C-Terminal Redox Domain of Arabidopsis APR1 Is a Non-Canonical Thioredoxin Domain with Glutaredoxin Function


Abstract
1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Size Exclusion Chromatography
2.3. Circular Dichroism (CD) Spectroscopy
2.4. Crystallization, Crystal Data Collection, Structure Determination, and Refinement
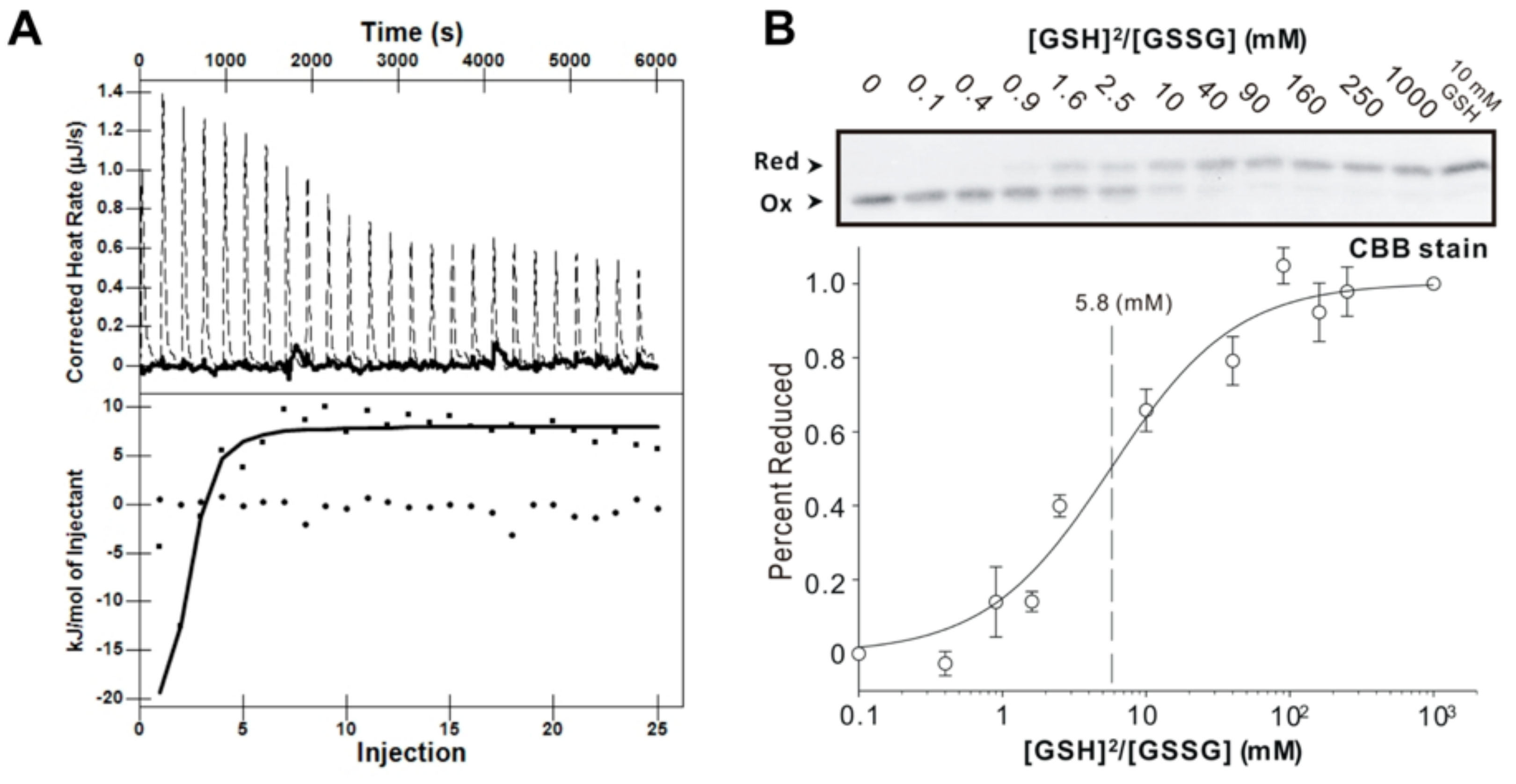
2.5. Isothermal Titration Calorimetry (ITC)
2.6. Redox Potential Assay
2.7. Molecular Modeling of Protein Complex
3. Results and Discussion
3.1. Biochemical Properties of AtAPR1 C-Terminal Redox Domain
3.2. Overall Structure of AtAPR1 Redox Domain
3.3. Structural Comparison of AtAPR1 Redox Domain
3.4. GSH Binding Ability of AtAPR1 C-Terminal Redox Domain
3.5. Redox Potential of AtAPR1 Redox Domain
3.6. Structural Model of GSH Bound to AtAPR1 Redox Domain
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brosnan, J.T.; Brosnan, M.E. The sulfur-containing amino acids: An overview. J. Nutr. 2006, 136, 1636S–1640S. [Google Scholar] [CrossRef] [PubMed]
- Fontecave, M. Iron-sulfur clusters: Ever-expanding roles. Nat. Chem. Biol. 2006, 2, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Davidian, J.-C.; Kopriva, S. Regulation of sulfate uptake and assimilation—The same or not the same? Molecular Plant 2010, 3, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Pinto, R.; Harrison, J.S.; Hsu, T.; Jacobs, W.R.; Leyh, T.S. Sulfite reduction in mycobacteria. J. Bacteriol. 2007, 189, 6714–6722. [Google Scholar] [CrossRef] [PubMed]
- Carroll, K.S.; Gao, H.; Chen, H.; Stout, C.D.; Leary, J.A.; Bertozzi, C.R. A conserved mechanism for sulfonucleotide reduction. PLoS Biol. 2005, 3, e250. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Gomes, V.; Gao, Y.; Chandramouli, K.; Johnson, M.K.; Knaff, D.B.; Leustek, T. The two-domain structure of 5’-adenylylsulfate (APS) reductase from Enteromorpha intestinalis is a requirement for efficient aps reductase activity. Biochemistry 2007, 46, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Loudet, O.; Saliba-Colombani, V.; Camilleri, C.; Calenge, F.; Gaudon, V.; Koprivova, A.; North, K.A.; Kopriva, S.; Daniel-Vedele, F. Natural variation for sulfate content in Arabidopsis thaliana is highly controlled by APR2. Nat. Genet. 2007, 39, 896–900. [Google Scholar] [CrossRef] [PubMed]
- Grant, K.; Carey, N.M.; Mendoza, M.; Schulze, J.; Pilon, M.; Pilon-Smits, E.A.; van Hoewyk, D. Adenosine 5’-phosphosulfate reductase (apr2) mutation in Arabidopsis implicates glutathione deficiency in selenate toxicity. Biochem. J. 2011, 438, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Hermsen, C.; Koprivova, A.; Matthewman, C.; Wesenberg, D.; Krauss, G.J.; Kopriva, S. Regulation of sulfate assimilation in physcomitrella patens: Mosses are different! Planta 2010, 232, 461–470. [Google Scholar] [CrossRef]
