
Gallic Acid Alleviates Acetaminophen-Induced Acute Liver Injury by Regulating Inflammatory and Oxidative Stress Signaling Proteins

Abstract

1. Introduction
2. Materials and Methods
2.1. Animal Housing
2.2. Experimental Design
2.3. Liver Injury Markers Analyses
2.4. Liver Histological Investigation
2.5. Oxidative Stress Markers Tests
2.6. Hepatic Cytokines Level Assay
2.7. qPCR Analysis
2.8. Functional Enrichment Analysis of GO and KEGG Pathways
2.9. Molecular Docking Validation
2.10. Western Blotting Assay
2.11. Statistical Analysis
3. Results
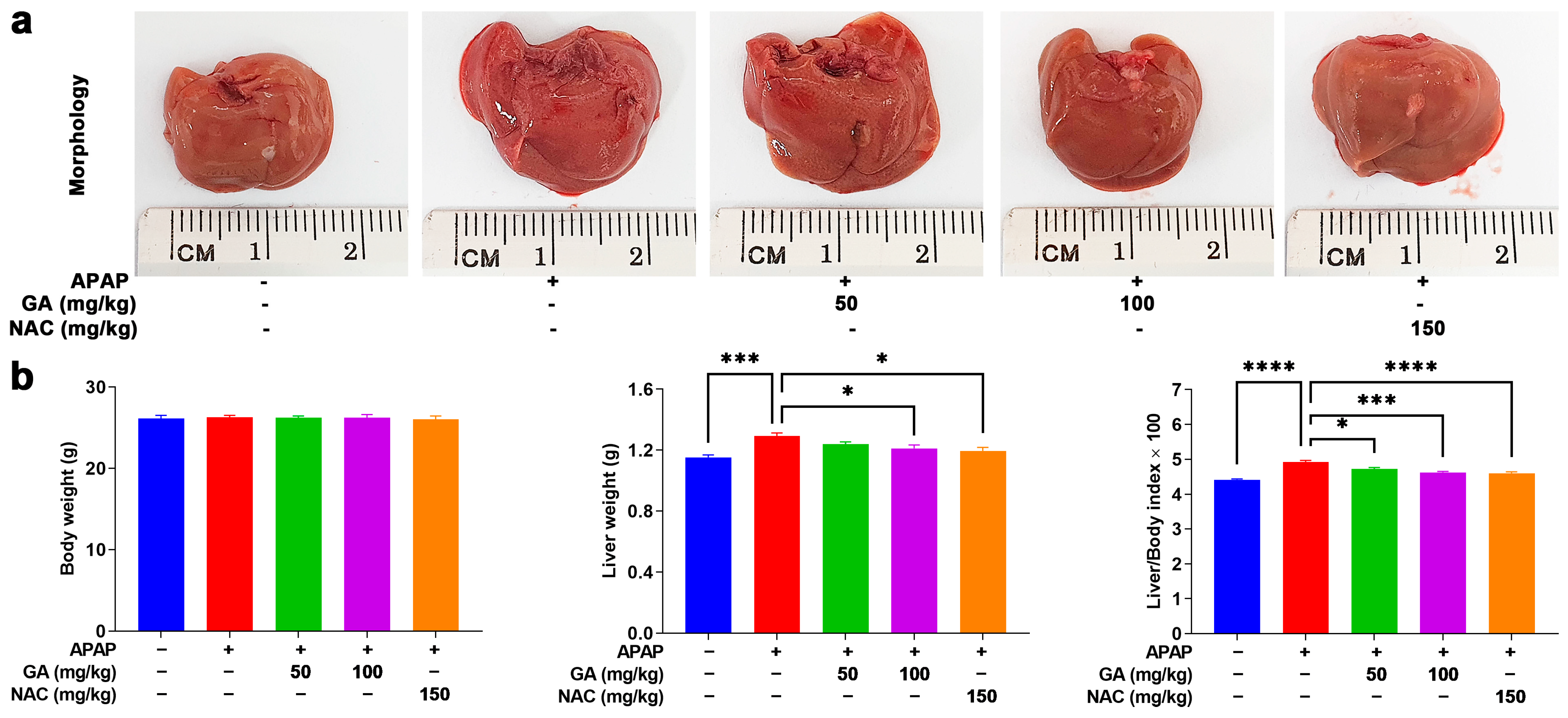
3.1. GA Treatment Mitigates Liver Burden in APAP-Exposed Mice
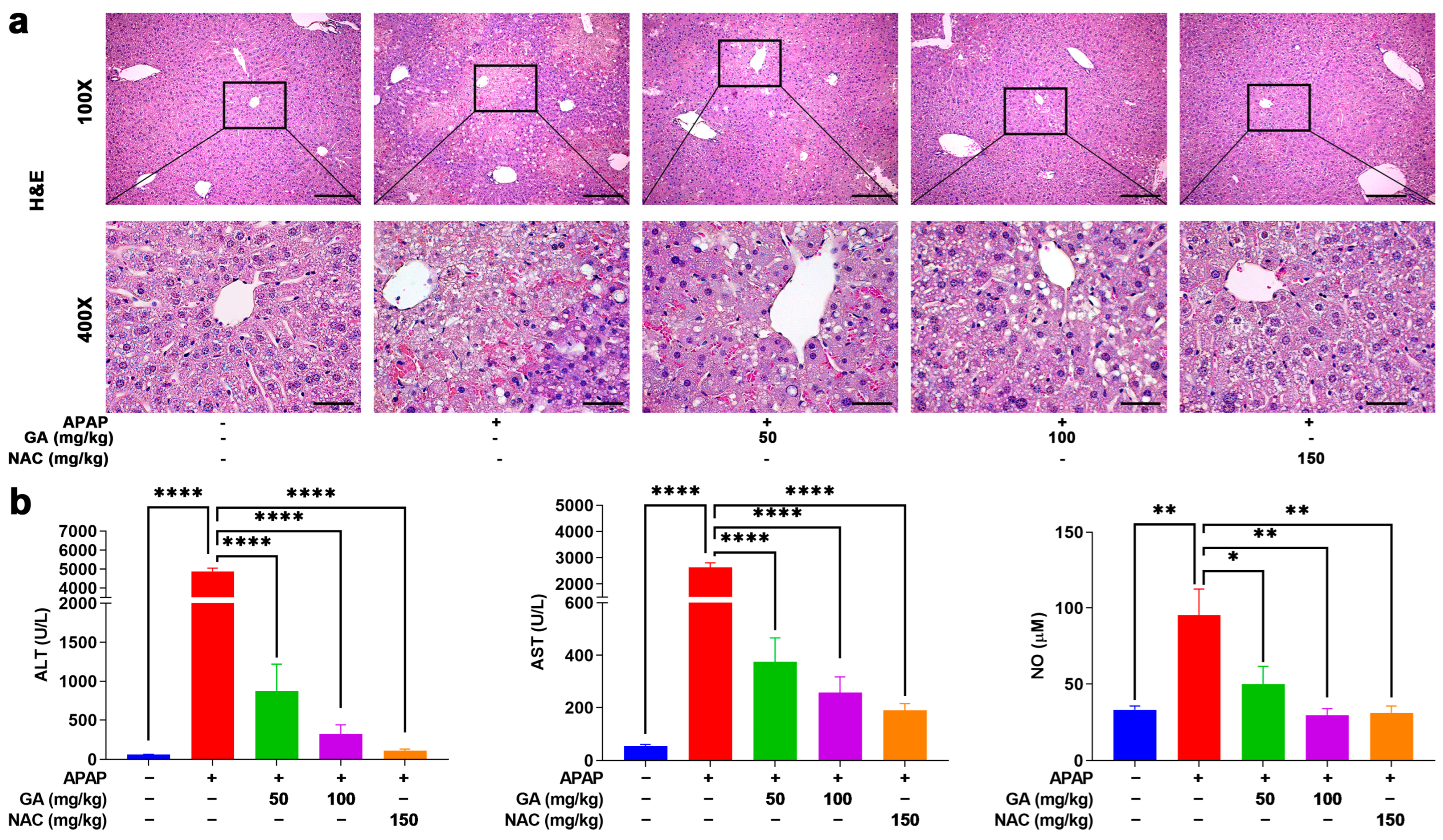
3.2. GA Treatment Attenuated the Liver Injury in APAP-Exposed Mice
3.3. GA Treatment Reduced Hepatic Cell Death in APAP-Induced Mice ALI
3.4. GA Treatment Attenuated APAP-Induced Hepatic Oxidative Stress
3.5. GA Suppresses APAP-Triggered Hepatic Inflammation
3.6. Integrated Pathway Analysis Reveals Multi-Modal Mechanisms of GA Against APAP-Induced Hepatotoxicity
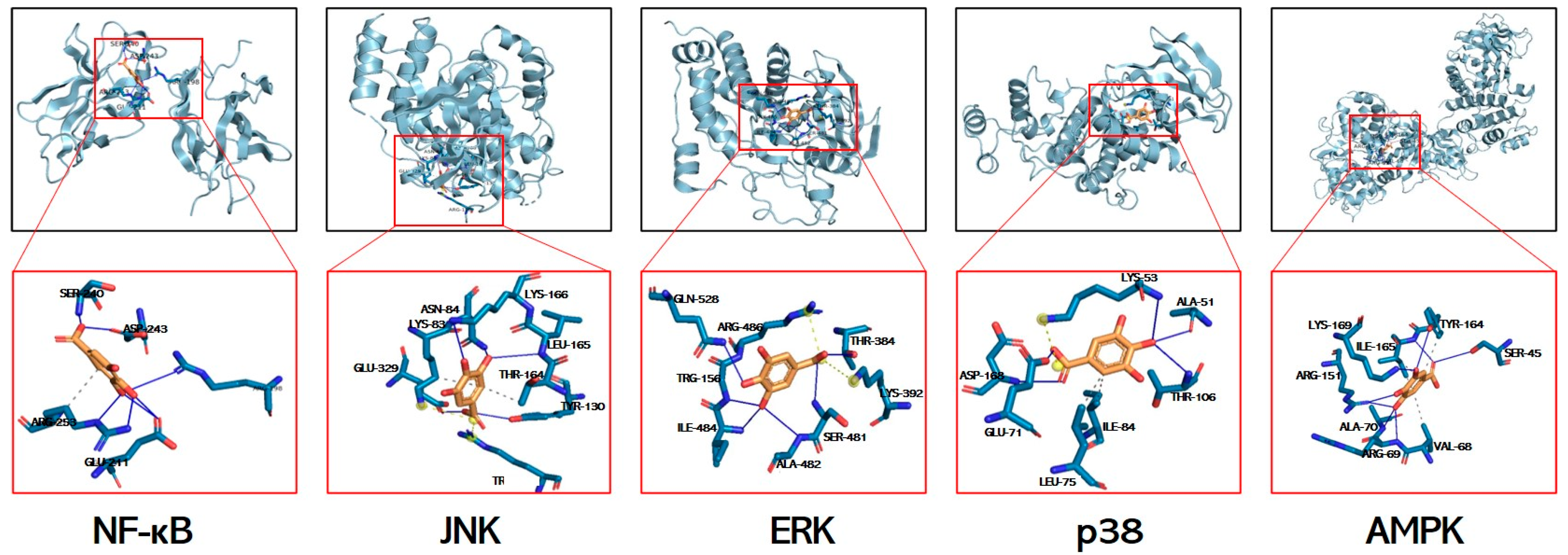
3.7. Molecular Docking Simulations of GA Interactions with Key Signaling Mediators
3.8. GA Treatment Reduced Phosphorylation of NF-κB and MAPKs
