Exercise Training Promotes Cardiac Hydrogen Sulfide Biosynthesis and Mitigates Pyroptosis to Prevent High-Fat Diet-Induced Diabetic Cardiomyopathy

 , , ,
, , ,  ,
, 
Abstract
1. Introduction
2. Methods
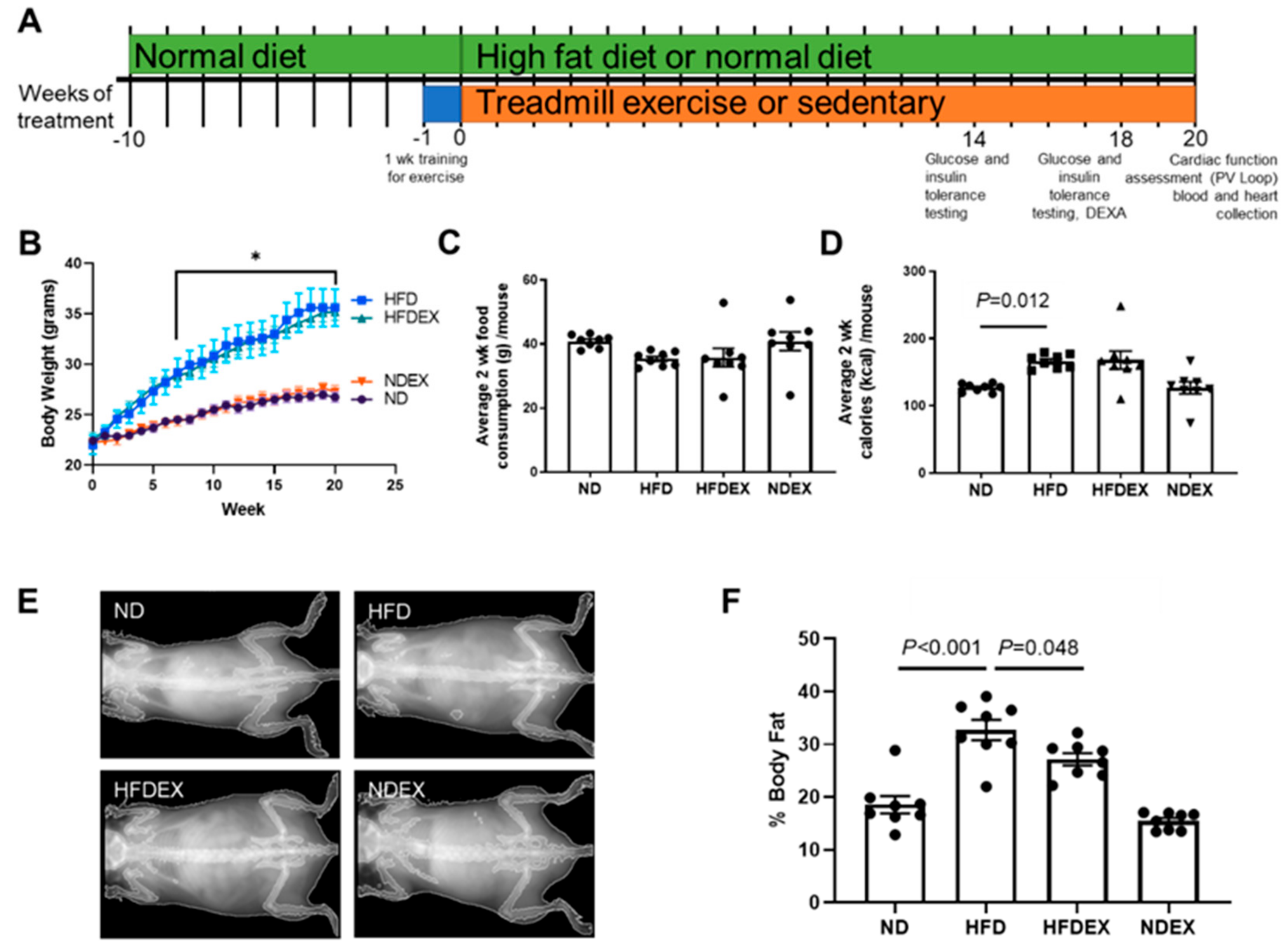
2.1. Animal Models and Treatment
2.2. Exercise Training (EX) Regimen
2.3. Dual Energy X-ray Absorbance (DEXA)
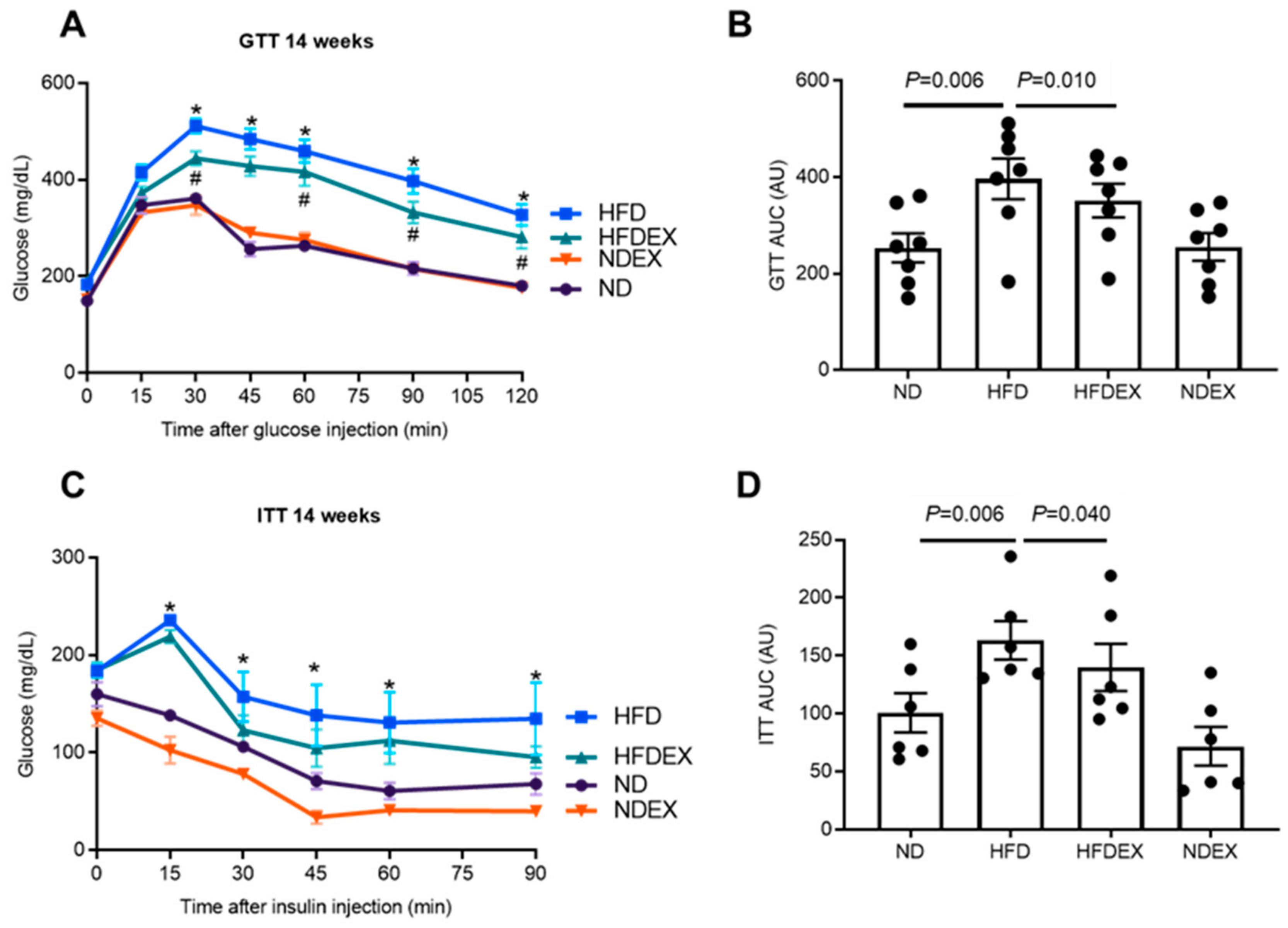
2.4. Glucose and Insulin Tolerance Tests (GTT, ITT)
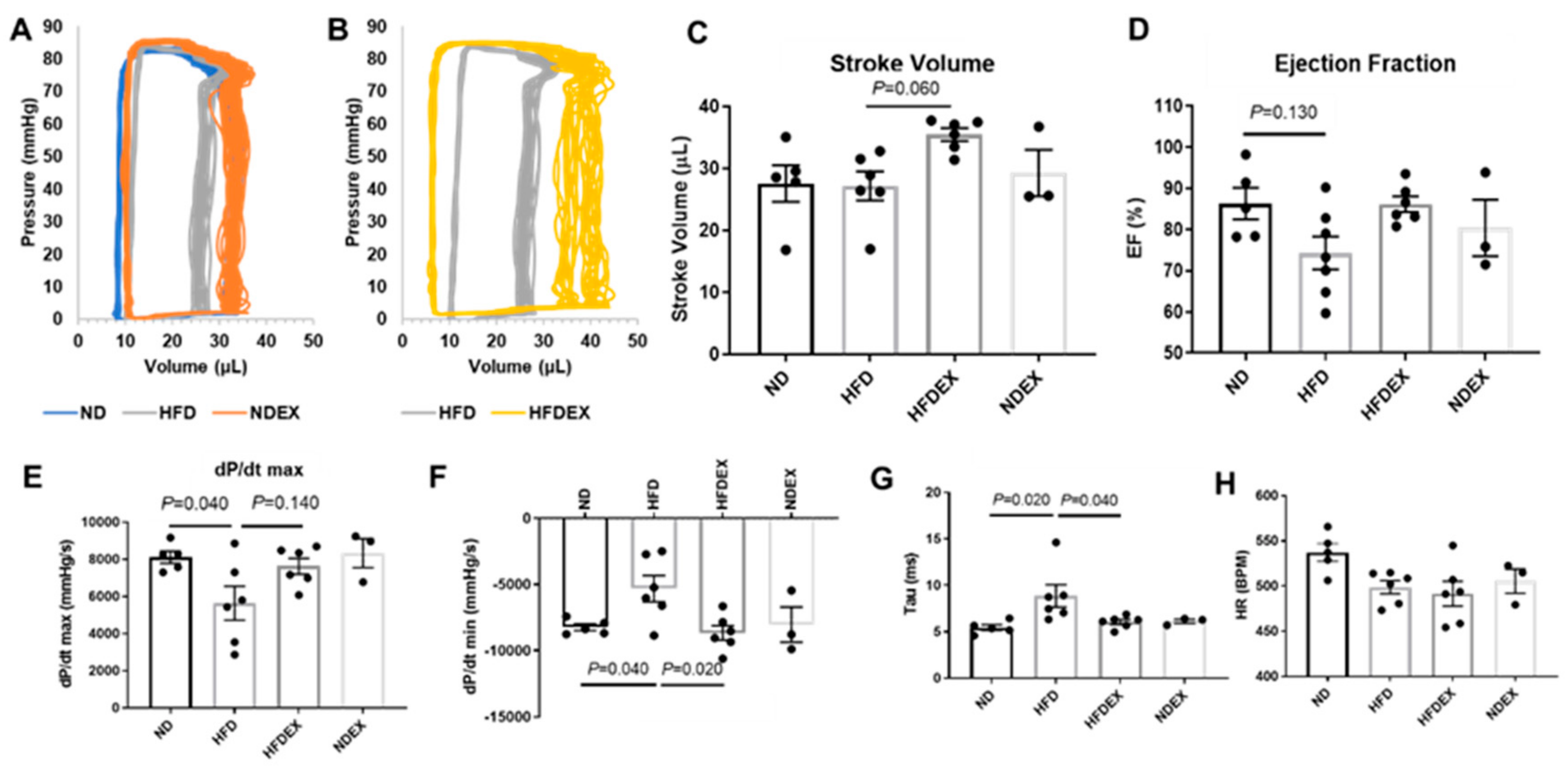
2.5. Left Ventricle Hemodynamics
2.6. Western Blotting
2.7. Gene Expression Assays
2.8. Electron Microscopy and Histology
2.9. Hydrogen Sulfide Measurement
2.10. Statistical Analyses
3. Results
3.1. High-Fat Diet Induces Obesity and Type 2 Diabetes (T2DM) Phenotype in Mice
3.2. Exercise Prevents High-Fat Diet (HFD)-Induced Cardiac Dysfunction in Mice
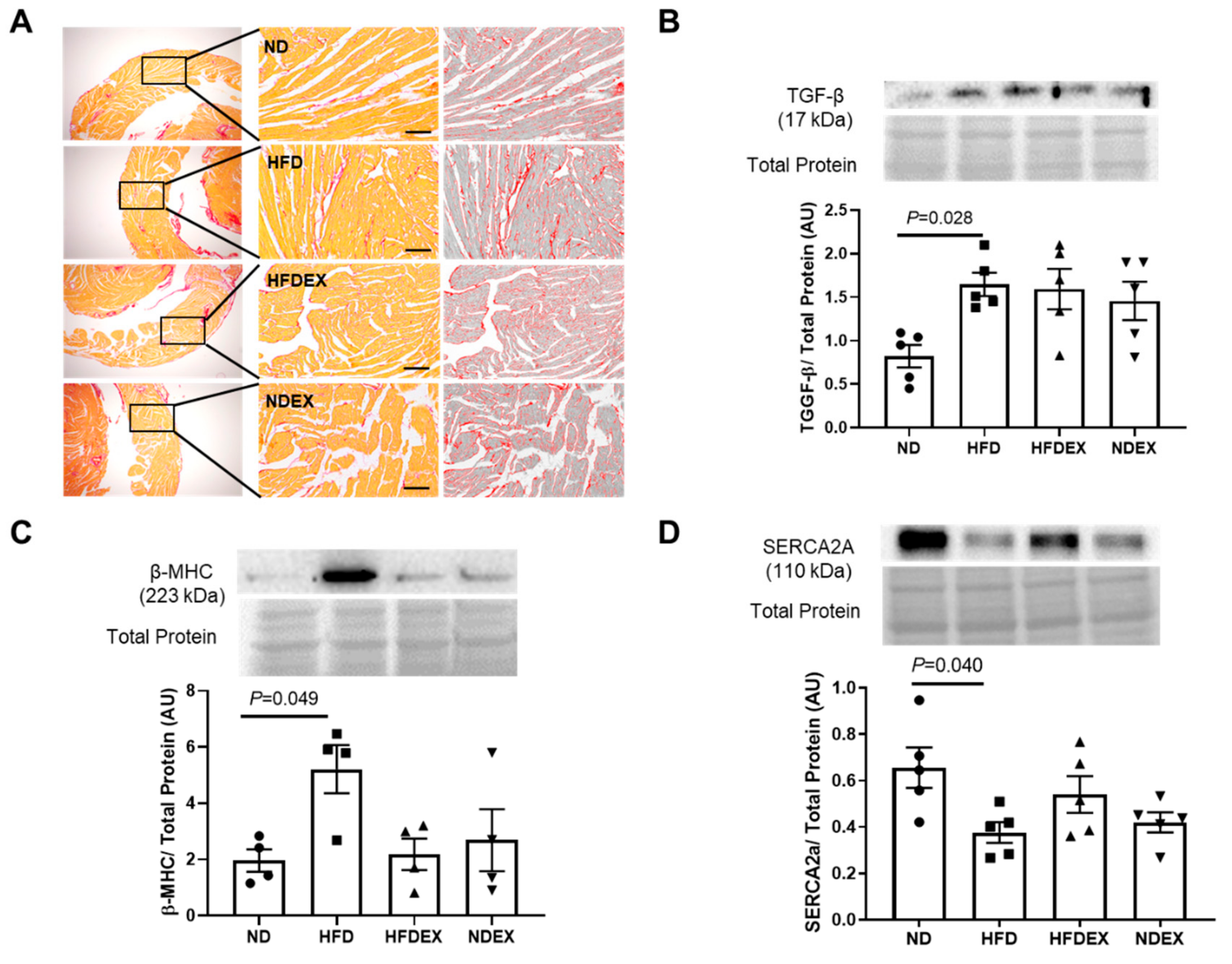
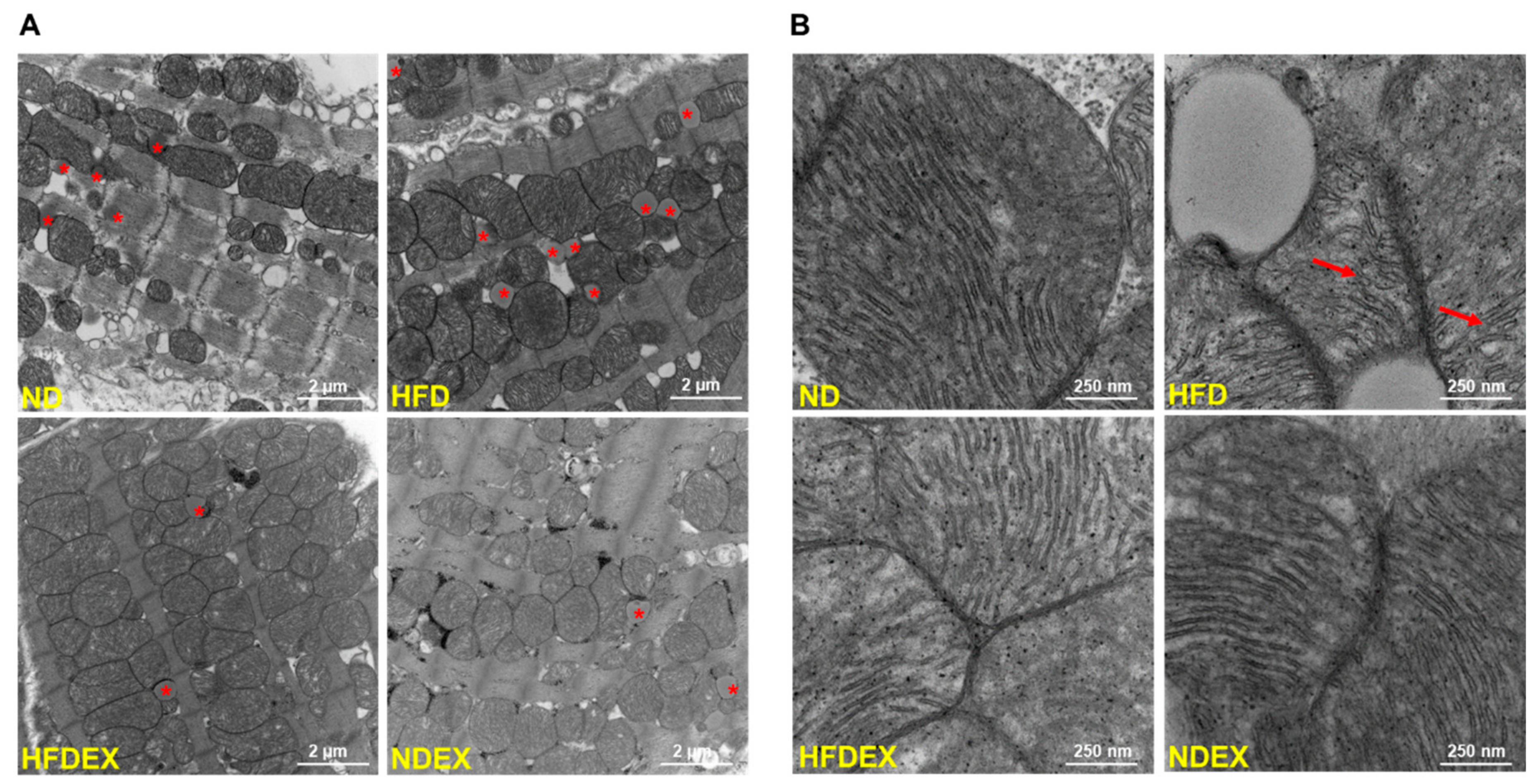
3.3. Obesity-Induced Structural and Metabolic Cardiac Remodeling
3.4. Exercise Prevents HFD-Induced Cardiac Pyroptosis
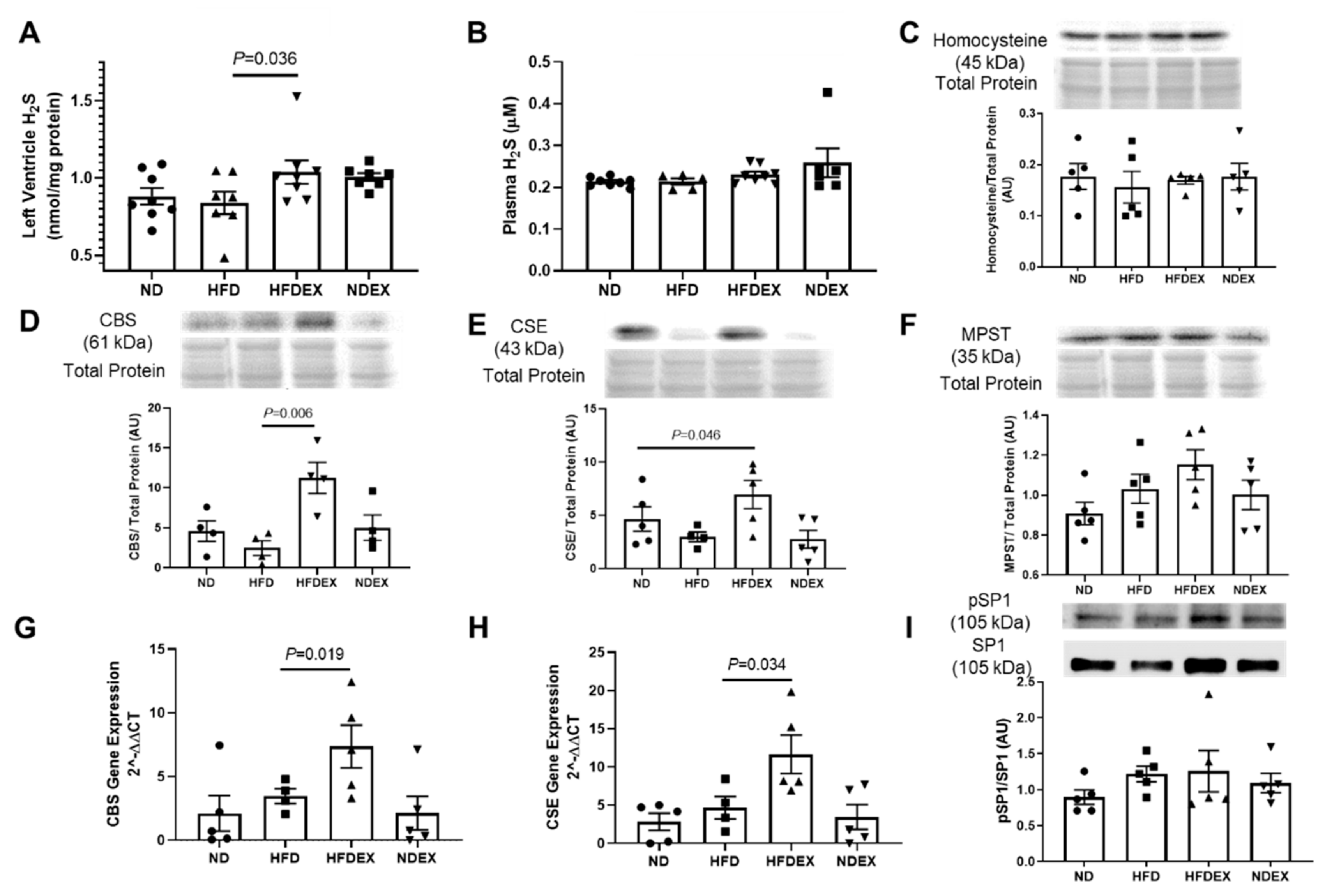
3.5. Exercise Training Induces Biosynthesis of Cardioprotective Hydrogen Sulfide
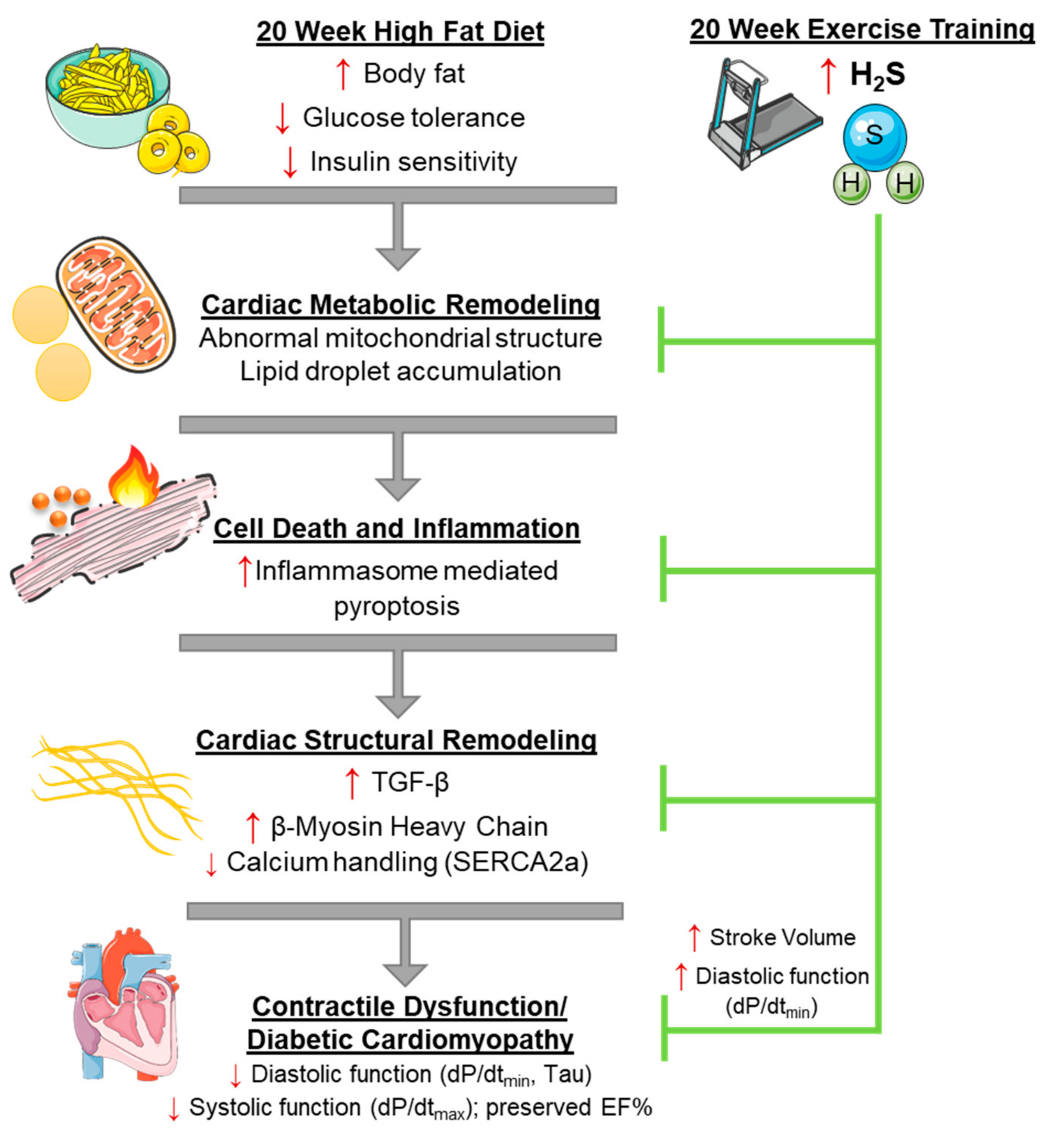
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Baena-Díez, J.M.; Byram, A.O.; Grau, M.; Gómez-Fernández, C.; Vidal-Solsona, M.; Ledesma-Ulloa, G.; González-Casafont, I.; Vasquez-Lazo, J.; Subirana, I.; Schroder, H. Obesity is an independent risk factor for heart failure: Zona franca cohort study. Clin. Cardiol. 2010, 33, 760–764. [Google Scholar] [CrossRef] [PubMed]
- De las Fuentes, L.; Brown, A.L.; Mathews, S.J.; Waggoner, A.D.; Soto, P.F.; Gropler, R.J.; Dávila-Román, V.G. Metabolic syndrome is associated with abnormal left ventricular diastolic function independent of left ventricular mass. Eur. Heart J. 2007, 28, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Von