Water-soluble CoQ10 as A Promising Anti-aging Agent for Neurological Dysfunction in Brain Mitochondria

{kind=link}
Abstract
1. Introduction
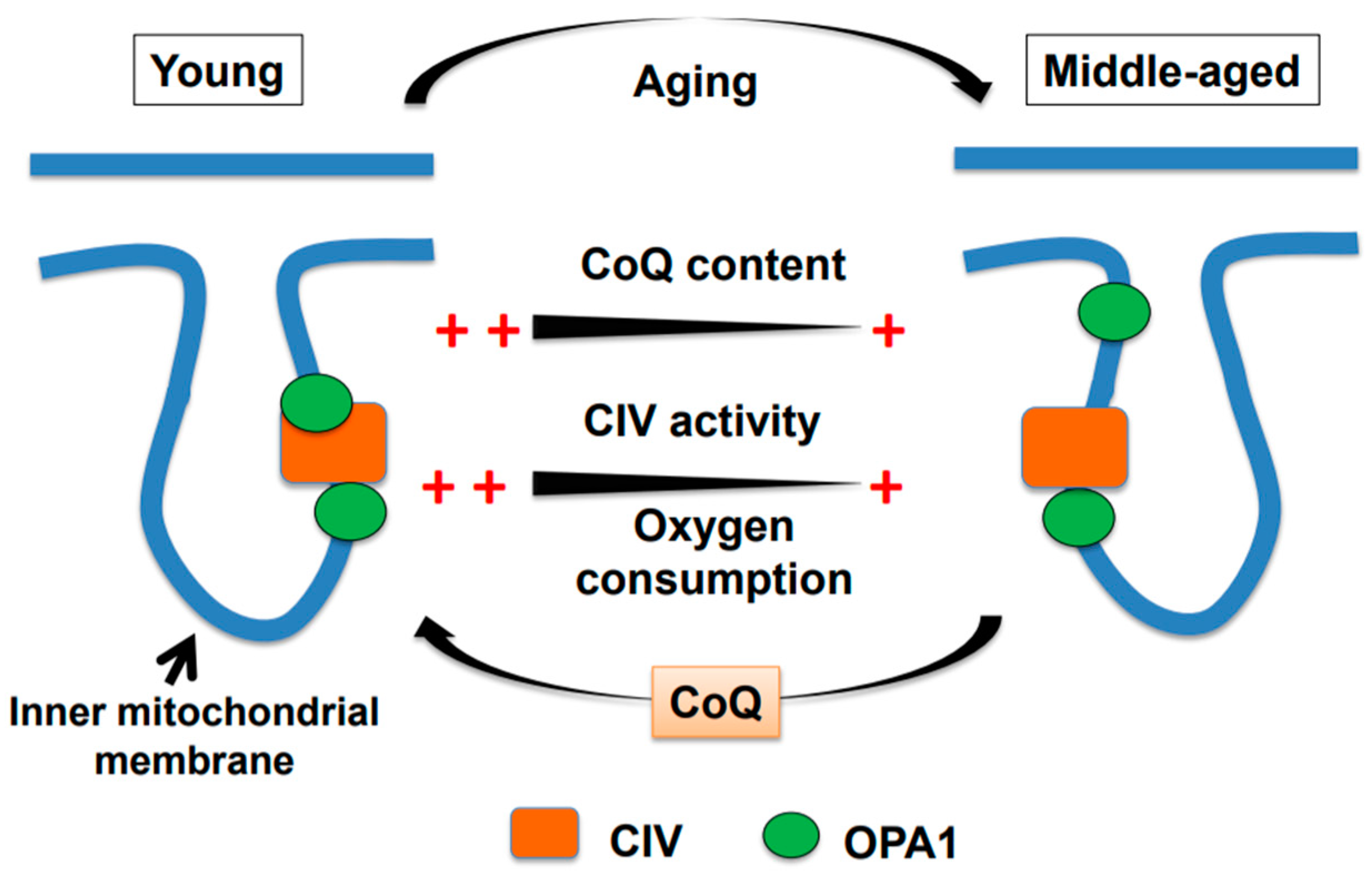
2. Age-associated Decline in Brain Mitochondrial Function and Motor Impairment
3. Water-soluble CoQ10 Rescues Brain Mitochondrial Dysfunction and Motor Impairment
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, T.; van Heek, P.; Leif, H.; Ohnishi, T.; Forche, E.; Kunze, B.; Jansen, R.; Trowitzsch-Kienast, W.; Hofle, G.; Reichenbach, H.; et al. Two binding sites of inhibitors in NADH: Ubiquinone oxidoreductase (complex I). Relationship of one site with the ubiquinone-binding site of bacterial glucose:ubiquinone oxidoreductase. Eur. J. Biochem. 1994, 219, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Brandt, U. Proton-translocation by membrane-bound NADH:ubiquinone-oxidoreductase (complex I) through redox-gated ligand conduction. Biochim. Biophys. Acta 1997, 1318, 79–91. [Google Scholar] [CrossRef]
- Guenebaut, V.; Vincentelli, R.; Mills, D.; Weiss, H.; Leonard, K.R. Three-dimensional structure of NADH-dehydrogenase from Neurospora crassa by electron microscopy and conical tilt reconstruction. J. Mol. Biol. 1997, 265, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, I.E. Molecular genetics of succinate:quinone oxidoreductase in eukaryotes. Prog. Nucleic Acid Res. Mol. Biol. 1998, 60, 267–315. [Google Scholar] [PubMed]
- Tsukihara, T.; Aoyama, H.; Yamashita, E.; Tomizaki, T.; Yamaguchi, H.; Shinzawa-Itoh, K.; Nakashima, R.; Yaono, R.; Yoshikawa, S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science 1996, 272, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Brandt, U. The chemistry and mechanics of ubihydroquinone oxidation at center P (Qo) of the cytochrome bc1 complex. Biochim. Biophys. Acta 1998, 1365, 261–268. [Google Scholar] [CrossRef]
- Varotsis, C.; Zhang, Y.; Appelman, E.H.; Babcock, G.T. Resolution of the reaction sequence during the reduction of O2 by cytochrome oxidase. Proc. Natl. Acad. Sci. USA 1993, 90, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Yu, C.A.; Kim, H.; Xia, J.Z.; Kachurin, A.M.; Zhang, L.; Yu, L.; Deisenhofer, J. Crystal structure of the cytochrome bc1 complex from bovine heart mitochondria. Science 1997, 277, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Takahashi, M. Exogenous administration of coenzyme Q10 restores mitochondrial oxygen consumption in the aged mouse brain. Mech. Ageing Dev. 2013, 134, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.C.; Battersby, B.J.; Moyes, C.D. Inter-tissue differences in mitochondrial enzyme activity, RNA and DNA in rainbow trout (Oncorhynchus mykiss). J. Exp. Biol. 1998, 201, 3377–3384. [Google Scholar]
