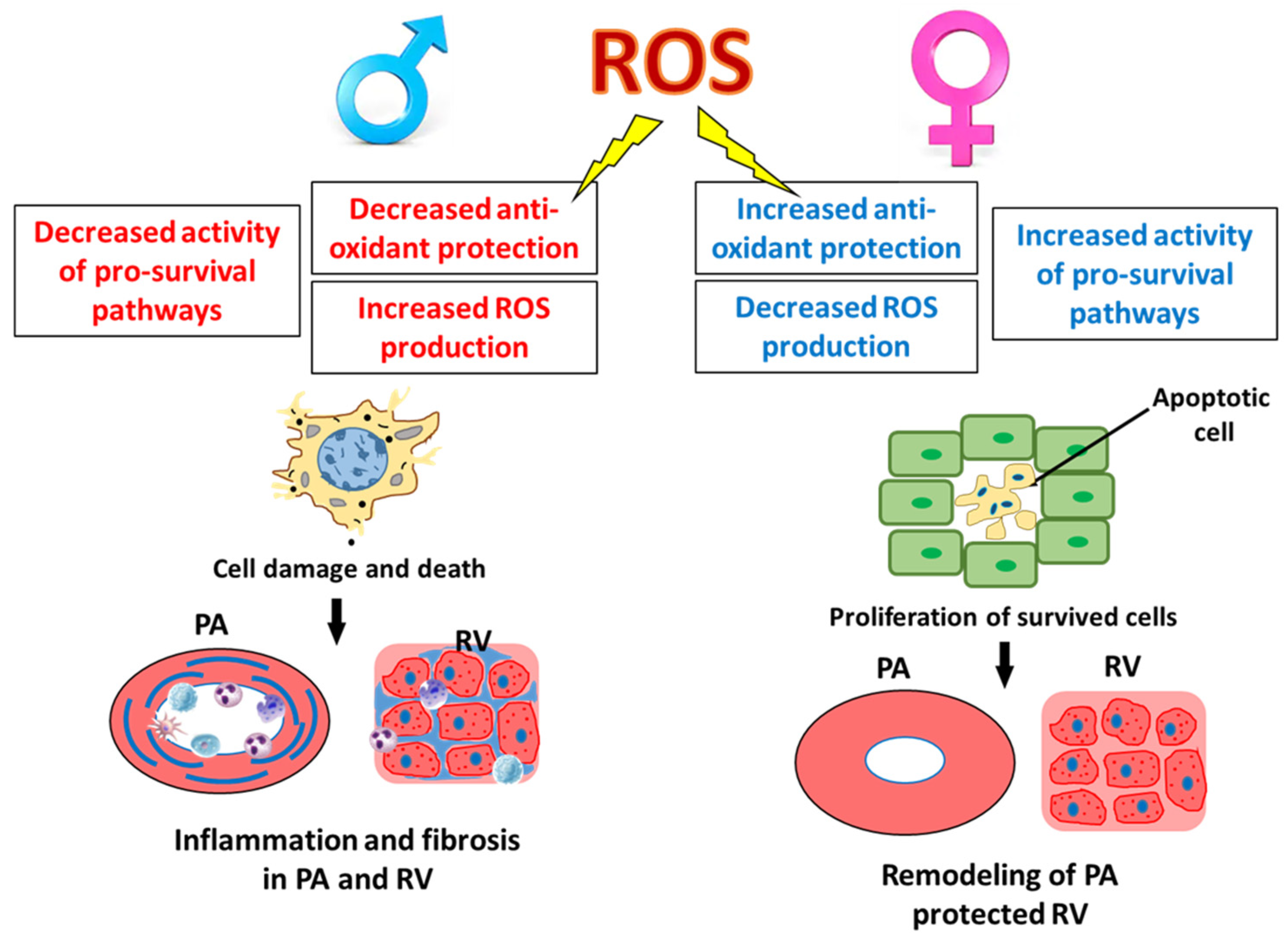
Role of Gender in Regulation of Redox Homeostasis in Pulmonary Arterial Hypertension

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
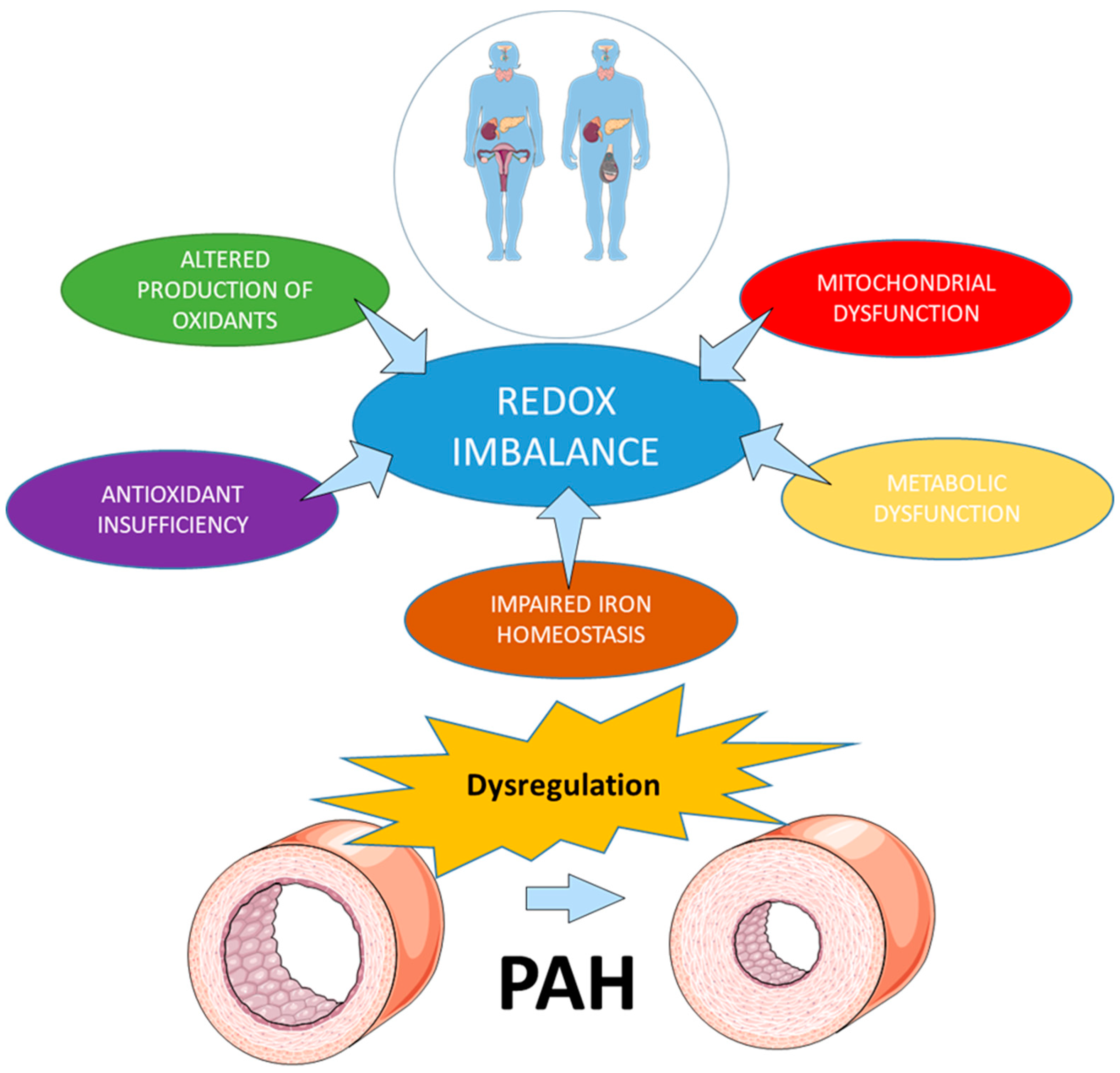
2. Redox Homeostasis in Health and Disease
3. The Role of Sex Hormones in Redox Homeostasis and Pulmonary Arterial Hypertension
4. Sources of ROS in PAH and the Role of Gender
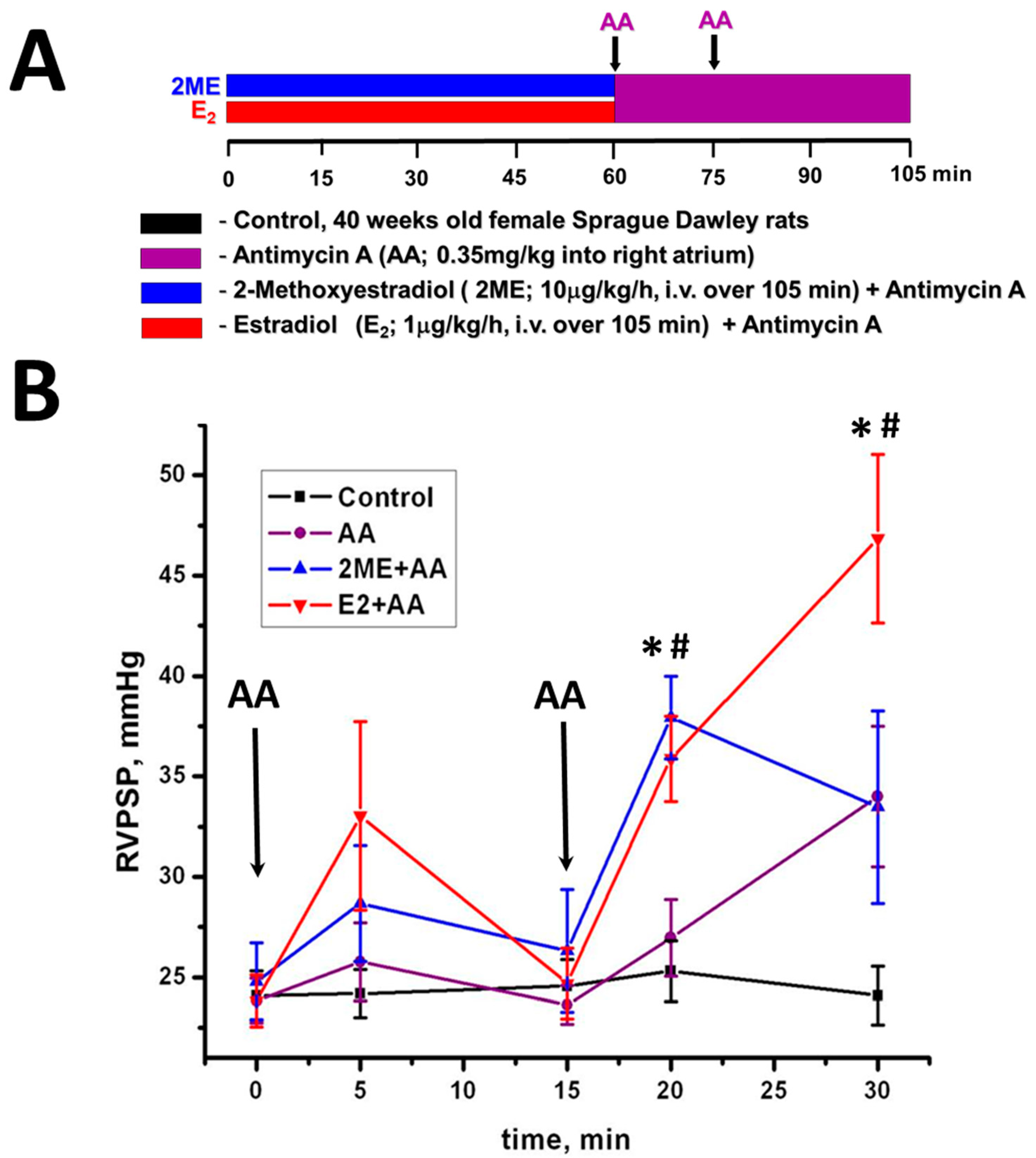
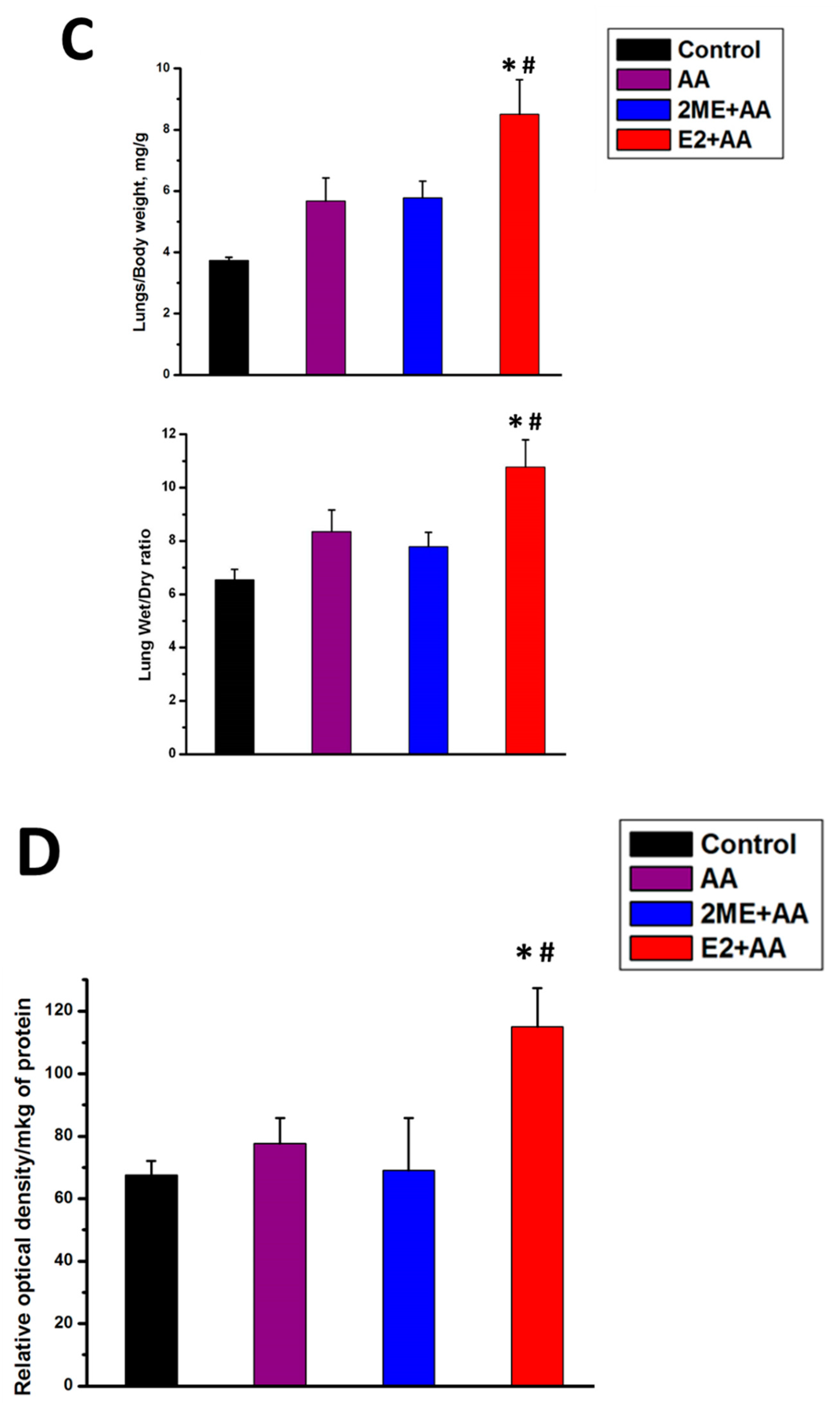
4.1. Mitochondrial ROS
4.2. NADPH Oxidases
4.3. Free Heme, Heme-Containing Proteins, and Free Iron
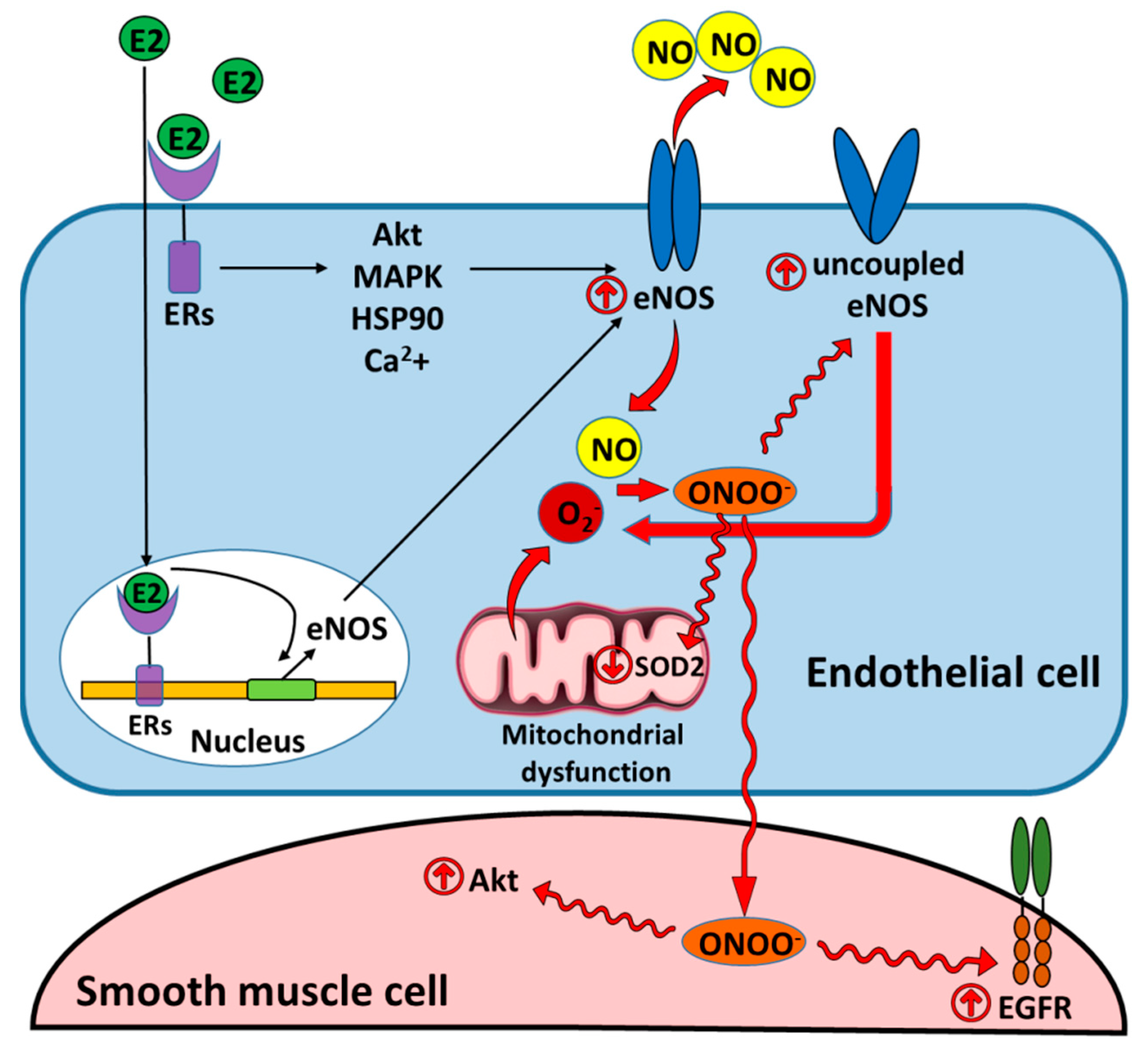
5. Nitric Oxide Synthase
6. The Antioxidant System in PAH and the Role of Gender
6.1. Gender Differences in SOD and Catalase
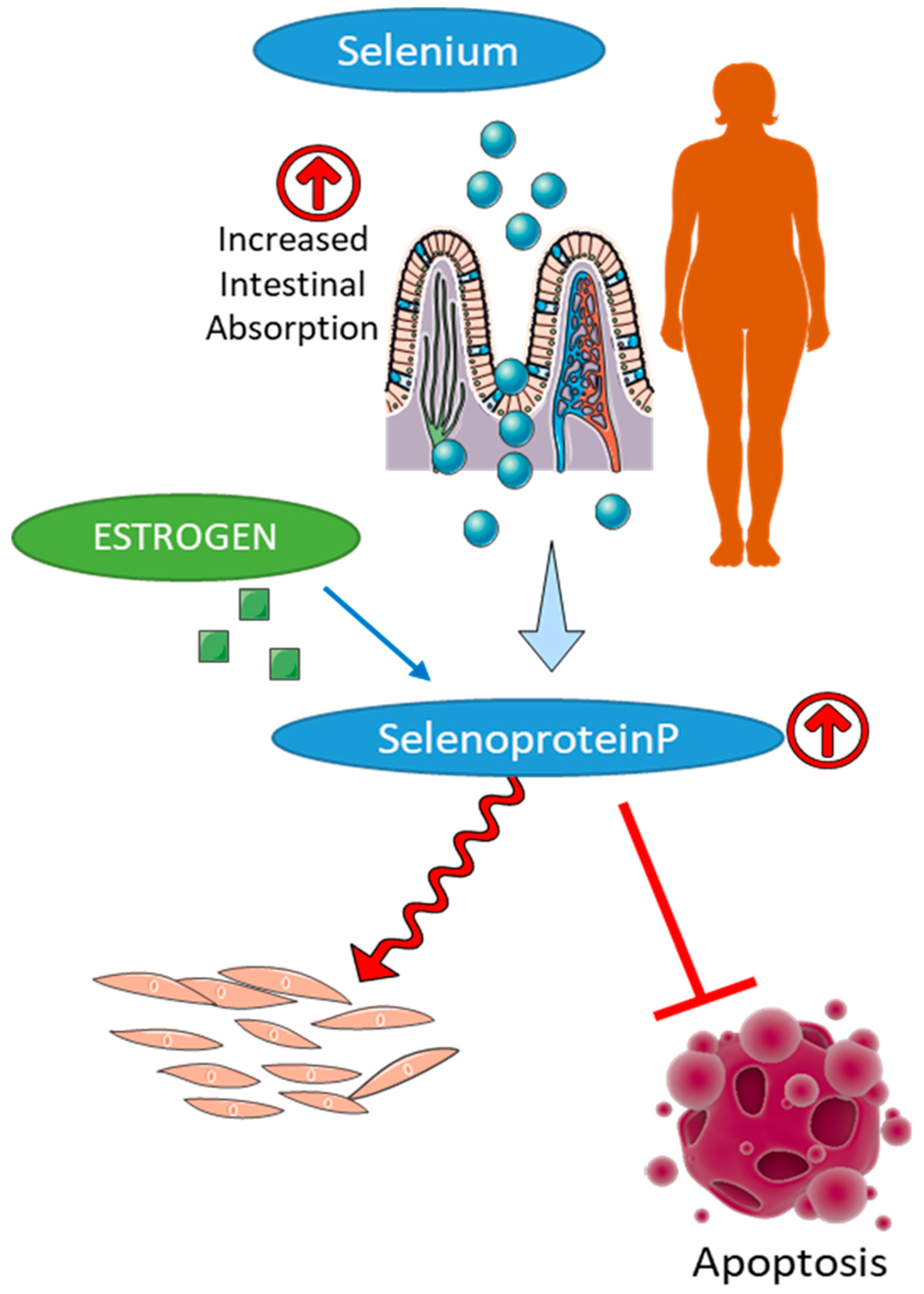
6.2. Selenium in Gender-Specific Redox Homeostasis
6.3. Gender Differences and Role of Heme Oxygenase in PAH
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thenappan, T.; Shah, S.J.; Rich, S.; Gomberg-Maitland, M. A united states-based registry for pulmonary arterial hypertension: 1982–2006. Eur. Respir. J. 2007, 30, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- McGoon, M.; Miller, D. REVEAL: A contemporary US pulmonary arterial hypertension registry. Eur. Respir. J. 2012, 21, 8–18. [Google Scholar] [CrossRef]
- Mair, K.M.; Johansen, A.K.; Wright, A.F.; Wallace, E.; MacLean, M.R. Pulmonary arterial hypertension: Basis of sex differences in incidence and treatment response. Br. J. Pharmacol. 2014, 171, 567–579. [Google Scholar] [CrossRef]
- Escribano-Subias, P.; Blanco, I.; Lopez-Meseguer, M.; Lopez-Guarch, C.J.; Roman, A.; Morales, P.; Castillo-Palma, M.J.; Segovia, J.; Gomez-Sanchez, M.A.; Barbera, J.A.; et al. Survival in pulmonary hypertension in Spain: Insights from the Spanish registry. Eur. Respir. J. 2012, 40, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Kander, M.C.; Cui, Y.; Liu, Z. Gender difference in oxidative stress: A new look at the mechanisms for cardiovascular diseases. J. Cell. Mol. Med. 2017, 21, 1024–1032. [Google Scholar] [CrossRef]
- Ruszkiewicz, J.A.; Miranda-Vizuete, A.; Tinkov, A.A.; Skalnaya, M.G.; Skalny, A.V.; Tsatsakis, A.; Aschner, M. Sex-Specific Differences in Redox Homeostasis in Brain Norm and Disease. J. Mol. Neurosci. 2019, 67, 312–342. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Ohashi, N.; Hayashidani, S.; Suematsu, N.; Tsuchihashi, M.; Tamai, H.; Takeshita, A. Greater oxidative stress in healthy young men compared with premenopausal women. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 438–442. [Google Scholar] [CrossRef]
- Diaz-Castro, J.; Pulido-Moran, M.; Moreno-Fernandez, J.; Kajarabille, N.; de Paco, C.; Garrido-Sanchez, M.; Prados, S.; Ochoa, J.J. Gender specific differences in oxidative stress and inflammatory signaling in healthy term neonates and their mothers. Pediatr. Res. 2016, 80, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Vassalle, C.; Maffei, S.; Boni, C.; Zucchelli, G.C. Gender-related differences in oxidative stress levels among elderly patients with coronary artery disease. Fertil. Steril. 2008, 89, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Boardman, H.M.; Hartley, L.; Eisinga, A.; Main, C.; Roque i Figuls, M.; Bonfill Cosp, X.; Gabriel Sanchez, R.; Knight, B. Hormone therapy for preventing cardiovascular disease in post-menopausal women. Cochrane Database Syst. Rev. 2015. [Google Scholar] [CrossRef]
- Main, C.; Knight, B.; Moxham, T.; Gabriel Sanchez, R.; Sanchez Gomez, L.M.; Roque i Figuls, M.; Bonfill Cosp, X. Hormone therapy for preventing cardiovascular disease in post-menopausal women. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Sauer, H.; Wartenberg, M.; Hescheler, J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell. Physiol. Biochem. 2001, 11, 173–186. [Google Scholar] [CrossRef]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Rafikov, R.; Nair, V.; Sinari, S.; Babu, H.; Sullivan, J.C.; Yuan, J.X.; Desai, A.A.; Rafikova, O. Gender Difference in Damage Mediated Signaling Contributes to Pulmonary Arterial Hypertension. Antioxid. Redox Signal. 2019. [Google Scholar] [CrossRef]
- Kazama, H.; Ricci, J.E.; Herndon, J.M.; Hoppe, G.; Green, D.R.; Ferguson, T.A. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 2008, 29, 21–32. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Zeh, H.J.; Lotze, M.T. High-mobility group box 1, oxidative stress, and disease. Antioxid. Redox Signal. 2011, 14, 1315–1335. [Google Scholar] [CrossRef]
- Jog, N.R.; Caricchio, R. Differential regulation of cell death programs in males and females by Poly (ADP-Ribose) Polymerase-1 and 17beta estradiol. Cell Death Dis. 2013, 4, e758. [Google Scholar] [CrossRef] [PubMed]
- Casimir, G.J.; Duchateau, J. Gender differences in inflammatory processes could explain poorer prognosis for males. J. Clin. Microbiol. 2011, 49, 478–479. [Google Scholar] [CrossRef] [PubMed]
- Rafikova, O.; Rafikov, R.; Meadows, M.L.; Kangath, A.; Jonigk, D.; Black, S.M. The sexual dimorphism associated with pulmonary hypertension corresponds to a fibrotic phenotype. Pulm. Circ. 2015, 5, 184–197. [Google Scholar] [CrossRef]
- Rafikova, O.; Rafikov, R.; Kumar, S.; Sharma, S.; Aggarwal, S.; Schneider, F.; Jonigk, D.; Black, S.M.; Tofovic, S.P. Bosentan inhibits oxidative and nitrosative stress and rescues occlusive pulmonary hypertension. Free Radic. Biol. Med. 2013, 56, 28–43. [Google Scholar] [CrossRef]
- Cassuto, J.; Dou, H.; Czikora, I.; Szabo, A.; Patel, V.S.; Kamath, V.; Belin de Chantemele, E.; Feher, A.; Romero, M.J.; Bagi, Z. Peroxynitrite disrupts endothelial caveolae leading to eNOS uncoupling and diminished flow-mediated dilation in coronary arterioles of diabetic patients. Diabetes 2014, 63, 1381–1393. [Google Scholar] [CrossRef] [PubMed]
- Bowers, R.; Cool, C.; Murphy, R.C.; Tuder, R.M.; Hopken, M.W.; Flores, S.C.; Voelkel, N.F. Oxidative stress in severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2004, 169, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Rafikov, R.; Rafikova, O.; Aggarwal, S.; Gross, C.; Sun, X.; Desai, J.; Fulton, D.; Black, S.M. Asymmetric dimethylarginine induces endothelial nitric-oxide synthase mitochondrial redistribution through the nitration-mediated activation of Akt1. J. Biol. Chem. 2013, 288, 6212–6226. [Google Scholar] [CrossRef]
