Unbiased Identification of Proteins Covalently Modified by Complex Mixtures of Peroxidized Lipids Using a Combination of Electrophoretic Mobility Band Shift with Mass Spectrometry


Abstract
1. Introduction
2. Materials and Methods
2.1. Lipids and Cell Culture
2.2. Isolation of HUVEC Proteomes
2.3. Electrophoretic Mobility Band Shift Analysis (EMSA)
2.4. In-Gel Protein Digestion and Nano-LC-MS/MS Analysis
2.5. Data Analysis
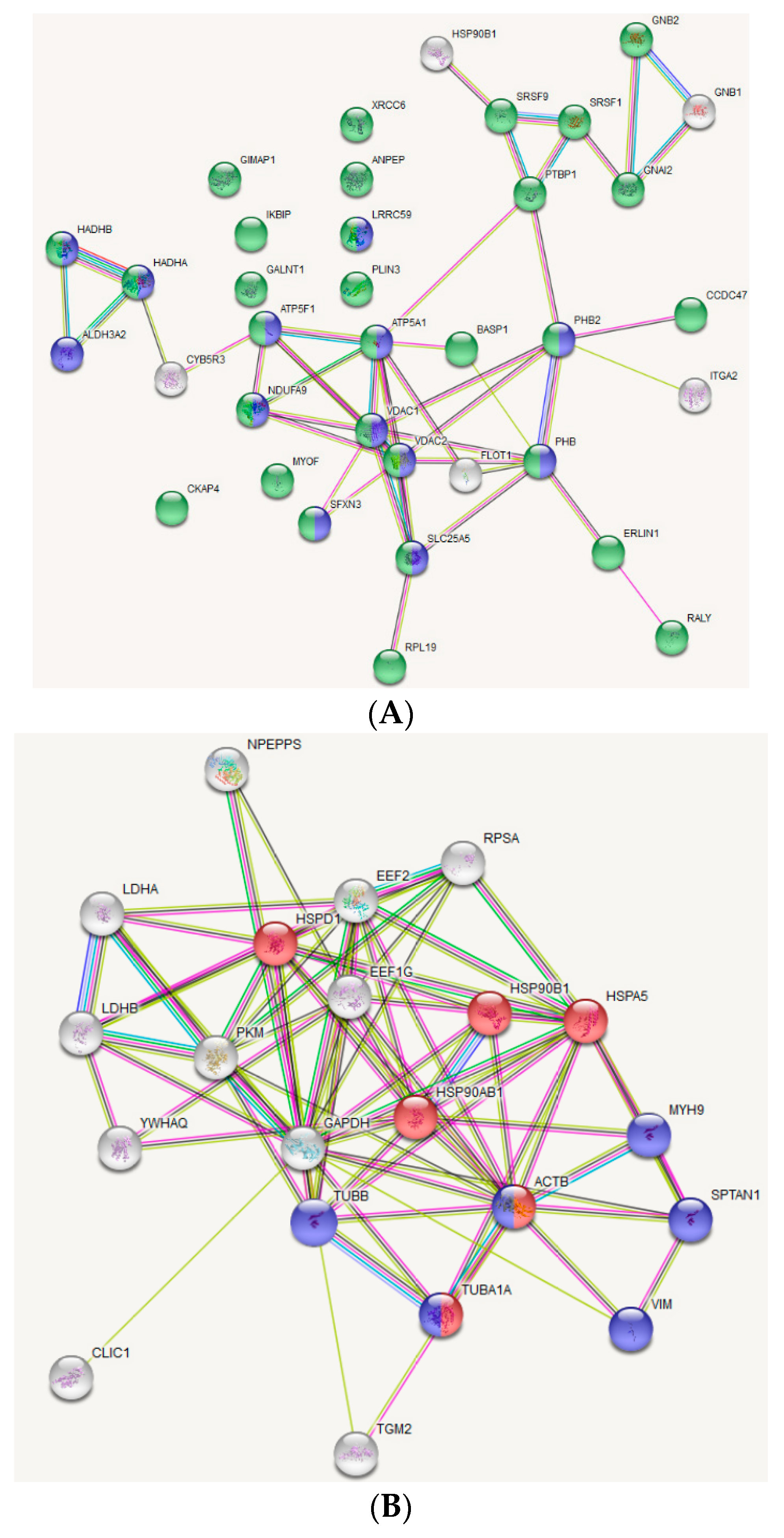
2.6. Protein-Protein Interaction Network Construction and Functional Annotation
2.7. Western Blotting
3. Results
3.1. Incubation with OxPLs Changes the Electrophoretic Mobility of Certain Proteins
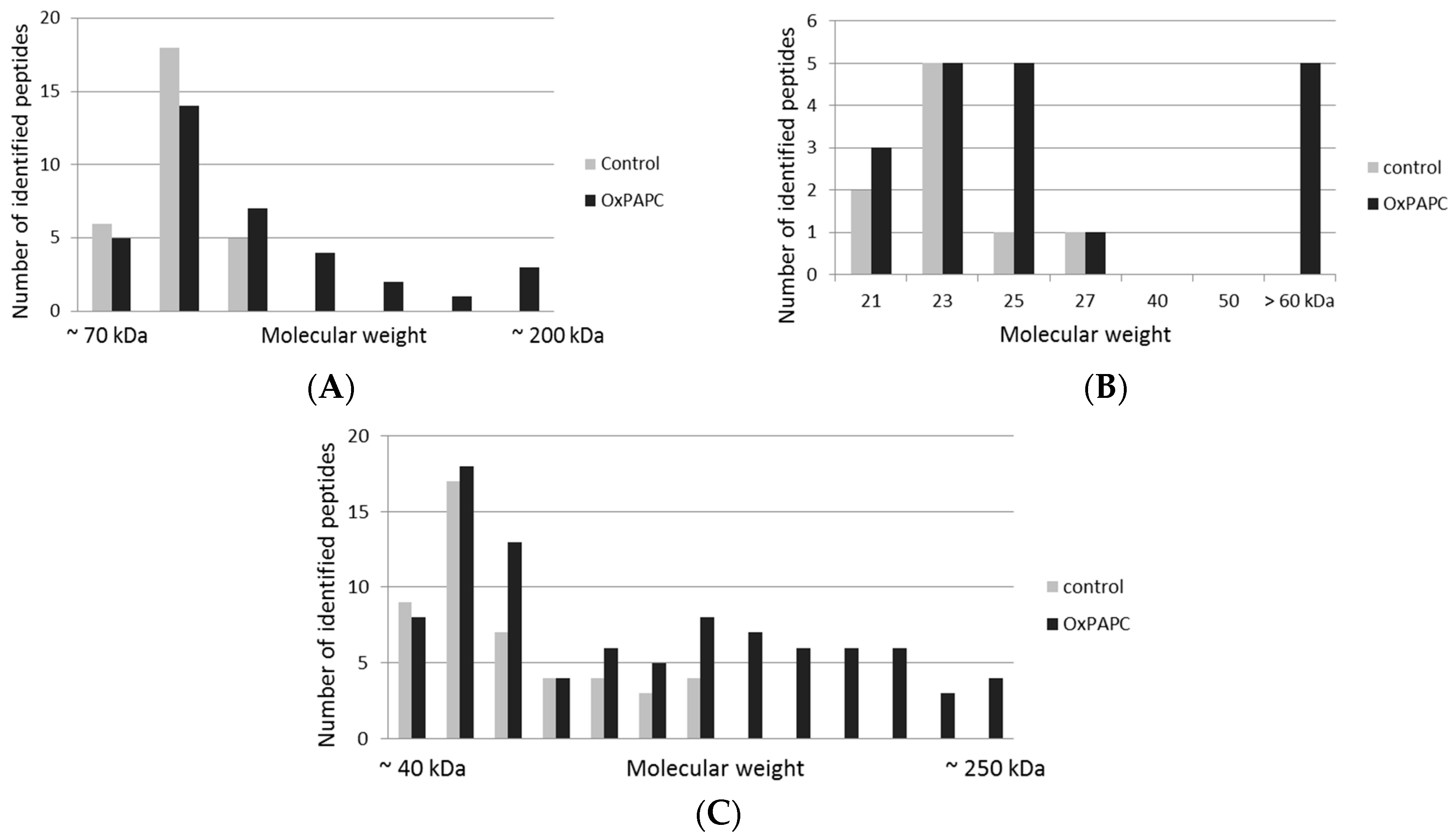
3.2. Protein Targets of OxPAPC Identified by LC-MS/MS
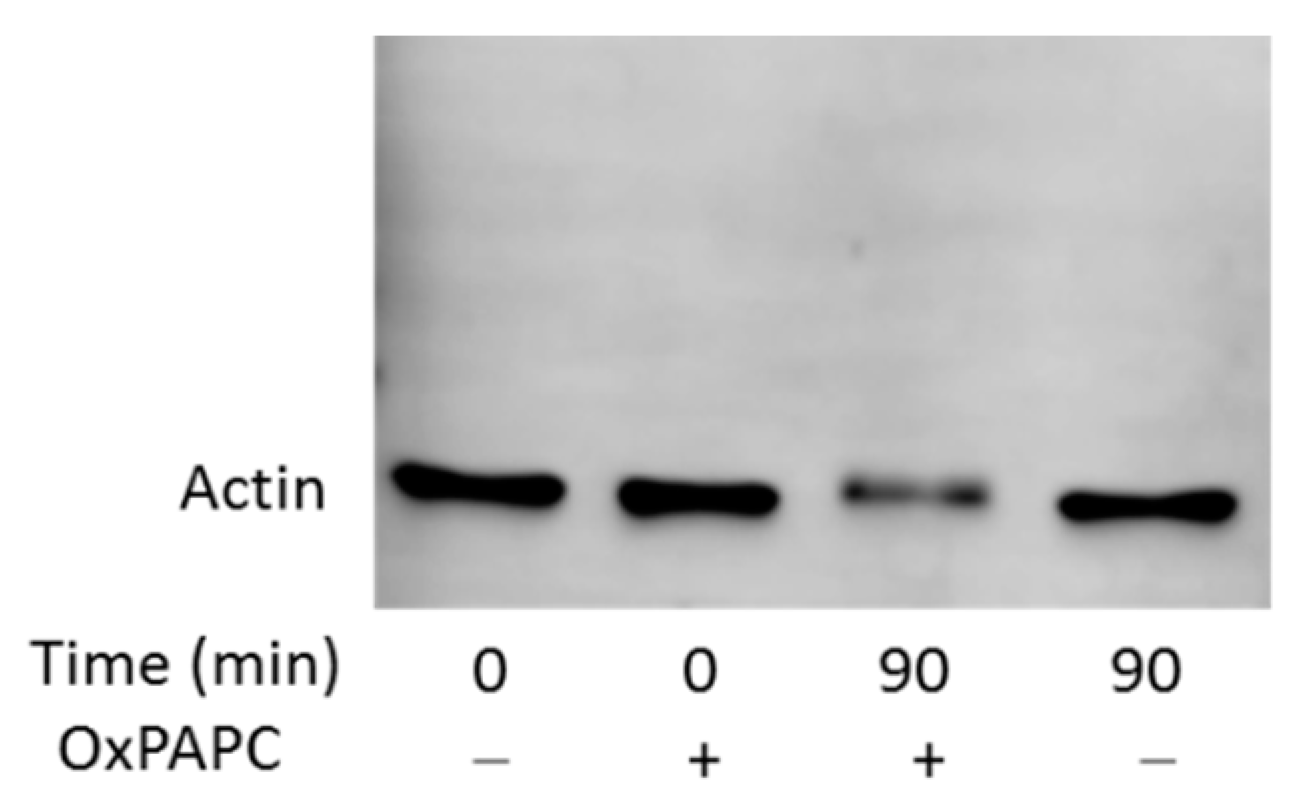
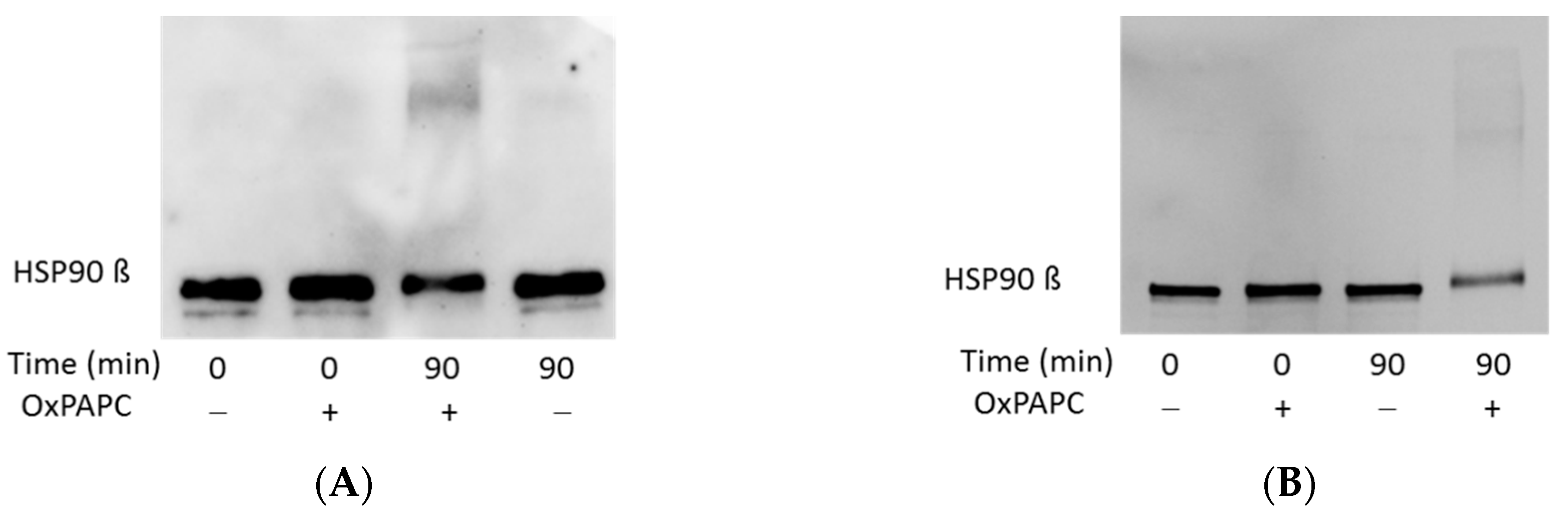
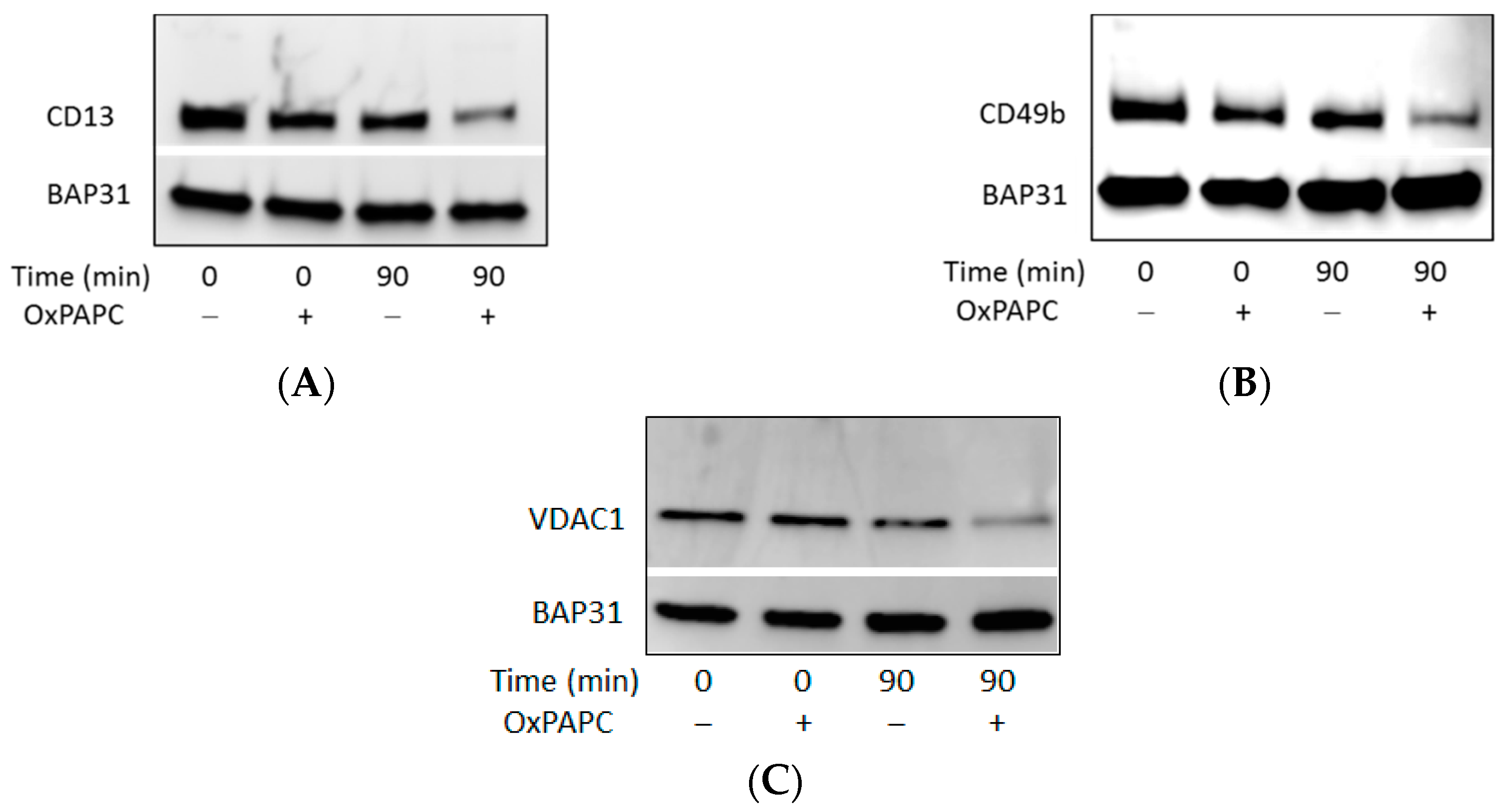
3.3. Confirmation of OxPAPC-Induced Protein Modifications by Western Blotting
3.4. OxPAPC Modifies Proteins in Live HUVECs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BAP31 | B-cell receptor-associated protein 31 |
| emPAI | Exponentially Modified Protein Abundance Index |
| EMSA | Electrophoretic mobility band shift analysis |
| HNE | 4-hydroxynonenal |
| HRP | horseradish peroxidase |
| HSP90β | heat shock protein HSP 90-beta |
| HUVEC | human umbilical vein endothelial cell |
| KODA-PC | 9-keto-12-oxo-10-dodecenoate-phosphatidylcholine |
| LOP | lipid oxidation product |
| MS | mass spectrometry |
| OxPAPC | oxidized palmitoyl-arachidonoyl-phosphatidylcholine |
| oxPL | oxidized phospholipid |
| PAPC | palmitoyl-arachidonoyl-phosphatidylcholine |
| PAPE | 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine |
| PAPS | 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phospho-l-serine |
| PBS | Phosphate-buffered saline |
| PC | phosphatidylcholine |
| PE | phosphoethanolamine |
| PBS-PI | phosphate buffered containing protease inhibitor cocktail |
| RIPA buffer, | Radioimmunoprecipitation assay buffer |
| PL | phospholipid |
