
Selective Inhibition of Vascular Smooth Muscle Cell Function by COVID-19 Antiviral Drugs: Impact of Heme Oxygenase-1


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Cell Proliferation and DNA Synthesis
2.4. Cell Migration
2.5. Cell Viability
2.6. Western Blotting
2.7. Quantitative Real-Time PCR (qRT-PCR) Analysis
2.8. HO Activity
2.9. HO-1 Promoter Activity
2.10. Chromatin Immunoprecipitation (ChIP) Assay
2.11. Intracellular Reactive Oxygen Species (ROS) Measurement
2.12. Small Interfering RNA (siRNA) Transfection
2.13. Statistical Analysis
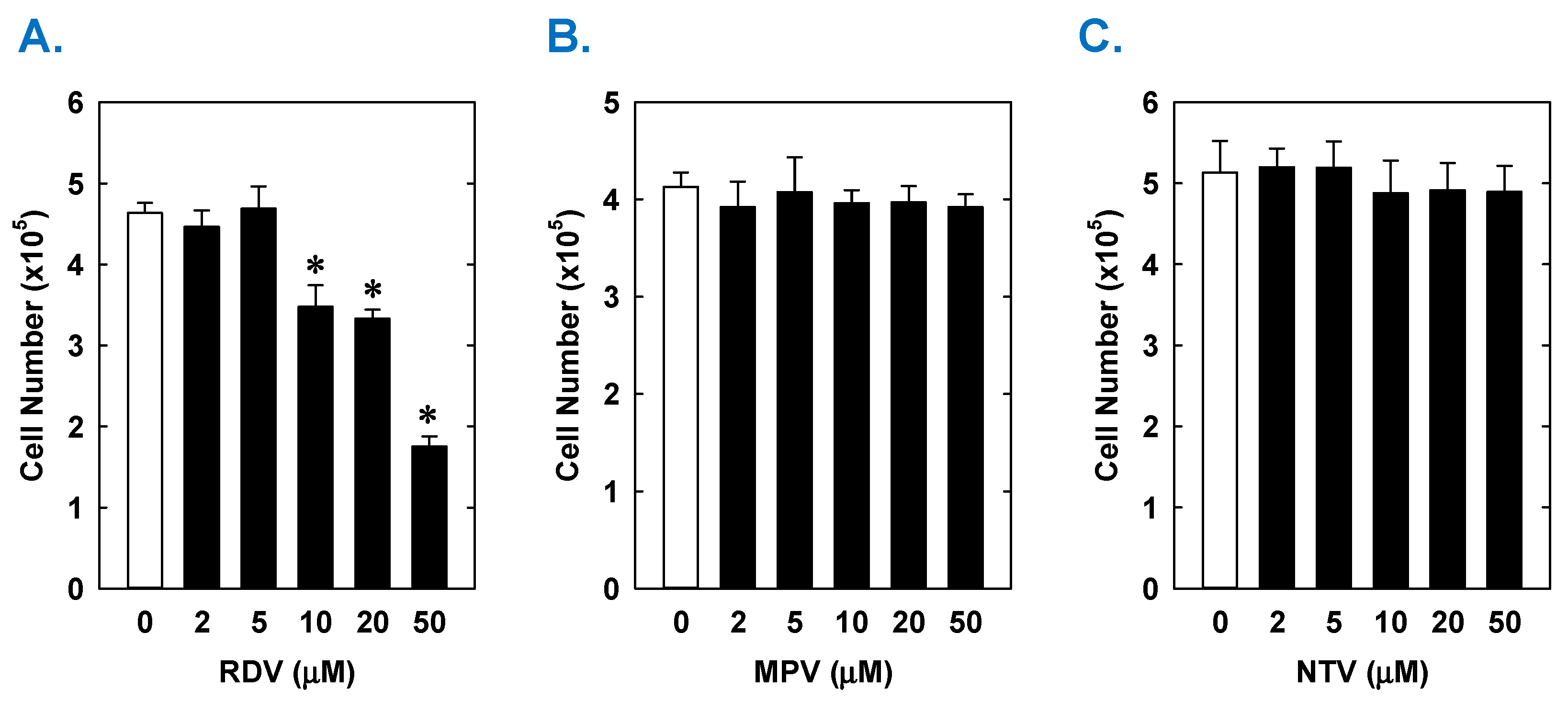