- Rafikova, O.; Rafikov, R.; Kangath, A.; Qu, N.; Aggarwal, S.; Sharma, S.; Desai, J.; Fields, T.; Ludewig, B.; Yuan, J.X.; et al. Redox regulation of epidermal growth factor receptor signaling during the development of pulmonary hypertension. Free Radic. Biol. Med. 2016, 95, 96–111. [Google Scholar] [CrossRef]
- Bjornstrom, L.; Sjoberg, M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef]
- Frump, A.L.; Goss, K.N.; Vayl, A.; Albrecht, M.; Fisher, A.; Tursunova, R.; Fierst, J.; Whitson, J.; Cucci, A.R.; Brown, M.B.; et al. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: Effects of endogenous and exogenous sex hormones. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L873–L890. [Google Scholar] [CrossRef] [PubMed]
- Lahm, T.; Albrecht, M.; Fisher, A.J.; Selej, M.; Patel, N.G.; Brown, J.A.; Justice, M.J.; Brown, M.B.; Van Demark, M.; Trulock, K.M.; et al. 17beta-Estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am. J. Respir. Crit. Care Med. 2012, 185, 965–980. [Google Scholar] [CrossRef]
- Umar, S.; Iorga, A.; Matori, H.; Nadadur, R.D.; Li, J.; Maltese, F.; van der Laarse, A.; Eghbali, M. Estrogen rescues preexisting severe pulmonary hypertension in rats. Am. J. Respir. Crit. Care Med. 2011, 184, 715–723. [Google Scholar] [CrossRef]
- Farhat, M.Y.; Chen, M.F.; Bhatti, T.; Iqbal, A.; Cathapermal, S.; Ramwell, P.W. Protection by oestradiol against the development of cardiovascular changes associated with monocrotaline pulmonary hypertension in rats. Br. J. Pharmacol. 1993, 110, 719–723. [Google Scholar] [CrossRef]
- Ahn, B.H.; Park, H.K.; Cho, H.G.; Lee, H.A.; Lee, Y.M.; Yang, E.K.; Lee, W.J. Estrogen and enalapril attenuate the development of right ventricular hypertrophy induced by monocrotaline in ovariectomized rats. J. Korean Med. Sci. 2003, 18, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Tofovic, S.P. Estrogens and development of pulmonary hypertension: Interaction of estradiol metabolism and pulmonary vascular disease. J. Cardiovasc. Pharmacol. 2010, 56, 696–708. [Google Scholar] [CrossRef]
- Austin, E.D.; Cogan, J.D.; West, J.D.; Hedges, L.K.; Hamid, R.; Dawson, E.P.; Wheeler, L.A.; Parl, F.F.; Loyd, J.E.; Phillips, J.A. Alterations in estrogen metabolism: Implications for higher penetrance of FPAH in females. Eur. Respir. J. 2009, 34, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Lahm, T.; West, J.; Tofovic, S.P.; Johansen, A.K.; Maclean, M.R.; Alzoubi, A.; Oka, M. Gender, sex hormones and pulmonary hypertension. Pulm. Circ. 2013, 3, 294–314. [Google Scholar] [CrossRef] [PubMed]
- Tofovic, S.P.; Jackson, E.K. Estrogens in Men: Another Layer of Complexity of Estradiol Metabolism in Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2016, 193, 1087–1090. [Google Scholar] [CrossRef]
- Henderson, B.E.; Feigelson, H.S. Hormonal carcinogenesis. Carcinogenesis 2000, 21, 427–433. [Google Scholar] [CrossRef]
- Boucherat, O.; Vitry, G.; Trinh, I.; Paulin, R.; Provencher, S.; Bonnet, S. The cancer theory of pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 285–299. [Google Scholar] [CrossRef]
- Kawut, S.M.; Archer-Chicko, C.L.; DeMichele, A.; Fritz, J.S.; Klinger, J.R.; Ky, B.; Palevsky, H.I.; Palmisciano, A.J.; Patel, M.; Pinder, D. Anastrozole in pulmonary arterial hypertension. A randomized, double-blind, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 2017, 195, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, K.; Kim, K.H.; Bender, J.R. Minireview: Estrogen receptor-mediated rapid signaling. Endocrinology 2006, 147, 5557–5563. [Google Scholar] [CrossRef]
- Miller, V.M.; Duckles, S.P. Vascular actions of estrogens: Functional implications. Pharmacol. Rev. 2008, 60, 210–241. [Google Scholar] [CrossRef]
- Bleier, L.; Drose, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim Biophys Acta 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Rafikov, R.; Sun, X.; Rafikova, O.; Meadows, M.L.; Desai, A.A.; Khalpey, Z.; Yuan, J.X.; Fineman, J.R.; Black, S.M. Complex I dysfunction underlies the glycolytic switch in pulmonary hypertensive smooth muscle cells. Redox Biol. 2015, 6, 278–286. [Google Scholar] [CrossRef]
- Rafikova, O.; Srivastava, A.; Desai, A.A.; Rafikov, R.; Tofovic, S.P. Recurrent inhibition of mitochondrial complex III induces chronic pulmonary vasoconstriction and glycolytic switch in the rat lung. Respir. Res. 2018, 19, 69. [Google Scholar] [CrossRef]
- Chignalia, A.Z.; Schuldt, E.Z.; Camargo, L.L.; Montezano, A.C.; Callera, G.E.; Laurindo, F.R.; Lopes, L.R.; Avellar, M.C.; Carvalho, M.H.; Fortes, Z.B.; et al. Testosterone induces vascular smooth muscle cell migration by NADPH oxidase and c-Src-dependent pathways. Hypertension 2012, 59, 1263–1271. [Google Scholar] [CrossRef]
- Pingili, A.K.; Kara, M.; Khan, N.S.; Estes, A.M.; Lin, Z.; Li, W.; Gonzalez, F.J.; Malik, K.U. 6beta-hydroxytestosterone, a cytochrome P450 1B1 metabolite of testosterone, contributes to angiotensin II-induced hypertension and its pathogenesis in male mice. Hypertension 2015, 65, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Navarro, G.; Mauvais-Jarvis, F. Androgen excess produces systemic oxidative stress and predisposes to beta-cell failure in female mice. PLoS ONE 2010, 5, e11302. [Google Scholar] [CrossRef]
- Lu, J.P.; Monardo, L.; Bryskin, I.; Hou, Z.F.; Trachtenberg, J.; Wilson, B.C.; Pinthus, J.H. Androgens induce oxidative stress and radiation resistance in prostate cancer cells though NADPH oxidase. Prostate Cancer Prostatic Dis. 2010, 13, 39–46. [Google Scholar] [CrossRef]
- Costa, T.J.; Ceravolo, G.S.; dos Santos, R.A.; de Oliveira, M.A.; Araújo, P.X.; Giaquinto, L.R.; Tostes, R.C.; Akamine, E.H.; Fortes, Z.B.; Dantas, A.P. Association of testosterone with estrogen abolishes the beneficial effects of estrogen treatment by increasing ROS generation in aorta endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H723–H732. [Google Scholar] [CrossRef]
- Gonzalez, F.; Nair, K.S.; Daniels, J.K.; Basal, E.; Schimke, J.M.; Blair, H.E. Hyperandrogenism sensitizes leukocytes to hyperglycemia to promote oxidative stress in lean reproductive-age women. J. Clin. Endocrinol. Metab. 2012, 97, 2836–2843. [Google Scholar] [CrossRef]
- Tomás-Zapico, C.; Álvarez-García, Ó.; Sierra, V.; Vega-Naredo, I.; Caballero, B.; García, J.J.; Acuña-Castroviejo, D.; Rodríguez, M.I.; Tolivia, D.; Rodríguez-Colunga, M.J. Oxidative damage in the livers of senescence-accelerated mice: a gender-related response. Can. J. Physiol. Pharmacol. 2006, 84, 213–220. [Google Scholar] [CrossRef]
