Lipid Peroxidation-Derived Aldehydes, 4-Hydroxynonenal and Malondialdehyde in Aging-Related Disorders




Abstract
{kind=link}
{kind=link}
{kind=link}
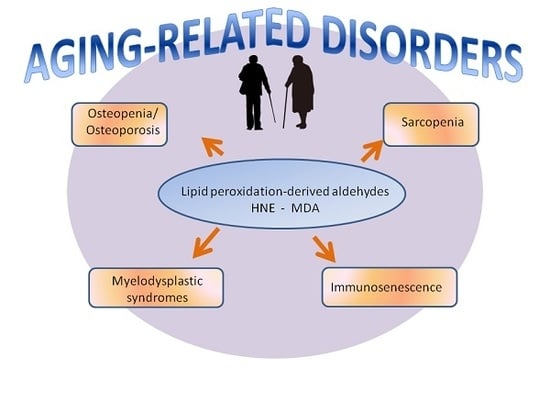
1. Introduction
Aldehyde–Protein Adduct in Human Disease
2. Osteopenia/Osteoporosis
3. Sarcopenia
4. Immunosenescence
5. Myelodysplastic Syndromes
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALDH | aldehyde dehydrogenase |
| AML | acute myeloid leukemia |
| apoB | apolipoprotein B |
| CMV | cytomegalovirus |
| DNPH | dinitrophenylhydrazine |
| HNE | 4-hydroxynonenal |
| HSC | hematopoietic stem cell |
| IkB | inihibitor of κ light gene enhancer in B cells |
| IGF-1 | insulin-like growth factor 1 |
| IOL | iron overload |
| JNK | c-jun N-terminal kinase |
| LDL | low-density lipoprotein |
| LIF | leukemia-inhibitory factor |
| LLC | lipid-laden multilocular and adipose cell |
| LPO | lipid peroxidation |
| MAPK | mitogen-activated protein kinase |
| MDA | malondialdehyde |
| MDS | myelodysplastic syndromes |
| MEL | murine erythroleukemia |
| NK-κB | nuclear factor of κ light gene enhancer in B cells |
| OSM | oncostatin M |
| PC | protein carbonyl |
| PPAR-γ | peroxysome proliferator-activated receptor γ |
| SCF | stem cell factor |
| TNF-α | tumor necrosis factor alpha |
| TREC | T-cell receptor excision circle |
| TSAT | transferrin saturation |
References
- Granic, A.; Mendonça, N.; Hill, T.R.; Jagger, C.; Stevenson, E.J.; Mathers, J.C.; Sayer, A.A. Nutrition in the Very Old. Nutrients 2018, 10, 269. [Google Scholar] [CrossRef] [PubMed]
- Kontis, V.; Bennett, J.E.; Mathers, C.D.; Li, G.; Foreman, K.; Ezzati, M. Future life expectancy in 35 industrialised countries: Projections with a Bayesian model ensemble. Lancet 2017, 389, 1323–1335. [Google Scholar] [CrossRef]
- Bektas, A.; Schurman, S.H.; Sen, R.; Ferrucci, L. Aging, inflammation and the environment. Exp. Gerontol. 2018, 105, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Vina, J.; Borras, C.; Miquel, J. Theories of ageing. IUBMB Life 2007, 59, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Orr, W.C. The redox stress hypothesis of aging. Free Radic. Biol. Med. 2012, 52, 539–555. [Google Scholar] [CrossRef] [PubMed]
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 1973, 134, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial aging and age-related dysfunction of mitochondria. BioMed. Res. Int. 2014, 2014, 238463. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and Biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G.; Pizzimenti, S.; Ciamporcero, E.S.; Daga, M.; Ullio, C.; Arcaro, A.; Cetrangolo, G.P.; Ferretti, C.; Dianzani, C.; Lepore, A.; et al. Role of 4-hydroxynonenal-protein adducts in human diseases. Antioxid. Redox Signal. 2015, 22, 1681–1702. [Google Scholar] [CrossRef] [PubMed]
- Domingues, R.M.; Domingues, P.; Melo, T.; Pérez-Sala, D.; Reis, A.; Spickett, C.M. Lipoxidation adducts with peptides and proteins: Deleterious modifications or signaling mechanisms? J. Proteom. 2013, 92, 110–131. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Zollern, H. Methods for determination of aldehydic lipid peroxidation products. Free Radic. Biol. Med. 1989, 7, 197–203. [Google Scholar] [CrossRef]