Bibra, H.; St John Sutton, M. Diastolic dysfunction in diabetes and the metabolic syndrome: Promising potential for diagnosis and prognosis. Diabetologia 2010, 53, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Alpert, M.A. Obesity cardiomyopathy: Pathophysiology and evolution of the clinical syndrome. Am. J. Med. Sci. 2001, 321, 225–236. [Google Scholar] [CrossRef]
- Badole, S.L.; Jangam, G.B. Animal Models of Diabetic Cardiomyopathy; Elsevier Inc.: Amsterdam, The Netherlands, 2014; ISBN 9780128005798. [Google Scholar]
- Fuentes-Antras, J.; Picatoste, B.; Gomez-Hernandez, A.; Egido, J.; Tunon, J.; Lorenzo, O. Updating experimental models of diabetic cardiomyopathy. J. Diabetes Res. 2015, 2015, 656795. [Google Scholar] [CrossRef]
- Carbone, S.; Mauro, A.G.; Mezzaroma, E.; Kraskauskas, D.; Marchetti, C.; Buzzetti, R.; Van Tassell, B.W.; Abbate, A.; Toldo, S. A high-sugar and high-fat diet impairs cardiac systolic and diastolic function in mice. Int. J. Cardiol. 2015, 198, 66–69. [Google Scholar] [CrossRef]
- Li, S.; Culver, B.; Ren, J. Benefit and risk of exercise on myocardial function in diabetes. Pharmacol. Res. 2003, 48, 127–132. [Google Scholar] [CrossRef]
- Wood, P.D.; Stefanick, M.L.; Williams, P.T.; Haskell, W.L. The Effects on Plasma Lipoproteins of a Prudent Weight-Reducing Diet, with or without Exercise, in Overweight Men and Women. N. Engl. J. Med. 1991, 325, 461–466. [Google Scholar] [CrossRef]
- Lund, J.; Hafstad, A.D.; Boardman, N.T.; Rossvoll, L.; Rolim, N.P.; Ahmed, M.S.; Florholmen, G.; Attramadal, H.; Wisløff, U.; Larsen, T.S.; et al. Exercise training promotes cardioprotection through oxygen-sparing action in high fat-fed mice. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H823–H829. [Google Scholar] [CrossRef]