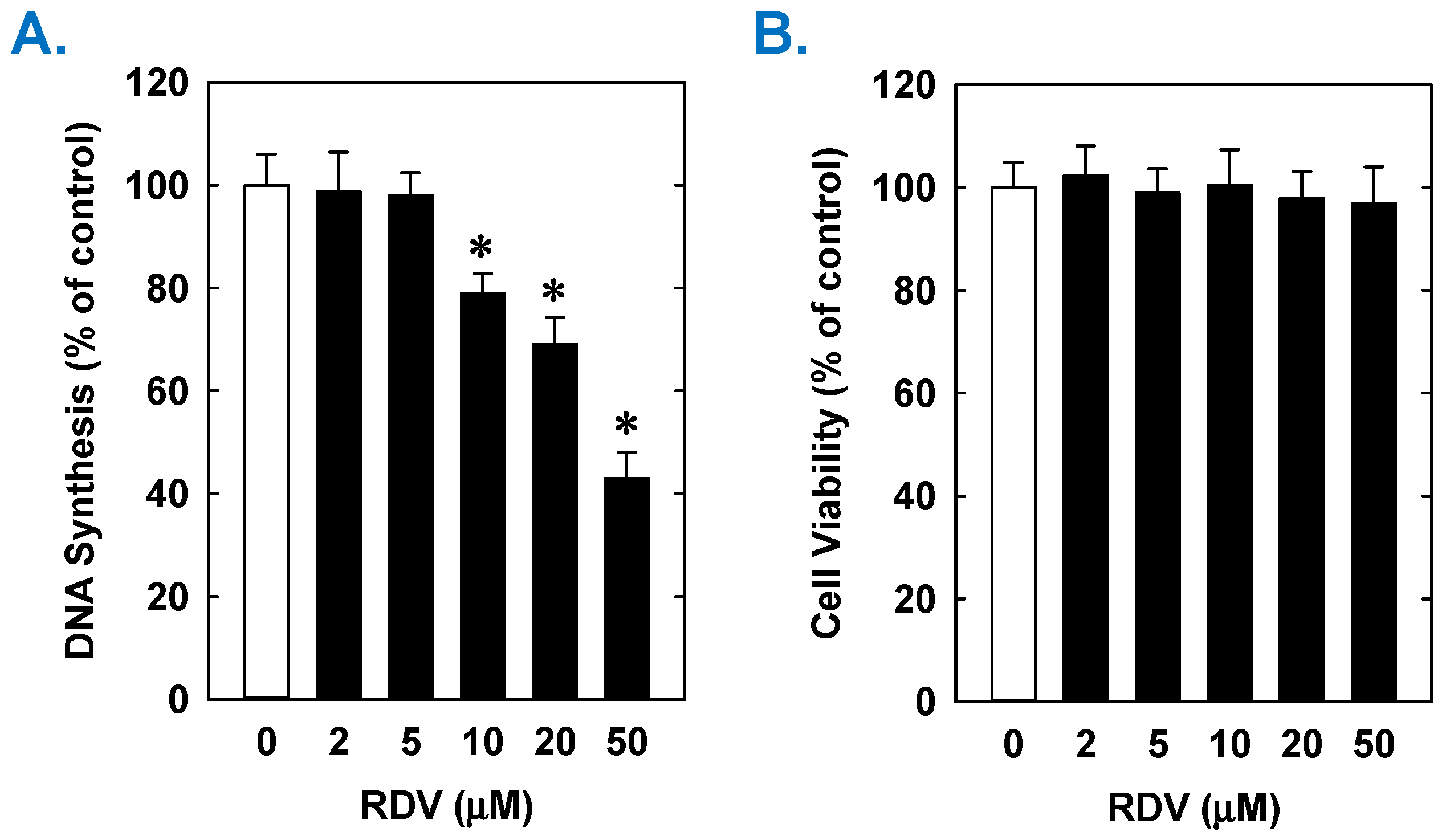
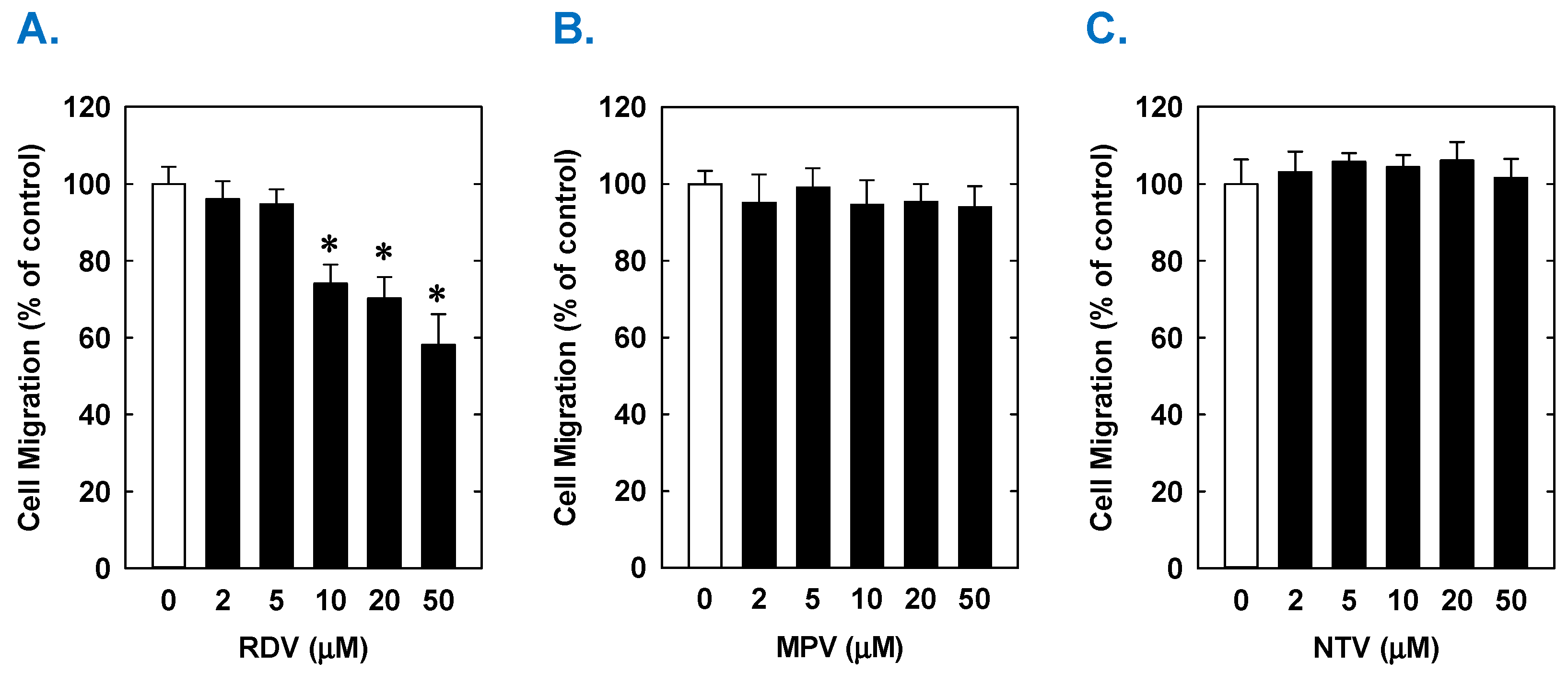
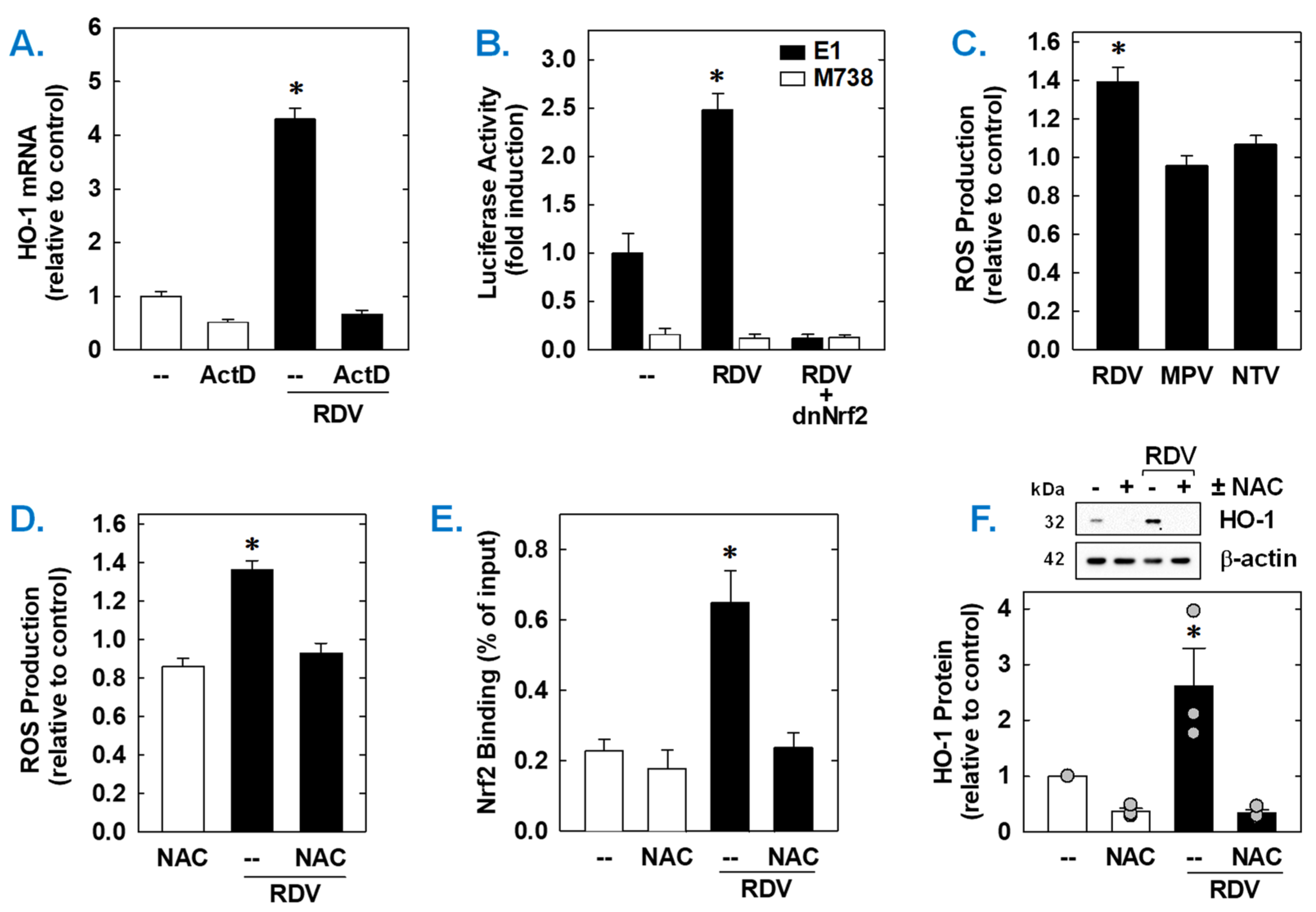
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard. 2023. Available online: https://data.who.int/dashboards/covid19/deaths?n=0 (accessed on 21 March 2025).