- Papac-Milicevic, N.; Busch, C.J.; Binder, C.J. Malondialdehyde Epitopes as Targets of Immunity and the Implications for Atherosclerosis. Adv. Immunol. 2016, 131, 1–59. [Google Scholar] [CrossRef] [PubMed]
- Nègre-Salvayre, A.; Garoby-Salom, S.; Swiader, A.; Rouahi, M.; Pucelle, M.; Salvayre, R. Proatherogenic effects of 4-hydroxynonenal. Free Radic. Biol. Med. 2017, 111, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Galli, A.; Pignalosa, P.; Frulloni, L.; Grappone, C.; Milani, S.; Pederzoli, P.; Cavallini, G.; Surrenti, C. Collagen type I synthesized by pancreatic periacinar stellate cells (PSC) co-localizes with lipid peroxidation-derived aldehydes in chronic alcoholic pancreatitis. J. Pathol. 2000, 192, 81–89. [Google Scholar] [CrossRef]
- Gil, L.; Siems, W.; Mazurek, B.; Gross, J.; Schroeder, P.; Voss, P.; Grune, T. Age-associated analysis of oxidative stress parameters in human plasma and erythrocytes. Free Radic. Res. 2006, 40, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Perluigi, M.; Allan Butterfield, D. Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Manolagas, S.C. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 2000, 2, 115–137. [Google Scholar] [CrossRef]
- Dawson-Hughes, B.; Looker, A.C.; Tosteson, A.N.; Johansson, H.; Kanis, J.A.; Melton, L.J., III. The potential impact of the National Osteoporosis Foundation guidance on treatment eligibility in the USA: An update in NHANES 2005–2008. Osteoporos. Int. 2012, 23, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Choksi, P.; Jepsen, K.J.; Clines, G.A. The challenges of diagnosing osteoporosis and the limitations of currently available tools. Clin. Diabetes Endocrinol. 2018, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.X.; Lv, B.; Wang, Y. Protein Phosphatase 2A Mediates Oxidative Stress Induced Apoptosis in Osteoblasts. Mediat. Inflamm. 2015, 2015, 804260. [Google Scholar] [CrossRef] [PubMed]
- Sendur, O.F.; Turan, Y.; Tastaban, E.; Serter, M. Antioxidant status in patients with osteoporosis: A controlled study. Joint Bone Spine 2009, 76, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Han, L.; Martin-Millan, M.; Plotkin, L.I.; Stewart, S.A.; Roberson, P.K.; Kousteni, S.; O’Brien, C.A.; Bellido, T.; Parfitt, A.M.; et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J. Biol. Chem. 2007, 282, 27285–27297. [Google Scholar] [CrossRef] [PubMed]
- Maggio, D.; Barabani, M.; Pierandrei, M.; Polidori, M.C.; Catani, M.; Mecocci, P.; Senin, U.; Pacifici, R.; Cherubini, A. Marked decrease in plasma antioxidants in aged osteoporotic women: Results of a cross-sectional study. J. Clin. Endocrinol. Metab. 2003, 88, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Lean, J.M.; Jagger, C.J.; Kirstein, B.; Fuller, K.; Chambers, T.J. Hydrogen peroxide is essential for estrogen-deficiency bone loss and osteoclast formation. Endocrinology 2005, 146, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Domazetovic, V.; Marcucci, G.; Iantomasi, T.; Brandi, M.L.; Vincenzini, M.T. Oxidative stress in bone remodeling: Role of antioxidants. Clin. Cases Miner. Bone Metab. 2017, 14, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Han, Y.; Guan, Y.; Zhang, L.; Bai, C.; Li, Y. Aluminum induces osteoblast apoptosis through the oxidative stress-mediated JNK signaling pathway. Biol. Trace Elem. Res. 2012, 150, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Pantano, C.; Reynaert, N.L.; van der Vliet, A.; Janssen-Heininger, Y.M. Redox-sensitive kinases of the nuclear factor-κB signaling pathway. Antioxid. Redox Signal. 