
Ferroptosis Resistance: Redundant Antioxidant Networks Are a Barrier to Cancer Therapy

Abstract
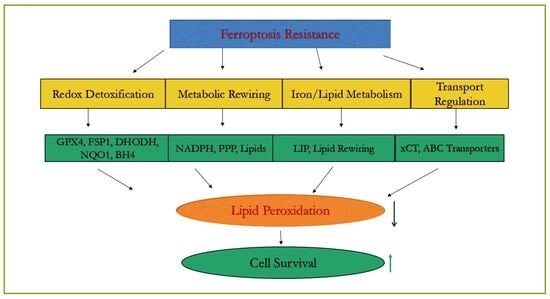
1. Introduction
2. Redox Detoxification Networks
2.1. System-GSH-GPX4 Axis
2.2. FSP1-CoQ10H2 Pathway
2.3. Dihydroorotate Dehydrogenase (DHODH)-CoQ10H2 Axis
2.4. GCH1-BH4 Axis
2.5. NQO1-NADPH Axis

{kind=link}
{kind=link}
{kind=link}
| Pathway | Cellular Localization | NRF2 Dependence | Biochemical Consequence | Reference(s) |
|---|---|---|---|---|
| GSH-GPX4 | Cytosol and Mitochondria | Yes (Indirect/Direct) | Catalyzes reduction of phospholipid hydroperoxides to non-toxic alcohols using GSH. | [46,47] |
| FSP1-CoQ10 | Plasma/Extramitochondrial Membranes | Partial (Context-dependent) | Utilizes NAD(P)H to continuously regenerate reduced CoQ10H2, an active lipid radical-trapping antioxidant. | [25,27,45] |
| DHODH-CoQ10 | Inner Mitochondrial Membrane | No | Regenerates mitochondrial CoQ10H2 to inhibit lipid peroxidation. | [30,31,33] |
| NQO1-NADPH | Cytosol | Yes | Drives multi-substrate quinone detoxification and supports extramitochondrial quinone reduction. | [38,42,43] |
3. Metabolic Rewiring and Lipid Remodeling
3.1. Iron Metabolism in Ferroptosis Resistance
3.2. Lipid Metabolism in Ferroptosis Resistance
4. Miscellaneous
4.1. ABC Transporter-Mediated Resistance
4.2. Tumor Heterogeneity in Ferroptosis Resistance
4.3. Distinct Cellular Differentiation States and Phenotypes
4.4. Metabolic Heterogeneity
4.5. Tumor Microenvironment in Ferroptosis Resistance
5. Future Directions and Therapeutic Considerations
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2021, 17, 2054–2081. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Singh, M.; Arora, H.L.; Naik, R.; Joshi, S.; Sonawane, K.; Sharma, N.K.; Sinha, B.K. Ferroptosis in Cancer: Mechanism and Therapeutic Potential. Int. J. Mol. Sci. 2025, 26, 3852. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, G.; Chen, X. Mechanism of ferroptosis resistance in cancer cells. Cancer Drug Resist. 2024, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, X.; Xia, R.; Zhang, H.S. Ferroptosis resistance in cancer: Recent advances and future perspectives. Biochem. Pharmacol. 2024, 219, 115933. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Meng, Y.; Li, D.; Yao, L.; Le, J.; Liu, Y.; Sun, Y.; Zeng, F.; Chen, X.; Deng, G. Ferroptosis in cancer: From molecular mechanisms to therapeutic strategies. Signal Transduct. Target. Ther. 2024, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.; Ling, V.; Low, C.; Wang, Y.Z.; Gout, P.W. Potential use of the anti-inflammatory drug, sulfasalazine, for targeted therapy of pancreatic cancer. Curr. Oncol. 2010, 17, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Yang, X.R.; Chung, W.Y.; Dennison, A.R.; Zhou, J. Targeted therapy for hepatocellular carcinoma. Signal Transduct. Target. Ther. 2020, 5, 146. [Google Scholar] [CrossRef] [PubMed]
- Grillone, A.; Riva, E.R.; Mondini, A.; Forte, C.; Calucci, L.; Innocenti, C.; de Julian Fernandez, C.; Cappello, V.; Gemmi, M.; Moscato, S.; et al. Active Targeting of Sorafenib: Preparation, Characterization, and In Vitro Testing of Drug-Loaded Magnetic Solid Lipid Nanoparticles. Adv. Healthc. Mater. 2015, 4, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.H.; Ye, Q.F.; Miao, X.Y.; Liu, X.; Huang, S.Q.; Xiong, L.; Wen, Y.; Zhang, Z.J. Current status of sorafenib nanoparticle delivery systems in the treatment of hepatocellular carcinoma. Theranostics 2021, 11, 5464–5490. [Google Scholar] [CrossRef] [PubMed]