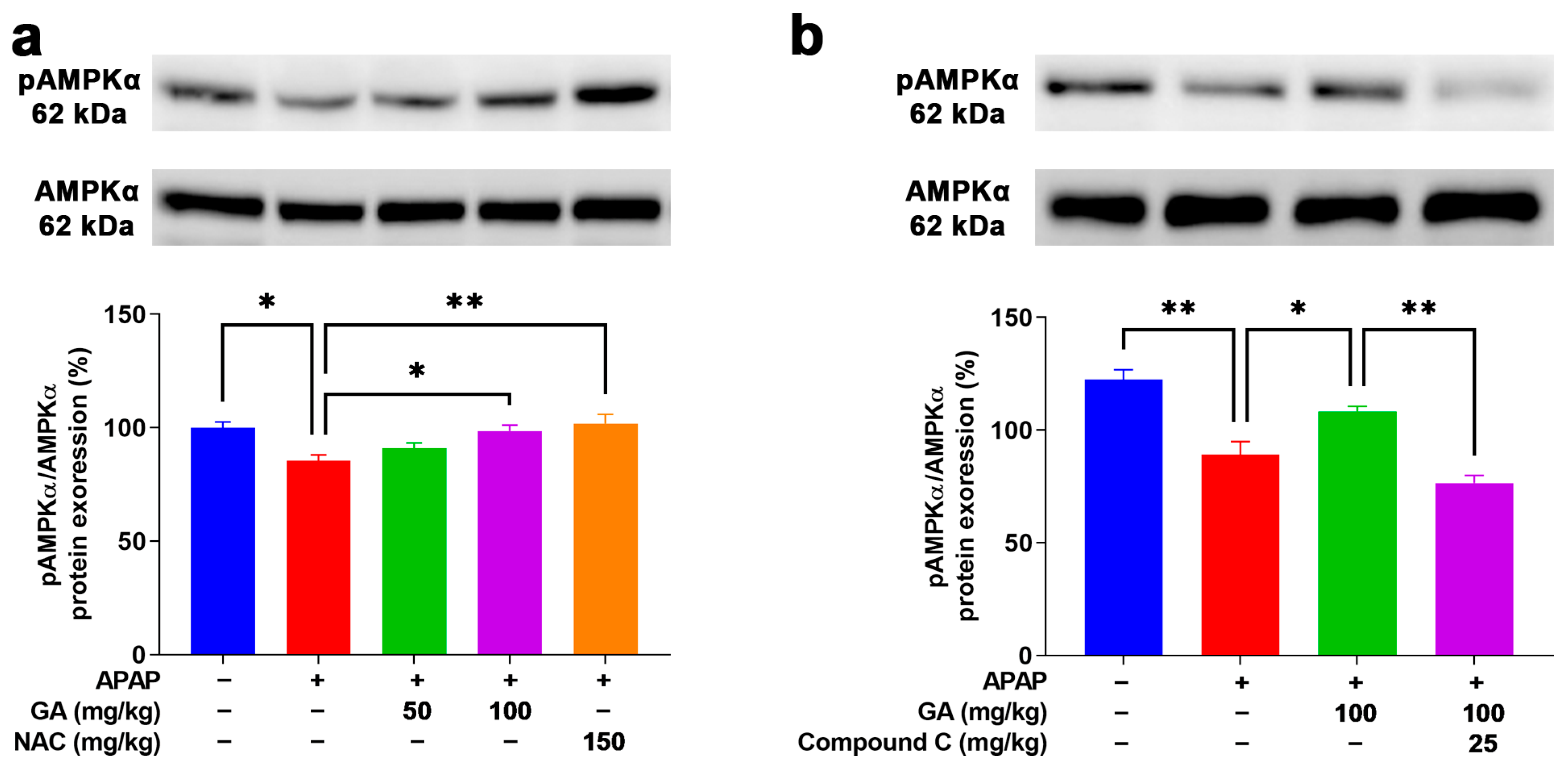
3.9. GA Enhanced AMPK Phosphorylation to Mitigate Oxidative Stress
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GA | Gallic acid; |
| NAC | N-acetylcysteine; |
| APAP | Acetaminophen; |
| UGT | Uridine diphosphate glucuronosyltransferase; |
| ALI | Acute liver injury; |
| ALF | Acute liver failure; |
| NAPQI | N-acetyl-p-benzoquinone imine; |
| GSH | Glutathione; |
| MAPK | Mitogen-activated protein kinase; |
| JNK | c-jun-N-terminal kinase; |
| ERK | Extracellular signal-regulated kinase; |
| H&E | Hematoxylin and eosin; |
| PBS | Phosphate-buffered saline; |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling; |
| DAB | Diaminobenzidine; |
| MDA | Malondialdehyde; |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase; |
| GO | Gene Ontology; |
| KEGG | Kyoto Encyclopedia of Genes and Genomes; |
| T-PER | Tissue protein extraction reagent; |
| SEM | Standard error of the mean; |
| DAMPs | Damage-associated molecular patterns. |
References
- McGill, M.R.; Jaeschke, H. Metabolism and disposition of acetaminophen: Recent advances in relation to hepatotoxicity and diagnosis. Pharm. Res. 2013, 30, 2174–2187. [Google Scholar] [CrossRef] [PubMed]