2006, 8, 1791–1806. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Han, L.; Ambrogini, E.; Bartell, S.M.; Manolagas, S.C. Oxidative stress stimulates apoptosis and activates NF-κB in osteoblastic cells via a PKCβ/p66shc signaling cascade: Counter regulation by estrogens or androgens. Mol. Endocrinol. 2010, 24, 2030–2037. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.Y.; Zhuang, J.S.; Wu, Q.; Liu, Z.Y.; Liao, C.R.; Luo, S.G.; Chen, J.T.; Zhong, Z.M. Advanced oxidation protein products induce pre-osteoblast apoptosis through a nicotinamide adenine dinucleotide phosphate oxidase-dependent, mitogen-activated protein kinases-mediated intrinsic apoptosis pathway. Aging Cell 2018, e12764. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, B.; Sun, J.; Jiang, Y.; Zhang, H.; Zhang, P.; Fei, B.; Xu, Y. Iron-induced oxidative stress stimulates osteoclast differentiation via NF-κB signaling pathway in mouse model. Metabolism 2018, 83, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Rachner, T.D.; Khosla, S.; Hofbauer, L.C. Osteoporosis: Now and the future. Lancet 2011, 377, 1276–1287. [Google Scholar] [CrossRef]
- Muzio, G.; Maggiora, M.; Paiuzzi, E.; Oraldi, M.; Canuto, R.A. Aldehyde dehydrogenases and cell proliferation. Free Radic. Biol. Med. 2012, 52, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Khan, K.; Pal, S.; Porwal, K.; China, S.P.; Barbhuyan, T.K.; Baghel, K.S.; Rawat, T.; Sanyal, S.; Bhadauria, S.; et al. The thiocarbamate disulphide drug, disulfiram induces osteopenia in rats by inhibition of osteoblast function due to suppression of acetaldehyde dehydrogenase activity. Toxicol. Sci. 2014, 139, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, H.; Hao, W.; Fujita, Y.; Funayama, A.; Miyauchi, Y.; Hashimoto, K.; Miyamoto, K.; Iwasaki, R.; Sato, Y.; Kobayashi, T.; et al. Aldehyde-stress resulting from Aldh2 mutation promotes osteoporosis due to impaired osteoblastogenesis. J. Bone Miner. Res. 2012, 27, 2015–2023. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Pal, S.; China, S.P.; Porwal, K.; Dev, K.; Shrivastava, R.; Raju, K.S.; Rashid, M.; Trivedi, A.K.; Sanyal, S.; et al. Pharmacological activation of aldehyde dehydrogenase 2 promotes osteoblast differentiation via bone morphogenetic protein-2 and induces bone anabolic effect. Toxicol. Appl. Pharmacol. 2017, 316, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Akpolat, V.; Bilgin, H.M.; Celik, M.Y.; Erdemoglu, M.; Isik, B. An evaluation of nitric oxide, folate, homocysteine levels and lipid peroxidation in postmenopausal osteoporosis. Adv. Clin. Exp. Med. 2013, 22, 403–409. [Google Scholar] [PubMed]
- Wu, Q.; Zhong, Z.M.; Pan, Y.; Zeng, J.H.; Zheng, S.; Zhu, S.Y.; Chen, J.T. Advanced Oxidation Protein Products as a Novel Marker of Oxidative Stress in Postmenopausal Osteoporosis. Med. Sci. Monit. 2015, 21, 2428–2432. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Jentoft, J.P.; Baeyens, J.M.; Bauer, Y.; Boirie, T.; Cederholm, F.; Landi, F.C.; Martin, J.P.; Michel, Y.; Rolland, S.M.; Schneider, E.; et al. European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Ageing 2010, 39, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Diz, J.B.; Leopoldino, A.A.; Moreira, B.S.; Henschke, N.; Dias, R.C.; Pereira, L.S.; Oliveira, V.C. Prevalence of sarcopenia in older Brazilians: A systematic review and meta-analysis. Geriatr. Gerontol. Int. 2017, 17, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Zanandrea, V.; Giua, R.; Costanzo, L.; Vellas, B.; Zamboni, M.; Cesari, M. Interventions against sarcopenia in older persons. Curr. Pharm. Des. 2014, 20, 5983–6006. [Google Scholar] [PubMed]
