Protective Effect of Glutathione against Oxidative Stress-induced Cytotoxicity in RAW 264.7 Macrophages through Activating the Nuclear Factor Erythroid 2-Related Factor-2/Heme Oxygenase-1 Pathway


 ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Glutathione Treatment
2.2. Cell Viability Assay
2.3. Apoptosis Assay Using a Fluorescence Microscope
2.4. Apoptosis Analysis using a Flow Cytometer
2.5. Colony Formation Assay
2.6. HO-1 Activity Assay
2.7. Measurement of ROS Level
2.8. Western Blot Analysis
2.9. Determination of 8-hydroxy-2’-deoxyguanosine (8-OHdG)
2.10. Comet Assay for DNA Damage
2.11. Measurement of MMP (Δψm)
2.12. Detection of ATP Levels
2.13. Colorimetric Assay of Caspase-3 Activity
2.14. Statistical Analysis
3. Results
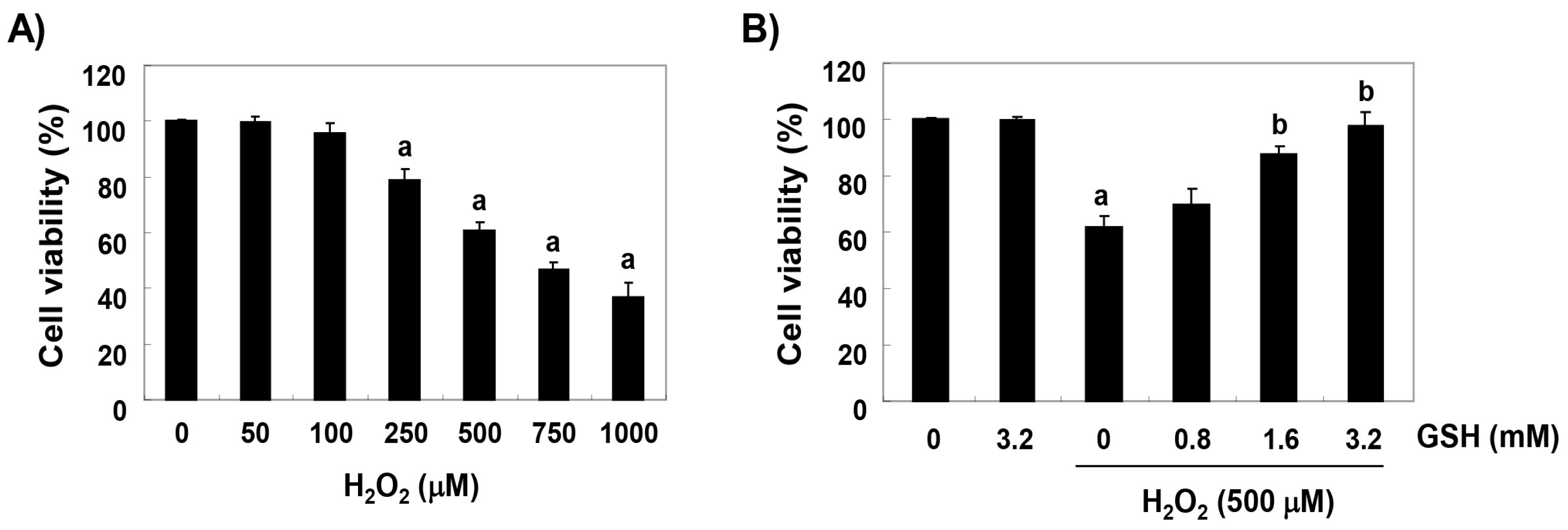
3.1. Glutathione Inhibits H2O2-Induced Cytotoxicity in RAW 264.7 Cells
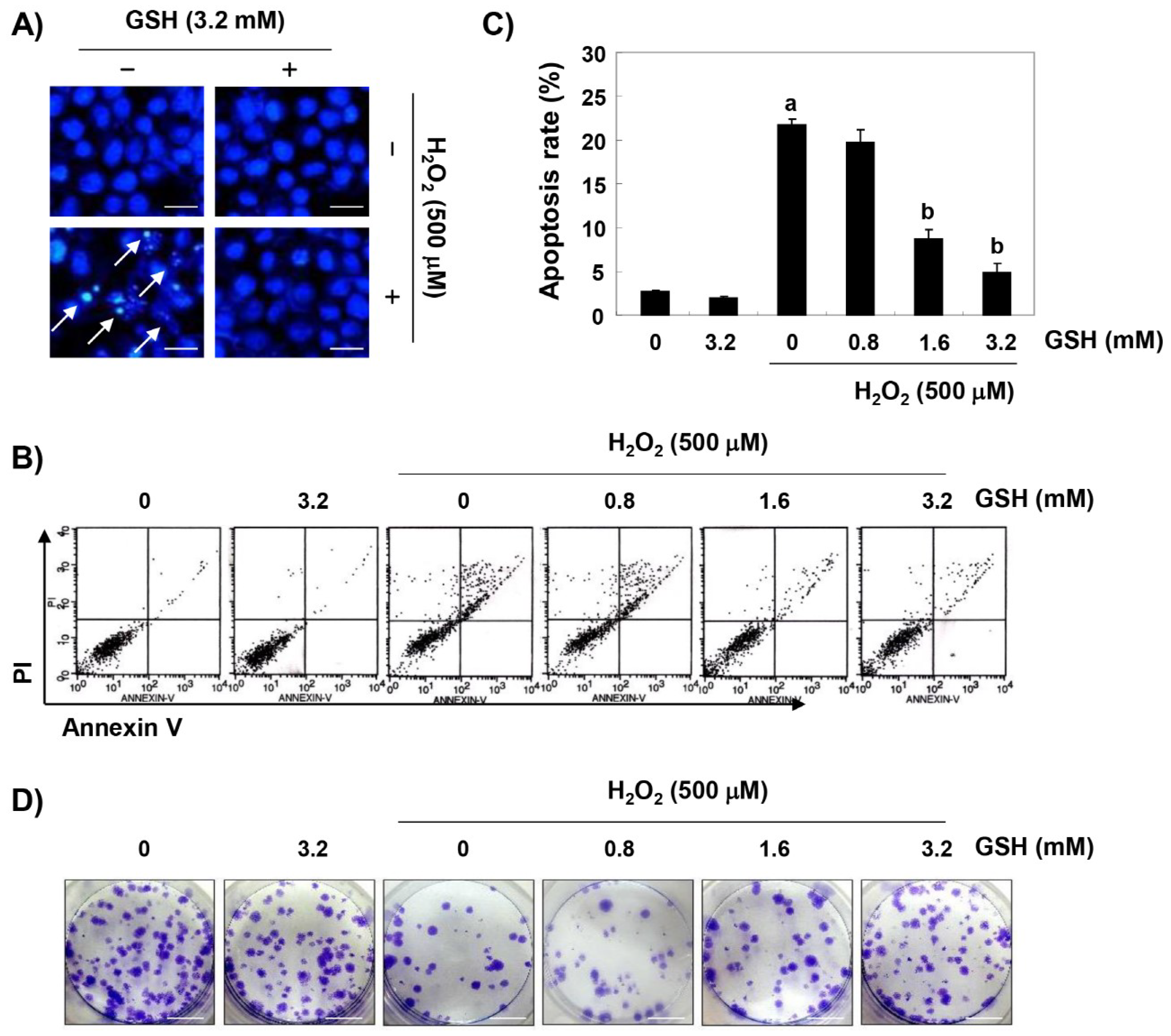
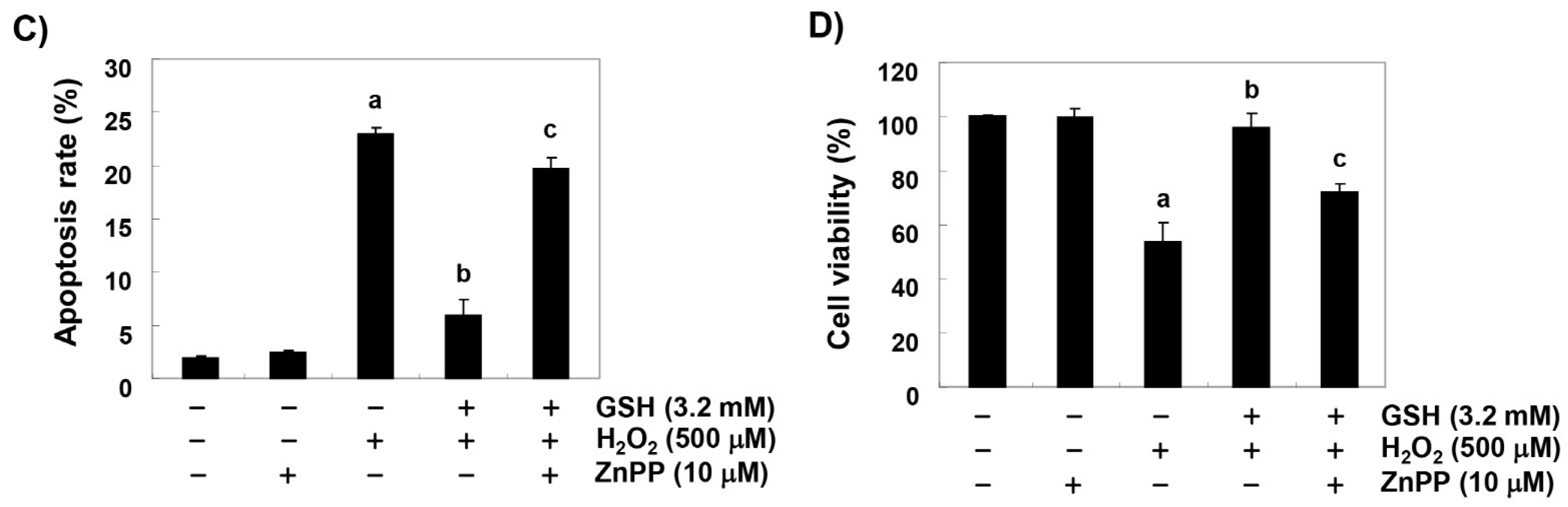
3.2. Glutathione Suppresses H2O2-Induced Apoptosis in RAW 264.7 Cells
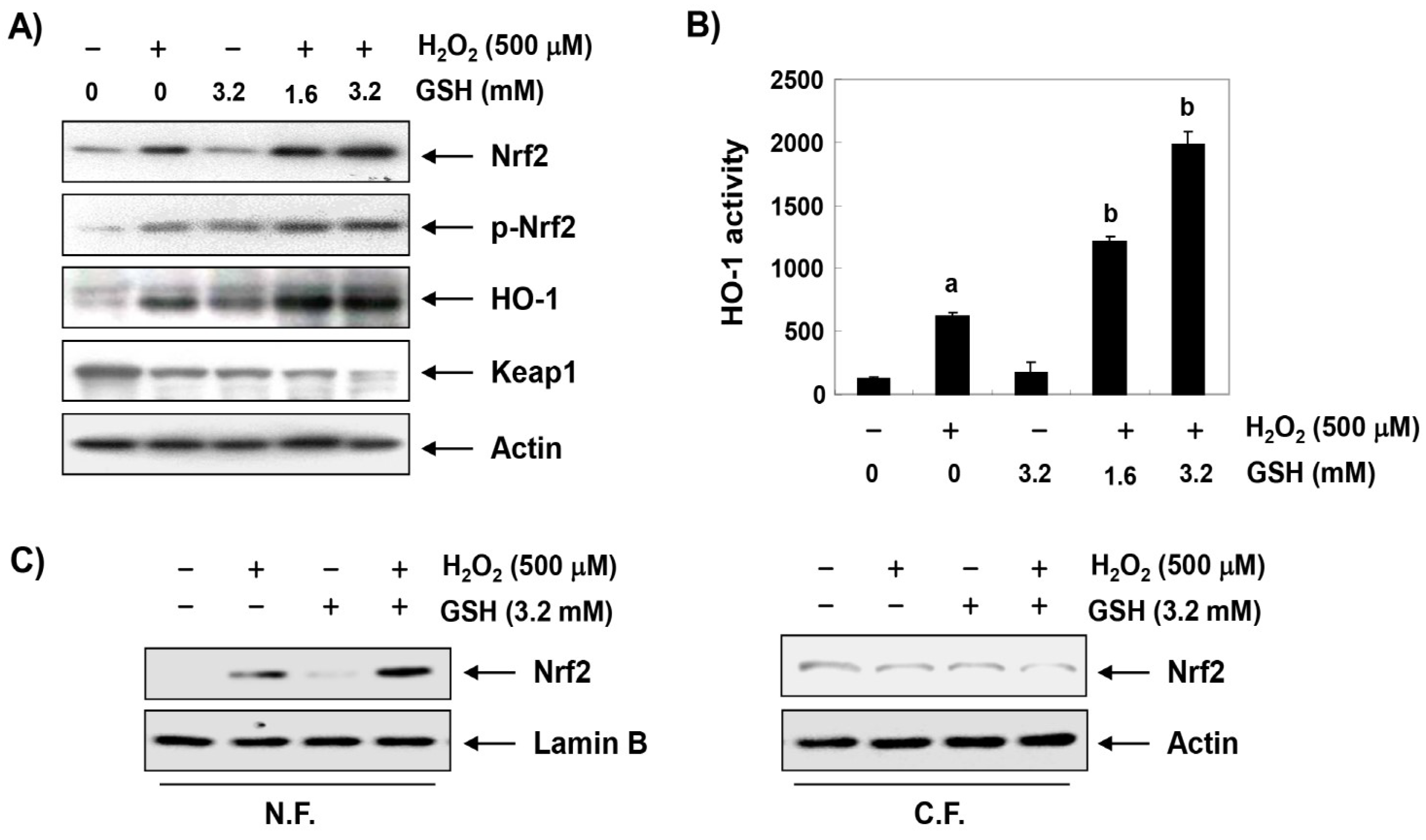
3.3. Glutathione Activates the Nrf2/HO-1 Signaling Pathway in H2O2-Treated RAW 264.7 Cells
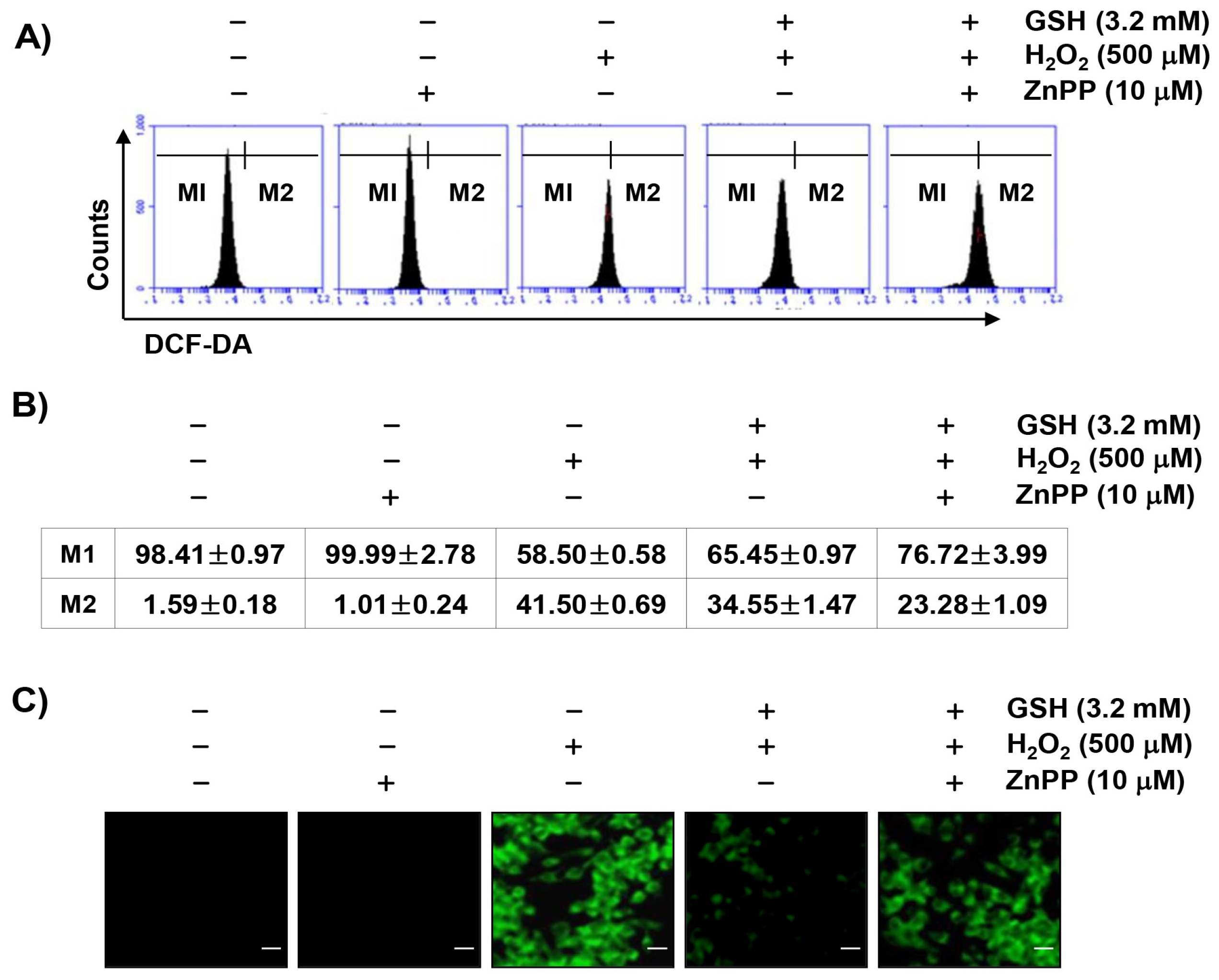
3.4. The Inhibition of H2O2-Induced ROS Generation by Glutathione is Attenuated by ZnPP in RAW 264.7 Cells
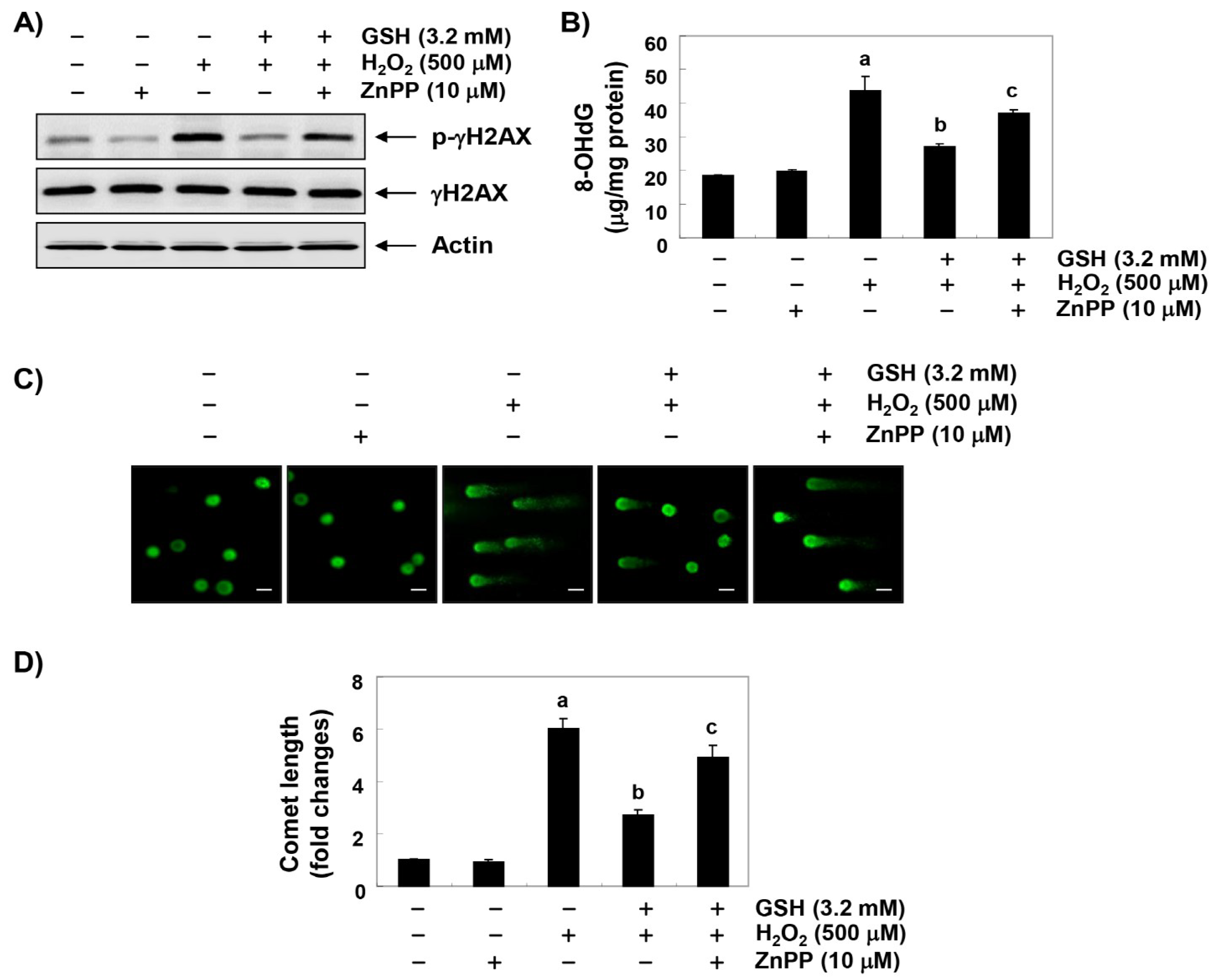
3.5. The Blockade of H2O2-Induced DNA Damage by Glutathione is Reduced by ZnPP in RAW 264.7 Cells
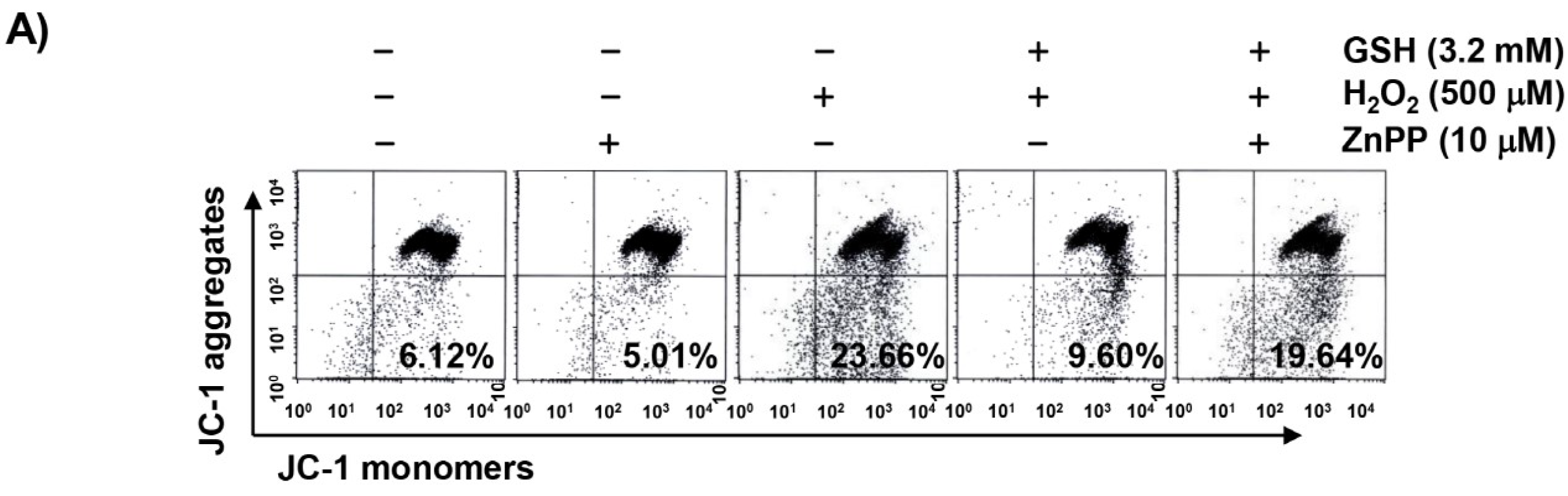
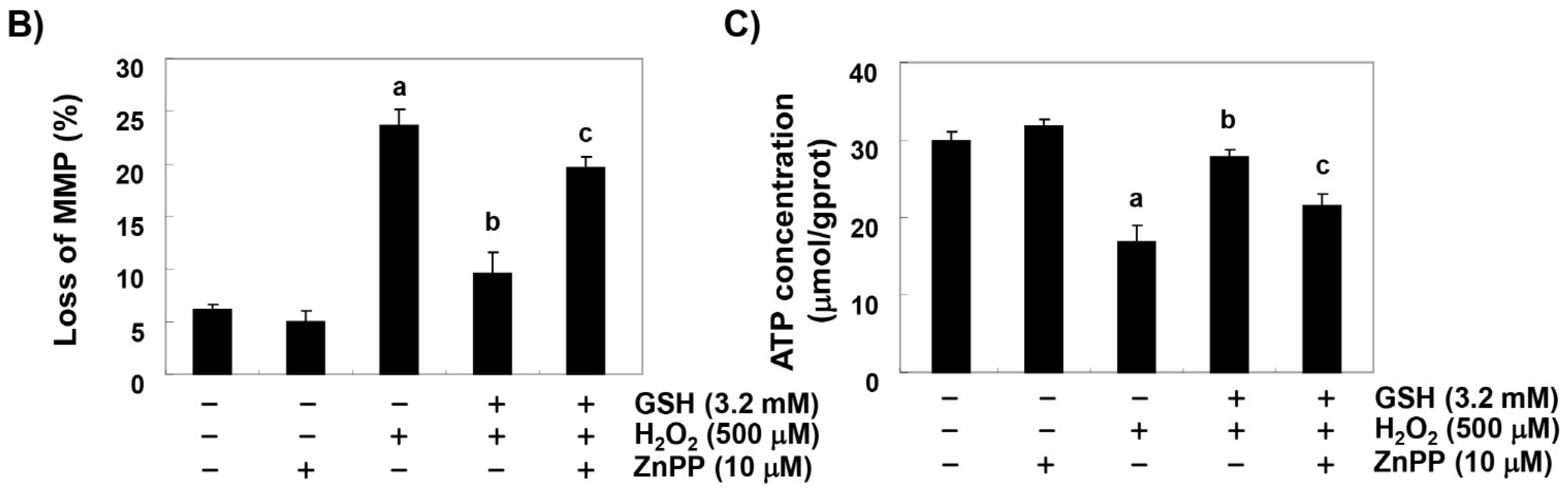
3.6. The Reduction of H2O2-Induced Mitochondrial Dysfunction by Glutathione is Diminished by ZnPP in RAW 264.7 Cells
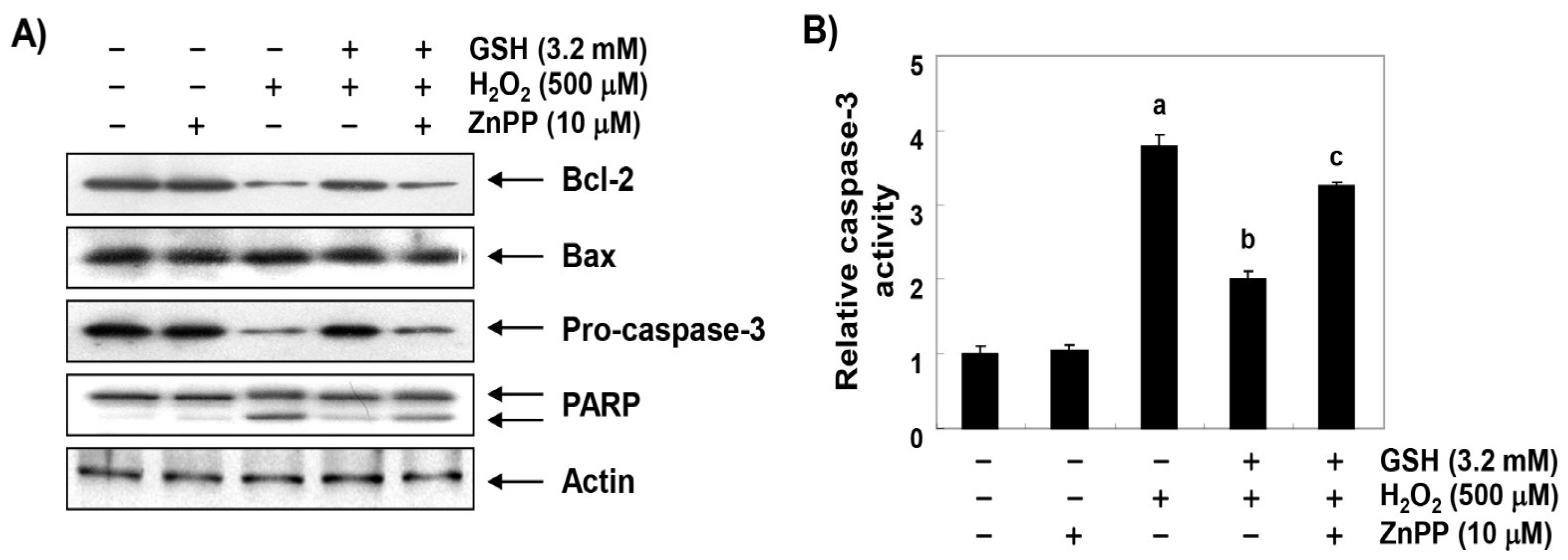