- Epp, R.A.; Susser, S.E.; Morissette, M.P.; Kehler, D.S.; Jassal, D.S.; Duhamel, T.A. Exercise training prevents the development of cardiac dysfunction in the low-dose streptozotocin diabetic rats fed a high-fat diet. Can. J. Physiol. Pharmacol. 2012, 91, 80–89. [Google Scholar] [CrossRef]
- Cai, L.; Kang, Y.J. Cell Death and Diabetic Cardiomyopathy. Cardiovasc. Toxicol. 2003, 3, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Gustafsson, Å.B. Role of apoptosis in cardiovascular disease. Apoptosis 2009, 14, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Mauro, A.G.; Cutter, Z.; Abbate, A. Inflammasome, pyroptosis, and cytokines in myocardial ischemia-reperfusion injury. Am. J. Physiol. Circ. Physiol. 2018, 315, H1553–H1568. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, M.; Zhang, Y.; An, F. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS ONE 2014, 9, 1–14. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, Y.; Zhang, M.; An, F. Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting NLRP3 inflammasome and MAPK pathways in a type 2 diabetes rat model. Cardiovasc. Drugs Ther. 2014, 28, 33–43. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–189. [Google Scholar] [CrossRef]
- Shi, H.; Zhang, Z.; Wang, X.; Li, R.; Hou, W.; Bi, W.; Zhang, X. Inhibition of autophagy induces IL-1β release from ARPE-19 cells via ROS mediated NLRP3 inflammasome activation under high glucose stress. Biochem. Biophys. Res. Commun. 2015, 463, 1071–1076. [Google Scholar] [CrossRef]
- Giordano, A.; Murano, I.; Mondini, E.; Perugini, J.; Smorlesi, A.; Severi, I.; Barazzoni, R.; Scherer, P.E.; Cinti, S. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J. Lipid Res. 2013, 54, 2423–2436. [Google Scholar] [CrossRef]
- Kar, S.; Kambis, T.N.; Mishra, P.K. Hydrogen sulfide-mediated regulation of cell death signaling ameliorates adverse cardiac remodeling and diabetic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1237–H1252. [Google Scholar] [CrossRef]