- Lee, B.R.; Koprivova, A.; Kopriva, S. The key enzyme of sulfate assimilation, adenosine 5’-phosphosulfate reductase, is regulated by hy5 in Arabidopsis. Plant J. 2011, 67, 1042–1054. [Google Scholar] [CrossRef]
- McCallum, J.; Thomas, L.; Shaw, M.; Pither-Joyce, M.; Leung, S.; Cumming, M.; McManus, M.T. Genotypic variation in the sulfur assimilation and metabolism of onion (Allium cepa l.) I. Plant composition and transcript accumulation. Phytochemistry 2011, 72, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.; Leung, S.; Cumming, M.; Shaw, M.; Albert, N.; McCallum, J.; McManus, M.T. Genotypic variation in sulphur assimilation and metabolism of onion (Allium cepa l.). II: Characterisation of ATP sulphurylase activity. Phytochemistry 2011, 72, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Vauclare, P.; Kopriva, S.; Fell, D.; Suter, M.; Sticher, L.; von Ballmoos, P.; Krahenbuhl, U.; den Camp, R.O.; Brunold, C. Flux control of sulphate assimilation in Arabidopsis thaliana: Adenosine 5’-phosphosulphate reductase is more susceptible than atp sulphurylase to negative control by thiols. Plant J. 2002, 31, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Koprivova, A.; Kopriva, S. Lessons from investigation of regulation of APS reductase by salt stress. Plant Signal Behav. 2008, 3, 567–569. [Google Scholar] [CrossRef] [PubMed]
- Tsakraklides, G.; Martin, M.; Chalam, R.; Tarczynski, M.C.; Schmidt, A.; Leustek, T. Sulfate reduction is increased in transgenic Arabidopsis thaliana expressing 5’-adenylylsulfate reductase from Pseudomonas aeruginosa. Plant J. 2002, 32, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.N.; Tarczynski, M.C.; Shen, B.; Leustek, T. The role of 5’-adenylylsulfate reductase in controlling sulfate reduction in plants. Photosynth. Res. 2005, 86, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Randewig, D.; Hamisch, D.; Herschbach, C.; Eiblmeier, M.; Gehl, C.; Jurgeleit, J.; Skerra, J.; Mendel, R.R.; Rennenberg, H.; Hansch, R. Sulfite oxidase controls sulfur metabolism under SO2 exposure in Arabidopsis thaliana. Plant Cell Environ. 2012, 35, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Prior, A.; Uhrig, J.F.; Heins, L.; Wiesmann, A.; Lillig, C.H.; Stoltze, C.; Soll, J.; Schwenn, J.D. Structural and kinetic properties of adenylyl sulfate reductase from Catharanthus roseus cell cultures. Biochimica Biophysica Acta 1999, 1430, 25–38. [Google Scholar] [CrossRef]
- Bick, J.A.; Aslund, F.; Chen, Y.; Leustek, T. Glutaredoxin function for the carboxyl-terminal domain of the plant-type 5’-adenylylsulfate reductase. Proc. Natl. Acad. Sci. USA 1998, 95, 8404–8409. [Google Scholar] [CrossRef]
- Bick, J.A.; Leustek, T. Plant sulfur metabolism—The reduction of sulfate to sulfite. Curr. Opin. Plant Biol. 1998, 1, 240–244. [Google Scholar] [CrossRef]