3.7. The Inhibitory Effect of Glutathione on the Change of Apoptosis Markers Genes by H2O2 is Reversed by ZnPP in RAW 264.7 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2018; Volume 80, pp. 50–64. [Google Scholar]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Reichert, A.S. How to get rid of mitochondria: Crosstalk and regulation of multiple mitophagy pathways. Biol. Chem. 2017, 399, 29–45. [Google Scholar] [CrossRef]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ROS generation and its regulation: Mechanisms involved in H2O2 signaling. Antioxid. Redox Signal. 2011, 14, 459–468. [Google Scholar] [CrossRef]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Lyublinskaya, O.G.; Ivanova, J.S.; Pugovkina, N.A.; Kozhukharova, I.V.; Kovaleva, Z.V.; Shatrova, A.N.; Aksenov, N.D.; Zenin, V.V.; Kaulin, Y.A.; Gamaley, I.A.; et al. Redox environment in stem and differentiated cells: A quantitative approach. Redox Biol. 2017, 12, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; LLeonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef]
- Cui, L.; Li, Z.; Chang, X.; Cong, G.; Hao, L. Quercetin attenuates vascular calcification by inhibiting oxidative stress and mitochondrial fission. Vascul. Pharmacol. 2017, 88, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S. Reactive oxygen species in mitochondria-mediated cell death. Drug Metab. Rev. 2007, 39, 443–455. [Google Scholar] [CrossRef]
- Gomez-Cabrera, M.C.; Salvador-Pascual, A.; Cabo, H.; Ferrando, B.; Viña, J. Redox modulation of mitochondriogenesis in exercise. Does antioxidant supplementation blunt the benefits of exercise training? Free Radic. Biol. Med. 2015, 86, 37–46. [Google Scholar] [CrossRef]
- Monostori, P.; Wittmann, G.; Karg, E.; Túri, S. Determination of glutathione and glutathione disulfide in biological samples: An in-depth review. J. Chromatogr. B 2009, 877, 3331–3346. [Google Scholar] [CrossRef]
