Regulation of Smooth Muscle Cell Proliferation by NADPH Oxidases in Pulmonary Hypertension


Abstract
1. Introduction
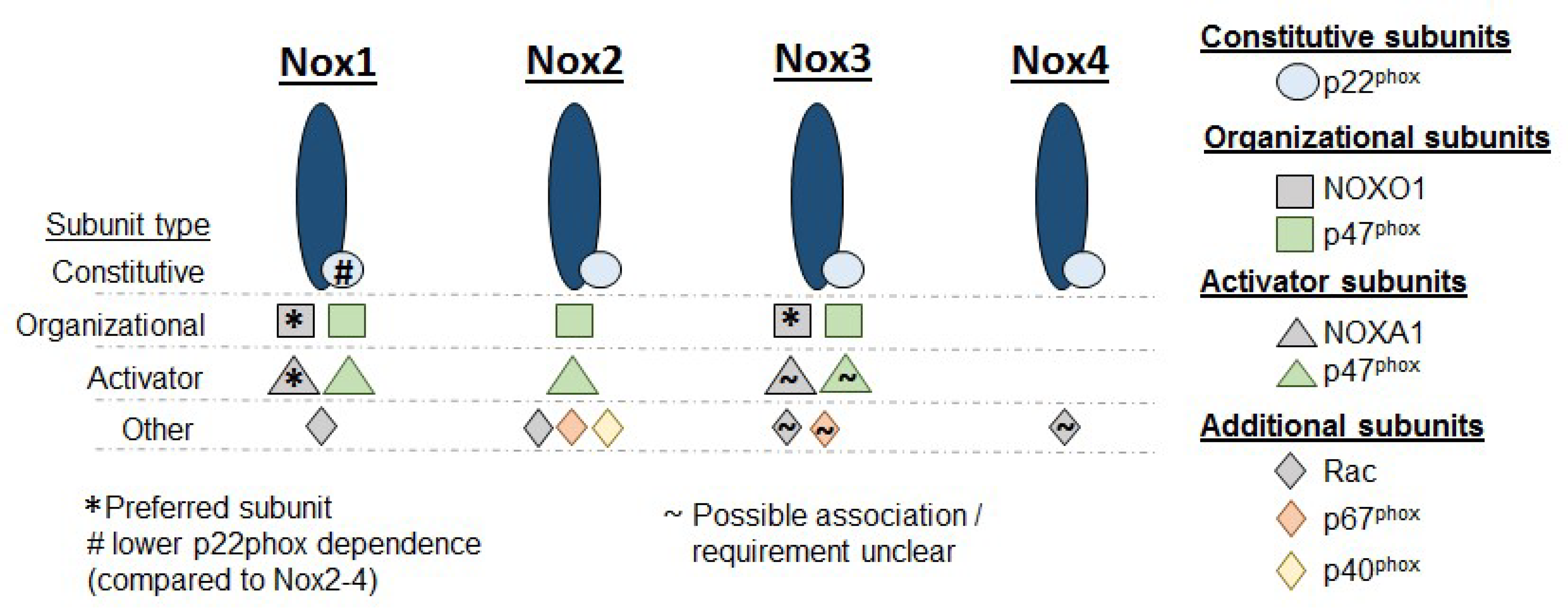
2. NADPH Oxidases
2.1. Noxs and the Regulation of Smooth Muscle Proliferation in Pulmonary Hypertension
2.1.1. Nox1
2.1.2. Nox2
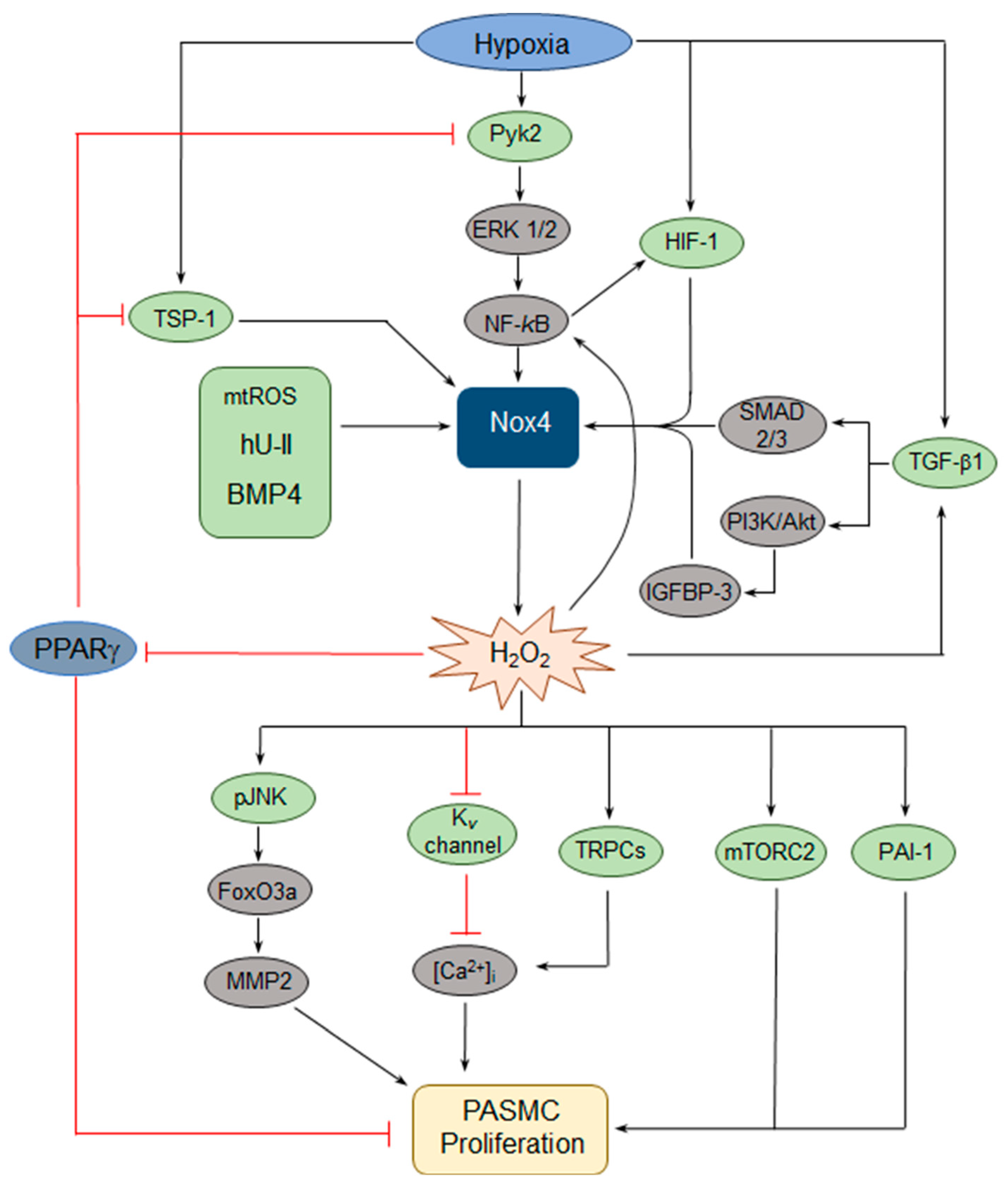
2.1.3. Nox4
2.1.4. Nox3
2.1.5. Nox5
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2018, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Tabima, D.M.; Frizzell, S.; Gladwin, M.T. Reactive oxygen and nitrogen species in pulmonary hypertension. Free Radic. Biol. Med. 2012, 52, 1970–1986. [Google Scholar] [CrossRef] [PubMed]
- Nozik-Grayck, E.; Stenmark, K.R. Role of reactive oxygen species in chronic hypoxia-induced pulmonary hypertension and vascular remodeling. Adv. Exp. Med. Biol. 2007, 618, 101–112. [Google Scholar] [PubMed]
- Fulton, D.J.R.; Li, X.; Bordan, Z.; Haigh, S.; Bentley, A.; Chen, F.; Barman, S.A. Reactive oxygen and nitrogen species in the development of pulmonary hypertension. Antioxidants 2017, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Freund-Michel, V.; Guibert, C.; Dubois, M.; Courtois, A.; Marthan, R.; Savineau, J.-P.; Muller, B. Reactive oxygen species as therapeutic targets in pulmonary hypertension. Ther. Adv. Respir. Dis. 2013, 7, 175–200. [Google Scholar] [CrossRef] [PubMed]
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of vascular nitric oxide and reactive oxygen species and their regulation. Physiol. Rev. 2019, 99, 311–379. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Gross, C.M.; Sharma, S.; Fineman, J.R.; Black, S.M. Reactive oxygen species in pulmonary vascular remodeling. Compr. Physiol. 2013, 3, 1011–1034. [Google Scholar] [PubMed]
- Chanock, S.J.; el Benna, J.; Smith, R.M.; Babior, B.M. The respiratory burst oxidase. J. Biol. Chem. 1994, 269, 24519–24522. [Google Scholar] [PubMed]
- Iyer, G.Y.N.; Islam, M.F.; Quastel, J.H. Biochemical aspects of phagocytosis. Nature 1961, 192, 535–541. [Google Scholar] [CrossRef]
- Clark, R.A.; Leidal, K.G.; Pearson, D.W.; Nauseef, W.M. NADPH oxidase of human neutrophils. Subcellular localization and characterization of an arachidonate-activatable superoxide-generating system. J. Biol. Chem. 1987, 262, 4065–4074. [Google Scholar] [PubMed]
- Heyworth, P.G.; Cross, A.R.; Curnutte, J.T. Chronic granulomatous disease. Curr. Opin. Immunol. 2003, 15, 578–584. [Google Scholar] [CrossRef]
- Isogai, Y.; Iizuka, T.; Shiro, Y. The mechanism of electron donation to molecular oxygen by phagocytic cytochrome b558. J. Biol. Chem. 1995, 270, 7853–7857. [Google Scholar] [CrossRef] [PubMed]