- Demuynck, R.; Efimova, I.; Catanzaro, E.; Krysko, D.V. Ferroptosis: Friend or foe in cancer immunotherapy? Oncoimmunology 2023, 12, 2182992. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Cai, D.; Gou, S.; Bai, Y.; Lei, H.; Li, Y.; Chen, Y.; Zhao, Y.; Shen, J.; Wu, X.; et al. The dynamic role of ferroptosis in cancer immunoediting: Implications for immunotherapy. Pharmacol. Res. 2025, 214, 107674. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal 2013, 18, 522–555. [Google Scholar] [CrossRef] [PubMed]
- Gochenauer, G.E.; Robinson, M.B. Dibutyryl-cAMP (dbcAMP) up-regulates astrocytic chloride-dependent L-[3H]glutamate transport and expression of both system xc(-) subunits. J. Neurochem. 2001, 78, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Warren, B.A.; Rhoderick, J.F.; Bridges, R.J. Differentiation of substrate and non-substrate inhibitors of transport system xc(-): An obligate exchanger of L-glutamate and L-cystine. Neuropharmacology 2004, 46, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Seiler, A.; Perisic, T.; Kolle, P.; Banjac Canak, A.; Forster, H.; Weiss, N.; Kremmer, E.; Lieberman, M.W.; Bannai, S.; et al. System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J. Biol. Chem. 2010, 285, 22244–22253. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Sato, H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): Cystine supplier and beyond. Amino Acids 2012, 42, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Kusumi, R.; Hamashima, S.; Kobayashi, S.; Sasaki, S.; Komiyama, Y.; Izumikawa, T.; Conrad, M.; Bannai, S.; Sato, H. The ferroptosis inducer erastin irreversibly inhibits system x(c)- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci. Rep. 2018, 8, 968. [Google Scholar] [CrossRef] [PubMed]
- Li, F.J.; Long, H.Z.; Zhou, Z.W.; Luo, H.Y.; Xu, S.G.; Gao, L.C. System X(c) (-)/GSH/GPX4 axis: An important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front. Pharmacol. 2022, 13, 910292. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.H.; Chen, X.; Cai, L.Y.; Nan, X.W.; Chen, J.H.; Chen, X.X.; Yang, Y.; Xing, Z.H.; Wei, M.N.; Li, Y.; et al. Erastin Reverses ABCB1-Mediated Docetaxel Resistance in Ovarian Cancer. Front. Oncol. 2019, 9, 1398. [Google Scholar] [CrossRef] [PubMed]
- Perera, L.; Brown, S.M.; Silver, B.B.; Tokar, E.J.; Sinha, B.K. Ferroptosis Inducers Erastin and RSL3 Enhance Adriamycin and Topotecan Sensitivity in ABCB1/ABCG2-Expressing Tumor Cells. Int. J. Mol. Sci. 2025, 26, 635. [Google Scholar] [CrossRef] [PubMed]
- Silver, B.B.; Murphy, C.; Tokar, E.J.; Sinha, B.K. Ferroptosis Suppressor Protein 1 (FSP1)-CoQ10-NADPH-Axis Is Responsible for Erastin Resistance in MCF-7 Breast Cancer Cells. Antioxidants 2026, 15, 239. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Bao, L.; Li, M.; Chang, K.; Yi, X. Erastin synergizes with cisplatin via ferroptosis to inhibit ovarian cancer growth in vitro and in vivo. J. Obstet. Gynaecol. Res. 2021, 47, 2481–2491. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Tahsin, T.; McPhail, D.K.; Champion, J.D.; Alzahrani, M.A.M.; Hilditch, M.L.; Faris-Orr, A.; Calver, B.L.; Cronin, J.G.; Mareque-Rivas, J.C.; Sexton, D.W.; et al. Targeting NRF2 and FSP1 to Overcome Ferroptosis Resistance in TSC2-Deficient and Cancer Cells. Cancers 2025, 17, 2714. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Chen, X.; Chen, L.; Lu, Y.; Wu, Y.; Deng, A.; Pan, F.; Huang, H.; Liu, Y.; Li, Y.; et al. DHODH-mediated mitochondrial redox homeostasis: A novel ferroptosis regulator and promising therapeutic target. Redox Biol. 2025, 85, 103788. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Roh, J.L. Dihydroorotate Dehydrogenase in Mitochondrial Ferroptosis and Cancer Therapy. Cells 2025, 14, 1889. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, J.; Wang, J.; Ren, C.; Tang, P.; Ouyang, L.; Wang, Y. Recent advances of human dihydroorotate dehydrogenase inhibitors for cancer therapy: Current development and future perspectives. Eur. J. Med. Chem. 2022, 232, 114176. [Google Scholar] [CrossRef] [PubMed]