- Castelblanco, M.; Lugrin, J.; Ehirchiou, D.; Nasi, S.; Ishii, I.; So, A.; Martinon, F.; Busso, N. Hydrogen sulfide inhibits NLRP3 inflammasome activation and reduces cytokine production both in vitro and in a mouse model of inflammation. J. Biol. Chem. 2018, 293, 2546–2557. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Das, A.; Mezzaroma, E.; Chau, V.Q.; Marchetti, C.; Durrant, D.; Samidurai, A.; Van Tassell, B.W.; Yin, C.; Ockaili, R.A.; et al. Induction of microrna-21 with exogenous hydrogen sulfide attenuates myocardial ischemic and inflammatory injury in mice. Circ. Cardiovasc. Genet. 2014, 7, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zeng, J.; Gu, Q. Exercise restores bioavailability of hydrogen sulfide and promotes autophagy influx in livers of mice fed with high-fat diet. Can. J. Physiol. Pharmacol. 2017, 95, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Shahshahan, H.R.; Kambis, T.N.; Yadav, S.K.; Li, Z.; Lefer, D.J.; Mishra, P.K. Hydrogen Sulfide Ameliorates Homocysteine-Induced Cardiac Remodeling and Dysfunction. Front. Physiol. 2019, 10, 598. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Kassiri, Z.; Virag, J.A.I.; de Castro Bras, L.E.; Scherrer-Crosbie, M. Guidelines for Measuring Cardiac Physiology in Mice. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H733–H752. [Google Scholar] [CrossRef]
- Brooks, H.L.; Lindsey, M.L. Guidelines for authors and reviewers on antibody use in physiology studies. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H724–H732. [Google Scholar] [CrossRef]
- Predmore, B.L.; Kondo, K.; Bhushan, S.; Zlatopolsky, M.A.; King, A.L.; Aragon, J.P.; Grinsfelder, D.B.; Condit, M.E.; Lefer, D.J. The polysulfide diallyl trisulfide protects the ischemic myocardium by preservation of endogenous hydrogen sulfide and increasing nitric oxide bioavailability. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H2410–H2418. [Google Scholar] [CrossRef]