- Larsen, F.S.; Wendon, J. Understanding paracetamol-induced liver failure. Intensive Care Med. 2014, 40, 888–890. [Google Scholar] [CrossRef] [PubMed]
- Im, Y.J.; Jeon, J.Y.; Kim, E.Y.; Kim, Y.; Oh, D.J.; Yoo, J.S.; Shin, D.H.; Chae, S.W.; Kim, M.G. An assessment of the pharmacokinetics of a sustained-release formulation of a tramadol/acetaminophen combination in healthy subjects. Clin. Ther. 2015, 37, 376–389. [Google Scholar] [CrossRef]
- Wang, M.; Sun, J.; Yu, T.; Wang, M.; Jin, L.; Liang, S.; Luo, W.; Wang, Y.; Li, G.; Liang, G. Diacerein protects liver against APAP-induced injury via targeting JNK and inhibiting JNK-mediated oxidative stress and apoptosis. Biomed. Pharmacother. 2022, 149, 112917. [Google Scholar] [CrossRef]
- Manthripragada, A.D.; Zhou, E.H.; Budnitz, D.S.; Lovegrove, M.C.; Willy, M.E. Characterization of acetaminophen overdose-related emergency department visits and hospitalizations in the United States. Pharmacoepidemiol. Drug Saf. 2011, 20, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; He, D.; Li, W.; Wang, M.; Zhao, S.; Liu, L.; Cao, Z.; Liu, R.; Huang, Y.; Li, H.; et al. Hepatoprotective Effect of Citrus aurantium L. Against APAP-induced Liver Injury by Regulating Liver Lipid Metabolism and Apoptosis. Int. J. Biol. Sci. 2020, 16, 752–765. [Google Scholar] [CrossRef]