- Rajamohamed, R.; Veerappapillai, S. Identification of novel dihydroorotate dehydrogenase (DHODH) inhibitors for cancer: Computational drug repurposing strategy. BMC Pharmacol. Toxicol. 2025, 26, 168. [Google Scholar] [CrossRef] [PubMed]
- Amos, A.; Amos, A.; Wu, L.; Xia, H. The Warburg effect modulates DHODH role in ferroptosis: A review. Cell Commun. Signal 2023, 21, 100. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Wang, X.; Qin, B.; Hu, R.; Miao, R.; Zhou, Y.; Wang, L.; Liu, T. Targeting NQO1 induces ferroptosis and triggers anti-tumor immunity in immunotherapy-resistant KEAP1-deficient cancers. Drug Resist. Updat. 2024, 77, 101160. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhong, B.; Zhao, L.; Hou, Y.; Ai, N.; Lu, J.J.; Ge, W.; Chen, X. Fighting drug-resistant lung cancer by induction of NAD(P)H:quinone oxidoreductase 1 (NQO1)-mediated ferroptosis. Drug Resist. Updat. 2023, 70, 100977. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Arutla, V.; Srivenugopal, K.S. Human NQO1 as a Selective Target. for Anticancer Therapeutics and Tumor Imaging. Cells 2024, 13, 1272. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.; Lu, L.; Pan, S.S.; Wei, X.Q.; Miao, R.R.; Liu, X.H.; Xue, M.; Lin, X.K.; Xu, H.L. Targeting NQO1/GPX4-mediated ferroptosis by plumbagin suppresses in vitro and in vivo glioma growth. Br. J. Cancer 2022, 127, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Song, C.; Huang, L.; Zhao, X. Artemisinin Protects Diabetic Cardiomyopathy by Inhibiting Ferroptosis via Upregulating Nrf2/NQO1 and GPX4 Pathways. Mol. Nutr. Food Res. 2026, 70, e70296. [Google Scholar] [CrossRef] [PubMed]
- Drolet, J.; Buchner-Duby, B.; Stykel, M.G.; Coackley, C.; Kang, J.X.; Ma, D.W.L.; Ryan, S.D. Docosahexanoic acid signals through the Nrf2-Nqo1 pathway to maintain redox balance and promote neurite outgrowth. Mol. Biol. Cell 2021, 32, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Seo, Y.; Roh, J.L. Ferroptosis and Nrf2 Signaling in Head and Neck Cancer: Resistance Mechanisms and Therapeutic Prospects. Antioxidants 2025, 14, 993. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, Y.; Zhao, H.; Tian, Z.; Cao, Q.; Li, Y.; Gu, Y.; Song, Q.; Hu, X.; Jin, M.; et al. Correlation of PD-L1 expression with CD8+ T cells and oxidative stress-related molecules NRF2 and NQO1 in esophageal squamous cell carcinoma. J. Pathol. Clin. Res. 2024, 10, e12390. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kim, M.J.; Han, T.H.; Lee, J.Y.; Kim, S.; Kim, H.; Oh, K.J.; Kim, W.K.; Han, B.S.; Bae, K.H.; et al. FSP1 confers ferroptosis resistance in KEAP1 mutant non-small cell lung carcinoma in NRF2-dependent and -independent manner. Cell Death Dis. 2023, 14, 567. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Zhang, H.; Lai, M.; Wei, J.; Qian, J.; Chen, X.; Wang, X.; Wang, Y. Sulfasalazine induces ferroptosis in osteosarcomas by regulating Nrf2/SLC7A11/GPX4 signaling axis. Sci. Rep. 2025, 15, 30197. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Kim, E.H.; Lee, J.; Roh, J.L. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic. Biol. Med. 2018, 129, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Qiang, Z.; Chai, D.; Peng, J.; Xia, Y.; Hu, R.; Jiang, H. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging 2020, 12, 12943–12959. [Google Scholar] [CrossRef] [PubMed]
- Anandhan, A.; Dodson, M.; Shakya, A.; Chen, J.; Liu, P.; Wei, Y.; Tan, H.; Wang, Q.; Jiang, Z.; Yang, K.; et al. NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci. Adv. 2023, 9, eade9585. [Google Scholar] [CrossRef] [PubMed]