- Marzetti, E.; Calvani, R.; Bernabei, R.; Leeuwenburgh, C. Apoptosis in skeletal myocytes: A potential target for interventions against sarcopenia and physical frailty—A mini-review. Gerontology 2012, 58, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Buonocore, D.; Rucci, S.; Vandoni, M.; Negro, M.; Marzatico, F. Oxidative system in aged skeletal muscle. Muscles Ligaments Tendons J. 2011, 1, 85–90. [Google Scholar] [PubMed]
- Dupont-Versteegden, E.E. Apoptosis in muscle atrophy: Relevance to sarcopenia. Exp. Gerontol. 2005, 40, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Aiken, J.; Bua, E.; Cao, Z.; Lopez, M.; Wanagat, J.; McKenzie, D.; McKiernan, S. Mitochondrial DNA deletion mutations and sarcopenia. Ann. N. Y. Acad. Sci. 2002, 959, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Moulin, M.; Ferreiro, A. Muscle redox disturbances and oxidative stress as pathomechanisms and therapeutic targets in early-onset myopathies. Semin. Cell Dev. Biol. 2017, 64, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Margioris, A.N. Sarcopenic obesity. Hormones 2018. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, C.; Friederich-Persson, M.; Palacios-Ramirez, R.; Nguyen Dinh Cat, A. Mitochondrial oxidative stress in obesity: Role of the mineralocorticoid receptor. J. Endocrinol. 2018, 238, R143–R159. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.C.; Lustgarten, M.S.; Liu, Y.; Muller, F.L.; Bhattacharya, A.; Liang, H.; Salmon, A.B.; Brooks, S.V.; Larkin, L.; Hayworth, C.R.; et al. Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junciton degeneration. FASEB J. 2010, 24, 1376–1390. [Google Scholar] [CrossRef] [PubMed]
- Vollaard, N.B.; Shearmann, J.P.; Cooper, C.E. Exercise induced oxidative stress: Myths, realities and physiological relevance. Sports Med. 2005, 35, 1045–1062. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Pannell, B.K. Redox characterization of functioning skeletal muscle. Front. Physiol. 2015, 6, 338. [Google Scholar] [CrossRef] [PubMed]
- Merry, T.L.; Steinberg, G.R.; Lynch, G.S.; McConell, G.K. Skeletal muscle uptake during contraction is regulated by nitric oxide and ROS independently of AMPK. Am. J. Physiol. Endocrinol. Metab. 2009, 298, E577–E585. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Talbert, E.E.; Adhihetty, P.J. Reactive oxygen and nitrogen species as intracellular signals in skeletal muscle. J. Physiol. 2011, 589, 2129–2138. [Google Scholar] [CrossRef] [PubMed]
- Bassel-Duby, R.; Olson, E.N. Signaling pathways in skeletal muscle remodeling. Annu. Rev. Biochem. 2006, 75, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Duarte, J.; Kavazis, A.N.; Talbert, E.E. Reactive oxygen species are signalling molecules for skeletal muscle adaptation. Exp. Physiol. 2010, 95, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.; Rodino-Klapac, L.R.; Mendell, J.R. Emerging drugs for Duchenne muscular dystrophy. Expert Opin. Emerg. Drugs 2012, 17, 261–277. [Google Scholar] [CrossRef]