- Kerksick, C.; Willoughby, D. The antioxidant role of glutathione and N-acetyl-cysteine supplements and exercise-induced oxidative stress. J. Int. Soc. Sports Nutr. 2005, 2, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Federici, G.; Bertini, E.; Piemonte, F. Analysis of glutathione: Implication in redox and detoxification. Clin. Chim. Acta 2003, 333, 19–39. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, L.; Yi, D.; Wu, G. N-acetylcysteine and intestinal health: A focus on its mechanism of action. Front. Biosci. (Landmark Ed.) 2015, 20, 872–891. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M. The incomplete clutathione puzzle: Just guessing at numbers and figures? Antioxid. Redox Signal. 2017, 27, 1130–1161. [Google Scholar] [CrossRef]
- Purohit, V.; Abdelmalek, M.F.; Barve, S.; Benevenga, N.J.; Halsted, C.H.; Kaplowitz, N.; Kharbanda, K.K.; Liu, Q.Y.; Lu, S.C.; McClain, C.J.; et al. Role of S-adenosylmethionine, folate, and betaine in the treatment of alcoholic liver disease: Summary of a symposium. Am. J. Clin. Nutr. 2007, 86, 14–24. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F.; DiNicolantonio, J.J. An increased need for dietary cysteine in support of glutathione synthesis may underlie the increased risk for mortality associated with low protein intake in the elderly. Age 2015, 37, 96. [Google Scholar] [CrossRef]
- Teskey, G.; Abrahem, R.; Cao, R.; Gyurjian, K.; Islamoglu, H.; Lucero, M.; Martinez, A.; Paredes, E.; Salaiz, O.; Robinson, B.; et al. Glutathione as a marker for human disease. Adv. Clin. Chem. 2018, 87, 141–159. [Google Scholar]