- Takahashi, K.; Ohsawa, I.; Shirasawa, T.; Takahashi, M. Early-onset motor impairment and increased accumulation of phosphorylated alpha-synuclein in the motor cortex of normal aging mice are ameliorated by coenzyme Q. Exp. Gerontol. 2016, 81, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Schagger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Fernandez-Silva, P.; Peleato, M.L.; Perez-Martos, A.; Enriquez, J.A. Respiratory active mitochondrial supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G.; Genova, M.L. Kinetics of integrated electron transfer in the mitochondrial respiratory chain: Random collisions vs. solid state electron channeling. Am. J. Physiol. Cell Physiol. 2007, 292, C1221–C1239. [Google Scholar] [CrossRef] [PubMed]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acin-Perez, R.; Latorre-Pellicer, A.; Colas, C.; Balsa, E.; Perales-Clemente, E.; Quiros, P.M.; Calvo, E.; Rodriguez-Hernandez, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.A.; Monette, J.S.; Chavez, J.D.; Maier, C.S.; Hagen, T.M. Supercomplexes of the mitochondrial electron transport chain decline in the aging rat heart. Arch. Biochem. Biophys. 2009, 490, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Schagger, H.; de Coo, R.; Bauer, M.F.; Hofmann, S.; Godinot, C.; Brandt, U. Significance of respirasomes for the assembly/stability of human respiratory chain complex I. J. Biol. Chem. 2004, 279, 36349–36353. [Google Scholar] [CrossRef] [PubMed]
- Wittig, I.; Karas, M.; Schagger, H. High resolution clear native electrophoresis for in-gel functional assays and fluorescence studies of membrane protein complexes. Mol. Cell. Proteom. 2007, 6, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Mourier, A.; Matic, S.; Ruzzenente, B.; Larsson, N.G.; Milenkovic, D. The respiratory chain supercomplex organization is independent of COX7a2l isoforms. Cell Metab. 2014, 20, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Ohsawa, I.; Shirasawa, T.; Takahashi, M. Optic atrophy 1 mediates coenzyme Q-responsive regulation of respiratory complex IV activity in brain mitochondria. Exp. Gerontol. 2017, 98, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Hoppins, S.; Lackner, L.; Nunnari, J. The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 2007, 76, 751–780. [Google Scholar] [CrossRef] [PubMed]
- Praefcke, G.J.; McMahon, H.T. The dynamin superfamily: Universal membrane tubulation and fission molecules? Nat. Rev. Mol. Cell Biol. 2004, 5, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [PubMed]
- Meeusen, S.; DeVay, R.; Block, J.; Cassidy-Stone, A.; Wayson, S.; McCaffery, J.M.; Nunnari, J. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell 2006, 127, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Civiletto, G.; Varanita, T.; Cerutti, R.; Gorletta, T.; Barbaro, S.; Marchet, S.; Lamperti, C.; Viscomi, C.; Scorrano, L.; Zeviani, M. Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab. 2015, 21, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Akepati, V.R.; Muller, E.C.; Otto, A.; Strauss, H.M.; Portwich, M.; Alexander, C. Characterization of OPA1 isoforms isolated from mouse tissues. J. Neurochem. 2008, 106, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Griparic, L.; Kanazawa, T.; van der Bliek, A.M. Regulation of the mitochondrial dynamin-like protein OPA1 by proteolytic cleavage. J. Cell Biol. 2007, 178, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef] [PubMed]