- Tay, M.Z.; Poh, C.M.; Renia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunology, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- McFadyen, J.D.; Stevens, H.; Peter, K. The emerging threat of (micro) thrombosis in COVID-19 and its therapeutic implications. Circ. Res. 2020, 127, 571–587. [Google Scholar] [CrossRef]
- Nazerian, Y.; Ghasemi, M.; Yassaghi, Y.; Nazerian, A.; Hashemi, S.M. Role of SARS-CoV-2-induced cytokine storm in multi-organ failure: Molecular pathways and potential therapeutic options. Int. Immunopharmacol. 2022, 113, 109428. [Google Scholar] [CrossRef]
- Morrow, A.J.; Sykes, R.; McIntosh, A.; Kamdar, A.; Bagot, C.; Bayes, H.K.; Blyth, K.G.; Briscoe, M.; Bulluck, H.; Carrick, D.; et al. A multisystem, cardio-renal investigation of post-COVID-19 illness. Nat. Med. 2022, 28, 1303–1313. [Google Scholar] [CrossRef]
- Onder, G.; Rezza, G.; Brusaferro, S. Case-fatality rate and characteristics of patients dying in relation to COVID-19. JAMA 2020, 323, 1775–1776. [Google Scholar] [CrossRef]
- Guo, Y.-R.; Cao, D.-Q.; Hong, Z.-S.; Tan, Y.-Y.; Chen, S.-D.; Jin, H.-J.; Tan, K.S.; Wang, D.-Y.; Yan, Y. The origin, transmission, and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—An update on the status. Mil. Med. Res. 2020, 7, 11. [Google Scholar] [CrossRef]
- Libby, P.; Luscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef]
- Ma, Z.; Yang, K.Y.; Huang, Y.; Liu, K.O. Endothelial contribution to COVID-19: An update on mechanisms and therapeutic implications. J. Mol. Cell. Cardiol. 2022, 164, 69–82. [Google Scholar] [CrossRef]
- Prasad, M.; Leon, M.; Lerman, L.O.; Lerman, A. Viral endothelial dysfunction: A unifying mechanism for COVID-19. Mayo Clin. Proc. 2021, 96, 3099–3108. [Google Scholar] [CrossRef]
- McCraken, I.R.; Saginc, G.; He, L.; Huseynov, A.; Daniels, A.; Fletcher, S.; Peghaire, C.; Kalna, V.; Andaloussi-Mae, M.; Muhl, L.; et al. Lack of evidence of angiotensin-converting enzyme 2 expression and replicative infection by SARS-CoV-2 in human endothelial cells. Circulation 2021, 143, 865–868. [Google Scholar] [CrossRef]
- Marchiano, S.; Hsiang, T.Y.; Khanna, A.; Higashi, T.; Whitmore, L.S.; Bargehr, J.; Davaapil, H.; Chang, J.; Smith, E.; Ong, L.P.; et al. SARS-CoV-2 infects human pluripotent stem cell-derived cardiomyocytes, impairing electrical and mechanical function. Stem. Cell Rep. 2021, 16, 478–492. [Google Scholar] [CrossRef]
- Suzuki, Y.J.; Nikolaienko, S.I.; Dibrova, V.A.; Dibrova, Y.V.; Vasylyk, V.M.; Novikov, M.Y.; Shults, N.V.; Gychka, S.G. SARS-CoV-2 spike protein-mediated cell signaling in lung vascular cells. Vasc. Pharmacol. 2021, 137, 106823. [Google Scholar] [CrossRef]
- Martinez-Salazar, B.; Holwerda, M.; Studle, C.; Piragyte, I.; Mercader, N.; Engelhardt, B.; Rieben, R.; Doring, Y. COVID-19 and the vasculature: Current aspects and long-term consequences. Front. Cell Devel. Biol. 2022, 10, 824851. [Google Scholar] [CrossRef]
- Tzankov, A.; Bhattacharyya, S.; Kotlo, K.; Tobacman, J.K. Increase in chondroitin sulfate and decline in arylsulfatase B may contribute to pathophysiology of COVID-19 respiratory failure. Pathobiology 2022, 89, 81–91. [Google Scholar] [CrossRef]
- Zanoli, L.; Gaudio, A.; Mikhailidis, D.P.; Katsiki, N.; Castellino, N.; Cicer, L.L.; Geraci, G.; Sessa, C.; Fiorito, L.; Marino, F.; et al. Vascular dysfunction of COVID-19 is partially reversed long-term. Circ. Res. 2022, 130, 1276–1285. [Google Scholar] [CrossRef]
- Sykes, R.A.; Neves, K.B.; Alves-Lopez, R.; Caputo, I.; Fallon, K.; Jamieson, N.B.; Kamdar, A.; Legrini, A.; Leslie, H.; McIntosh, A.; et al. Vascular mechanisms of post-COVID-19 conditions: Rho-kinase is a novel target for therapy. Eur. Heart J. 2023, 9, 371–386. [Google Scholar] [CrossRef]
- Islam, T.; Hasan, M.; Rhamna, M.S.; Islam, M.R. Comparative evaluation of authorized drugs for treating COVID-19 patients. Health Sci. Rep. 2022, 5, e671. [Google Scholar] [CrossRef]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for the inhibition of RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Schinazi, R.F.; Gotte, M. Molnupiravir promotes SARS-CoV-2 mutagenesis vi the RNA template. J. Biol. Chem. 2021, 297, 100770. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M(pro) inhibitor candidate for the treatment of COVID-19. Science 2021, 374, 1568–1593. [Google Scholar] [CrossRef]