- Thomas-Ahner, J.M.; Wulff, B.C.; Tober, K.L.; Kusewitt, D.F.; Riggenbach, J.A.; Oberyszyn, T.M. Gender differences in UVB-induced skin carcinogenesis, inflammation, and DNA damage. Cancer Res. 2007, 67, 3468–3474. [Google Scholar] [CrossRef] [PubMed]
- Ko, E.; Choi, H.; Kim, B.; Kim, M.; Park, K.N.; Bae, I.H.; Sung, Y.K.; Lee, T.R.; Shin, D.W.; Bae, Y.S. Testosterone stimulates Duox1 activity through GPRC6A in skin keratinocytes. J. Biol. Chem. 2014, 289, 28835–28845. [Google Scholar] [CrossRef]
- Lopes, R.A.; Neves, K.B.; Pestana, C.R.; Queiroz, A.L.; Zanotto, C.Z.; Chignalia, A.Z.; Valim, Y.M.; Silveira, L.R.; Curti, C.; Tostes, R.C. Testosterone induces apoptosis in vascular smooth muscle cells via extrinsic apoptotic pathway with mitochondria-generated reactive oxygen species involvement. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1485–H1494. [Google Scholar] [CrossRef]
- Huang, C.; Gu, H.; Zhang, W.; Herrmann, J.L.; Wang, M. Testosterone-down-regulated Akt pathway during cardiac ischemia/reperfusion: A mechanism involving BAD, Bcl-2 and FOXO3a. J. Surg. Res. 2010, 164, e1–e11. [Google Scholar] [CrossRef] [PubMed]
- Demarest, T.G.; McCarthy, M.M. Sex differences in mitochondrial (dys) function: Implications for neuroprotection. J. Bioenerg. Biomembr. 2015, 47, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Cavasin, M.A.; Tao, Z.Y.; Yu, A.L.; Yang, X.P. Testosterone enhances early cardiac remodeling after myocardial infarction, causing rupture and degrading cardiac function. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2043–H2050. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Maynard, K.B.; Champion, H.C.; Gleaves, L.; Penner, N.; West, J.; Newman, J.H. Testosterone negatively regulates right ventricular load stress responses in mice. Pulm. Circ. 2012, 2, 352–358. [Google Scholar] [CrossRef]
- Blacker, T.S.; Duchen, M.R. Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radic. Biol. Med. 2016, 100, 53–65. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Moulin, M.; Piquereau, J.; Lemaire, C.; Mericskay, M.; Veksler, V.; Garnier, A. Mitochondria: A central target for sex differences in pathologies. Clin. Sci. (Lond.) 2017, 131, 803–822. [Google Scholar] [CrossRef]
- Rutkai, I.; Dutta, S.; Katakam, P.V.; Busija, D.W. Dynamics of enhanced mitochondrial respiration in female compared with male rat cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1490–H1500. [Google Scholar] [CrossRef] [PubMed]
- Colom, B.; Alcolea, M.P.; Valle, A.; Oliver, J.; Roca, P.; Garcia-Palmer, F.J. Skeletal muscle of female rats exhibit higher mitochondrial mass and oxidative-phosphorylative capacities compared to males. Cell. Physiol. Biochem. 2007, 19, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Colom, B.; Oliver, J.; Roca, P.; Garcia-Palmer, F.J. Caloric restriction and gender modulate cardiac muscle mitochondrial H2O2 production and oxidative damage. Cardiovasc. Res. 2007, 74, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Lagranha, C.J.; Deschamps, A.; Aponte, A.; Steenbergen, C.; Murphy, E. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ. Res. 2010, 106, 1681–1691. [Google Scholar] [CrossRef]
- Borrás, C.; Gambini, J.; Gómez-Cabrera, M.C.; Sastre, J.; Pallardó, F.V.; Mann, G.E.; Viña, J. 17β-oestradiol up-regulates longevity-related, antioxidant enzyme expression via the ERK1 and ERK2 [MAPK]/NFκB cascade. Aging Cell 2005, 4, 113–118. [Google Scholar] [CrossRef]
- Liu, Z.; Gou, Y.; Zhang, H.; Zuo, H.; Zhang, H.; Liu, Z.; Yao, D. Estradiol improves cardiovascular function through up-regulation of SOD2 on vascular wall. Redox Biol. 2014, 3, 88–99. [Google Scholar] [CrossRef]
- Misiak, M.; Beyer, C.; Arnold, S. Gender-specific role of mitochondria in the vulnerability of 6-hydroxydopamine-treated mesencephalic neurons. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 1178–1188. [Google Scholar] [CrossRef]
- Miller, W.L. Steroid hormone synthesis in mitochondria. Mol. Cell. Endocrinol. 2013, 379, 62–73. [Google Scholar] [CrossRef]
- Robertson, C.L.; Puskar, A.; Hoffman, G.E.; Murphy, A.Z.; Saraswati, M.; Fiskum, G. Physiologic progesterone reduces mitochondrial dysfunction and hippocampal cell loss after traumatic brain injury in female rats. Neurobiol. Dis. 2006, 197, 235–243. [Google Scholar] [CrossRef]
- Gaignard, P.; Savouroux, S.; Liere, P.; Pianos, A.; Thérond, P.; Schumacher, M.; Slama, A.; Guennoun, R. Effect of sex differences on brain mitochondrial function and its suppression by ovariectomy and in aged mice. Endocrinology 2015, 156, 2893–2904. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Wong, S.; Li, M.; Liang, W.; Liesa, M.; Serra, C.; Jasuja, R.; Bartke, A.; Kirkland, J.L.; Shirihai, O.; et al. Testosterone plus low-intensity physical training in late life improves functional performance, skeletal muscle mitochondrial biogenesis, and mitochondrial quality control in male mice. PLoS ONE 2012, 7, e51180. [Google Scholar] [CrossRef]
- Yan, W.; Kang, Y.; Ji, X.; Li, S.; Li, Y.; Zhang, G.; Cui, H.; Shi, G. Testosterone Upregulates the Expression of Mitochondrial ND1 and ND4 and Alleviates the Oxidative Damage to the Nigrostriatal Dopaminergic System in Orchiectomized Rats. Oxid. Med. Cell. Longev. 2017, 2017, 1202459. [Google Scholar] [CrossRef]
- Traish, A.M.; Abdallah, B.; Yu, G. Androgen deficiency and mitochondrial dysfunction: Implications for fatigue, muscle dysfunction, insulin resistance, diabetes, and cardiovascular disease. Horm. Mol. Biol. Clin. Investig. 2011, 8, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Paulin, R.; Michelakis, E.D. The metabolic theory of pulmonary arterial hypertension. Circ. Res. 2014, 115, 148–164. [Google Scholar] [CrossRef]
- Sutendra, G.; Michelakis, E.D. The metabolic basis of pulmonary arterial hypertension. Cell Metab. 2014, 19, 558–573. [Google Scholar] [CrossRef]
- Morrell, N.W.; Adnot, S.; Archer, S.L.; Dupuis, J.; Jones, P.L.; MacLean, M.R.; McMurtry, I.F.; Stenmark, K.R.; Thistlethwaite, P.A.; Weissmann, N.; et al. Cellular and molecular basis of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009, 54, S20–S31. [Google Scholar] [CrossRef]
- Bonnet, S.; Michelakis, E.D.; Porter, C.J.; Andrade-Navarro, M.A.; Thebaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 2006, 113, 2630–2641. [Google Scholar] [CrossRef]
- Sutendra, G.; Dromparis, P.; Kinnaird, A.; Stenson, T.H.; Haromy, A.; Parker, J.M.; McMurtry, M.S.; Michelakis, E.D. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene 2013, 32, 1638–1650. [Google Scholar] [CrossRef] [PubMed]