- Zanna, C.; Ghelli, A.; Porcelli, A.M.; Karbowski, M.; Youle, R.J.; Schimpf, S.; Wissinger, B.; Pinti, M.; Cossarizza, A.; Vidoni, S.; et al. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain 2008, 131, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Büeler, H. Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson’s disease. Exp. Neurol. 2009, 218, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; Przedborski, S. Mitochondria: The next (neurode) generation. Neuron 2011, 70, 1033–1053. [Google Scholar] [CrossRef] [PubMed]
- Trancikova, A.; Tsika, E.; Moore, D.J. Mitochondrial dysfunction in genetic animal models of Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 896–919. [Google Scholar] [CrossRef] [PubMed]
- Hornykiewicz, O.; Kish, S.J. Biochemical pathophysiology of Parkinson’s disease. Adv. Neurol. 1987, 45, 19–34. [Google Scholar] [PubMed]
- Mizuno, Y.; Ohta, S.; Tanaka, M.; Takamiya, S.; Suzuki, K.; Sato, T.; Oya, H.; Ozawa, T.; Kagawa, Y. Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem. Biophys. Res. Commun. 1989, 163, 1450–1455. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D., Jr.; Parks, J.K.; Swerdlow, R.H. Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Ogasahara, S.; Engel, A.G.; Frens, D.; Mack, D. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc. Natl. Acad. Sci. USA 1989, 86, 2379–2382. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; DiMauro, S.; Hirano, M. Human coenzyme Q10 deficiency. Neurochem Res 2007, 32, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Artuch, R.; Salviati, L.; Jackson, S.; Hirano, M.; Navas, P. Coenzyme Q10 deficiencies in neuromuscular diseases. Adv. Exp. Med. Biol. 2009, 652, 117–128. [Google Scholar] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Feany, M.B. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Arawaka, S.; Hara, S.; Fukushima, S.; Koga, K.; Koyama, S.; Kato, T. Authentically phosphorylated alpha-synuclein at Ser129 accelerates neurodegeneration in a rat model of familial Parkinson’s disease. J. Neurosci. 2011, 31, 16884–16894. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Kato, T.; Arawaka, S. The role of Ser129 phosphorylation of alpha-synuclein in neurodegeneration of Parkinson’s disease: A review of in vivo models. Rev. Neurosci. 2013, 24, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Yang, R.; Guo, J.C.; Ren, H.M.; Zha, X.L.; Cheng, J.S.; Cai, D.F. Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport 2007, 18, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Bellocchio, E.E.; Reimer, R.J.; Fremeau, R.T., Jr.; Edwards, R.H. Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science 2000, 289, 957–960. [Google Scholar] [CrossRef] [PubMed]
- Takamori, S.; Rhee, J.S.; Rosenmund, C.; Jahn, R. Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 2000, 407, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Fujiyama, F. Complementary distribution of vesicular glutamate transporters in the central nervous system. Neurosci. Res. 2002, 42, 243–250. [Google Scholar] [CrossRef]