- Saito, C.; Lemasters, J.J.; Jaeschke, H. c-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicol. Appl. Pharmacol. 2010, 246, 8–17. [Google Scholar] [CrossRef]
- Jaeschke, H.; McGill, M.R.; Ramachandran, A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: Lessons learned from acetaminophen hepatotoxicity. Drug Metab. Rev. 2012, 44, 88–106. [Google Scholar] [CrossRef]
- Tian, W.; Zhao, J.; Choo, B.K.; Kim, I.S.; Ahn, D.; Tae, H.J.; Islam, M.S.; Park, B.Y. Camellia japonica diminishes acetaminophen-induced acute liver failure by attenuating oxidative stress in mice. Environ. Sci. Pollut. Res. Int. 2021, 28, 57192–57206. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Min, R.W.; Aghajan, M.; Kaplowitz, N. c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src. Hepatology 2016, 63, 1987–2003. [Google Scholar] [CrossRef]
- Ramachandran, A.; Jaeschke, H. Acetaminophen Hepatotoxicity. Semin. Liver Dis. 2019, 39, 221–234. [Google Scholar] [CrossRef]
- Yan, M.; Huo, Y.; Yin, S.; Hu, H. Mechanisms of acetaminophen-induced liver injury and its implications for therapeutic interventions. Redox Biol. 2018, 17, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Xin, J.; You, T.; Jiang, X.; Fu, D.; Wang, J.; Jiang, W.; Feng, X.; Wen, J.; Huang, Y.; Hu, C. Caveolin-1 Alleviates Acetaminophen-Induced Hepatotoxicity in Alcoholic Fatty Liver Disease by Regulating the Ang II/EGFR/ERK Axis. Int. J. Mol. Sci. 2022, 23, 7587. [Google Scholar] [CrossRef]
- Lee, E.H.; Baek, S.Y.; Park, J.Y.; Kim, Y.W. Rifampicin activates AMPK and alleviates oxidative stress in the liver as mediated with Nrf2 signaling. Chem. Biol. Interact. 2020, 315, 108889. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Hong, L.; Tian, Y.; Yin, C.; Zhu, C.; Feng, H. Corilagin alleviates acetaminophen-induced hepatotoxicity via enhancing the AMPK/GSK3beta-Nrf2 signaling pathway. Cell Commun. Signal. 2019, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Guan, H.; Zhao, X.; Xie, Q.; Xie, Z.; Cai, F.; Dang, R.; Li, M.; Wang, C. Dietary gallic acid as an antioxidant: A review of its food industry applications, health benefits, bioavailability, nano-delivery systems, and drug interactions. Food Res. Int. 2024, 180, 114068. [Google Scholar] [CrossRef]