| PLPC | 1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphatidylcholine |
| PUFA | polyunsaturated fatty acid |
| SDS | sodium dodecyl sulfate |
| VDAC-1 | voltage-dependent anion-selective channel protein 1 |
References
- Bochko, V.N.; Oskolkova, O.V.; Birukov, K.G.; Levonen, A.L.; Binder, C.J.; Stöckl, J. Generation and biological activities of oxidized phospholipids. Antioxid. Redox Signal. 2010, 12, 1009–1059. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Birukov, K.G.; Romanoski, C.E.; Springstead, J.R.; Lusis, A.J.; Berliner, J.A. Role of phospholipid oxidation products in atherosclerosis. Circ. Res. 2012, 111, 778–799. [Google Scholar] [CrossRef] [PubMed]
- Negre-Salvayre, A.; Coatrieux, C.; Ingueneau, C.; Salvayre, R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br. J. Pharmacol. 2008, 153, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Adibhatla, R.M.; Hatcher, J.F. Lipid oxidation and peroxidation in CNS health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 125–169. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, V.N. Inflammatory profile of oxidized phospholipids. Thromb. Haemost. 2007, 97, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, V.; Gesslbauer, B.; Mauerhofer, C.; Philippova, M.; Erne, P.; Oskolkova, O.V. Pleiotropic effects of oxidized phospholipids. Free Radic. Biol. Med. 2017, 111, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Ullery, J.C.; Marnett, L.J. Protein modification by oxidized phospholipids and hydrolytically released lipid electrophiles: Investigating cellular responses. Biochim. Biophys. Acta 2012, 1818, 2424–2435. [Google Scholar] [CrossRef] [PubMed]
- West, J.D.; Marnett, L.J. Endogenous reactive intermediates as modulators of cell signaling and cell death. Chem. Res. Toxicol. 2006, 19, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Kapahi, P.; Natoli, G.; Takahashi, T.; Chen, Y.; Karin, M.; Santoro, M.G. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature 2000, 403, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Golizeh, M.; Geib, T.; Sleno, L. Identification of 4-hydroxynonenal protein targets in rat, mouse and human liver microsomes by two-dimensional liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2016, 30, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Rauniyar, N.; Stevens, S.M.; Prokai-Tatrai, K.; Prokai, L. Characterization of 4-hydroxy-2-nonenal-modified peptides by liquid chromatography-tandem mass spectrometry using data-dependent acquisition: Neutral loss-driven MS3 versus neutral loss-driven electron capture dissociation. Anal. Chem. 2009, 81, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Andringa, K.K.; Udoh, U.S.; Landar, A.; Bailey, S.M. Proteomic analysis of 4-hydroxynonenal (4-HNE) modified proteins in liver mitochondria from chronic ethanol-fed rats. Redox Biol. 2014, 2, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.; Hernaez, M.L.; Diez, A.; Puyet, A.; Bautista, J.M. Combined proteomic approaches for the identification of specific amino acid residues modified by 4-hydroxy-2-nonenal under physiological conditions. J. Proteome Res. 2010, 9, 5770–5781. [Google Scholar] [CrossRef] [PubMed]
- Reed, T.T.; Pierce, W.M.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of HNE-bound proteins in early Alzheimer disease: Insights into the role of lipid peroxidation in the progression of AD. Brain Res. 2009, 1274, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cong, Y.; Quan, B.; Lan, T.; Chu, X.; Ye, Z.; Hou, X.; Wang, C. Chemoproteomic profiling of targets of lipid-derived electrophiles by bioorthogonal aminooxy probe. Redox Biol. 2017, 12, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Codreanu, S.G.; Zhang, B.; Sobecki, S.M.; Billheimer, D.D.; Liebler, D.C. Global analysis of protein damage by the lipid electrophile 4-hydroxy-2-nonenal. Mol. Cell. Proteomics 2009, 8, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Roe, M.R.; Xie, H.; Bandhakavi, S.; Griffin, T.J. Proteomic mapping of 4-hydroxynonenal protein modification sites by solid-phase hydrazide chemistry and mass spectrometry. Anal. Chem. 2007, 79, 3747–3756. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Stevens, J.F.; Maier, C.S. Design, synthesis, and application of a hydrazide-functionalized isotope-coded affinity tag for the quantification of oxylipid-protein conjugates. Anal. Chem. 2007, 79, 3342–3354. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.; Wu, J.; Han, B.; Chung, W.G.; Maier, C.S. New role for an old probe: Affinity labeling of oxylipid protein conjugates by N’-aminooxymethylcarbonylhydrazino d-biotin. Anal. Chem. 2006, 78, 6847–6854. [Google Scholar] [CrossRef] [PubMed]
- Vila, A.; Tallman, K.A.; Jacobs, A.T.; Liebler, D.C.; Porter, N.A.; Marnett, L.J. Identification of protein targets of 4-hydroxynonenal using click chemistry for ex vivo biotinylation of azido and alkynyl derivatives. Chem. Res. Toxicol. 2008, 21, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Codreanu, S.G.; Ullery, J.C.; Zhu, J.; Tallman, K.A.; Beavers, W.N.; Porter, N.A.; Marnett, L.J.; Zhang, B.; Liebler, D.C. Alkylation damage by lipid electrophiles targets functional protein systems. Mol. Cell. Proteomics 2014, 13, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Weerapana, E.; Blewett, M.M.; Cravatt, B.F. A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat. Methods 2014, 11, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Gugiu, B.G.; Mouillesseaux, K.; Duong, V.; Herzog, T.; Hekimian, A.; Koroniak, L.; Vondriska, T.M.; Watson, A.D. Protein targets of oxidized phospholipids in endothelial cells. J. Lipid Res. 2008, 49, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Tallman, K.A.; Kim, H.Y.; Ji, J.X.; Szapacs, M.E.; Yin, H.; McIntosh, T.J.; Liebler, D.C.; Porter, N.A. Phospholipid-protein adducts of lipid peroxidation: Synthesis and study of new biotinylated phosphatidylcholines. Chem. Res. Toxicol. 2007, 20, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Szapacs, M.E.; Kim, H.Y.; Porter, N.A.; Liebler, D.C. Identification of proteins adducted by lipid peroxidation products in plasma and modifications of apolipoprotein A1 with a novel biotinylated phospholipid probe. J. Proteome Res. 2008, 7, 4237–4246. [Google Scholar] [CrossRef] [PubMed]