- Herzog, E.; Landry, M.; Buhler, E.; Bouali-Benazzouz, R.; Legay, C.; Henderson, C.E.; Nagy, F.; Dreyfus, P.; Giros, B.; El Mestikawy, S. Expression of vesicular glutamate transporters, VGLUT1 and VGLUT2, in cholinergic spinal motoneurons. Eur. J. Neurosci. 2004, 20, 1752–1760. [Google Scholar] [CrossRef] [PubMed]
- Tatton, W.G.; Lee, R.G. Evidence for abnormal long-loop reflexes in rigid Parkinsonian patients. Brain Res. 1975, 100, 671–676. [Google Scholar] [CrossRef]
- Logigian, E.; Hefter, H.; Reiners, K.; Freund, H.J. Does tremor pace repetitive voluntary motor behavior in Parkinson’s disease? Ann. Neurol. 1991, 30, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.G.; Martin, K.E.; Bradshaw, J.L.; Iansek, R. Could bradykinesia in Parkinson’s disease simply be compensation? J. Neurol. 1994, 241, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, N.; Hirose, Y.; Ohara, S.; Ono, T.; Watanabe, Y. A simple quantitative bradykinesia test in MPTP-treated mice. Res. Commun. Chem. Pathol. Pharmacol. 1985, 50, 435–441. [Google Scholar] [PubMed]
- Ogawa, N.; Mizukawa, K.; Hirose, Y.; Kajita, S.; Ohara, S.; Watanabe, Y. MPTP-induced parkinsonian model in mice: Biochemistry, pharmacology and behavior. Eur. Neurol. 1987, 26 (Suppl. 1), 16–23. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, K.; Kabuto, H.; Makino, H.; Ogawa, N. Pole test is a useful method for evaluating the mouse movement disorder caused by striatal dopamine depletion. J. Neurosci. Methods 1997, 73, 45–48. [Google Scholar] [CrossRef]
- Hwang, D.Y.; Fleming, S.M.; Ardayfio, P.; Moran-Gates, T.; Kim, H.; Tarazi, F.I.; Chesselet, M.F.; Kim, K.S. 3,4-dihydroxyphenylalanine reverses the motor deficits in Pitx3-deficient aphakia mice: Behavioral characterization of a novel genetic model of Parkinson’s disease. J. Neurosci. 2005, 25, 2132–2137. [Google Scholar] [CrossRef] [PubMed]
- Christmas, A.J.; Maxwell, D.R. A comparison of the effects of some benzodiazepines and other drugs on aggressive and exploratory behaviour in mice and rats. Neuropharmacology 1970, 9, 17–29. [Google Scholar] [CrossRef]
- Ganea, K.; Liebl, C.; Sterlemann, V.; Muller, M.B.; Schmidt, M.V. Pharmacological validation of a novel home cage activity counter in mice. J. Neurosci. Methods 2007, 162, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Halcomb, R.A.; Hegmann, J.P.; DeFries, J.C. Open-field behavior in mice: A diallel analysis of selected lines. Behav. Genet. 1975, 5, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Kitaoka, K.; Hattori, A.; Chikahisa, S.; Miyamoto, K.; Nakaya, Y.; Sei, H. Vitamin A deficiency induces a decrease in EEG delta power during sleep in mice. Brain Res. 2007, 1150, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, K.; Shibasaki, M.; Mizuno, K.; Ohkuma, S. Gabapentin blocks methamphetamine-induced sensitization and conditioned place preference via inhibition of alpha(2)/delta-1 subunits of the voltage-gated calcium channels. Neuroscience 2011, 176, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Gitler, A.D. Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 2016, 539, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Alexander, G.E.; DeLong, M.R.; Strick, P.L. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu. Rev. Neurosci. 1986, 9, 357–381. [Google Scholar] [CrossRef] [PubMed]