- Song, J.; Kang, S.M.; Lee, W.T.; Park, K.A.; Lee, K.M.; Lee, J.E. Glutathione protects brain endothelial cells from hydrogen peroxide-induced oxidative stress by increasing nrf2 expression. Exp. Neurobiol. 2014, 23, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Yamamoto, M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid. Redox Signal. 2005, 7, 385–394. [Google Scholar] [CrossRef]
- Lee, S.E.; Jeong, S.I.; Kim, G.D.; Yang, H.; Park, C.S.; Jin, Y.H.; Park, Y.S. Upregulation of heme oxygenase-1 as an adaptive mechanism for protection against crotonaldehyde in human umbilical vein endothelial cells. Toxicol. Lett. 2011, 201, 240–248. [Google Scholar] [CrossRef]
- Sun Jang, J.; Piao, S.; Cha, Y.N.; Kim, C. Taurine chloramine activates Nrf2, increases HO-1 expression and protects cells from death caused by hydrogen peroxide. J. Clin. Biochem. Nutr. 2009, 45, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Tomczyk, M.; Kraszewska, I.; Dulak, J.; Jazwa-Kusior, A. Modulation of the monocyte/macrophage system in heart failure by targeting heme oxygenase-1. Vascul. Pharmacol. 2019, 112, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Alaoui-Jamali, M.A.; Bismar, T.A.; Gupta, A.; Szarek, W.A.; Su, J.; Song, W.; Xu, Y.; Xu, B.; Liu, G.; Vlahakis, J.Z.; et al. A novel experimental heme oxygenase-1-targeted therapy for hormone-refractory prostate cancer. Cancer Res. 2009, 20, 8017–8024. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.Y.; Cha, H.J.; Choi, E.O.; Kim, C.H.; Kim, G.Y.; Yoo, Y.H.; Hwang, H.J.; Park, H.T.; Yoon, H.M.; Choi, Y.H. Activation of the Nrf2/HO-1 signaling pathway contributes to the protective effects of baicalein against oxidative stress-induced DNA damage and apoptosis in HEI193 Schwann cells. Int. J. Med. Sci. 2019, 16, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Jeong, S.I.; Yang, H.; Park, C.S.; Jin, Y.H.; Park, Y.S. Fisetin induces Nrf2-mediated HO-1 expression through PKC-δ and p38 in human umbilical vein endothelial cells. J. Cell. Biochem. 2011, 112, 2352–2360. [Google Scholar] [CrossRef]