- Stemmer, U.; Ramprecht, C.; Zenzmaier, E.; Stojčić, B.; Rechberger, G.; Kollroser, M.; Hermetter, A. Uptake and protein targeting of fluorescent oxidized phospholipids in cultured RAW 264.7 macrophages. Biochim. Biophys. Acta 2012, 1821, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Willard, B.; Podrez, E.A. Analysis of covalent modifications of proteins by oxidized phospholipids using a novel method of peptide enrichment. Anal. Chem. 2014, 86, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Oskolkova, O.V.; Afonyushkin, T.; Leitner, A.; Schlieffen, E.V.; Gargalovic, P.S.; Lusis, A.J.; Binder, B.R.; Bochkov, V.N. ATF4-dependent transcription is a key mechanism in VEGF up-regulation by oxidized phospholipids: critical role of oxidized sn-2 residues in activation of unfolded protein response. Blood 2008, 112, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef] [PubMed]
- Ishihama, Y.; Oda, Y.; Tabata, T.; Sato, T.; Nagasu, T.; Rappsilber, J.; Mann, M. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics 2005, 4, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Afonyushkin, T.; Oskolkova, O.V.; Philippova, M.; Resink, T.J.; Erne, P.; Binder, B.R.; Bochkov, V.N. Oxidized phospholipids regulate expression of ATF4 and VEGF in endothelial cells via NRF2-dependent mechanism: novel point of convergence between electrophilic and unfolded protein stress pathways. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Pecorelli, A.; Cervellati, C.; Cortelazzo, A.; Cervellati, F.; Sticozzi, C.; Mirasole, C.; Guerranti, R.; Trentini, A.; Zolla, L.; Savelli, V.; et al. Proteomic analysis of 4-hydroxynonenal and nitrotyrosine modified proteins in RTT fibroblasts. Int. J. Biochem. Cell Biol. 2016, 81, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Usatyuk, P.V.; Natarajan, V. Hydroxyalkenals and oxidized phospholipids modulation of endothelial cytoskeleton, focal adhesion and adherens junction proteins in regulating endothelial barrier function. Microvasc. Res. 2012, 83, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.; Chung, W.G.; Miranda, C.L.; Singhal, M.; Stevens, J.F.; Maier, C.S. Site-specific protein adducts of 4-hydroxy-2(E)-nonenal in human THP-1 monocytic cells: protein carbonylation is diminished by ascorbic acid. Chem. Res. Toxicol. 