- Kiritani, T.; Wickersham, I.R.; Seung, H.S.; Shepherd, G.M. Hierarchical connectivity and connection-specific dynamics in the corticospinal-corticostriatal microcircuit in mouse motor cortex. J. Neurosci. 2012, 32, 4992–5001. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, G.M. Corticostriatal connectivity and its role in disease. Nat. Rev. Neurosci. 2013, 14, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.P.; Lane, A.; Sleiman, P.M. The coenzyme Q10 status of the brain regions of Parkinson’s disease patients. Neurosci. Lett. 2008, 447, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Shults, C.W.; Beal, M.F.; Fontaine, D.; Nakano, K.; Haas, R.H. Absorption, tolerability, and effects on mitochondrial activity of oral coenzyme Q10 in parkinsonian patients. Neurology 1998, 50, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Marbois, B.N.; Clarke, C.F. The COQ7 gene encodes a protein in saccharomyces cerevisiae necessary for ubiquinone biosynthesis. J. Biol. Chem. 1996, 271, 2995–3004. [Google Scholar] [CrossRef] [PubMed]
- Felkai, S.; Ewbank, J.J.; Lemieux, J.; Labbe, J.C.; Brown, G.G.; Hekimi, S. CLK-1 controls respiration, behavior and aging in the nematode Caenorhabditis elegans. EMBO J. 1999, 18, 1783–1792. [Google Scholar] [CrossRef] [PubMed]
- Lakowski, B.; Hekimi, S. Determination of life-span in Caenorhabditis elegans by four clock genes. Science 1996, 272, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, T.; Larsen, P.L.; Clarke, C.F. A dietary source of coenzyme Q is essential for growth of long-lived Caenorhabditis elegans clk-1 mutants. Proc. Natl. Acad. Sci. USA 2001, 98, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Nakai, D.; Yuasa, S.; Takahashi, M.; Shimizu, T.; Asaumi, S.; Isono, K.; Takao, T.; Suzuki, Y.; Kuroyanagi, H.; Hirokawa, K.; et al. Mouse homologue of coq7/clk-1, longevity gene in Caenorhabditis elegans, is essential for coenzyme Q synthesis, maintenance of mitochondrial integrity, and neurogenesis. Biochem. Biophys. Res. Commun. 2001, 289, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Shimizu, T.; Moriizumi, E.; Shirasawa, T. Clk-1 deficiency induces apoptosis associated with mitochondrial dysfunction in mouse embryos. Mech. Ageing Dev. 2008, 129, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Beyer, R.E.; Burnett, B.A.; Cartwright, K.J.; Edington, D.W.; Falzon, M.J.; Kreitman, K.R.; Kuhn, T.W.; Ramp, B.J.; Rhee, S.Y.; Rosenwasser, M.J.; et al. Tissue coenzyme Q (ubiquinone) and protein concentrations over the life span of the laboratory rat. Mech. Ageing Dev. 1985, 32, 267–281. [Google Scholar] [CrossRef]