- Chen, F.F.; Chang, Y.Y.; Cho, C.C.; Hsu, C.H. Crystallization of the c-terminal redox domain of the sulfur-assimilatory enzyme apr1 from Arabidopsis thaliana. Acta Crystallogr. F Struct. Biol. Commun. 2014, 70, 1211–1214. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Sreerama, N.; Woody, R.W. Estimation of protein secondary structure from circular dichroism spectra: Comparison of contin, selcon, and cdsstr methods with an expanded reference set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [PubMed]
- Long, F.; Vagin, A.A.; Young, P.; Murshudov, G.N. Balbes: A molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, M.; Maegawa, K.; Suzuki, M.; Ushioda, R.; Araki, K.; Matsumoto, Y.; Hoseki, J.; Nagata, K.; Inaba, K. Structural basis of an erad pathway mediated by the er-resident protein disulfide reductase ERdj5. Mol. Cell 2011, 41, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.Refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. Phenix: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. Molprobity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Macarthur, M.W.; Moss, D.S.; Thornton, J.M. Procheck—A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Hsu, C.H.; Shen, T.L.; Chang, C.F.; Chang, Y.Y.; Huang, L.Y. Solution structure of the oncogenic mien1 protein reveals a thioredoxin-like fold with a redox-active motif. PLoS ONE 2012, 7, e52292. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. Nih image to imagej: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Rost, J.; Rapoport, S. Reduction-potential of glutathione. Nature 1964, 201, 185. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.H.; Kim, H.Y.; Hwang, K.Y. The gsh- and gssg-bound structures of glutaredoxin from clostridium oremlandii. Arch. Biochem. Biophys. 2014, 564, 20–25. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. Gromacs: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. Ligplot: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Suter, M.; von Ballmoos, P.; Kopriva, S.; den Camp, R.O.; Schaller, J.; Kuhlemeier, C.; Schurmann, P.; Brunold, C. Adenosine 5’-phosphosulfate sulfotransferase and adenosine 5’-phosphosulfate reductase are identical enzymes. J. Biol. Chem. 2000, 275, 930–936. [Google Scholar] [CrossRef]
- Kopriva, S.; Koprivova, A. Plant adenosine 5’-phosphosulphate reductase: The past, the present, and the future. J. Exp. Bot. 2004, 55, 1775–1783. [Google Scholar] [CrossRef]
- Rinaldelli, M.; Ravera, E.; Calderone, V.; Parigi, G.; Murshudov, G.N.; Luchinat, C. Simultaneous use of solution nmr and X-ray data in REFMAC5 for joint refinement/detection of structural differences. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 958–967. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Rosenström, P. Dali server: Conservation mapping in 3d. Nucleic. Acids Res. 2010, 38, W545–W549. [Google Scholar] [CrossRef]