- Ayer, A.; Zarjou, A.; Agarwal, A.; Stocker, R. Heme oxygenases in cardiovascular health and disease. Physiol. Rev. 2016, 96, 1449–1508. [Google Scholar] [CrossRef]
- Durante, W.; Johnson, F.K.; Johnson, R.A. Role of carbon monoxide in cardiovascular function. J. Cell. Mol. Med. 2006, 10, 672–686. [Google Scholar] [CrossRef]
- Durante, W. Targeting heme oxygenase-1 in the arterial response to injury and disease. Antioxidants 2020, 9, 829. [Google Scholar] [CrossRef]
- Durante, W. Protective role of heme oxygenase-1 against inflammation in atherosclerosis. Front. Biosci. 2011, 16, 2372–2388. [Google Scholar] [CrossRef]
- Espinosa, J.A.; Gonzalez, P.A.; Kalergis, A.M. Modulation of antiviral immunity by heme oxygenase-1. Am. J. Pathol. 2017, 187, 487–493. [Google Scholar] [CrossRef]
- Wagener, F.A.D.T.G.; Pickkers, P.; Peterson, S.J.; Immenschuh, S.; Abraham, N.G. Targeting the heme-heme oxygenase system to prevent severe complications following COVID-19 infections. Antioxidants 2020, 96, 540. [Google Scholar] [CrossRef]
- Singh, D.; Wasan, H.; Reeta, K.H. Heme oxygenase-1 modulation: A potential therapeutic target for COVID-19 and associated complications. Free Radic. Biol. Med. 2020, 161, 263–271. [Google Scholar] [CrossRef]
- Rossi, M.; Piagnerelli, M.; Van Meerhaeghe, A.; Boudjeltia, K.Z. Heme oxygenase-1 (HO-1) cytoprotective pathway: A potential treatment strategy against coronavirus disease 2019 (COVID-19)-induced cytokine storm. Med. Hypotheses 2020, 144, 110242. [Google Scholar] [CrossRef]
- Liu, X.M.; Durante, Z.E.; Peyton, K.J.; Durante, W. Heme oxygenase-1-derived bilirubin counteracts HIV protease inhibitor-mediated endothelial dysfunction. Free Radic. Biol. Med. 2016, 94, 218–229. [Google Scholar] [CrossRef]
- Ben-Romano, R.; Rudich, A.; Etzion, S.; Potashnik, R.; Kagan, E.; Greenbaum, U.; Bashan, N. Nelfinavir induces adipocyte insulin resistance through induction of oxidative stress: Differential protective effect of antioxidant agents. Antiviral Ther. 2002, 11, 1051–1060. [Google Scholar] [CrossRef]
- Muhl, H.; Paulukat, J.; Hofler, S.; Hellmuth, M.; Franzen, R.; Pfeilschifter, J. The HIV protease inhibitor ritonavir synergizes with butyrate for the induction of apoptotic cell death and mediates expression of heme oxygenase-1 in DLD-1 colon carcinoma cells. Br. J. Pharmacol. 2004, 143, 890–898. [Google Scholar] [CrossRef]
- Laurence, J.; Elhadad, S.; Gostynska, S.; Yu, Z.; Terry, H.; Varshney, R.; Fung, K.-M.; Choi, M.E.; Ahamed, J. HIV protease inhibitor ritonavir induces renal fibrosis and dysfunction: Role of platelet-derived TGF-β1 and intervention via antioxidant pathway. AIDS 2020, 34, 989–1000. [Google Scholar] [CrossRef]
- Durante, W.; Schini, V.B.; Catovsky, S.; Kroll, M.H.; Vanhoutte, P.M.; Schafer, A.I. Plasmin potentiates induction of nitric oxide synthesis by interleukin-1 beta in vascular smooth muscle cells. Am. J. Physiol. 1993, 264, H617–H624. [Google Scholar] [CrossRef]
- Peyton, K.J.; Reyna, S.V.; Chapman, G.B.; Ensenat, D.; Liu, X.M.; Wang, H.; Schafer, A.I.; Durante, W. Heme oxygenase-derived carbon monoxide is an autocrine inhibitor of vascular smooth muscle cell growth. Blood 2002, 99, 4443–4448. [Google Scholar] [CrossRef]
- Peyton, K.J.; Yu, Y.; Yates, B.; Shebib, A.R.; Liu, X.M.; Wang, H.; Durante, W. Compound C inhibits vascular smooth muscle cell proliferation and migration in an AMPK-activated protein kinase-independent fashion. J. Pharmacol. Exp. Ther. 2011, 338, 476–484. [Google Scholar] [CrossRef]
- Peyton, K.J.; Shebib, A.R.; Azam, A.M.; Liu, X.M.; Tulis, D.A.; Durante, W. Bilirubin inhibits neointima formation and vascular smooth muscle cell proliferation and migration. Front. Pharmacol. 2012, 3, 48. [Google Scholar] [CrossRef]