- Dunham-Snary, K.J.; Wu, D.; Sykes, E.A.; Thakrar, A.; Parlow, L.R.G.; Mewburn, J.D.; Parlow, J.L.; Archer, S.L. Hypoxic Pulmonary Vasoconstriction: From Molecular Mechanisms to Medicine. Chest 2017, 151, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Docherty, C.K.; Nilsen, M.; MacLean, M.R. Influence of 2-Methoxyestradiol and Sex on Hypoxia-Induced Pulmonary Hypertension and Hypoxia-Inducible Factor-1-alpha. J. Am. Heart Assoc. 2019, 8, e011628. [Google Scholar] [CrossRef]
- Tarnopolsky, M.A. Sex differences in exercise metabolism and the role of 17-beta estradiol. Med. Sci. Sports Exerc. 2008, 40, 648–654. [Google Scholar] [CrossRef]
- Maher, A.C.; Akhtar, M.; Vockley, J.; Tarnopolsky, M.A. Women have higher protein content of beta-oxidation enzymes in skeletal muscle than men. PLoS ONE 2010, 5, e12025. [Google Scholar] [CrossRef]
- Foster, D.W. Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. J. Clin. Investig. 2012, 122, 1958–1959. [Google Scholar] [CrossRef]
- Fang, Y.H.; Piao, L.; Hong, Z.; Toth, P.T.; Marsboom, G.; Bache-Wiig, P.; Rehman, J.; Archer, S.L. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: Exploiting Randle’s cycle. J. Mol. Med. (Berl.) 2012, 90, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.; Cook, G.A.; Heimberg, M. Regulation by oestrogen of carnitine palmitoyltransferase in hepatic mitochondria. Biochem. J. 1986, 237, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.E.; Febbraio, M.A. Effect of ovarian hormones on mitochondrial enzyme activity in the fat oxidation pathway of skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E803–E808. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.; Li, X.; Bordan, Z.; Haigh, S.; Bentley, A.; Chen, F.; Barman, S. Reactive oxygen and nitrogen species in the development of pulmonary hypertension. Antioxidants 2017, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Barman, S.A.; Chen, F.; Su, Y.; Dimitropoulou, C.; Wang, Y.; Catravas, J.D.; Han, W.; Orfi, L.; Szantai-Kis, C.; Keri, G.; et al. NADPH oxidase 4 is expressed in pulmonary artery adventitia and contributes to hypertensive vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1704–1715. [Google Scholar] [CrossRef]
- Yu, W.; Ji, W.; Mi, L.; Lin, C. Mechanisms of Nacetylcysteine in reducing monocrotalineinduced pulmonary hypertension in rats: Inhibiting the expression of Nox1 in pulmonary vascular smooth muscle cells. Mol. Med. Rep. 2017, 16, 6148–6155. [Google Scholar] [CrossRef]
- Csiszar, A.; Labinskyy, N.; Olson, S.; Pinto, J.T.; Gupte, S.; Wu, J.M.; Hu, F.; Ballabh, P.; Podlutsky, A.; Losonczy, G.; et al. Resveratrol prevents monocrotaline-induced pulmonary hypertension in rats. Hypertension 2009, 54, 668–675. [Google Scholar] [CrossRef]
- Veit, F.; Pak, O.; Egemnazarov, B.; Roth, M.; Kosanovic, D.; Seimetz, M.; Sommer, N.; Ghofrani, H.A.; Seeger, W.; Grimminger, F.; et al. Function of NADPH oxidase 1 in pulmonary arterial smooth muscle cells after monocrotaline-induced pulmonary vascular remodeling. Antioxid. Redox Signal. 2013, 19, 2213–2231. [Google Scholar] [CrossRef]
- Ghouleh, I.A.; Sahoo, S.; Meijles, D.N.; Amaral, J.H.; de Jesus, D.S.; Sembrat, J.; Rojas, M.; Goncharov, D.A.; Goncharova, E.A.; Pagano, P.J. Endothelial Nox1 oxidase assembly in human pulmonary arterial hypertension; driver of Gremlin1-mediated proliferation. Clin. Sci. (Lond.) 2017, 131, 2019–2035. [Google Scholar] [CrossRef]
- Peng, J.J.; Liu, B.; Xu, J.Y.; Peng, J.; Luo, X.J. NADPH oxidase: Its potential role in promotion of pulmonary arterial hypertension. Naunyn Schmiedebergs Arch. Pharmacol. 2017, 390, 331–338. [Google Scholar] [CrossRef]
- Liu, J.Q.; Zelko, I.N.; Erbynn, E.M.; Sham, J.S.; Folz, R.J. Hypoxic pulmonary hypertension: Role of superoxide and NADPH oxidase (gp91phox). Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L2–L10. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, R.E.; Graves, A.S.; Kleinhenz, D.J.; Rupnow, H.L.; Reed, A.L.; Fan, T.H.; Mitchell, P.O.; Sutliff, R.L.; Hart, C.M. The role of NADPH oxidase in chronic intermittent hypoxia-induced pulmonary hypertension in mice. Am. J. Respir. Cell Mol. Biol. 2009, 40, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Rathore, R.; Zheng, Y.M.; Niu, C.F.; Liu, Q.H.; Korde, A.; Ho, Y.S.; Wang, Y.X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 1223–1231. [Google Scholar] [CrossRef]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX Activation by Subunit Interaction and Underlying Mechanisms in Disease. Front. Cell. Neurosci. 2016, 10, 301. [Google Scholar] [CrossRef] [PubMed]
- Sturrock, A.; Cahill, B.; Norman, K.; Huecksteadt, T.P.; Hill, K.; Sanders, K.; Karwande, S.V.; Stringham, J.C.; Bull, D.A.; Gleich, M.; et al. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L661–L673. [Google Scholar] [CrossRef]
- Mittal, M.; Roth, M.; Konig, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef]
- Goncharov, D.A.; Kudryashova, T.V.; Ziai, H.; Ihida-Stansbury, K.; DeLisser, H.; Krymskaya, V.P.; Tuder, R.M.; Kawut, S.M.; Goncharova, E.A. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 2014, 129, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Veith, C.; Kraut, S.; Wilhelm, J.; Sommer, N.; Quanz, K.; Seeger, W.; Brandes, R.P.; Weissmann, N.; Schroder, K. NADPH oxidase 4 is not involved in hypoxia-induced pulmonary hypertension. Pulm. Circ. 2016, 6, 397–400. [Google Scholar] [CrossRef]
- Iwata, K.; Ikami, K.; Matsuno, K.; Yamashita, T.; Shiba, D.; Ibi, M.; Matsumoto, M.; Katsuyama, M.; Cui, W.; Zhang, J.; et al. Deficiency of NOX1/nicotinamide adenine dinucleotide phosphate, reduced form oxidase leads to pulmonary vascular remodeling. Arterioscler. Thrombosis Vasc. Biol. 2014, 34, 110–119. [Google Scholar] [CrossRef]
- Juan, S.H.; Chen, J.J.; Chen, C.H.; Lin, H.; Cheng, C.F.; Liu, J.C.; Hsieh, M.H.; Chen, Y.L.; Chao, H.H.; Chen, T.H.; et al. 17beta-estradiol inhibits cyclic strain-induced endothelin-1 gene expression within vascular endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1254–H1261. [Google Scholar] [CrossRef]
- Wassmann, S.; Laufs, U.; Stamenkovic, D.; Linz, W.; Stasch, J.P.; Ahlbory, K.; Rosen, R.; Bohm, M.; Nickenig, G. Raloxifene improves endothelial dysfunction in hypertension by reduced oxidative stress and enhanced nitric oxide production. Circulation 2002, 105, 2083–2091. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.A.; Drummond, G.R.; Mast, A.E.; Schmidt, H.H.; Sobey, C.G. Effect of gender on NADPH-oxidase activity, expression, and function in the cerebral circulation: Role of estrogen. Stroke 2007, 38, 2142–2149. [Google Scholar] [CrossRef] [PubMed]