- Aggeli, I.K.; Gaitanaki, C.; Beis, I. Involvement of JNKs and p38-MAPK/MSK1 pathways in H2O2-induced upregulation of heme oxygenase-1 mRNA in H9c2 cells. Cell. Signal. 2006, 18, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Rothfuss, A.; Speit, G. Overexpression of heme oxygenase-1 (HO-1) in V79 cells results in increased resistance to hyperbaric oxygen (HBO)-induced DNA damage. Environ. Mol. Mutagen. 2002, 40, 258–265. [Google Scholar] [CrossRef]
- Zhu, X.F.; Li, W.; Ma, J.Y.; Shao, N.; Zhang, Y.J.; Liu, R.M.; Wu, W.B.; Lin, Y.; Wang, S.M. Knockdown of heme oxygenase-1 promotes apoptosis and autophagy and enhances the cytotoxicity of doxorubicin in breast cancer cells. Oncol. Lett. 2015, 10, 2974–2980. [Google Scholar] [CrossRef]
- Hirai, K.; Sasahira, T.; Ohmori, H.; Fujii, K.; Kuniyasu, H. Inhibition of heme oxygenase-1 by zinc protoporphyrin IX reduces tumor growth of LL/2 lung cancer in C57BL mice. Int. J. Cancer 2007, 120, 500–505. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Valero, T. Mitochondrial biogenesis: Pharmacological approaches. Curr. Pharm. Des. 2014, 20, 5507–5509. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.M.; Chen, B.; Lv, J.M.; Lei, Q.; Pan, Y.J.; Yang, Q. Knockdown of IRF6 attenuates hydrogen dioxide-induced oxidative stress via inhibiting mitochondrial dysfunction in HT22 cells. Cell. Mol. Neurobiol. 2016, 36, 1077–1086. [Google Scholar] [CrossRef]