- Prevost, C.T.; Gansereit, W.B.; Kashatus, D.F. NRF2 regulates lipid droplet dynamics to prevent lipotoxicity. iScience 2025, 28, 112925. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Dogon, G.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis. Int. J. Mol. Sci. 2022, 24, 449. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.; et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front. Aging Neurosci. 2016, 8, 308. [Google Scholar] [CrossRef] [PubMed]
- Loftus, L.V.; Rolle, L.T.A.; Wang, B.; Pienta, K.J.; Amend, S.R. Dysregulation of Labile Iron Predisposes Chemotherapy Resistant Cancer Cells to Ferroptosis. Int. J. Mol. Sci. 2025, 26, 4193. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wei, H.; Lyu, M.; Yu, Z.; Chen, J.; Lyu, X.; Zhuang, F. Correction for: Iron retardation in lysosomes protects senescent cells from ferroptosis. Aging 2024, 16, 11475–11476. [Google Scholar] [CrossRef] [PubMed]
- Caneque, T.; Baron, L.; Muller, S.; Carmona, A.; Colombeau, L.; Versini, A.; Solier, S.; Gaillet, C.; Sindikubwabo, F.; Sampaio, J.L.; et al. Activation of lysosomal iron triggers ferroptosis in cancer. Nature 2025, 642, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Liu, G.; Li, Q.; Hu, Y.; Wang, H. Effects on Iron Metabolism and System Xc-/GPX4 Pathway from Hydroquinone Suggest Ferroptosis of Jurkat Cells. Toxics 2024, 12, 644. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, A.M.; Sacco, A.; Giorgio, E.; Petriaggi, L.; Elzanowska, J.; Cruz, A.R.; Rocha, L.; Pereira, C.E.; Strano Moraes, M.C.; Palazzo, L.; et al. Expulsion of iron-rich ferritin via CD63-mediated exosome drives ferroptosis resistance in ovarian cancer cells. Front. Cell Dev. Biol. 2025, 13, 1532097. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xue, Z.; Huang, T.; Che, X.; Wu, G. Lipid metabolism in ferroptosis and ferroptosis-based cancer therapy. Front. Oncol. 2022, 12, 941618. [Google Scholar] [CrossRef] [PubMed]
- Sokol, K.H.; Lee, C.J.; Rogers, T.J.; Waldhart, A.; Ellis, A.E.; Madireddy, S.; Daniels, S.R.; House, R.R.J.; Ye, X.; Olesnavich, M.; et al. Lipid availability influences ferroptosis sensitivity in cancer cells by regulating polyunsaturated fatty acid trafficking. Cell Chem. Biol. 2025, 32, 408–422.e406. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Klycheva, K.V.; Razumovskaya, A.V.; Shatsillo, A.D.; Mastykina, M.D.; Kulagin, T.A.; Silkina, M.O.; Nikulin, S.V. ELOVL5 Regulates Ferroptosis in Breast Cancer Cells. Dokl. Biochem. Biophys. 2025, 525, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Conrad, M. Ferroptosis: When metabolism meets cell death. Physiol. Rev. 2025, 105, 651–706. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhu, T.; Wang, X.; Xiong, F.; Hu, Z.; Qiao, X.; Yuan, X.; Wang, D. ACSL3 and ACSL4, Distinct Roles in Ferroptosis and Cancers. Cancers 2022, 14, 5896. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yu, H.; Gao, R.; Liu, M.; Xie, W. A comprehensive review of the family of very-long-chain fatty acid elongases: Structure, function, and implications in physiology and pathology. Eur. J. Med. Res. 2023, 28, 532. [Google Scholar] [CrossRef] [PubMed]
- Nitta, S.; Kandori, S.; Tanaka, K.; Sakka, S.; Siga, M.; Nagumo, Y.; Negoro, H.; Kojima, T.; Mathis, B.J.; Shimazui, T.; et al. ELOVL5-mediated fatty acid elongation promotes cellular proliferation and invasion in renal cell carcinoma. Cancer Sci. 2022, 113, 2738–2752. [Google Scholar] [CrossRef] [PubMed]
- Danielli, M.; Perne, L.; Jarc Jovicic, E.; Petan, T. Lipid droplets and polyunsaturated fatty acid trafficking: Balancing life and death. Front. Cell Dev. Biol. 2023, 11, 1104725. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Melino, G. Stearoyl CoA Desaturase Regulates Ferroptosis in Ovarian Cancer Offering New Therapeutic Perspectives. Cancer Res. 2019, 79, 5149–5150. [Google Scholar] [CrossRef] [PubMed]