- Charles, A.-L.; Meyer, A.; Dal-Ros, S.; Auger, C.; Keller, N.; Ramamoorthy, T.G.; Zoll, J.; Metzger, D.; Schini-Kerth, V.; Geny, B. Polyphenols prevent ageing-related impairment in skeletal muscle mitochondrial function through decreased reactive oxygen species production. Exp. Physiol. 2013, 98, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Visser, S.B.; Kritchevsky, B.H.; Goodpaster, A.B.; Newman, M.; Nevitt, E. Leg muscle mass and composition in relation to lower extremity performance in men and women aged 70–79: The health, aging and body composition study. J. Am. Geriatr. Soc. 2002, 50, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Navratil, M.; Thompson, L.V.; Arriaga, E.A. Estimating relative carbonyl levels in muscle microstructures by fluorescence imaging. Anal. Bioanal. Chem. 2008, 391, 2591–2598. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Xie, H.; Meany, D.L.; Thompson, L.V.; Arriaga, E.A.; Griffin, T.J. Quantitative proteomic profiling of muscle type-dependent and age-dependent protein carbonylation in rat skeletal muscle mitochondria. J. Gerontol. A Biol. Sci. Med.Sci. 2008, 63, 1137–1152. [Google Scholar] [CrossRef] [PubMed]
- Braga, M.; Sinha Hikim, A.P.; Datta, S.; Ferrini, M.G.; Brown, D.; Kovacheva, E.L.; Gonzalez-Cadavid, N.F.; Sinha-Hikim, I. Involvement of oxidative stress and caspase 2-mediated intrinsic pathway signaling in age-related increase in muscle cell apoptosis in mice. Apoptosis 2008, 13, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Ohsawa, I.; Nishimaki, K.; Kumamoto, S.; Maruyama, I.; Suzuki, Y.; Ohta, S. Preventive effects of Chlorella on skeletal muscle atrophy in muscle-specific mitochondrial aldehyde dehydrogenase 2 activity-deficient mice. BMC Complement. Altern. Med. 2014, 14, 390. [Google Scholar] [CrossRef] [PubMed]
- Beltran Valls, M.R.; Wilkinson, D.J.; Narici, M.V.; Smith, K.; Phillips, B.E.; Caporossi, D.; Atherton, P.J. Protein carbonylation and heat shock proteins in human skeletal muscle: Relationships to age and sarcopenia. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 70, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Coto Montes, A.; Boga, J.A.; Bermejo Millo, C.; Rubio González, A.; Potes Ochoa, Y.; Vega Naredo, I.; Martínez Reig, M.; Romero Rizos, L.; Sánchez Jurado, P.M.; Solano, J.J.; et al. Potential early biomarkers of sarcopenia among independent older adults. Maturitas 2017, 104, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Bellanti, F.; Romano, A.D.; Lo Buglio, A.; Castriotta, V.; Guglielmi, G.; Greco, A.; Serviddio, G.; Vendemiale, G. Oxidative stress is increased in sarcopenia and associated with cardiovascular disease risk in sarcopenic obesity. Maturitas 2018, 109, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Gueugneau, M.; Coudy-Gandilhon, C.; Gourbeyre, O.; Chambon, C.; Combaret, L.; Polge, C.; Taillandier, D.; Attaix, D.; Friguet, B.; Maier, A.B.; et al. Proteomics of muscle chronological ageing in post-menopausal women. BMC Genom. 2014, 15, 1165. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Ventura, M.T.; Casciaro, M.; Gangemi, S.; Buquicchio, R. Immunosenescence in aging: Between immune cells depletion and cytokines. Clin. Mol. Allergy 2017, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Goronzy, J.J.; Weyand, C.M. T cell development and receptor diversity during aging. Curr. Opin. Immunol. 2005, 17, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Taub, D.D.; Longo, D.L. Insight into thymic aging and regeneration. Immunol. Rev. 2005, 205, 72–93. [Google Scholar] [CrossRef] [PubMed]