- Animal Models of Diabetic Complications Consortium. Validation of Models of Cardiovascular Disease in Diabetes. Available online: https://www.diacomp.org/shared/document.aspx?id=26&docType=Protocol (accessed on 6 April 2003).
- Fang, C.X.; Dong, F.; Thomas, D.P.; Ma, H.; He, L.; Ren, J. Hypertrophic cardiomyopathy in high-fat diet-induced obesity: Role of suppression of forkhead transcription factor and atrophy gene transcription. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1206–H1215. [Google Scholar] [CrossRef]
- Pulinilkunnil, T.; Kienesberger, P.C.; Nagendran, J.; Sharma, N.; Young, M.E.; Dyck, J.R.B. Cardiac-specific adipose triglyceride lipase overexpression protects from cardiac steatosis and dilated cardiomyopathy following diet-induced obesity. Int. J. Obes. 2014, 38, 205. [Google Scholar] [CrossRef]
- Nunes, S.; Soares, E.; Fernandes, J.; Viana, S.; Carvalho, E.; Pereira, F.C.; Reis, F. Early cardiac changes in a rat model of prediabetes: Brain natriuretic peptide overexpression seems to be the best marker. Cardiovasc. Diabetol. 2013, 12, 44. [Google Scholar] [CrossRef]
- Margulies, K.B.; Anstrom, K.J.; Hernandez, A.F.; Redfield, M.M.; Shah, M.R.; Braunwald, E.; Cappola, T.P. GLP-1 Agonist Therapy for Advanced Heart Failure With Reduced Ejection Fraction. Circ. Heart Fail. 2014, 7, 673–679. [Google Scholar] [CrossRef]
- Wencker, D.; Chandra, M.; Nguyen, K.; Miao, W.; Garantziotis, S.; Factor, S.M.; Shirani, J.; Armstrong, R.C.; Kitsis, R.N. A mechanistic role for cardiac myocyte apoptosis in heart failure. J. Clin. Investig. 2003, 111, 1497–1504. [Google Scholar] [CrossRef]
- Olivetti, G.; Abbi, R.; Quaini, F.; Kajstura, J.; Cheng, W.; Nitahara, J.A.; Quaini, E.; Di Loreto, C.; Beltrami, C.A.; Krajewski, S.; et al. Apoptosis in the Failing Human Heart. N. Engl. J. Med. 1997, 336, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278. [Google Scholar] [CrossRef]