2010, 23, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.K.; Rogowska-Wrzesinska, A.; Roepstorff, P.; Bulteau, A.L.; Friguet, B. Protein modification and replicative senescence of WI-38 human embryonic fibroblasts. Aging Cell. 2010, 9, 252–272. [Google Scholar] [CrossRef] [PubMed]
- Vladykovskaya, E.; Sithu, S.D.; Haberzettl, P.; Wickramasinghe, N.S.; Merchant, M.L.; Hill, B.G.; McCracken, J.; Agarwal, A.; Dougherty, S.; Gordon, S.A.; et al. Lipid peroxidation product 4-hydroxy-trans-2-nonenal causes endothelial activation by inducing endoplasmic reticulum stress. J. Biol. Chem. 2012, 287, 11398–11409. [Google Scholar] [CrossRef] [PubMed]
- Beavers, W.N.; Rose, K.L.; Galligan, J.J.; Mitchener, M.M.; Rouzer, C.A.; Tallman, K.A.; Lamberson, C.R.; Wang, X.; Hill, S.; Ivanova, P.T.; et al. Protein Modification by Endogenously Generated Lipid Electrophiles: Mitochondria as the Source and Target. ACS Chem. Biol. 2017, 12, 2062–2069. [Google Scholar] [CrossRef] [PubMed]
- Landar, A.; Zmijewski, J.W.; Dickinson, D.A.; Le, G.C.; Johnson, M.S.; Milne, G.L.; Zanoni, G.; Vidari, G.; Morrow, J.D.; Darley-Usmar, V.M. Interaction of electrophilic lipid oxidation products with mitochondria in endothelial cells and formation of reactive oxygen species. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1777–H1787. [Google Scholar] [CrossRef] [PubMed]
- Reed, T.T. Lipid peroxidation and neurodegenerative disease. Free Radic. Biol. Med. 2011, 51, 1302–1319. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Accession | Reported OxPL Target * |
|---|---|---|
| 14-3-3 protein theta | 1433T_HUMAN | |
| 40S ribosomal protein SA | RSSA_HUMAN | |
| 60 kDa heat shock protein, mitochondrial | CH60_HUMAN | |
| 78 kDa glucose-regulated protein | GRP78_HUMAN | [23,26] |
| Actin, cytoplasmic 1 | ACTB_HUMAN | [23] |