- Alon, A.; Grossman, I.; Gat, Y.; Kodali, V.K.; DiMaio, F.; Mehlman, T.; Haran, G.; Baker, D.; Thorpe, C.; Fass, D. The dynamic disulphide relay of quiescin sulphydryl oxidase. Nature 2012, 488, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Gat, Y.; Vardi-Kilshtain, A.; Grossman, I.; Major, D.T.; Fass, D. Enzyme structure captures four cysteines aligned for disulfide relay. Protein Sci. 2014, 23, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Haq, I. Thermodynamics of drug-DNA interactions. Arch. Biochem. Biophys. 2002, 403, 1–15. [Google Scholar] [CrossRef]
- Foyer, C.H.; Halliwell, B. The presence of glutathione and glutathione reductase in chloroplasts: A proposed role in ascorbic acid metabolism. Planta 1976, 133, 21–25. [Google Scholar] [CrossRef]
- Garcia Donoso, C.; Guerra Tapia, A.; Iglesias Diez, L. Nodular lesion and generalized pruritus on plantar. Cutaneous filariasis type onchocerca volvulus without ocular presentation. Rev. Clin. Esp. 2000, 200, 449–450. [Google Scholar]
- Collin, V.; Issakidis-Bourguet, E.; Marchand, C.; Hirasawa, M.; Lancelin, J.M.; Knaff, D.B.; Miginiac-Maslow, M. The arabidopsis plastidial thioredoxins: New functions and new insights into specificity. J. Biol. Chem. 2003, 278, 23747–23752. [Google Scholar] [CrossRef]
- Hirasawa, M.; Schurmann, P.; Jacquot, J.P.; Manieri, W.; Jacquot, P.; Keryer, E.; Hartman, F.C.; Knaff, D.B. Oxidation-reduction properties of chloroplast thioredoxins, ferredoxin:Thioredoxin reductase, and thioredoxin f-regulated enzymes. Biochemistry 1999, 38, 5200–5205. [Google Scholar] [CrossRef]
- Kim, S.G.; Chi, Y.H.; Lee, J.S.; Schlesinger, S.R.; Zabet-Moghaddam, M.; Chung, J.S.; Knaff, D.B.; Kim, S.T.; Lee, S.Y.; Kim, S.K. Redox properties of a thioredoxin-like arabidopsis protein, attdx. Biochimica Biophysica Acta 2010, 1804, 2213–2221. [Google Scholar] [CrossRef]
- Chibani, K.; Tarrago, L.; Schurmann, P.; Jacquot, J.P.; Rouhier, N. Biochemical properties of poplar thioredoxin z. FEBS Lett. 2011, 585, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Dangoor, I.; Peled-Zehavi, H.; Levitan, A.; Pasand, O.; Danon, A. A small family of chloroplast atypical thioredoxins. Plant Physiol. 2009, 149, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Aslund, F.; Berndt, K.D.; Holmgren, A. Redox potentials of glutaredoxins and other thiol-disulfide oxidoreductases of the thioredoxin superfamily determined by direct protein-protein redox equilibria. J. Biol. Chem. 1997, 272, 30780–30786. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Manzaneque, M.T.; Tamarit, J.; Belli, G.; Ros, J.; Herrero, E. Grx5 is a mitochondrial glutaredoxin required for the activity of iron/sulfur enzymes. Mol. Biol. Cell 2002, 13, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.L. Thioredoxin—a fold for all reasons. Structure 1995, 3, 245–250. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. Ligplot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Parameters | |