- Mishra, P.K.; Adameová, A.; Hill, J.A.; Baines, C.P.; Kang, P.M.; Downey, J.; Narula, J.; Takahashi, M.; Abbate, A.; Piristine, H.C.; et al. Guidelines for evaluating myocardial cell death. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H891–H922. [Google Scholar] [CrossRef] [PubMed]
- Hexeberg, S.; Hessevik, I.; Hexeberg, E. Intravenous lipid infusion results in myocardial lipid droplet accumulation combined with reduced myocardial performance in heparinized rabbits. Acta Physiol. Scand. 1995, 153, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen, P.; Schrauwen-Hinderling, V.; Hoeks, J.; Hesselink, M.K.C. Mitochondrial dysfunction and lipotoxicity. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 266–271. [Google Scholar] [CrossRef]
- Son, N.H.; Park, T.S.; Yamashita, H.; Yokoyama, M.; Huggins, L.A.; Okajima, K.; Homma, S.; Szabolcs, M.J.; Huang, L.S.; Goldberg, I.J. Cardiomyocyte expression of PPARγ leads to cardiac dysfunction in mice. J. Clin. Investig. 2007, 117, 2791–2801. [Google Scholar] [CrossRef]
- George, A.K.; Singh, M.; Homme, R.P.; Majumder, A.; Tyagi, S. Role of Hydrogen Sulfide (H2S) on Homocysteine Mediated Glutamate Excitotoxicity, Endoplasmic Reticulum Stress and Pyroptosis in Retina. FASEB J. 2018, 32, 748.5. [Google Scholar]
- Ren, L.; Wang, Q.; Chen, Y.; Ma, Y.; Wang, D. Involvement of MicroRNA-133a in the Protective Effect of Hydrogen Sulfide against Ischemia/Reperfusion-Induced Endoplasmic Reticulum Stress and Cardiomyocyte Apoptosis. Pharmacology 2019, 103, 1–9. [Google Scholar] [CrossRef]
- Bazzano, L.A.; Reynolds, K.; Holder, K.N.; He, J. Effect of Folic Acid Supplementation on Risk of Cardiovascular Diseases. JAMA 2006, 296, 2720. [Google Scholar] [CrossRef] [PubMed]