- Wassmann, K.; Wassmann, S.; Nickenig, G. Progesterone antagonizes the vasoprotective effect of estrogen on antioxidant enzyme expression and function. Circ. Res. 2005, 97, 1046–1054. [Google Scholar] [CrossRef]
- Chignalia, A.Z.; Oliveira, M.A.; Debbas, V.; Dull, R.O.; Laurindo, F.R.; Touyz, R.M.; Carvalho, M.H.; Fortes, Z.B.; Tostes, R.C. Testosterone induces leucocyte migration by NADPH oxidase-driven ROS- and COX2-dependent mechanisms. Clin. Sci. (Lond.) 2015, 129, 39–48. [Google Scholar] [CrossRef]
- Dantas, A.P.; Franco Mdo, C.; Silva-Antonialli, M.M.; Tostes, R.C.; Fortes, Z.B.; Nigro, D.; Carvalho, M.H. Gender differences in superoxide generation in microvessels of hypertensive rats: Role of NAD(P)H-oxidase. Cardiovasc. Res. 2004, 61, 22–29. [Google Scholar] [CrossRef]
- Wegiel, B.; Nemeth, Z.; Correa-Costa, M.; Bulmer, A.C.; Otterbein, L.E. Heme oxygenase-1: A metabolic nike. Antioxid. Redox Signal. 2014, 20, 1709–1722. [Google Scholar] [CrossRef]
- Busch, A.W.; Montgomery, B.L. Interdependence of tetrapyrrole metabolism, the generation of oxidative stress and the mitigative oxidative stress response. Redox Biol. 2015, 4, 260–271. [Google Scholar] [CrossRef]
- Cooper, C.E. Nitric oxide and iron proteins. Biochim. Biophys. Acta Bioenerg. 1999, 1411, 290–309. [Google Scholar] [CrossRef]
- Helms, C.; Kim-Shapiro, D.B. Hemoglobin-mediated nitric oxide signaling. Free Radic. Biol. Med. 2013, 61, 464–472. [Google Scholar] [CrossRef]
- Quintela-Carvalho, G.; Luz, N.F.; Celes, F.S.; Zanette, D.L.; Andrade, D.; Menezes, D.; Tavares, N.M.; Brodskyn, C.I.; Prates, D.B.; Gonçalves, M.S. Heme Drives Oxidative Stress-Associated Cell Death in Human Neutrophils Infected with Leishmania infantum. Front. Immunol. 2017, 8, 1620. [Google Scholar] [CrossRef]
- Ríos-González, B.B.; Román-Morales, E.M.; Pietri, R.; López-Garriga, J. Hydrogen sulfide activation in hemeproteins: The sulfheme scenario. J. Inorg. Biochem. 2014, 133, 78–86. [Google Scholar] [CrossRef]
- Mense, S.M.; Zhang, L. Heme: A versatile signaling molecule controlling the activities of diverse regulators ranging from transcription factors to MAP kinases. Cell Res. 2006, 16, 681. [Google Scholar] [CrossRef]
- Cochran, A.G.; Schultz, P.G. Peroxidase activity of an antibody-heme complex. J. Am. Chem. Soc. 1990, 112, 9414–9415. [Google Scholar] [CrossRef]
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2888. [Google Scholar] [CrossRef]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M. Oxygen free radicals and iron in relation to biology and medicine: some problems and concepts. Arch. Biochem. Biophys. 1986, 246, 501–514. [Google Scholar] [CrossRef]
- Valko, M.; Izakovic, M.; Mazur, M.; Rhodes, C.J.; Telser, J. Role of oxygen radicals in DNA damage and cancer incidence. Mol. Cell. Biochem. 2004, 266, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Huang, J.; Wu, J.M.; Fallon, J.T.; Gewitz, M.H. Hematological disorders and pulmonary hypertension. World J. Cardiol. 2016, 8, 703. [Google Scholar] [CrossRef]
- Rafikova, O.; Williams, E.R.; McBride, M.L.; Zemskova, M.; Srivastava, A.; Nair, V.; Desai, A.A.; Langlais, P.R.; Zemskov, E.; Simon, M. Hemolysis-induced Lung Vascular Leakage Contributes to the Development of Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2018, 59, 334–345. [Google Scholar] [CrossRef]
- Moraes, J.; Barcellos-de-Souza, P.; Rodrigues, G.; Nascimento-Silva, V.; Silva, S.; Assreuy, J.; Arruda, M.; Barja-Fidalgo, C. Heme modulates smooth muscle cell proliferation and migration via NADPH oxidase: A counter-regulatory role for heme oxygenase system. Atherosclerosis 2012, 224, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Balla, J.; Vercellotti, G.M.; Jeney, V.; Yachie, A.; Varga, Z.; Eaton, J.W.; Balla, G. Heme, heme oxygenase and ferritin in vascular endothelial cell injury. Mol. Nutr. Food Res. 2005, 49, 1030–1043. [Google Scholar] [CrossRef]
- Gáll, T.; Pethő, D.; Nagy, A.; Hendrik, Z.; Méhes, G.; Potor, L.; Gram, M.; Åkerström, B.; Smith, A.; Nagy, P. Heme induces endoplasmic reticulum stress (HIER stress) in human aortic smooth muscle cells. Front. Physiol. 2018, 9, 1595. [Google Scholar] [CrossRef]
- Lenna, S.; Farina, A.G.; Martyanov, V.; Christmann, R.B.; Wood, T.A.; Farber, H.W.; Scorza, R.; Whitfield, M.L.; Lafyatis, R.; Trojanowska, M. Increased expression of endoplasmic reticulum stress and unfolded protein response genes in peripheral blood mononuclear cells from patients with limited cutaneous systemic sclerosis and pulmonary arterial hypertension. Arthritis Rheum. 2013, 65, 1357–1366. [Google Scholar] [CrossRef]
- Koyama, M.; Furuhashi, M.; Ishimura, S.; Mita, T.; Fuseya, T.; Okazaki, Y.; Yoshida, H.; Tsuchihashi, K.; Miura, T. Reduction of endoplasmic reticulum stress by 4-phenylbutyric acid prevents the development of hypoxia-induced pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1314–H1323. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Stuehr, D.J. Regulation of sGC via hsp90, cellular heme, sGC agonists, and NO: New pathways and clinical perspectives. Antioxid. Redox Signal. 2017, 26, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, A.; Miller, F.; Worwood, M.; Beamish, M.; Wardrop, C. Ferritin in the serum of normal subjects and patients with iron deficiency and iron overload. Br. Med. J. 1972, 4, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Domellof, M.; Dewey, K.G.; Lonnerdal, B.; Cohen, R.J.; Hernell, O. The diagnostic criteria for iron deficiency in infants should be reevaluated. J. Nutr. 2002, 132, 3680–3686. [Google Scholar] [CrossRef]
- Zacharski, L.R.; Ornstein, D.L.; Woloshin, S.; Schwartz, L.M. Association of age, sex, and race with body iron stores in adults: analysis of NHANES III data. Am. Heart J. 2000, 140, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Milman, N.; Kirchhoff, M.; Jørgensen, T. Iron status markers, serum ferritin and hemoglobin in 1359 Danish women in relation to menstruation, hormonal contraception, parity, and postmenopausal hormone treatment. Ann. Hematol. 1992, 65, 96–102. [Google Scholar] [CrossRef]