- Martyn, K.D.; Fredericka, L.M.; von Loehneysen, K.; Dinauer, M.C.; Knaus, U.G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006, 18, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Takac, I.; Schroder, K. No superoxide--no stress? Nox4, the good NADPH oxidase! Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1255–1257. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M. Biological roles for the NOX family NADPH oxidases. J. Biol. Chem. 2008, 283, 16961–16965. [Google Scholar] [CrossRef] [PubMed]
- Groemping, Y.; Rittinger, K. Activation and assembly of the NADPH oxidase: A structural perspective. Biochem. J. 2005, 386 Pt 3, 401–416. [Google Scholar] [CrossRef]
- Nauseef, W.M. Assembly of the phagocyte NADPH oxidase. Histochem. Cell Biol. 2004, 122, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Dinauer, M.C.; Pierce, E.A.; Bruns, G.A.; Curnutte, J.T.; Orkin, S.H. Human neutrophil cytochrome b light chain (p22-phox). Gene structure, chromosomal location, and mutations in cytochrome-negative autosomal recessive chronic granulomatous disease. J. Clin. Investig. 1990, 86, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Parkos, C.A.; Dinauer, M.C.; Jesaitis, A.J.; Orkin, S.H.; Curnutte, J.T. Absence of both the 91kD and 22kD subunits of human neutrophil cytochrome b in two genetic forms of chronic granulomatous disease. Blood 1989, 73, 1416–1420. [Google Scholar] [PubMed]
- Han, C.-H.; Freeman, J.L.R.; Lee, T.; Motalebi, S.A.; Lambeth, J.D. Regulation of the neutrophil respiratory burst oxidase. Identification of an activation domain in p67(phox). J. Biol. Chem. 1998, 273, 16663–16668. [Google Scholar] [CrossRef] [PubMed]
- Hilenski, L.L.; Clempus, R.E.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Nakagawa, K.; Yamasaki, T.; Nakamura, K.-i.; Takeya, R.; Kuribayashi, F.; Imajoh-Ohmi, S.; Igarashi, K.; Shibata, Y.; Sueishi, K.; et al. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells 2005, 10, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Altenhofer, S.; Kleikers, P.W.M.; Radermacher, K.A.; Scheurer, P.; Hermans, J.J.R.; Schiffers, P.; Ho, H.; Wingler, K.; Schmidt, H.H.H.W. The NOX toolbox: Validating the role of NADPH oxidases in physiology and disease. Cell. Mol. Life Sci. 2012, 69, 2327–2343. [Google Scholar] [CrossRef] [PubMed]
- Veit, F.; Pak, O.; Egemnazarov, B.; Roth, M.; Kosanovic, D.; Seimetz, M.; Sommer, N.; Ghofrani, H.A.; Seeger, W.; Grimminger, F.; et al. Function of NADPH oxidase 1 in pulmonary arterial smooth muscle cells after monocrotaline-induced pulmonary vascular remodeling. Antioxid. Redox Signal. 2013, 19, 2213–2231. [Google Scholar] [CrossRef] [PubMed]