- Rios, E.C.S.; Szczesny, B.; Soriano, F.G.; Olah, G.; Szabo, C. Hydrogen sulfide attenuates cytokine production through the modulation of chromatin remodeling. Int. J. Mol. Med. 2015, 35, 1741–1746. [Google Scholar] [CrossRef] [PubMed]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen sulfide mediates cardioprotection through nrf2 signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Xiao, T.; Long, J.; Liu, M.; Li, Z.; Liu, S.; Yang, J. Hydrogen sulfide alleviates myocardial fibrosis in mice with alcoholic cardiomyopathy by downregulating autophagy. Int. J. Mol. Med. 2017, 40, 1781–1791. [Google Scholar] [CrossRef]
- Contreras, C.; González-García, I.; Martínez-Sánchez, N.; Seoane-Collazo, P.; Jacas, J.; Morgan, D.A.; Serra, D.; Gallego, R.; Gonzalez, F.; Casals, N.; et al. Central ceramide-induced hypothalamic lipotoxicity and ER stress regulate energy balance. Cell Rep. 2014, 9, 366–377. [Google Scholar] [CrossRef]
- Menu, P.; Mayor, A.; Zhou, R.; Tardivel, A.; Ichijo, H.; Mori, K.; Tschopp, J. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. 2012, 3, e261. [Google Scholar] [CrossRef]
- Khadir, A.; Kavalakatt, S.; Abubaker, J.; Cherian, P.; Madhu, D.; Al-Khairi, I.; Abu-Farha, M.; Warsame, S.; Elkum, N.; Dehbi, M.; et al. Physical exercise alleviates ER stress in obese humans through reduction in the expression and release of GRP78 chaperone. Metabolism 2016, 65, 1409–1420. [Google Scholar] [CrossRef]
- Barr, L.A.; Shimizu, Y.; Lambert, J.P.; Nicholson, C.K.; Calvert, J.W. Hydrogen sulfide attenuates high fat diet-induced cardiac dysfunction via the suppression of endoplasmic reticulum stress. Nitric Oxide Biol. Chem. 2015, 46, 145–156. [Google Scholar] [CrossRef]
- Polhemus, D.J.; Li, Z.; Pattillo, C.B.; Gojon, G.; Gojon, G.; Giordano, T.; Krum, H. A Novel Hydrogen Sulfide Prodrug, SG1002, Promotes Hydrogen Sulfide and Nitric Oxide Bioavailability in Heart Failure Patients. Cardiovasc. Ther. 2015, 33, 216–226. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | ND (n = 8) | HFD (n = 8) | HFDEX (n = 8) | NDEX (n = 8) |