- Liu, X.M.; Peyton, K.J.; Durante, W. Physiologic cyclic strain promotes endothelial cell survival via the induction of heme oxygenase-1. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1634–H1643. [Google Scholar] [CrossRef]
- Liu, X.M.; Peyton, K.J.; Ensenat, D.E.; Wang, H.; Schafer, A.I.; Alam, J.; Durante, W. Endoplasmic reticulum stress stimulates heme oxygenase-1 gene expression in vascular smooth muscle cells. Role in cell survival. J. Biol. Chem. 2005, 280, 872–877. [Google Scholar] [CrossRef]
- Liu, X.M.; Peyton, K.J.; Ensenat, D.; Wang, H.; Hannink, M.; Alam, J.; Durante, W. Nitric oxide stimulates heme oxygenase-1 gene transcription via the Nrf2/ARE complex to promote vascular smooth muscle cell survival. Cardiovasc. Res. 2007, 75, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Durante, W.; Peyton, K.J.; Schafer, A.I. Platelet-derived growth factor stimulates heme oxygenase-1 gene expression and carbon monoxide production in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2666–2672. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, J.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomized, double-blind, placebo-controlled multicenter trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Painter, W.P.; Holman, W.; Bush, J.A.; Almazedi, F.; Malik, H.; Eraut, N.C.J.E.; Morin, M.J.; Szewczyk, L.J.; Painter, G.R. Human safety, tolerability, and pharmacokinetics of molnupiravir, a novel broad-spectrum oral antiviral agent with activity against SARS-CoV-2. Antimicrob. Agents Chemother. 2021, 65, e02428-20. [Google Scholar] [CrossRef]
- Hau, R.K.; Wright, S.H.; Cherrington, N.J. PF-07321332 (Nirmatrelvir) does not interact with human ENT1 or ENT2: Implications for COVID-19 patients. Clin. Transl. Sci. 2022, 15, 1599–1605. [Google Scholar] [CrossRef]
- Duckers, H.J.; Boehm, M.; True, A.L.; Yet, S.F.; San, H.; Park, J.L.; Clinton Webb, R.; Lee, M.E.; Nagel, G.J.; Nagel, E.G. Heme oxygenase-1 protects against vascular constriction and proliferation. Nat. Med. 2001, 7, 693–698. [Google Scholar] [CrossRef]
- Tulis, D.A.; Durante, W.; Peyton, K.J.; Evans, A.J.; Schafer, A.I. Heme oxygenase-1 attenuates vascular remodeling following balloon injury in rat carotid arteries. Atherosclerosis 2001, 155, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.I.; Gangopadhyay, A.; Kelly, E.E.; Pagano, P.J.; Zuckerbraun, B.S.; Bauer, P.M. HO-1 and CO decrease platelet-derived growth factor-induced vascular smooth muscle cell migration via inhibition of Nox1. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 98–104. [Google Scholar] [CrossRef]
- Fledderus, J.O.; Boon, R.A.; Volger, O.L.; Hurttila, H.; Yla-Herttuala, S.; Pannekoek, H.; Levonen, A.-L.; Horrevoets, A.J.G. KLF2 primes the antioxidant transcription factor Nrf2 for activation in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1339–1346. [Google Scholar] [CrossRef]
- Di Francesco, L.; Totani, L.; Dovizio, M.; Piccoli, A.; Di Francesco, A.; Salvatore, T.; Pandolfi, A.; Evangelista, V.; Dercho, R.A.; Seta, F.; et al. Induction of prostacyclin by steady laminar shear stress suppresses tumor necrosis factor-α biosynthesis via heme oxygenase-1 in human endothelial cells. Circ. Res. 2009, 104, 506–513. [Google Scholar] [CrossRef]
- Vinals, M.; Martinez-Gonzalez, J.; Badimon, J.J.; Badimon, L. HDL-induced prostacyclin release in smooth muscle cells is dependent on cyclooxygenase-2 (Cox-2). Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2381–3488. [Google Scholar] [CrossRef] [PubMed]
- Futaki, N.; Takahashi, S.; Yokoyama, M.; Arai, I.; Higuchi, S.; Otomo, S. NS-398, a new anti-inflammatory agent, selectively inhibits prostacyclin G/H synthase/cyclooxygenase (COX-2) activity in vitro. Prostaglandins 1994, 47, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Merches, K.; Breunig, L.; Fender, J.; Brand, T.; Bätz, V.; Idel, S.; Kollipara, L.; Reinders, Y.; Sickmann, A.; Mally, A.; et al. The potential of remdesivir to affect function, metabolism and proliferation of cardiac and kidney cells in vitro. Arch. Toxicol. 2022, 96, 2341–2360. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, X.; Guo, X.; Li, W.; Zhang, T.; Chai, D.; Liu, Y.; Chen, L.; Ai, X.; Zhou, T.; et al. Remdesivir alleviates skin fibrosis by suppressing TGF-β1 signaling pathway. PLoS ONE 2024, 19, e0305927. [Google Scholar] [CrossRef]