- Luo, T.; Shen, X.Y.; Li, S.; Ouyang, T.; Mai, Q.A.; Wang, H.Q. The protective effect of jatrorrhizine against oxidative stress in primary rat cortical neurons. CNS Neurol. Disord. Drug Targ. 2017, 16, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.T.; Sun, X.Q.; Wu, C.; Chen, J.L.; Yuan, J.J.; Pang, Q.F.; Wang, Z.P. Heme oxygnease-1 induction by methylene blue protects RAW264.7 cells from hydrogen peroxide-induced injury. Biochem. Pharmacol. 2018, 148, 265–277. [Google Scholar] [CrossRef]
- Tian, X.; He, W.; Yang, R.; Liu, Y. Dl-3-n-butylphthalide protects the heart against ischemic injury and H9c2 cardiomyoblasts against oxidative stress: Involvement of mitochondrial function and biogenesis. J. Biomed. Sci. 2017, 24, 38. [Google Scholar] [CrossRef]
- Yu, L.; Gong, B.; Duan, W.; Fan, C.; Zhang, J.; Li, Z.; Xue, X.; Xu, Y.; Meng, D.; Li, B.; et al. Melatonin ameliorates myocardial ischemia/reperfusion injury in type 1 diabetic rats by preserving mitochondrial function: Role of AMPK-PGC-1α-SIRT3 signaling. Sci. Rep. 2017, 7, 41337. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.Y.; Lu, H.B.; Kong, N.; Li, J.Y.; Liu, M.; Yang, C.Y.; Yang, P. Levocarnitine protects H9c2 rat cardiomyocytes from H2O2-induced mitochondrial dysfunction and apoptosis. Int. J. Med. Sci. 2014, 11, 1107–1115. [Google Scholar] [CrossRef]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef]
- Schultz, D.R.; Harrington, W.J., Jr. Apoptosis: Programmed cell death at a molecular level. Semin. Arthr. Rheum. 2003, 32, 345–369. [Google Scholar] [CrossRef]
- Kiraz, Y.; Adan, A.; Kartal Yandim, M.; Baran, Y. Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biol. 2016, 37, 8471–8486. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, A.B.; Gottlieb, R.A. Bcl-2 family members and apoptosis, taken to heart. Am. J. Physiol. Cell. Physiol. 2007, 292, C45–C51. [Google Scholar] [CrossRef]