| Chloride intracellular channel protein 1 | CLIC1_HUMAN | |
| Elongation factor 1-gamma | EF1G_HUMAN | |
| Elongation factor 2 | EF2_HUMAN | |
| Endoplasmin | ENPL_HUMAN | [26] |
| Glyceraldehyde-3-phosphate dehydrogenase | G3P_HUMAN | |
| Heat shock protein HSP 90-beta | HS90B_HUMAN | |
| L-lactate dehydrogenase A chain | LDHA_HUMAN | |
| L-lactate dehydrogenase B chain | LDHB_HUMAN | |
| Myosin-9 | MYH9_HUMAN | [23] |
| Protein-glutamine gamma-glutamyltransferase 2 | TGM2_HUMAN | |
| Puromycin-sensitive aminopeptidase | PSA_HUMAN | |
| Pyruvate kinase PKM | KPYM_HUMAN | [26] |
| Spectrin alpha chain, non-erythrocytic 1 | SPTN1_HUMAN | |
| Tubulin alpha-1A chain | TBA1A_HUMAN | |
| Tubulin beta chain | TBB5_HUMAN | [23] |
| Vimentin | VIME_HUMAN | [23,26] |
| Protein | Accession | Reported OxPL Target * |
|---|---|---|
| ADP/ATP Translocase 2 | ADT2_HUMAN | |
| Aminopeptidase N | AMPN_HUMAN | |
| ATP synthase F(0) complex subunit B1 | AT5F1_HUMAN | |
| ATP synthase subunit alpha, mitochondrial | ATPA_HUMAN | [23] |
| Brain acid soluble protein 1 | BASP1_HUMAN | |
| Coiled-coil domain-containing protein 47 | CCD47_HUMAN | |
| Cytoskeleton-associated protein 4 | CKAP4_HUMAN | [23] |
| Endoplasmin | ENPL_HUMAN | [26] |
| Erlin 1 | ERLN1_HUMAN | |
| Fatty aldehyde dehydrogenase | AL3A2_HUMAN | |
| Flotillin 1 | FLOT1_HUMAN | |
| GTPase IMAP family member 1 | GIMA1_HUMAN | |
| Guanine nucleotide-binding protein G (i) subunit alpha-2 | GNAI2_HUMAN | [23] |
| Guanine nucleotide-binding protein G(I)/G(S)/G(T) subunit beta-1 | GBB1_HUMAN | [26] |
| Guanine nucleotide-binding protein G(I)/G(S)/G(T) subunit beta-2 | GBB2_HUMAN | |
| Inhibitor of nuclear factor kappa-B kinase-interacting protein | IKIP_HUMAN | |
| Integrin alpha-2 | ITA2_HUMAN | |
| Leucine-rich repeat-containing protein 59 | LRC59_HUMAN | |
| Myoferlin | MYOF_HUMAN | |
| NADH-cytochrome b5 reductase 3 | NB5R3_HUMAN | |
| NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 9 | NDUA9_HUMAN | |
| Perilipin 3 | PLIN3_HUMAN | |
| Polypeptide N-acetylgalactosaminyltransferase 1 | GALT1_HUMAN | |