- Paton, C.M.; Ntambi, J.M. Biochemical and physiological function of stearoyl-CoA desaturase. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E28–E37. [Google Scholar] [CrossRef] [PubMed]
- Hoy, A.J.; Nagarajan, S.R.; Butler, L.M. Tumour fatty acid metabolism in the context of therapy resistance and obesity. Nat. Rev. Cancer 2021, 21, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Bergeron, K.F.; Lingrand, M.; Mounier, C. Unveiling the MUFA-Cancer Connection: Insights from Endogenous and Exogenous Perspectives. Int. J. Mol. Sci. 2023, 24, 9921. [Google Scholar] [CrossRef] [PubMed]
- Tracz-Gaszewska, Z.; Dobrzyn, P. Stearoyl-CoA Desaturase 1 as a Therapeutic Target. for the Treatment of Cancer. Cancers 2019, 11, 948. [Google Scholar] [CrossRef] [PubMed]
- Sen, U.; Coleman, C.; Sen, T. Stearoyl coenzyme A desaturase-1: Multitasker in cancer, metabolism, and ferroptosis. Trends Cancer 2023, 9, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wu, Z.; Si, Y.; Tang, W.; Xu, X.; Cheng, Y.; Lin, J. Identification of LPCAT1 expression as a potential prognostic biomarker guiding treatment choice in acute myeloid leukemia. Oncol. Lett. 2021, 21, 105. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hu, Y.; Zheng, H.; Li, M.; Liu, Y.; Feng, R.; Li, X.; Zhang, S.; Tang, M.; Yang, M.; et al. LPCAT1-mediated membrane phospholipid remodelling promotes ferroptosis evasion and tumour growth. Nat. Cell Biol. 2024, 26, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zhuang, L.; Gan, B. Rewiring cancer cell death: LPCAT1 shapes lipid composition and ferroptosis resistance. Cell Death Differ. 2024, 31, 1101–1103. [Google Scholar] [CrossRef] [PubMed]
- Park, V.S.; Pope, L.E.; Ingram, J.P.; Alchemy, G.A.; Purkal, J.J.; Murray, M.B.; Jin, S.; Andino-Frydman, E.Y.; Singh, S.; Chen, A.; et al. Lipid Composition Alters Ferroptosis Sensitivity. Cancer Res. 2025, 85, 4380–4397. [Google Scholar] [CrossRef] [PubMed]
- Xin, S.; Mueller, C.; Pfeiffer, S.; Kraft, V.A.N.; Merl-Pham, J.; Bao, X.; Feederle, R.; Jin, X.; Hauck, S.M.; Schmitt-Kopplin, P.; et al. MS4A15 drives ferroptosis resistance through calcium-restricted lipid remodeling. Cell Death Differ. 2022, 29, 670–686. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Wang, L.; Wu, Y.; Yu, Y.; Yao, Y.; Yang, H.; Hao, C. Lipid metabolism in ferroptosis: Mechanistic insights and therapeutic potential. Front. Immunol. 2025, 16, 1545339. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Johnson, E.; Lewinson, O. ABC transporters: The power to change. Nat. Rev. Mol. Cell Biol. 2009, 10, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Frye, W.J.E.; Huff, L.M.; Gonzalez Dalmasy, J.M.; Salazar, P.; Carter, R.M.; Gensler, R.T.; Esposito, D.; Robey, R.W.; Ambudkar, S.V.; Gottesman, M.M. The multidrug resistance transporter P-glycoprotein confers resistance to ferroptosis inducers. Cancer Drug Resist. 2023, 6, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.M.; Sinha, B.K.; Cannon, R.E. A Role for iNOS in Erastin Mediated Reduction of P-Glycoprotein Transport Activity. Cancers 2024, 16, 1733. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gan, X.; Cheng, X.; Jia, Y.; Wang, G.; Tang, X.; Du, H.; Li, X.; Liu, X.; Xing, X.; et al. ABCC2 induces metabolic vulnerability and cellular ferroptosis via enhanced glutathione efflux in gastric cancer. Clin. Transl. Med. 2024, 14, e1754. [Google Scholar] [CrossRef] [PubMed]