- Kalen, A.; Appelkvist, E.L.; Dallner, G. Age-related changes in the lipid compositions of rat and human tissues. Lipids 1989, 24, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Battino, M.; Gorini, A.; Villa, R.F.; Genova, M.L.; Bovina, C.; Sassi, S.; Littarru, G.P.; Lenaz, G. Coenzyme Q content in synaptic and non-synaptic mitochondria from different brain regions in the ageing rat. Mech. Ageing Dev. 1995, 78, 173–187. [Google Scholar] [CrossRef]
- Sikorska, M.; Lanthier, P.; Miller, H.; Beyers, M.; Sodja, C.; Zurakowski, B.; Gangaraju, S.; Pandey, S.; Sandhu, J.K. Nanomicellar formulation of coenzyme Q10 (Ubisol-Q10) effectively blocks ongoing neurodegeneration in the mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model: Potential use as an adjuvant treatment in Parkinson’s disease. Neurobiol. Aging 2014, 35, 2329–2346. [Google Scholar] [CrossRef] [PubMed]
- Gupta, B.K.; Kumar, S.; Kaur, H.; Ali, J.; Baboota, S. Attenuation of Oxidative Damage by Coenzyme Q10 Loaded Nanoemulsion Through Oral Route for the Management of Parkinson’s Disease. Rejuvenation Res. 2018, 21, 232–248. [Google Scholar] [CrossRef] [PubMed]
- Saiki, R.; Lunceford, A.L.; Shi, Y.; Marbois, B.; King, R.; Pachuski, J.; Kawamukai, M.; Gasser, D.L.; Clarke, C.F. Coenzyme Q10 supplementation rescues renal disease in Pdss2kd/kd mice with mutations in prenyl diphosphate synthase subunit 2. Am. J. Physiol. Renal Physiol. 2008, 295, F1535–F1544. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Corzo, L.; Luna-Sanchez, M.; Doerrier, C.; Ortiz, F.; Escames, G.; Acuna-Castroviejo, D.; Lopez, L.C. Ubiquinol-10 ameliorates mitochondrial encephalopathy associated with CoQ deficiency. Biochim. Biophys. Acta 2014, 1842, 893–901. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, S.; Somayajulu, M.; Sikorska, M.; Borowy-Borowski, H.; Pandey, S. Paraquat induces oxidative stress and neuronal cell death; neuroprotection by water-soluble Coenzyme Q10. Toxicol. Appl. Pharmacol. 2004, 201, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Bergamini, C.; Moruzzi, N.; Sblendido, A.; Lenaz, G.; Fato, R. A water soluble CoQ10 formulation improves intracellular distribution and promotes mitochondrial respiration in cultured cells. PLoS ONE 2012, 7, e33712. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Shimizu, T.; Shirasawa, T. Reversal of slow growth and heartbeat through the restoration of mitochondrial function in clk-1-deficient mouse embryos by exogenous administration of coenzyme Q10. Exp. Gerontol. 2012, 47, 425–431. [Google Scholar] [CrossRef] [PubMed]
- van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef] [PubMed]
- Kar, R.; Mishra, N.; Singha, P.K.; Venkatachalam, M.A.; Saikumar, P. Mitochondrial remodeling following fission inhibition by 15d-PGJ2 involves molecular changes in mitochondrial fusion protein OPA1. Biochem. Biophys. Res. Commun. 2010, 399, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Kar, R.; Singha, P.K.; Venkatachalam, M.A.; McEwen, D.G.; Saikumar, P. Inhibition of mitochondrial division through covalent modification of Drp1 protein by 15 deoxy-Delta(12,14)-prostaglandin J2. Biochem. Biophys. Res. Commun. 2010, 395, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Kim, K.Y.; Lindsey, J.D.; Dai, Y.; Heo, H.; Nguyen, D.H.; Ellisman, M.H.; Weinreb, R.N.; Ju, W.K. A selective inhibitor of drp1, mdivi-1, increases retinal ganglion cell survival in acute ischemic mouse retina. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2837–2843. [Google Scholar] [CrossRef] [PubMed]