|---|---|
| Crystal | AtAPR1 redox domain |
| Space Group | P43212 |
| Unit cell parameters (Å) | a = b = 58.2; c= 86.7 |
| Monomers per asymmetric unit cell | 1 |
| Data collection | |
| Wavelength (Å) | 1.00 |
| Resolution range (Å) | 24.2–2.70 (2.80–2.70) |
| Unique no. of reflections | 4467 (432) |
| Total no. of reflections | 57471 (5450) |
| I/σ a | 32.4 (4.7) |
| Rmergea,b | 0.079 (0.660) |
| Completeness (%) | 99.6 (100.0) |
| Refinement statistics | |
| Resolution (Å) | 2.70 |
| Rwork (%)/Rfree (%) c | 17.93/25.04 |
| RMSD | |
| Bonds (Å) | 0.008 |
| Angles (o) | 1.19 |
| Mean B-factor (Å2) | 72.0 |
| Protein | 72.0 |
| Water | 66.7 |
| Ramachandran plot (%) | |
| Favored | 97.0 |
| Allowed | 3.0 |
| Outliers | 0.0 |
| Ligand | Kd (µM) | n | ∆H (KJ/mol) | –T∆S (KJ/mol) | ∆G (KJ/mol) |
|---|---|---|---|---|---|
| GSH | 2.84 ± 0.29 | 0.90 ± 0.03 | −9.45 ± 2.17 | −23.92 ± 6.15 | −31.56 ± 0.13 |
| GSSG | ND | ND | ND | ND | ND |
| Protein | Organism | Localization/type | Midpoint Redox Potentials (Em,7.0, mV) | References |
|---|---|---|---|---|
| Trx-m2 | A. thaliana | Chloroplastic thioredoxin | −368 | [48] |
| Trx-f1 | A. thaliana | Chloroplastic thioredoxin | −351 | [48] |
| Trx-m | S. oleracea | Chloroplastic thioredoxin | −300 | [49] |
| AtTDX | A. thaliana | Cytosolic and nuclear thioredoxin | −260 | [50] |
| Trx-z | P. trichocarpa | Chloroplastic thioredoxin | −251 | [51] |
| AtACHT4 | A. thaliana | Chloroplastic thioredoxin | −240 | [52] |
| Grx1 | E. coli | Cytosolic glutaredoxin | −230 | [53] |
| AtAPR1C | A. thaliana | Chloroplastic glutaredoxin | −188 | This study |
| Grx3 | E. coli | Cytosolic glutaredoxin | −180 | [53] |
| Grx5 | S. cerevisiae | Mitochondrial glutaredoxin | −175 | [54] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, F.-F.; Chien, C.-Y.; Cho, C.-C.; Chang, Y.-Y.; Hsu, C.-H. C-Terminal Redox Domain of Arabidopsis APR1 Is a Non-Canonical Thioredoxin Domain with Glutaredoxin Function. Antioxidants 2019, 8, 461. https://doi.org/10.3390/antiox8100461
Chen F-F, Chien C-Y, Cho C-C, Chang Y-Y, Hsu C-H. C-Terminal Redox Domain of Arabidopsis APR1 Is a Non-Canonical Thioredoxin Domain with Glutaredoxin Function. Antioxidants. 2019; 8(10):461. https://doi.org/10.3390/antiox8100461
Chicago/Turabian StyleChen, Fang-Fang, Chia-Yu Chien, Chao-Cheng Cho, Yu-Yung Chang, and Chun-Hua Hsu. 2019. "C-Terminal Redox Domain of Arabidopsis APR1 Is a Non-Canonical Thioredoxin Domain with Glutaredoxin Function" Antioxidants 8, no. 10: 461. https://doi.org/10.3390/antiox8100461
APA StyleChen, F.-F., Chien, C.-Y., Cho, C.-C., Chang, Y.-Y., & Hsu, C.-H. (2019). C-Terminal Redox Domain of Arabidopsis APR1 Is a Non-Canonical Thioredoxin Domain with Glutaredoxin Function. Antioxidants, 8(10), 461. https://doi.org/10.3390/antiox8100461