- de Souza, I.; Monteiro, L.K.S.; Guedes, C.B.; Silva, M.M.; Andrade-Tomaz, M.; Contieri, B.; Latancia, M.T.; Mendes, D.; Porchia, B.; Lazarini, M.; et al. High levels of NRF2 sensitize temozolomide-resistant glioblastoma cells to ferroptosis via ABCC1/MRP1 upregulation. Cell Death Dis. 2022, 13, 591. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Chen, K.; Lu, Y.; Zhang, D.; Cheng, Y.; Li, L.; Huang, W.; He, G.; Liao, H.; Cai, L.; et al. ABCC5 facilitates the acquired resistance of sorafenib through the inhibition of SLC7A11-induced ferroptosis in hepatocellular carcinoma. Neoplasia 2021, 23, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Proietto, M.; Crippa, M.; Damiani, C.; Pasquale, V.; Sacco, E.; Vanoni, M.; Gilardi, M. Tumor heterogeneity: Preclinical models, emerging technologies, and future applications. Front. Oncol. 2023, 13, 1164535. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hu, J.; Yang, Y.; Zhang, H.; Liu, Y.; Fang, Y.; Qu, L.; Lin, A.; Luo, P.; Jiang, A.; et al. Drug resistance in cancer: Molecular mechanisms and emerging treatment strategies. Mol. Biomed. 2025, 6, 111. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Chen, M.; Gao, Y.; Huang, D.; Cao, H.; Peng, Y.; Guo, N.; Wang, F.; Zhang, S. Ferroptosis and Tumor Drug Resistance: Current Status and Major Challenges. Front. Pharmacol. 2022, 13, 879317. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Han, L.; Wang, J.; Song, L. Ferroptosis: The dawn of reversing drug resistance in digestive cancers. Genes Dis. 2026, 13, 101873. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Li, Y.; Xu, Y.; Liu, X.; Kang, X.; Zhu, J.; Long, S.; Han, Y.; Xue, C.; Sun, Z.; et al. SLC7A11 protects luminal A breast cancer cells against ferroptosis induced by CDK4/6 inhibitors. Redox Biol. 2024, 76, 103304. [Google Scholar] [CrossRef] [PubMed]
- Karner, E.R.; Wang, M.; Goel, H.L.; Mercurio, A.M. GATA3 promotes ferroptosis resistance by repressing integrin beta1 signaling. Proc. Natl. Acad. Sci. USA 2025, 122, e2427304122. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Shen, H.; Xu, J.; Fang, Z.; Wo, G.; Ma, Y.; Yang, K.; Wang, Y.; Yu, Q.; Tang, J.H. GATA3 mediates doxorubicin resistance by inhibiting CYB5R2-catalyzed iron reduction in breast cancer cells. Drug Resist. Updat. 2023, 69, 100974. [Google Scholar] [CrossRef] [PubMed]
- Querzoli, P.; Pedriali, M.; Rinaldi, R.; Secchiero, P.; Rossi, P.G.; Kuhn, E. GATA3 as an Adjunct Prognostic Factor in Breast Cancer Patients with Less Aggressive Disease: A Study with a Review of the Literature. Diagnostics 2021, 11, 604. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Lu, X.; Yang, H.; Huang, L.; Huang, W.; Tang, Y.; Liu, S.; Wang, H.; Zhang, Y. Metabolic heterogeneity protects metastatic mucosal melanomas cells from ferroptosis. Int. J. Mol. Med. 2022, 50, 124. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Zhang, L.; Li, S.; Tang, L. Metabolic reprogramming in melanoma therapy. Cell Death Discov. 2025, 11, 308. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Liang, Y.; Zhang, C. Ferroptosis Regulated by Hypoxia in Cells. Cells 2023, 12, 1050. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Hu, W.; Qian, D.; Bai, X.; He, H.; Li, L.; Sun, S. The Mechanisms of Ferroptosis Under Hypoxia. Cell. Mol. Neurobiol. 2023, 43, 3329–3341. [Google Scholar] [CrossRef] [PubMed]
- Bae, T.; Hallis, S.P.; Kwak, M.K. Hypoxia, oxidative stress, and the interplay of HIFs and NRF2 signaling in cancer. Exp. Mol. Med. 2024, 56, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.S.; Vizzerra, A.D.; Liou, H.; Flores, C.E.; Snider, A.J.; Snider, J.M.; Warfel, N.A. Hypoxia induced lipid droplet accumulation promotes resistance to ferroptosis in prostate cancer. Oncotarget 2025, 16, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.R.; Damaghi, M.; Marunaka, Y.; Spugnini, E.P.; Fais, S.; Gillies, R.J. Causes, consequences, and therapy of tumors acidosis. Cancer Metastasis Rev. 2019, 38, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Wojtkowiak, J.W.; Verduzco, D.; Schramm, K.J.; Gillies, R.J. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol. Pharm. 2011, 8, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.C.; Chen, L.; Na, R.; Yoo, K.; Ran, Q. The Gpx4NIKO Mouse Is a Versatile Model for Testing Interventions Targeting Ferroptotic Cell Death of Spinal Motor Neurons. Neurotox. Res. 2022, 40, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zheng, M.; Lester, K.L.; Han, Z. Light-induced Nrf2(-/-) mice as atrophic age-related macular degeneration model and treatment with nanoceria laden injectable hydrogel. Sci. Rep. 2019, 9, 14573. [Google Scholar] [CrossRef] [PubMed]
- Shakya, A.; McKee, N.W.; Dodson, M.; Chapman, E.; Zhang, D.D. Anti-Ferroptotic Effects of Nrf2: Beyond the Antioxidant Response. Mol. Cells 2023, 46, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lu, T.; Liu, Y.; Liu, Y.; Bai, S.; Chen, Q.; Zhao, B.; Wu, X. Establishment of SLC7A11-knockout mouse and its preliminary investigation in melanoma. Vitr. Cell. Dev. Biol. Anim. 2023, 59, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.K. Ferroptosis in Toxicology: Present and Future. Int. J. Mol. Sci. 2025, 26, 6658. [Google Scholar] [CrossRef] [PubMed]
- Sritharan, S.; Sivalingam, N. A comprehensive review on time-tested anticancer drug doxorubicin. Life Sci. 2021, 278, 119527. [Google Scholar] [CrossRef] [PubMed]
- Mimnaugh, E.G.; Kennedy, K.A.; Trush, M.A.; Sinha, B.K. Adriamycin-enhanced membrane lipid peroxidation in isolated rat nuclei. Cancer Res. 1985, 45, 3296–3304. [Google Scholar] [PubMed]
- Sinha, B.K.; Mimnaugh, E.G. Free radicals and anticancer drug resistance: Oxygen free radicals in the mechanisms of drug cytotoxicity and resistance by certain tumors. Free Radic. Biol. Med. 1990, 8, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Olson, F.; Mayhew, E.; Maslow, D.; Rustum, Y.; Szoka, F. Characterization, toxicity and therapeutic efficacy of adriamycin encapsulated in liposomes. Eur. J. Cancer Clin. Oncol. 1982, 18, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Kipps, E.; Young, K.; Starling, N. Liposomal irinotecan in gemcitabine-refractory metastatic pancreatic cancer: Efficacy, safety and place in therapy. Ther. Adv. Med. Oncol. 2017, 9, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, C.; Li, B.; Zheng, C.; Liu, G.; Liu, Z.; Zhang, L.; Xu, P. Discovery of ML210-Based glutathione peroxidase 4 (GPX4) degrader inducing ferroptosis of human cancer cells. Eur. J. Med. Chem. 2023, 254, 115343. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, J.M.; Doubravsky, C.E.; Wehri, E.; Li, Z.; Roberts, M.A.; Deol, K.K.; Lange, M.; Lasheras-Otero, I.; Momper, J.D.; Dixon, S.J.; et al. Identification of structurally diverse FSP1 inhibitors that sensitize cancer cells to ferroptosis. Cell Chem. Biol. 2023, 30, 1090–1103 e1097. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, C.R.; Guo, H.; Kyani, A.; Madak, J.T.; Arabzada, Z.; Neamati, N. The Dihydroorotate Dehydrogenase Inhibitor Brequinar Is Synergistic with ENT1/2 Inhibitors. ACS Pharmacol. Transl. Sci. 2020, 3, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- Ogihara, K.; Kikuchi, E.; Okazaki, S.; Hagiwara, M.; Takeda, T.; Matsumoto, K.; Kosaka, T.; Mikami, S.; Saya, H.; Oya, M. Sulfasalazine could modulate the CD44v9-xCT system and enhance cisplatin-induced cytotoxic effects in metastatic bladder cancer. Cancer Sci. 2019, 110, 1431–1441. [Google Scholar] [CrossRef] [PubMed]