- Barman, S.A.; Chen, F.; Su, Y.; Dimitropoulou, C.; Wang, Y.; Catravas, J.D.; Han, W.; Orfi, L.; Szantai-Kis, C.; Keri, G.; et al. NADPH oxidase 4 is expressed in pulmonary artery adventitia and contributes to hypertensive vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1704–1715. [Google Scholar] [CrossRef] [PubMed]
- Hood, K.Y.; Montezano, A.C.; Harvey, A.P.; Nilsen, M.; MacLean, M.R.; Touyz, R.M. Nicotinamide adenine dinucleotide phosphate oxidase-mediated redox signaling and vascular remodeling by 16a-hydroxyestrone in human pulmonary artery cells: Implications in pulmonary arterial hypertension. Hypertension 2016, 68, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Iwata, K.; Ikami, K.; Matsuno, K.; Yamashita, T.; Shiba, D.; Ibi, M.; Matsumoto, M.; Katsuyama, M.; Cui, W.; Zhang, J.; et al. Deficiency of NOX1/nicotinamide adenine dinucleotide phosphate, reduced form oxidase leads to pulmonary vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Hood, K.Y.; Mair, K.M.; Harvey, A.P.; Montezano, A.C.; Touyz, R.M.; MacLean, M.R. Serotonin signaling through the 5-HT1B receptor and NADPH oxidase 1 in pulmonary arterial hypertension. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1361–1370. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Reeve, H.L.; Michelakis, E.; Puttagunta, L.; Waite, R.; Nelson, D.P.; Dinauer, M.C.; Weir, E.K. O2 sensing is preserved in mice lacking the gp91 phox subunit of NADPH oxidase. Proc. Natl. Acad. Sci. USA 1999, 96, 7944–7949. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Q.; Zelko, I.N.; Erbynn, E.M.; Sham, J.S.K.; Folz, R.J. Hypoxic pulmonary hypertension: Role of superoxide and NADPH oxidase (gp91phox). Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L2–L10. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Labinskyy, N.; Olson, S.; Pinto, J.T.; Gupte, S.; Wu, J.M.; Hu, F.; Ballabh, P.; Podlutsky, A.; Losonczy, G.; et al. Resveratrol prevents monocrotaline-induced pulmonary hypertension in rats. Hypertension 2009, 54, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Muzaffar, S.; Angelini, G.D.; Jeremy, J.Y. Acute hypoxia simultaneously induces the expression of gp91phox and endothelial nitric oxide synthase in the porcine pulmonary artery. Thorax 2005, 60, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Rathore, R.; Zheng, Y.-M.; Niu, C.-F.; Liu, Q.-H.; Korde, A.; Ho, Y.S.; Wang, Y.X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCε signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, R.E.; Graves, A.S.; Kleinhenz, D.J.; Rupnow, H.L.; Reed, A.L.; Fan, T.-H.M.; Mitchell, P.O.; Sutliff, R.L.; Hart, C.M. The role of NADPH oxidase in chronic intermittent hypoxia-induced pulmonary hypertension in mice. Am. J. Respir. Cell Mol. Biol. 2009, 40, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Seta, F.; Rahmani, M.; Turner, P.V.; Funk, C.D. Pulmonary oxidative stress is increased in cyclooxygenase-2 knockdown mice with mild pulmonary hypertension induced by monocrotaline. PLoS ONE 2011, 6, e23439. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Roth, M.; König, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.-C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Amara, N.; Goven, D.; Prost, F.; Muloway, R.; Crestani, B.; Boczkowski, J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax 2010, 65, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Sorescu, D.; Weiss, D.; Lassègue, B.; Clempus, R.E.; Szöcs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, R.E.; Bland, J.M.; Kleinhenz, D.J.; Mitchell, P.O.; Walp, E.R.; Sutliff, R.L.; Hart, C.M. Rosiglitazone attenuates chronic hypoxia-induced pulmonary hypertension in a mouse model. Am. J. Respir. Cell Mol. Biol. 2010, 42, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Goncharov, D.A.; Kudryashova, T.V.; Ziai, H.; Ihida-Stansbury, K.; DeLisser, H.; Krymskaya, V.P.; Tuder, R.M.; Kawut, S.M.; Goncharova, E.A. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 2014, 129, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Wedgwood, S.; Lakshminrusimha, S.; Czech, L.; Schumacker, P.T.; Steinhorn, R.H. Increased p22(phox)/Nox4 expression is involved in remodeling through hydrogen peroxide signaling in experimental persistent pulmonary hypertension of the newborn. Antioxid. Redox Signal. 2013, 18, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.E.; Fallon, M.B.; Krowka, M.J.; Brown, R.S.; Trotter, J.F.; Peter, I.; Tighiouart, H.; Knowles, J.A.; Rabinowitz, D.; Benza, R.L.; et al. Genetic risk factors for portopulmonary hypertension in patients with advanced liver disease. Am. J. Respir. Crit. Care Med. 2009, 179, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Bijli, K.M.; Kang, B.-Y.; Sutliff, R.L.; Hart, C.M. Proline-rich tyrosine kinase 2 downregulates peroxisome proliferator-activated receptor gamma to promote hypoxia-induced pulmonary artery smooth muscle cell proliferation. Pulm. Circ. 2016, 6, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Green, D.E.; Kang, B.-Y.; Murphy, T.C.; Hart, C.M. Peroxisome proliferator-activated receptor gamma (PPARγ) regulates thrombospondin-1 and Nox4 expression in hypoxia-induced human pulmonary artery smooth muscle cell proliferation. Pulm. Circ. 2012, 2, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Bijli, K.M.; Kleinhenz, J.M.; Murphy, T.C.; Kang, B.