- Milman, N.; Clausen, J.; Byg, K.-E. Iron status in 268 Danish women aged 18–30 years: Influence of menstruation, contraceptive method, and iron supplementation. Ann. Hematol. 1998, 77, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.B.; Hurrell, R.F. Nutritional iron deficiency. Lancet 2007, 370, 511–520. [Google Scholar] [CrossRef]
- Ruiter, G.; Lankhorst, S.; Boonstra, A.; Postmus, P.; Zweegman, S.; Westerhof, N.; Van Der Laarse, W.; Vonk-Noordegraaf, A. Iron deficiency is common in idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2011, 37, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Viethen, T.; Gerhardt, F.; Dumitrescu, D.; Knoop-Busch, S.; ten Freyhaus, H.; Rudolph, T.K.; Baldus, S.; Rosenkranz, S. Ferric carboxymaltose improves exercise capacity and quality of life in patients with pulmonary arterial hypertension and iron deficiency: A pilot study. Int. J. Cardiol. 2014, 175, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Hugh Rushton, D.; Barth, J. What is the evidence for gender differences in ferritin and haemoglobin? Crit. Rev. Oncol. Hematol. 2010, 73, 1–9. [Google Scholar] [CrossRef]
- Ruiter, G.; Manders, E.; Happé, C.M.; Schalij, I.; Groepenhoff, H.; Howard, L.S.; Wilkins, M.R.; Bogaard, H.J.; Westerhof, N.; van der Laarse, W.J.; et al. Intravenous Iron Therapy in Patients with Idiopathic Pulmonary Arterial Hypertension and Iron Deficiency. Pulm. Circ. 2015, 5, 466–472. [Google Scholar] [CrossRef]
- Rhodes, C.J.; Wharton, J.; Howard, L.; Gibbs, J.S.R.; Vonk-Noordegraaf, A.; Wilkins, M.R. Iron deficiency in pulmonary arterial hypertension: A potential therapeutic target. Eur. Respir. J. 2011, 38, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Wrighting, D.M.; Andrews, N.C. Interleukin-6 induces hepcidin expression through STAT3. Blood 2006, 108, 3204–3209. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J.; Howard, L.S.; Busbridge, M.; Ashby, D.; Kondili, E.; Gibbs, J.S.R.; Wharton, J.; Wilkins, M.R. Iron deficiency and raised hepcidin in idiopathic pulmonary arterial hypertension: clinical prevalence, outcomes, and mechanistic insights. J. Am. Coll. Cardiol. 2011, 58, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J.; Wharton, J.; Howard, L.S.; Gibbs, J.S.R.; Wilkins, M.R. Red cell distribution width outperforms other potential circulating biomarkers in predicting survival in idiopathic pulmonary arterial hypertension. Heart 2011, 97, 1054–1060. [Google Scholar] [CrossRef]
- Zuckerbraun, B.S.; Stoyanovsky, D.A.; Sengupta, R.; Shapiro, R.A.; Ozanich, B.A.; Rao, J.; Barbato, J.E.; Tzeng, E. Nitric oxide-induced inhibition of smooth muscle cell proliferation involves S-nitrosation and inactivation of RhoA. Am. J. Physiol. Cell Physiol. 2007, 292, C824–C831. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, M.J.; Channon, K.M. Synthesis and recycling of tetrahydrobiopterin in endothelial function and vascular disease. Nitric Oxide 2011, 25, 81–88. [Google Scholar] [CrossRef]
- Zhao, Y.Y.; Zhao, Y.D.; Mirza, M.K.; Huang, J.H.; Potula, H.H.; Vogel, S.M.; Brovkovych, V.; Yuan, J.X.; Wharton, J.; Malik, A.B. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J. Clin. Investig. 2009, 119, 2009–2018. [Google Scholar] [CrossRef]
- Zhao, Y.Y.; Malik, A.B. A novel insight into the mechanism of pulmonary hypertension involving caveolin-1 deficiency and endothelial nitric oxide synthase activation. Trends Cardiovasc. Med. 2009, 19, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Fayard, E.; Tintignac, L.A.; Baudry, A.; Hemmings, B.A. Protein kinase B/Akt at a glance. J. Cell Sci. 2005, 118, 5675–5678. [Google Scholar] [CrossRef]
- Fulton, D.; Gratton, J.P.; McCabe, T.J.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.F.; Papapetropoulos, A.; Sessa, W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [CrossRef]
- Iwakiri, Y.; Tsai, M.H.; McCabe, T.J.; Gratton, J.P.; Fulton, D.; Groszmann, R.J.; Sessa, W.C. Phosphorylation of eNOS initiates excessive NO production in early phases of portal hypertension. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H2084–H2090. [Google Scholar] [CrossRef]
- Garat, C.V.; Crossno, J.T., Jr.; Sullivan, T.M.; Reusch, J.E.; Klemm, D.J. Inhibition of phosphatidylinositol 3-kinase/Akt signaling attenuates hypoxia-induced pulmonary artery remodeling and suppresses CREB depletion in arterial smooth muscle cells. J. Cardiovasc. Pharmacol. 2013, 62, 539. [Google Scholar] [CrossRef]
- Tang, H.; Chen, J.; Drennan, A.R.; Fraidenburg, D.R.; Song, S.; Sysol, J.R.; Smith, K.A.; Machado, R.F.; Makino, A.; Yuan, J.X. Akt/mTOR Signaling Contributes to the Development of Pulmonary Arterial Hypertension. In Proceedings of the American Thoracic Society 2014 International Conference, San Diego, CA, USA, 16–21 May 2014. [Google Scholar]
- Krymskaya, V.P.; Snow, J.; Cesarone, G.; Khavin, I.; Goncharov, D.A.; Lim, P.N.; Veasey, S.C.; Ihida-Stansbury, K.; Jones, P.L.; Goncharova, E.A. mTOR is required for pulmonary arterial vascular smooth muscle cell proliferation under chronic hypoxia. FASEB J. 2011, 25, 1922–1933. [Google Scholar] [CrossRef]
- Huang, A.; Kaley, G. Gender-specific regulation of cardiovascular function: Estrogen as key player. Microcirculation 2004, 11, 9–38. [Google Scholar] [CrossRef]
- Simoncini, T.; Mannella, P.; Fornari, L.; Caruso, A.; Varone, G.; Genazzani, A.R. Genomic and non-genomic effects of estrogens on endothelial cells. Steroids 2004, 69, 537–542. [Google Scholar] [CrossRef]
- Malorni, W.; Campesi, I.; Straface, E.; Vella, S.; Franconi, F. Rodox features of the cell: A gender perspective. Antioxid. Redox Signal. 2007, 9, 1779–1802. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M.G.; Vanetti, C.; Decimo, I.; Di Chio, M.; Martano, G.; Garrone, G.; Bifari, F.; Vicentini, L.M. Sex-specific eNOS activity and function in human endothelial cells. Sci. Rep. 2017, 7, 9612. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Guo, H.; Xu, X.; Lu, Z.; Fassett, J.; Hu, X.; Xu, Y.; Tang, Q.; Hu, D.; Somani, A. Exacerbated pulmonary arterial hypertension and right ventricular hypertrophy in animals with loss of function of extracellular superoxide dismutase. Hypertension 2011, 58, 303–309. [Google Scholar] [CrossRef]
- DeMarco, V.G.; Whaley-Connell, A.T.; Sowers, J.R.; Habibi, J.; Dellsperger, K.C. Contribution of oxidative stress to pulmonary arterial hypertension. World J. Cardiol. 2010, 2, 316. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Marsboom, G.; Kim, G.H.; Zhang, H.J.; Toth, P.T.; Svensson, E.C.; Dyck, J.R.; Gomberg-Maitland, M.; Thébaud, B.; Husain, A.N. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation 2010, 121, 2661–2671. [Google Scholar] [CrossRef]