- Cleren, C.; Yang, L.; Lorenzo, B.; Calingasan, N.Y.; Schomer, A.; Sireci, A.; Wille, E.J.; Beal, M.F. Therapeutic effects of coenzyme Q10 (CoQ10) and reduced CoQ10 in the MPTP model of Parkinsonism. J. Neurochem. 2008, 104, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Camacho, J.D.; Bernier, M.; Lopez-Lluch, G.; Navas, P. Coenzyme Q10 Supplementation in Aging and Disease. Front. Physiol. 2018, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Zaki, N.M. Strategies for oral delivery and mitochondrial targeting of CoQ10. Drug Deliv. 2016, 23, 1868–1881. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Pathak, K.; Misra, A. Formulation and characterization of nanoemulsion-based drug delivery system of risperidone. Drug Dev. Ind. Pharm. 2009, 35, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lluch, G.; Del Pozo-Cruz, J.; Sanchez-Cuesta, A.; Cortes-Rodriguez, A.B.; Navas, P. Bioavailability of coenzyme Q10 supplements depends on carrier lipids and solubilization. Nutrition 2019, 57, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Salami, A.; Mora, R.; Dellepiane, M.; Manini, G.; Santomauro, V.; Barettini, L.; Guastini, L. Water-soluble coenzyme Q10 formulation (Q-TER®) in the treatment of presbycusis. Acta Otolaryngol. 2010, 130, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, S.; Fattirolli, F.; Guarducci, L.; Cellai, T.; Baldasseroni, S.; Tarantini, F.; Di Bari, M.; Masotti, G.; Marchionni, N. Coenzyme Q10 terclatrate and creatine in chronic heart failure: A randomized, placebo-controlled, double-blind study. Clin. Cardiol. 2011, 34, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Guastini, L.; Mora, R.; Dellepiane, M.; Santomauro, V.; Giorgio, M.; Salami, A. Water-soluble coenzyme Q10 formulation in presbycusis: Long-term effects. Acta Otolaryngol. 2011, 131, 512–517. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takahashi, M.; Takahashi, K. Water-soluble CoQ10 as A Promising Anti-aging Agent for Neurological Dysfunction in Brain Mitochondria. Antioxidants 2019, 8, 61. https://doi.org/10.3390/antiox8030061
Takahashi M, Takahashi K. Water-soluble CoQ10 as A Promising Anti-aging Agent for Neurological Dysfunction in Brain Mitochondria. Antioxidants. 2019; 8(3):61. https://doi.org/10.3390/antiox8030061
Chicago/Turabian StyleTakahashi, Mayumi, and Kazuhide Takahashi. 2019. "Water-soluble CoQ10 as A Promising Anti-aging Agent for Neurological Dysfunction in Brain Mitochondria" Antioxidants 8, no. 3: 61. https://doi.org/10.3390/antiox8030061
APA StyleTakahashi, M., & Takahashi, K. (2019). Water-soluble CoQ10 as A Promising Anti-aging Agent for Neurological Dysfunction in Brain Mitochondria. Antioxidants, 8(3), 61. https://doi.org/10.3390/antiox8030061