-Y.; Adesina, S.E.; Sutliff, R.L.; Hart, C.M. Peroxisome proliferator-activated receptor γ depletion stimulates Nox4 expression and human pulmonary artery smooth muscle cell proliferation. Free Radic. Biol. Med. 2015, 80, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Bijli, K.M.; Ramirez, A.; Murphy, T.C.; Kleinhenz, J.; Hart, C.M. Hypoxia downregulates PPARγ via an ERK1/2-NF-kB-Nox4-dependent mechanism in human pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2013, 63, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Murphy, T.C.; Nanes, M.S.; Hart, C.M. PPARγ regulates hypoxia-induced Nox4 expression in human pulmonary artery smooth muscle cells through NF-kB. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L559–L566. [Google Scholar] [CrossRef] [PubMed]
- Wedgwood, S.; Lakshminrusimha, S.; Schumacker, P.T.; Steinhorn, R.H. Cyclic stretch stimulates mitochondrial reactive oxygen species and Nox4 signaling in pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L196–L203. [Google Scholar] [CrossRef] [PubMed]
- Bonello, S.; Zähringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Görlach, A. Reactive oxygen species activate the HIF-1α promoter via a functional NFkB site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Petry, A.; Hess, J.; Görlach, A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol. Biol. Cell 2010, 21, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Flügel, D.; Becht, S.; BelAiba, R.S.; Bonello, S.; Hess, J.; Kietzmann, T.; Görlach, A. The hypoxia-inducible factor-2α is stabilized by oxidative stress involving NOX4. Antioxid. Redox Signal. 2010, 13, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Sturrock, A.; Cahill, B.; Norman, K.; Huecksteadt, T.P.; Hill, K.; Sanders, K.; Karwande, S.V.; Stringham, J.C.; Bull, D.A.; Gleich, M.; et al. Transforming growth factor-β1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L661–L673. [Google Scholar] [CrossRef] [PubMed]
- Ismail, S.; Sturrock, A.; Wu, P.; Cahill, B.; Norman, K.; Huecksteadt, T.; Sanders, K.; Kennedy, T.; Hoidal, J. NOX4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: The role of autocrine production of transforming growth factor-β1 and insulin-like growth factor binding protein-3. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L489–L499. [Google Scholar] [CrossRef] [PubMed]
- Green, D.E.; Murphy, T.C.; Kang, B.-Y.; Kleinhenz, J.M.; Szyndralewiez, C.; Page, P.; Sutliff, R.L.; Hart, C.M. The Nox4 inhibitor GKT137831 attenuates hypoxia-induced pulmonary vascular cell proliferation. Am. J. Respir. Cell Mol. Biol. 2012, 47, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, T.; BelAiba, R.S.; Bonello, S.; Pfeilschifter, J.; Hess, J.; Görlach, A. Human urotensin II is a novel activator of NADPH oxidase in human pulmonary artery smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Petry, A.; Burger, M.; Hess, J.; Görlach, A. NOX4 mediates activation of FoxO3a and matrix metalloproteinase-2 expression by urotensin-II. Mol. Biol. Cell 2011, 22, 4424–4434. [Google Scholar] [CrossRef] [PubMed]