- Ramiro-Diaz, J.M.; Nitta, C.H.; Maston, L.D.; Codianni, S.; Giermakowska, W.; Resta, T.C.; Bosc, L.V.G. NFAT is required for spontaneous pulmonary hypertension in superoxide dismutase 1 knockout mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L613–L625. [Google Scholar] [CrossRef]
- Villegas, L.R.; Kluck, D.; Field, C.; Oberley-Deegan, R.E.; Woods, C.; Yeager, M.E.; El Kasmi, K.C.; Savani, R.C.; Bowler, R.P.; Nozik-Grayck, E. Superoxide dismutase mimetic, MnTE-2-PyP, attenuates chronic hypoxia-induced pulmonary hypertension, pulmonary vascular remodeling, and activation of the NALP3 inflammasome. Antioxid. Redox Signal. 2013, 18, 1753–1764. [Google Scholar] [CrossRef]
- Xu, M.; Xu, M.; Han, L.; Yuan, C.; Mei, Y.; Zhang, H.; Chen, S.; Sun, K.; Zhu, B. Role for functional SOD2 polymorphism in pulmonary arterial hypertension in a chinese population. Int. J. Environ. Res. Public Health 2017, 14, 266. [Google Scholar] [CrossRef] [PubMed]
- Lakshminrusimha, S.; Russell, J.A.; Wedgwood, S.; Gugino, S.F.; Kazzaz, J.A.; Davis, J.M.; Steinhorn, R.H. Superoxide dismutase improves oxygenation and reduces oxidation in neonatal pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2006, 174, 1370–1377. [Google Scholar] [CrossRef]
- Bravard, A.; Sabatier, L.; Hoffschir, F.; Ricoul, M.; Luccioni, C.; Dutrillaux, B. SOD2: A new type of tumor-suppressor gene? Int. J. Cancer 1992, 51, 476–480. [Google Scholar] [CrossRef]
- Li, N.; Oberley, T.D.; Oberley, L.W.; Zhong, W. Overexpression of manganese superoxide dismutase in DU145 human prostate carcinoma cells has multiple effects on cell phenotype. Prostate 1998, 35, 221–233. [Google Scholar] [CrossRef]
- Ryan, J.; Dasgupta, A.; Huston, J.; Chen, K.-H.; Archer, S.L. Mitochondrial dynamics in pulmonary arterial hypertension. J. Mol. Med. 2015, 93, 229–242. [Google Scholar] [CrossRef]
- Ball, M.K.; Waypa, G.B.; Mungai, P.T.; Nielsen, J.M.; Czech, L.; Dudley, V.J.; Beussink, L.; Dettman, R.W.; Berkelhamer, S.K.; Steinhorn, R.H.; et al. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1alpha. Am. J. Respir. Crit. Care Med. 2014, 189, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.A.; Yuan, J.X. Hypoxia-inducible factor-1alpha in pulmonary arterial smooth muscle cells and hypoxia-induced pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Farha, S.; Asosingh, K.; Xu, W.; Sharp, J.; George, D.; Comhair, S.; Park, M.; Tang, W.H.; Loyd, J.E.; Theil, K.; et al. Hypoxia-inducible factors in human pulmonary arterial hypertension: A link to the intrinsic myeloid abnormalities. Blood 2011, 117, 3485–3493. [Google Scholar] [CrossRef] [PubMed]
- Nozik-Grayck, E.; Suliman, H.B.; Majka, S.; Albietz, J.; Van Rheen, Z.; Roush, K.; Stenmark, K.R. Lung EC-SOD overexpression attenuates hypoxic induction of Egr-1 and chronic hypoxic pulmonary vascular remodeling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L422–L430. [Google Scholar] [CrossRef] [PubMed]
- Adesina, S.E.; Kang, B.-Y.; Bijli, K.M.; Ma, J.; Cheng, J.; Murphy, T.C.; Hart, C.M.; Sutliff, R.L. Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free Radic. Biol. Med. 2015, 87, 36–47. [Google Scholar] [CrossRef]
- Rafikova, O.; Salah, E.M.; Tofovic, S.P. Renal and metabolic effects of tempol in obese ZSF1 rats--distinct role for superoxide and hydrogen peroxide in diabetic renal injury. Metab. Clin. Exp. 2008, 57, 1434–1444. [Google Scholar] [CrossRef]
- Wedgwood, S.; Lakshminrusimha, S.; Fukai, T.; Russell, J.A.; Schumacker, P.T.; Steinhorn, R.H. Hydrogen peroxide regulates extracellular superoxide dismutase activity and expression in neonatal pulmonary hypertension. Antioxid. Redox Signal. 2011, 15, 1497–1506. [Google Scholar] [CrossRef]
- Wedgwood, S.; Steinhorn, R.H.; Bunderson, M.; Wilham, J.; Lakshminrusimha, S.; Brennan, L.A.; Black, S.M. Increased hydrogen peroxide downregulates soluble guanylate cyclase in the lungs of lambs with persistent pulmonary hypertension of the newborn. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L660–L666. [Google Scholar] [CrossRef]
- Strehlow, K.; Rotter, S.; Wassmann, S.; Adam, O.; Grohé, C.; Laufs, K.; Böhm, M.; Nickenig, G. Modulation of antioxidant enzyme expression and function by estrogen. Circ. Res. 2003, 93, 170–177. [Google Scholar] [CrossRef]
- Glorieux, C.; Zamocky, M.; Sandoval, J.M.; Verrax, J.; Calderon, P.B. Regulation of catalase expression in healthy and cancerous cells. Free Radic. Biol. Med. 2015, 87, 84–97. [Google Scholar] [CrossRef]
- Bellanti, F.; Matteo, M.; Rollo, T.; De Rosario, F.; Greco, P.; Vendemiale, G.; Serviddio, G. Sex hormones modulate circulating antioxidant enzymes: Impact of estrogen therapy. Redox Biol. 2013, 1, 340–346. [Google Scholar] [CrossRef]
- Guerra, R.C.; Zuniga-Munoz, A.; Guarner Lans, V.; Diaz-Diaz, E.; Tena Betancourt, C.A.; Perez-Torres, I. Modulation of the activities of catalase, cu-zn, mn superoxide dismutase, and glutathione peroxidase in adipocyte from ovariectomised female rats with metabolic syndrome. Int. J. Endocrinol. 2014, 2014, 175080. [Google Scholar] [CrossRef]
- Azevedo, R.B.; Lacava, Z.G.; Miyasaka, C.K.; Chaves, S.B.; Curi, R. Regulation of antioxidant enzyme activities in male and female rat macrophages by sex steroids. Braz. J. Med. Biol. Res. 2001, 34, 683–687. [Google Scholar] [CrossRef]
- Sheng-Huang, C.; Chieh-Hsin, C.; Mu-Chun, Y.; Wen-Tung, H.; Chia-Ying, H.; Ya-Ting, H.; Wan-Ling, S.U.; Jiuan-Jen, S.; Chih-Yang, H.; Jer-Yuh, L. Effects of estrogen on glutathione and catalase levels in human erythrocyte during menstrual cycle. Biomed. Rep. 2015, 3, 266–268. [Google Scholar] [CrossRef]
- Petit, E.; Courtin, A.; Kloosterboer, H.J.; Rostene, W.; Forgez, P.; Gompel, A. Progestins induce catalase activities in breast cancer cells through PRB isoform: correlation with cell growth inhibition. J. Steroid Biochem. Mol. Biol. 2009, 115, 153–160. [Google Scholar] [CrossRef]
- Kim, J.; Kil, I.S.; Seok, Y.M.; Yang, E.S.; Kim, D.K.; Lim, D.G.; Park, J.W.; Bonventre, J.V.; Park, K.M. Orchiectomy attenuates post-ischemic oxidative stress and ischemia/reperfusion injury in mice. A role for manganese superoxide dismutase. J. Biol. Chem. 2006, 281, 20349–20356. [Google Scholar] [CrossRef] [PubMed]
- Choobineh, H.; Gilani, M.A.S.; Pasalar, P.; Jahanzad, I.; Ghorbani, R.; Hassanzadeh, G. The effects of testosterone on oxidative stress markers in mice with spinal cord injuries. Int. J. Fertil. Steril. 2016, 10, 87. [Google Scholar]