| Polypyrimidine tract-binding protein 1 | PTBP1_HUMAN | |
| Prohibitin | PHB_HUMAN | |
| Prohibitin 2 | PHB2_HUMAN | [26] |
| RNA-binding protein Raly | RALY_HUMAN | |
| Serine/arginine-rich splicing factor 1 | SRSF1_HUMAN | |
| Serine/arginine-rich splicing factor 9 | SRSF9_HUMAN | |
| Sideroflexin-3 | SFXN3_HUMAN | |
| Trifunctional enzyme subunit alpha, mitochondrial | ECHA_HUMAN | |
| Trifunctional enzyme subunit beta, mitochondria | ECHB_HUMAN | [23] |
| Vesicle-trafficking protein SEC22b | SC22B_HUMAN | |
| Voltage-dependent anion-selective channel protein 1 | VDAC1_HUMAN | [26] |
| Voltage-dependent anion-selective channel protein 2 | VDAC2_HUMAN | [26] |
| X-ray repair cross-complementing protein 6 | XRCC6_HUMAN | |
| 60S ribosomal protein L19 | RL19_HUMAN |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gesslbauer, B.; Kuerzl, D.; Valpatic, N.; Bochkov, V.N. Unbiased Identification of Proteins Covalently Modified by Complex Mixtures of Peroxidized Lipids Using a Combination of Electrophoretic Mobility Band Shift with Mass Spectrometry. Antioxidants 2018, 7, 116. https://doi.org/10.3390/antiox7090116
Gesslbauer B, Kuerzl D, Valpatic N, Bochkov VN. Unbiased Identification of Proteins Covalently Modified by Complex Mixtures of Peroxidized Lipids Using a Combination of Electrophoretic Mobility Band Shift with Mass Spectrometry. Antioxidants. 2018; 7(9):116. https://doi.org/10.3390/antiox7090116
Chicago/Turabian StyleGesslbauer, Bernd, David Kuerzl, Niko Valpatic, and Valery N. Bochkov. 2018. "Unbiased Identification of Proteins Covalently Modified by Complex Mixtures of Peroxidized Lipids Using a Combination of Electrophoretic Mobility Band Shift with Mass Spectrometry" Antioxidants 7, no. 9: 116. https://doi.org/10.3390/antiox7090116
APA StyleGesslbauer, B., Kuerzl, D., Valpatic, N., & Bochkov, V. N. (2018). Unbiased Identification of Proteins Covalently Modified by Complex Mixtures of Peroxidized Lipids Using a Combination of Electrophoretic Mobility Band Shift with Mass Spectrometry. Antioxidants, 7(9), 116. https://doi.org/10.3390/antiox7090116