- Narang, V.S.; Pauletti, G.M.; Gout, P.W.; Buckley, D.J.; Buckley, A.R. Sulfasalazine-induced reduction of glutathione levels in breast cancer cells: Enhancement of growth-inhibitory activity of Doxorubicin. Chemotherapy 2007, 53, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.; Bates, S.E. Tariquidar (XR9576): A P-glycoprotein drug efflux pump inhibitor. Expert Rev. Anticancer Ther. 2007, 7, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.K.; Wang, Y.J.; Gupta, P.; Chen, Z.S. Multidrug Resistance Proteins (MRPs) and Cancer Therapy. AAPS J. 2015, 17, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Ru, Q.; Li, Y.; Chen, L.; Wu, Y.; Min, J.; Wang, F. Iron homeostasis and ferroptosis in human diseases: Mechanisms and therapeutic prospects. Signal Transduct. Target. Ther. 2024, 9, 271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Luo, Y.L.; Xiang, Y.; Bai, X.Y.; Qiang, R.R.; Zhang, X.; Yang, Y.L.; Liu, X.L. Ferroptosis inhibitors: Past, present and future. Front. Pharmacol. 2024, 15, 1407335. [Google Scholar] [CrossRef] [PubMed]
- Essandoh, K.; Yang, L.; Wang, X.; Huang, W.; Qin, D.; Hao, J.; Wang, Y.; Zingarelli, B.; Peng, T.; Fan, G.C. Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochim. Biophys. Acta 2015, 1852, 2362–2371. [Google Scholar] [CrossRef] [PubMed]


| Molecular Regulator | Primary Biological Function | Impact on Ferroptosis | Mechanism | References |
|---|---|---|---|---|
| ELOVL5 | Long-chain PUFA elongation | ↑ Sensitivity | Expands the pool of oxidizable PUFAs available for membrane incorporation. | [61,62] |
| ACSL4 | PUFA activation and phospholipid incorporation | ↑ Sensitivity | Enriches PUFA-containing phospholipids that undergo lipid peroxidation. | [68] |
| ACSL3 | MUFA activation | ↓ Sensitivity | Promotes MUFA incorporation, displacing oxidizable PUFAs. | [70] |
| SCD1 | MUFA biosynthesis | ↓ Sensitivity | Increases membrane MUFA content and limits lipid peroxidation. | [70,76] |
| LPCAT1 | Phospholipid remodeling (Lands cycle) | ↓ Sensitivity | Incorporates saturated fatty acids into membrane phospholipids, increasing membrane saturation and reducing PUFA-dependent lipid peroxidation. | [78,79] |
| Resistance Node | Role in Ferroptosis Resistance | Targeting Strategy | Representative Agents | Clinical Status | References |
|---|---|---|---|---|---|
| GPX4 | Detoxifies phospholipid hydroperoxides | GPX4 inhibition | RSL3, ML210 | Preclinical | [115,116] |
| FSP1 | Regenerates CoQ10H2 independently of GPX4 | FSP1 inhibition | iFSP1, FSEN1 | Preclinical | [25,27,117] |
| DHODH | Mitochondrial antioxidant defense | DHODH inhibition | Brequinar | Clinical drug repurposing | [118] |
| SLC7A11 (xCT) | Maintains cystine uptake and GSH synthesis | xCT inhibition | Sulfasalazine | Clinical drug repurposing | [119,120] |
| ABC transporters | Efflux ferroptosis inducers | Transport inhibition | Tariquidar, Cyclosporine, PSC833 | Preclinical | [121,122] |
| Lipid remodeling enzymes | Reduce oxidizable phospholipids | ACSL4 activation, SCD1 inhibition | Experimental | Preclinical | [66,71] |
| Iron metabolism | Restricts labile iron pool | Iron-based therapies | Artesunate, DHA, Iron nanoparticles | Clinical/Preclinical | [11,123,124] |
| CD63-mediated exosomes | Export ferritin-bound iron | Exosome inhibition | GW4869, CD63 antibody | Experimental | [125] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Sinha, B.K. Ferroptosis Resistance: Redundant Antioxidant Networks Are a Barrier to Cancer Therapy. Antioxidants 2026, 15, 860. https://doi.org/10.3390/antiox15070860
Sinha BK. Ferroptosis Resistance: Redundant Antioxidant Networks Are a Barrier to Cancer Therapy. Antioxidants. 2026; 15(7):860. https://doi.org/10.3390/antiox15070860
Chicago/Turabian StyleSinha, Birandra K. 2026. "Ferroptosis Resistance: Redundant Antioxidant Networks Are a Barrier to Cancer Therapy" Antioxidants 15, no. 7: 860. https://doi.org/10.3390/antiox15070860
APA StyleSinha, B. K. (2026). Ferroptosis Resistance: Redundant Antioxidant Networks Are a Barrier to Cancer Therapy. Antioxidants, 15(7), 860. https://doi.org/10.3390/antiox15070860