- Herndler-Brandstetter, D.; Cioca, D.P.; Grubeck-Loebensteiner, B. Immunizations in the elderly: Do they live up to their promise? Wien. Med. Wochenschr. 2006, 156, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Gruver, A.L.; Hudson, L.L.; Sempowski, G.D. Immunosenescence of ageing. J. Pathol. 2007, 211, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Wikby, A.; Ferguson, F.; Forsey, R.; Thompson, J.; Strindhall, J.; Lofgren, S.; Nilsson, B.O.; Ernerudh, J.; Pawelec, G.; Johansson, B. An immune risk phenotype, cognitive impairment, and survival in very late life: Impact of allostatic load in Swedish octogenarian and nonagenarian humans. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 556–565. [Google Scholar] [CrossRef] [PubMed]
- De Mello-Coelho, V.; Cutler, R.G.; Bunbury, A.; Tammara, A.; Mattson, M.P.; Taub, D.D. Age-associated alterations in the levels of cytotoxic lipid molecular species and oxidative stress in the murine thymus are reduced by growth hormone treatment. Mech. Ageing Dev. 2017, 167, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Langhi, L.G.; Andrade, L.R.; Shimabukuro, M.K.; van Ewijk, W.; Taub, D.D.; Borojevic, R.; de Mello Coelho, V. Lipid-laden multilocular cells in the aging thymus are phenotypically heterogeneous. PLoS ONE 2015, 10, e0141516. [Google Scholar] [CrossRef] [PubMed]
- Taub, D.D.; Murphy, W.J.; Longo, D.L. Rejuvenation of the aging thymus: Growth hormone- and ghrelin-mediated signaling pathways. Curr. Opin. Pharmacol. 2010, 10, 408–424. [Google Scholar] [CrossRef] [PubMed]
- De Mello-Coelho, V.; Bunbury, A.; Rangel, L.B.; Giri, B.; Weeraratna, A.; Morin, P.J.; Bernier, M.; Taub, D.D. Fat-storing multilocular cells expressing CCR5 increase in the thymus with advancing age: Potential role for CCR5 ligands on the differentiation and migration of preadipocytes. Int. J. Med. Sci. 2010, 7, 1–14. [Google Scholar] [CrossRef]
- Gautam, N.; Das, S.; Mahapatra, S.K.; Chakraborti, S.P.; Kundu, P.K.; Roy, S. Age associated oxidative damage in lymphocytes. Oxid. Med. Cell. Longev. 2010, 3, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Ma, X. Epidemiology of myelodysplastic syndromes. Am. J. Med. 2012, 125, S2–S5. [Google Scholar] [CrossRef] [PubMed]
- Shumilov, E.; Flach, J.; Kohlmann, A.; Banz, Y.; Bonadies, N.; Fiedler, M.; Pabst, T.; Bacher, U. Current status and trends in the diagnostics of AML and MDS. Blood Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Della Porta, M.G.; Pascutto, C.; Invernizzi, R.; Boni, M.; Travaglino, E.; Passamonti, F.; Arcaini, L.; Maffioli, M.; Bernasconi, P.; et al. Prognostic factors and life expectancy in myelodysplastic syndromes classified according to WHO criteria: A basis for clinical decision-making. J. Clin. Oncol. 2005, 23, 7594–7603. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, M.J.; Bowen, D.T. Oxidative stress and the myelodysplastic syndromes. Int. J. Hematol. 2003, 77, 342–350. [Google Scholar] [CrossRef] [PubMed]