- Kulikov, A.V.; Shilov, E.S.; Mufazalov, I.A.; Gogvadze, V.; Nedospasov, S.A.; Zhivotovsky, B. Cytochrome c: The Achilles’ heel in apoptosis. Cell. Mol. Life Sci. 2012, 69, 1787–1797. [Google Scholar] [CrossRef]
- Imahashi, K.; Schneider, M.D.; Steenbergen, C.; Murphy, E. Transgenic expression of Bcl-2 modulates energy metabolism, prevents cytosolic acidification during ischemia, and reduces ischemia/reperfusion injury. Circ. Res. 2004, 95, 734–741. [Google Scholar] [CrossRef]
- Yin, Y.; Lu, L.; Wang, D.; Shi, Y.; Wang, M.; Huang, Y.; Chen, D.; Deng, C.; Chen, J.; Lv, P.; et al. Astragalus polysaccharide inhibits autophagy and apoptosis from peroxide-induced injury in C2C12 myoblasts. Cell. Biochem. Biophys. 2015, 73, 433–439. [Google Scholar] [CrossRef]
- Siu, P.M.; Wang, Y.; Always, S.E. Apoptotic signaling induced by H2O2-mediated oxidative stress in differentiated C2C12 myotubes. Life Sci. 2009, 84, 468–481. [Google Scholar] [CrossRef]
- Haramizu, S.; Asano, S.; Butler, D.C.; Stanton, D.A.; Hajira, A.; Mohamed, J.S.; Always, S.E. Dietary resveratrol confers apoptotic resistance to oxidative stress in myoblasts. J. Nutr. Biochem. 2017, 50, 103–115. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, W.J.; Lee, M.H.; Kim, S.Y.; Seo, D.H.; Kim, H.S.; Gelinsky, M.; Kim, T.J. Anti-skeletal muscle atrophy effect of Oenothera odorata root extract via reactive oxygen species-dependent signaling pathways in cellular and mouse model. Biosci. Biotechnol. Biochem. 2016, 80, 80–88. [Google Scholar] [CrossRef]
- Chen, H.; Tang, X.; Zhou, B.; Zhou, Z.; Xu, N.; Wang, Y. A ROS-mediated mitochondrial pathway and Nrf2 pathway activation are involved in BDE-47 induced apoptosis in Neuro-2a cells. Chemosphere 2017, 184, 679–686. [Google Scholar] [CrossRef]