|---|---|---|---|---|
| Body Weight (g) | 26.7 ± 0.57 | 35.6 ± 2.13 * | 35.2 ± 1.04 * | 27.2 ± 0.43 |
| Heart Weight (mg) | 124 ± 5.92 | 137 ± 6.82 | 146 ± 7.01 | 125 ± 3.43 |
| HW/TL (mg/mm) | 6.87 ± 0.35 | 7.81 ± 0.31 | 8.23 ± 0.38 | 7.08 ± 0.18 |
| Fasting Glucose (mg/dL) | 162 ± 9.13 | 213 ± 13.5 * | 182 ± 13.5 # | 136 ± 10.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kar, S.; Shahshahan, H.R.; Hackfort, B.T.; Yadav, S.K.; Yadav, R.; Kambis, T.N.; Lefer, D.J.; Mishra, P.K. Exercise Training Promotes Cardiac Hydrogen Sulfide Biosynthesis and Mitigates Pyroptosis to Prevent High-Fat Diet-Induced Diabetic Cardiomyopathy. Antioxidants 2019, 8, 638. https://doi.org/10.3390/antiox8120638
Kar S, Shahshahan HR, Hackfort BT, Yadav SK, Yadav R, Kambis TN, Lefer DJ, Mishra PK. Exercise Training Promotes Cardiac Hydrogen Sulfide Biosynthesis and Mitigates Pyroptosis to Prevent High-Fat Diet-Induced Diabetic Cardiomyopathy. Antioxidants. 2019; 8(12):638. https://doi.org/10.3390/antiox8120638
Chicago/Turabian StyleKar, Sumit, Hamid R. Shahshahan, Bryan T. Hackfort, Santosh K. Yadav, Roopali Yadav, Tyler N. Kambis, David J. Lefer, and Paras K. Mishra. 2019. "Exercise Training Promotes Cardiac Hydrogen Sulfide Biosynthesis and Mitigates Pyroptosis to Prevent High-Fat Diet-Induced Diabetic Cardiomyopathy" Antioxidants 8, no. 12: 638. https://doi.org/10.3390/antiox8120638
APA StyleKar, S., Shahshahan, H. R., Hackfort, B. T., Yadav, S. K., Yadav, R., Kambis, T. N., Lefer, D. J., & Mishra, P. K. (2019). Exercise Training Promotes Cardiac Hydrogen Sulfide Biosynthesis and Mitigates Pyroptosis to Prevent High-Fat Diet-Induced Diabetic Cardiomyopathy. Antioxidants, 8(12), 638. https://doi.org/10.3390/antiox8120638