- Bjork, J.A.; Wallace, K.B. Remdesivir; molecular and functional measures of mitochondrial safety. Toxic. Appl. Pharmacol. 2021, 433, 115783. [Google Scholar] [CrossRef]
- Xu, Y.; Barauskas, O.; Kim, C.; Babusis, D.; Murakami, E.; Kornyeyev, D.; Lee, G.; Stepan, G.; Perron, M.; Bannister, R.; et al. Off-target in vitro profiling demonstrates that remdesivir is a highly selective antiviral agent. Antimicrob. Agents Chemother. 2021, 65, e02237. [Google Scholar] [CrossRef]
- Lee, C.M.; Kang, M.-A.; Bae, J.S.; Park, K.; Yang, Y.-H.; Lee, J.; Jang, K.Y.; Park, S.-H. An in vitro study on anti-carcinogenic effect of remdesivir in human ovarian cancer cells via generation of reactive oxygen species. Hum. Exp. Toxicol. 2022, 41, 1–10. [Google Scholar] [CrossRef]
- Ali, F.; Zakkar, M.; Karu, K.; Liddington, E.A.; Hamdulay, S.S.; Boyle, J.J.; Zloh, M.; Bauer, A.; Haskard, D.O.; Evans, P.C.; et al. Induction of the cytoprotective enzyme heme oxygenase-1 by statins is enhanced in vascular endothelium exposed to laminar shear stress and impaired by disturbed blood flow. J. Biol. Chem. 2009, 284, 1882–1892. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, M.M.; Darbani, R.; Rabet, O.; Ghorbanihaghjo, A.; Rashtchizadeh, N.; Raeisi, S.; Khordadmehr, M. Effects of remdesivir on liver enzymes, oxidative stress and liver histopathology in rats. Int. J. Ecotoxicol. Ecobiol. 2024, 9, 148–159. [Google Scholar] [CrossRef]
- Nugnes, R.; Orlo, E.; Russo, C.; Lavorgna, M.; Isidori, M. Comprehensive eco-geno-toxicity and environmental risk of common antiviral drugs in aquatic environments post pandemic. J. Hazard Mater. 2024, 480, 135947. [Google Scholar] [CrossRef]
- Kong, K.; Chang, Y.; Qiao, H.; Zhao, C.; Chen, X.; Rong, K.; Zhang, P.; Jin, M.; Zhang, J.; Li, H.; et al. Paxlovid accelerates cartilage degeneration and senescence through activating endoplasmic reticulum stress and interfering redox homeostasis. J. Transl. Med. 2022, 20, 549. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A. Electron donor-acceptor capacity of selected pharmaceuticals against COVID-19. Antioxidants 2021, 10, 979. [Google Scholar] [CrossRef]
- Yasri, S.; Wiwanitki, V. Molnupiravir, favipiravir and other antiviral drugs with proposed potentials for management of COVID-19: A concern on antioxidant aspect. Int. J. Biochem. Mol. Biol. 2022, 13, 1–4. [Google Scholar]
- Bartolini, D.; Stabile, A.M.; Bastianelli, S.; Giustarini, D.; Pierucci, S.; Busti, C.; Vacca, C.; Gidari, A.; Francisci, D.; Castronari, R.; et al. SARS-CoV2 infection impairs the metabolism and redox function of cellular glutathione. Red. Biol. 2021, 45, 102041. [Google Scholar] [CrossRef]
- Kwok, M.; Lee, C.; Li, H.S.; Deng, R.; Tsoi, C.; Ding, Q.; Tsang, S.Y.; Leung, K.T.; Yan, B.P.; Poon, E.N. Remdesivir induces persistent mitochondrial and structural damage in human induced pluripotent stem cell-derived cardiomyocytes. Cardiovasc. Res. 2021, 118, 2652–2664. [Google Scholar] [CrossRef]
- DeFoor, N.; Paul, S.; Li, S.; Basso, E.K.G.; Stevenson, V.; Browning, J.L.; Prater, A.K.; Brindley, S.; Tao, G.; Pickrell, A.A. Remdesivir increases mtDNA copy number causing mild alterations to oxidative phosphorylation. Sci. Rep. 2023, 13, 15339. [Google Scholar] [CrossRef]
- Fisar, Z.; Luptak, M.; Hroudova, J. Little in vitro effect of remdesivir on mitochondrial respiration and monoamine oxidase activity in isolated mitochondria. Tox. Lett. 2021, 350, 143–151. [Google Scholar] [CrossRef]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in keap1 are required for keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventative agents and oxidative stress. Mol. Cell Biol. 2003, 23, 137–151. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Zuckerbraun, B.S.; Haga, M.; Liu, F.; Song, R.; Usheva, A.; Stachulak, C.; Bodyak, N.; Smith, R.N.; Csizmadia, E.; et al. Carbon monoxide suppresses artherosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat. Med. 2003, 9, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Ollinger, R.; Bilban, M.; Erat, A.; Froio, A.; McDaid, J.; Tyagi, S.; Csizmadia, E.; Graca-Souza, A.V.; Liloia, A.; Soares, M.P.; et al. Bilirubin: A natural inhibitor of vascular smooth muscle proliferation. Circulation 2005, 112, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Levonen, A.L.; Inkala, M.; Heikura, S.; Jauhiainen, S.; Jyrkkanen, H.K.; Kansanen, E.; Maatta, K.; Romppanen, E.; Turunen, P.; Rutanen, J.; et al. Nrf2 gene transfer induces antioxidant enzymes and suppresses smooth muscle cell growth in vitro and reduces oxidative stress in rabbit aorta in vivo. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Ashino, T.; Yamamoto, M.; Yoshida, T.; Numazawa, S. Redox-sensitive transcription factor Nrf2 regulates vascular smooth muscle cell migration and neointimal hyperplasia. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 760–768. [Google Scholar] [CrossRef]
- Yin, J.; Xia, W.; Wu, M.; Zhang, Y.; Huang, S.; Zhang, A.; Jia, Z. Inhibition of mitochondrial complex I activity attenuates neointimal hyperplasia by inhibiting smooth muscle cell proliferation and migration. Chem. Biol. Interact. 2019, 304, 73–82. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, J.; Jiang, Y. Transcription factor Nrf2 as a potential therapeutic agent for COVID-19. Cell Stress Chap. 2023, 28, 11–20. [Google Scholar] [CrossRef]
- Olagnier, D.; Farahani, E.; Thyrsted, J.; Blay-Cadanet, J.; Herengt, A.; Idorn, M.; Hait, A.; Hernaez, B.; Knudson, A.; Iversen, M.B.; et al. SARS-CoV2-mediated suppression of Nrf2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumurate. Nat. Commun. 2020, 11, 4938. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Haas de Mello, A.; Morris, D.R.; Jones-Hall, Y.L.; Ivanciuc, T.; Sattler, R.A.; Paessler, S.; Menachery, V.D.; Garafolo, R.P.; Casola, A. SARS-CoV-2 inhibits Nrf2-mediated antioxidant responses in airway epithelial cells and in the lung of a murine model of infection. Microbiol. Spectr. 2023, 11, e0037823. [Google Scholar] [CrossRef]
- Ryter, S.; Choi, A.M.K. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl. Res. 2016, 167, 7–34. [Google Scholar] [CrossRef]
- Libby, P. Endothelial inflammation in COVID-19: Disrupted endothelial function underlies the multiorgan complications of COVID-19. Science 2024, 386, 972–973. [Google Scholar] [CrossRef]
- True, A.L.; Olive, M.; Boehm, M.; San, H.; Westrick, R.J.; Raghavachari, N.; Xu, X.; Lynn, E.G.; Sack, M.N.; Munson, P.J.; et al. Heme oxygenase-1 deficiency accelerates formation of arterial thrombosis through oxidative damage to endothelium, which is rescued by inhaled carbon monoxide. Circ. Res. 2007, 101, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Pierce, J.; Franklin, C.; Olson, R.M.; Morrison, A.R.; Amos-Landgraf, J. Translating animal models of SARS-CoV-2 infection to vascular, neurological and gastrointestinal manifestations of COVID-19. Dis. Models Mech. 2025, 18, dmm052086. [Google Scholar] [CrossRef]
- Poss, K.D.; Tonegawa, S. Heme oxygenase 1 is required for mammalian iron utilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peyton, K.J.; Durante, G.L.; Durante, W. Selective Inhibition of Vascular Smooth Muscle Cell Function by COVID-19 Antiviral Drugs: Impact of Heme Oxygenase-1. Antioxidants 2025, 14, 945. https://doi.org/10.3390/antiox14080945
Peyton KJ, Durante GL, Durante W. Selective Inhibition of Vascular Smooth Muscle Cell Function by COVID-19 Antiviral Drugs: Impact of Heme Oxygenase-1. Antioxidants. 2025; 14(8):945. https://doi.org/10.3390/antiox14080945
Chicago/Turabian StylePeyton, Kelly J., Giovanna L. Durante, and William Durante. 2025. "Selective Inhibition of Vascular Smooth Muscle Cell Function by COVID-19 Antiviral Drugs: Impact of Heme Oxygenase-1" Antioxidants 14, no. 8: 945. https://doi.org/10.3390/antiox14080945
APA StylePeyton, K. J., Durante, G. L., & Durante, W. (2025). Selective Inhibition of Vascular Smooth Muscle Cell Function by COVID-19 Antiviral Drugs: Impact of Heme Oxygenase-1. Antioxidants, 14(8), 945. https://doi.org/10.3390/antiox14080945