- De Souza, G.F.; Barbosa, M.C.; Santos, T.E.; Carvalho, T.M.; de Freitas, R.M.; Martins, M.R.; Gonçalves, R.P.; Pinheiro, R.F.; Magalhães, S.M. Increased parameters of oxidative stress and its relation to transfusion iron overload in patients with myelodysplastic syndromes. J. Clin. Pathol. 2013, 66, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Gan, Z.S.; Wang, Q.Q.; Li, J.H.; Wang, X.L.; Wang, Y.Z.; Du, H.H. Iron Reduces M1 Macrophage Polarization in RAW264.7 Macrophages Associated with Inhibition of STAT1. Mediat. Inflamm. 2017, 8570818. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, E.; Cianciulli, P.; Finelli, C.; Mecucci, C.; Voso, M.T.; Tura, S. Unraveling the mechanisms behind iron overload and ineffective hematopoiesis in myelodysplastic syndromes. Leuk. Res. 2017, 62, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Ivars, D.; Orero, M.T.; Javier, K.; Díaz-Vico, L.; García-Giménez, J.L.; Mena, S.; Tormos, C.; Egea, M.; Pérez, P.L.; Arrizabalaga, B.; et al. Oxidative imbalance in low/intermediate-1-risk myelodysplastic syndrome patients: The influence of iron overload. Clin. Biochem. 2017, 50, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Ghoti, H.; Amer, J.; Winder, A.; Rachmilewitz, E.; Fibach, E. Oxidative stress in red blood cells, platelets and polymorphonuclear leukocytes from patients with myelodysplastic syndromes. Eur. J. Haematol. 2007, 79, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Rassool, F.V.; Gaymes, T.J.; Omidvar, N.; Brady, N.; Beurlet, S.; Pla, M.; Reboul, M.; Lea, N.; Chomienne, C.; Thomas, N.S.; et al. Reactive oxygen species, DNA damage, and error-prone repair: A model for genomic instability with progression in myeloid leukemia? Cancer Res. 2007, 67, 8762–8771. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Gasparetto, M.; Jordan, C.; Pollyea, D.A.; Vasiliou, V. The effects of alcohol and aldehyde dehydrogenases on disorders of hematopoiesis. Adv. Exp. Med. Biol. 2015, 815, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Gasparetto, M.; Smith, C.A. ALDHs in normal and malignant hematopoietic cells: Potential new avenues for treatment of AML and other blood cancers. Chem. Biol. Interact. 2017, 276, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Semlitsch, T.; Tillian, H.M.; Zarkovic, N.; Borovic, S.; Purtscher, M.; Hohenwarter, O.; Schaur, R.J. Differential influence of the lipid peroxidation product 4-hydroxynonenal on the growth of human lymphatic leukaemia cells and human peripheral blood lymphocytes. Anticancer Res. 2002, 22, 1689–1697. [Google Scholar] [PubMed]
- Rinaldi, M.; Barrera, G.; Spinsanti, P.; Pizzimenti, S.; Ciafrè, S.A.; Parella, P.; Farace, M.G.; Signori, E.; Dianzani, M.U.; Fazio, V.M. Growth inhibition and differentiation induction in murine erythroleukemia cells by 4-hydroxynonenal. Free Radic. Res. 2001, 34, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Gasparetto, M.; Pei, S.; Minhajuddin, M.; Khan, N.; Pollyea, D.A.; Myers, J.R.; Ashton, J.M.; Becker, M.W.; Vasiliou, V.; Humphries, K.R.; et al. Targeted therapy for a subset of acute myeloid leukemias that lack expression of aldehyde dehydrogenase1A1. Haematologica 2017, 102, 1054–1065. [Google Scholar] [CrossRef] [PubMed]
- Koelling, T.M.; Yeager, A.M.; Hilton, J.; Haynie, D.T.; Wiley, J.M. Development and characterization of a cyclophosphamide-resistant subline of acute myeloid leukemia in the Lewis × Brown Norway hybrid rat. Blood 1990, 76, 1209–1213. [Google Scholar] [PubMed]