- Zhang, L.; Wu, S.; Ruan, Y.; Hong, L.; Xing, X.; Lai, W. Testosterone suppresses oxidative stress via androgen receptor-independent pathway in murine cardiomyocytes. Mol. Med. Rep. 2011, 4, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Sobocanec, S.; Balog, T.; Sverko, V.; Marotti, T. Sex-dependent antioxidant enzyme activities and lipid peroxidation in ageing mouse brain. Free Rad. Res. 2003, 37, 743–748. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Simon, R.G.J. The changing landscape of pulmonary arterial hypertension and implications for patient care. Eur. Respir. Rev. 2014, 23, 450–457. [Google Scholar] [CrossRef]
- Köhrle, J.; Brigelius-Flohé, R.; Böck, A.; Gärtner, R.; Meyer, O.; Flohé, L. Selenium in biology: Facts and medical perspectives. Biol. Chem. 2000, 381, 849–864. [Google Scholar] [CrossRef]
- Kikuchi, N.; Satoh, K.; Kurosawa, R.; Yaoita, N.; Elias-Al-Mamun, M.; Siddique, M.A.H.; Omura, J.; Satoh, T.; Nogi, M.; Sunamura, S. Selenoprotein P promotes the development of pulmonary arterial hypertension: possible novel therapeutic target. Circulation 2018, 138, 600–623. [Google Scholar] [CrossRef]
- Voorde, J.V.; Ackermann, T.; Pfetzer, N.; Sumpton, D.; Mackay, G.; Kalna, G.; Nixon, C.; Blyth, K.; Gottlieb, E.; Tardito, S. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci. Adv. 2019, 5, eaau7314. [Google Scholar] [CrossRef] [PubMed]
- Masri, F.A.; Comhair, S.A.; Dostanic-Larson, I.; Kaneko, F.T.; Dweik, R.A.; Arroliga, A.C.; Erzurum, S.C. Deficiency of lung antioxidants in idiopathic pulmonary arterial hypertension. Clin. Transl. Sci. 2008, 1, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, N.M.; Stewart, R.; Robinson, M.F. The metabolism of [75Se] selenomethionine in four women. Br. J. Nutr. 1976, 35, 373–382. [Google Scholar] [CrossRef]
- Janghorbani, M.; Christensen, M.; Nahapetian, A.; Young, V. Selenium metabolism in healthy adults: Quantitative aspects using the stable isotope 74SeO32−. Am. J. Clin. Nutr. 1982, 35, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Seale, L.A.; Ogawa-Wong, A.N.; Berry, M.J. Sexual dimorphism in selenium metabolism and selenoproteins. Free Radic. Biol. Med. 2018, 127, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Letsiou, S.; Nomikos, T.; Panagiotakos, D.; Pergantis, S.; Fragopoulou, E.; Pitsavos, C.; Stefanadis, C.; Antonopoulou, S. Gender-specific distribution of selenium to serum selenoproteins: Associations with total selenium levels, age, smoking, body mass index, and physical activity. Biofactors 2014, 40, 524–535. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, L.; Riese, C.; Renko, K.; Schweizer, U. Effect of age on sexually dimorphic selenoprotein expression in mice. Biol. Chem. 2007, 388, 1035–1041. [Google Scholar] [CrossRef]
- Riese, C.; Michaelis, M.; Mentrup, B.; Gotz, F.; Kohrle, J.; Schweizer, U.; Schomburg, L. Selenium-dependent pre-and posttranscriptional mechanisms are responsible for sexual dimorphic expression of selenoproteins in murine tissues. Endocrinology 2006, 147, 5883–5892. [Google Scholar] [CrossRef]
- Zhou, X.; Smith, A.M.; Failla, M.L.; Hill, K.E.; Yu, Z. Estrogen status alters tissue distribution and metabolism of selenium in female rats. J. Nutr. Biochem. 2012, 23, 532–538. [Google Scholar] [CrossRef]
- Arias-Loza, P.A.; Muehlfelder, M.; Pelzer, T. Estrogen and estrogen receptors in cardiovascular oxidative stress. Pflugers Arch. 2013, 465, 739–746. [Google Scholar] [CrossRef]
- Liang, O.D.; Mitsialis, S.A.; Chang, M.S.; Vergadi, E.; Lee, C.; Aslam, M.; Fernandez-Gonzalez, A.; Liu, X.; Baveja, R.; Kourembanas, S. Mesenchymal stromal cells expressing heme oxygenase-1 reverse pulmonary hypertension. Stem Cells 2011, 29, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Belhaj, A.; Dewachter, L.; Kerbaul, F.; Brimioulle, S.; Dewachter, C.; Naeije, R.; Rondelet, B. Heme oxygenase-1 and inflammation in experimental right ventricular failure on prolonged overcirculation-induced pulmonary hypertension. PLoS ONE 2013, 8, e69470. [Google Scholar] [CrossRef]
- Li, M.; Li, Z.; Sun, X.; Yang, L.; Fang, P.; Liu, Y.; Li, W.; Xu, J.; Lu, J.; Xie, M.; et al. Heme oxygenase-1/p21WAF1 mediates peroxisome proliferator-activated receptor-gamma signaling inhibition of proliferation of rat pulmonary artery smooth muscle cells. FEBS J. 2010, 277, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, G.; Han, D.; Zhang, Y.; Xu, J.; Lu, J.; Li, S.; Xie, X.; Liu, L.; Dong, L.; et al. Activation of PPAR-gamma ameliorates pulmonary arterial hypertension via inducing heme oxygenase-1 and p21(WAF1): An in vivo study in rats. Life Sci. 2014, 98, 39–43. [Google Scholar] [CrossRef]
- Macak-Safranko, Z.; Sobocanec, S.; Saric, A.; Balog, T.; Sverko, V.; Kusic, B.; Marotti, T. Cytochrome P450 gender-related differences in response to hyperoxia in young CBA mice. Exp. Toxicol. Pathol. 2011, 63, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Posa, A.; Kupai, K.; Menesi, R.; Szalai, Z.; Szabo, R.; Pinter, Z.; Palfi, G.; Gyongyosi, M.; Berko, A.; Pavo, I.; et al. Sexual dimorphism of cardiovascular ischemia susceptibility is mediated by heme oxygenase. Oxid. Med. Cell. Longev. 2013, 2013, 521563. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Hamid, R. Y Not? Sex Chromosomes May Modify Sexual Dimorphism in Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2018, 197, 858–859. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rafikov, R.; James, J.; McClain, N.; Tofovic, S.P.; Rafikova, O. Role of Gender in Regulation of Redox Homeostasis in Pulmonary Arterial Hypertension. Antioxidants 2019, 8, 135. https://doi.org/10.3390/antiox8050135
Rafikov R, James J, McClain N, Tofovic SP, Rafikova O. Role of Gender in Regulation of Redox Homeostasis in Pulmonary Arterial Hypertension. Antioxidants. 2019; 8(5):135. https://doi.org/10.3390/antiox8050135
Chicago/Turabian StyleRafikov, Ruslan, Joel James, Nolan McClain, Stevan P. Tofovic, and Olga Rafikova. 2019. "Role of Gender in Regulation of Redox Homeostasis in Pulmonary Arterial Hypertension" Antioxidants 8, no. 5: 135. https://doi.org/10.3390/antiox8050135
APA StyleRafikov, R., James, J., McClain, N., Tofovic, S. P., & Rafikova, O. (2019). Role of Gender in Regulation of Redox Homeostasis in Pulmonary Arterial Hypertension. Antioxidants, 8(5), 135. https://doi.org/10.3390/antiox8050135