- Yu, J.; Zhu, X.; Qi, X.; Che, J.; Cao, B. Paeoniflorin protects human EA.hy926 endothelial cells against gamma-radiation induced oxidative injury by activating the NF-E2-related factor 2/heme oxygenase-1 pathway. Toxicol. Lett. 2013, 218, 224–234. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, D.H.; Cha, H.-J.; Lee, H.; Hong, S.-H.; Park, C.; Park, S.-H.; Kim, G.-Y.; Kim, S.; Kim, H.-S.; Hwang, H.-J.; et al. Protective Effect of Glutathione against Oxidative Stress-induced Cytotoxicity in RAW 264.7 Macrophages through Activating the Nuclear Factor Erythroid 2-Related Factor-2/Heme Oxygenase-1 Pathway. Antioxidants 2019, 8, 82. https://doi.org/10.3390/antiox8040082
Kwon DH, Cha H-J, Lee H, Hong S-H, Park C, Park S-H, Kim G-Y, Kim S, Kim H-S, Hwang H-J, et al. Protective Effect of Glutathione against Oxidative Stress-induced Cytotoxicity in RAW 264.7 Macrophages through Activating the Nuclear Factor Erythroid 2-Related Factor-2/Heme Oxygenase-1 Pathway. Antioxidants. 2019; 8(4):82. https://doi.org/10.3390/antiox8040082
Chicago/Turabian StyleKwon, Da Hye, Hee-Jae Cha, Hyesook Lee, Su-Hyun Hong, Cheol Park, Shin-Hyung Park, Gi-Young Kim, Suhkmann Kim, Heui-Soo Kim, Hye-Jin Hwang, and et al. 2019. "Protective Effect of Glutathione against Oxidative Stress-induced Cytotoxicity in RAW 264.7 Macrophages through Activating the Nuclear Factor Erythroid 2-Related Factor-2/Heme Oxygenase-1 Pathway" Antioxidants 8, no. 4: 82. https://doi.org/10.3390/antiox8040082
APA StyleKwon, D. H., Cha, H.-J., Lee, H., Hong, S.-H., Park, C., Park, S.-H., Kim, G.-Y., Kim, S., Kim, H.-S., Hwang, H.-J., & Choi, Y. H. (2019). Protective Effect of Glutathione against Oxidative Stress-induced Cytotoxicity in RAW 264.7 Macrophages through Activating the Nuclear Factor Erythroid 2-Related Factor-2/Heme Oxygenase-1 Pathway. Antioxidants, 8(4), 82. https://doi.org/10.3390/antiox8040082