- Adesina, S.E.; Kang, B.-Y.; Bijli, K.M.; Ma, J.; Cheng, J.; Murphy, T.C.; Hart, C.M.; Sutliff, R.L. Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free Radic. Biol. Med. 2015, 87, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Ambalavanan, N.; Liu, H.; Sun, Y.; Jhala, N.; Bradley, W.E.; Dell’Italia, L.J.; Michalek, S.; Wu, H.; Steele, C.; et al. TLR4 regulates pulmonary vascular homeostasis and remodeling via redox signaling. Front. Biosci. 2016, 21, 397–409. [Google Scholar]
- Mittal, M.; Gu, X.Q.; Pak, O.; Pamenter, M.E.; Haag, D.; Fuchs, D.B.; Schermuly, R.T.; Ghofrani, H.A.; Brandes, R.P.; Seeger, W.; et al. Hypoxia induces Kv channel current inhibition by increased NADPH oxidase-derived reactive oxygen species. Free Radic. Biol. Med. 2012, 52, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Fu, X.; Tian, L.; Chen, Y.; Yang, K.; Chen, X.; Zhang, J.; Lu, W.; Wang, J. NOX4 mediates BMP4-induced upregulation of TRPC1 and 6 protein expressions in distal pulmonary arterial smooth muscle cells. PLoS ONE 2014, 9, e107135. [Google Scholar] [CrossRef] [PubMed]
- Heumuller, S.; Wind, S.; Barbosa-Sicard, E.; Schmidt, H.H.H.W.; Busse, R.; Schröder, K.; Brandes, R.P. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 2008, 51, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Altenhofer, S.; Radermacher, K.A.; Kleikers, P.W.M.; Wingler, K.; Schmidt, H.H.H.W. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef] [PubMed]
- Veith, C.; Kraut, S.; Wilhelm, J.; Sommer, N.; Quanz, K.; Seeger, W.; Brandes, R.P.; Weissmann, N.; Schröder, K. NADPH oxidase 4 is not involved in hypoxia-induced pulmonary hypertension. Pulm. Circ. 2016, 6, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Dorfmuller, P.; Chaumais, M.-C.; Giannakouli, M.; Durand-Gasselin, I.; Raymond, N.; Fadel, E.; Mercier, O.; Charlotte, F.; Montani, D.; Simonneau, G.; et al. Increased oxidative stress and severe arterial remodeling induced by permanent high-flow challenge in experimental pulmonary hypertension. Respir. Res. 2011, 12, 119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shan, P.; Srivastava, A.; Jiang, G.; Zhang, X.; Lee, P.J. An endothelial Hsp70-TLR4 axis limits Nox3 expression and protects against oxidant injury in lungs. Antioxid. Redox Signal. 2016, 24, 991–1012. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Li, K.; Yu, Y.; Huang, H.; Yu, Y.; Wang, Z.; Yan, J.; Pu, Y.; Li, Z.; Li, D.; et al. Genome-wide association study identifies loci and candidate genes for non-idiopathic pulmonary hypertension in Eastern Chinese Han population. BMC Pulm. Med. 2018, 18, 158. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Barman, S.; Yu, Y.; Haigh, S.; Wang, Y.; Dou, H.; Bagi, Z.; Han, W.; Su, Y.; Fulton, D.J.R. Caveolin-1 is a negative regulator of NADPH oxidase-derived reactive oxygen species. Free Radic. Biol. Med. 2014, 73, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, G.; Szyndralewiez, C.; Molango, S.; Carnesecchi, S.; Heitz, F.; Wiesel, P.; Wood, J.M. Therapeutic potential of NADPH oxidase 1/4 inhibitors. Br. J. Pharmacol. 2017, 174, 1647–1669. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Group 1—Pulmonary Arterial Hypertension (PAH) |
| Idiopathic |
| Heritable |
| Drug and toxin induced |
| Associated with connective tissue disease; infections; portal hypertension; congenital heart diseases |
| Long-term responders to Ca2+ channel blockers Overt pulmonary venous and/or capillary involvement Persistent pulmonary hypertension of the newborn |
| Group 2—Pulmonary Hypertension Due to Left Heart Disease |
| Failure with preserved ejection fraction Failure with reduced ejection fraction Valvular disease Congenital/acquired left heart inflow/outflow tract obstruction and congenital cardiomyopathies |
| Group 3—Pulmonary Hypertension Due to Lung Diseases and/or Hypoxia |
| Chronic obstructive pulmonary disease Restrictive lung disease Other pulmonary diseases with mixed restrictive and obstructive pattern Hypoxia without lung disease Developmental lung disease |
| Group 4—Pulmonary Hypertension Due to Pulmonary Artery Obstructions |
| Chronic thromboembolic pulmonary hypertension (CTEPH) Other obstructions |
| Group 5—Pulmonary Hypertension with Unclear/Multifactorial Mechanisms |
| Hematologic disorders Systemic/metabolic disorders Complex congenital heart disease Others |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huetsch, J.C.; Suresh, K.; Shimoda, L.A. Regulation of Smooth Muscle Cell Proliferation by NADPH Oxidases in Pulmonary Hypertension. Antioxidants 2019, 8, 56. https://doi.org/10.3390/antiox8030056
Huetsch JC, Suresh K, Shimoda LA. Regulation of Smooth Muscle Cell Proliferation by NADPH Oxidases in Pulmonary Hypertension. Antioxidants. 2019; 8(3):56. https://doi.org/10.3390/antiox8030056
Chicago/Turabian StyleHuetsch, John C., Karthik Suresh, and Larissa A. Shimoda. 2019. "Regulation of Smooth Muscle Cell Proliferation by NADPH Oxidases in Pulmonary Hypertension" Antioxidants 8, no. 3: 56. https://doi.org/10.3390/antiox8030056
APA StyleHuetsch, J. C., Suresh, K., & Shimoda, L. A. (2019). Regulation of Smooth Muscle Cell Proliferation by NADPH Oxidases in Pulmonary Hypertension. Antioxidants, 8(3), 56. https://doi.org/10.3390/antiox8030056