- Ran, D.; Schubert, M.; Pietsch, L.; Taubert, I.; Wuchter, P.; Eckstein, V.; Bruckner, T.; Zoeller, M.; Ho, A.D. Aldehyde dehydrogenase activity among primary leukemia cells is associated with stem cell features and correlates with adverse clinical outcomes. Exp. Hematol. 2009, 37, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Hassane, D.C.; Guzman, M.L.; Corbett, C.; Li, X.; Abboud, R.; Young, F.; Liesveld, J.L.; Carroll, M.; Jordan, C.T. Discovery of agents that eradicate leukemia stem cells using an in silico screen of public gene expression data. Blood 2008, 111, 5654–5662. [Google Scholar] [CrossRef] [PubMed]
- Pimková, K.; Chrastinová, L.; Suttnar, J.; Štikarová, J.; Kotlín, R.; Hermák, J.E. Plasma Levels of Aminothiols, Nitrite, Nitrate, and Malondialdehyde in Myelodysplastic Syndromes in the Context of Clinical Outcomes and as a Consequence of Iron Overload. Oxid. Med. Cell. Longev. 2014, 2014, 416028. [Google Scholar] [CrossRef] [PubMed]
- Gradinaru, D.; Margina, D.; Ilie, M.; Borsa, C.; Ionescu, C.; Prada, G.I. Correlation between erythropoietin serum levels and erythrocyte susceptibility to lipid peroxidation in elderly with type 2 diabetes. Acta Physiol. Hung. 2015, 102, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Esfahani, A.; Ghoreishi, Z.; Nikanfar, A.; Sanaat, Z.; Ghorbanihaghjo, A. Influence of chemotherapy on the lipid peroxidation and antioxidant status in patients with acute myeloid leukemia. Acta Med. Iran. 2012, 50, 454–458. [Google Scholar] [PubMed]
- Brioche, T.; Lemoine-Morel, S. Oxidative Stress, Sarcopenia, Antioxidant Strategies and Exercise: Molecular Aspects. Curr. Pharm. Des. 2016, 22, 2664–2678. [Google Scholar] [CrossRef] [PubMed]
- Espino, J.; Pariente, J.A.; Rodríguez, A.B. Oxidative stress and immunosenescence: Therapeutic effects of melatonin. Oxid. Med. Cell. Longev. 2012, 2012, 670294. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barrera, G.; Pizzimenti, S.; Daga, M.; Dianzani, C.; Arcaro, A.; Cetrangolo, G.P.; Giordano, G.; Cucci, M.A.; Graf, M.; Gentile, F. Lipid Peroxidation-Derived Aldehydes, 4-Hydroxynonenal and Malondialdehyde in Aging-Related Disorders. Antioxidants 2018, 7, 102. https://doi.org/10.3390/antiox7080102
Barrera G, Pizzimenti S, Daga M, Dianzani C, Arcaro A, Cetrangolo GP, Giordano G, Cucci MA, Graf M, Gentile F. Lipid Peroxidation-Derived Aldehydes, 4-Hydroxynonenal and Malondialdehyde in Aging-Related Disorders. Antioxidants. 2018; 7(8):102. https://doi.org/10.3390/antiox7080102
Chicago/Turabian StyleBarrera, Giuseppina, Stefania Pizzimenti, Martina Daga, Chiara Dianzani, Alessia Arcaro, Giovanni Paolo Cetrangolo, Giulio Giordano, Marie Angele Cucci, Maria Graf, and Fabrizio Gentile. 2018. "Lipid Peroxidation-Derived Aldehydes, 4-Hydroxynonenal and Malondialdehyde in Aging-Related Disorders" Antioxidants 7, no. 8: 102. https://doi.org/10.3390/antiox7080102
APA StyleBarrera, G., Pizzimenti, S., Daga, M., Dianzani, C., Arcaro, A., Cetrangolo, G. P., Giordano, G., Cucci, M. A., Graf, M., & Gentile, F. (2018). Lipid Peroxidation-Derived Aldehydes, 4-Hydroxynonenal and Malondialdehyde in Aging-Related Disorders. Antioxidants, 7(8), 102. https://doi.org/10.3390/antiox7080102