- Sohrabi, F.; Dianat, M.; Badavi, M.; Radan, M.; Mard, S.A. Gallic acid suppresses inflammation and oxidative stress through modulating Nrf2-HO-1-NF-kappaB signaling pathways in elastase-induced emphysema in rats. Environ. Sci. Pollut. Res. Int. 2021, 28, 56822–56834. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Kishimoto, Y.; Sasaki, M.; Sato, A.; Kamiya, T.; Kondo, K.; Iida, K. Terminalia bellirica (Gaertn.) Roxb. Extract and Gallic Acid Attenuate LPS-Induced Inflammation and Oxidative Stress via MAPK/NF-kappaB and Akt/AMPK/Nrf2 Pathways. Oxid. Med. Cell Longev. 2018, 2018, 9364364. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Ramachandran, A.; Jaeschke, H. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol. 2016, 10, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Tenorio, M.; Graciliano, N.G.; Moura, F.A.; Oliveira, A.C.M.; Goulart, M.O.F. N-Acetylcysteine (NAC): Impacts on Human Health. Antioxidants 2021, 10, 967. [Google Scholar] [CrossRef]
- Liu, J.; Luo, D.; Wu, Y.; Gao, C.; Lin, G.; Chen, J.; Wu, X.; Zhang, Q.; Cai, J.; Su, Z. The Protective Effect of Sonneratia apetala Fruit Extract on Acetaminophen-Induced Liver Injury in Mice. Evid. Based Complement. Altern. Med. 2019, 2019, 6919834. [Google Scholar] [CrossRef] [PubMed]
- Golestan Jahromi, M.; Nabavizadeh, F.; Vahedian, J.; Nahrevanian, H.; Dehpour, A.R.; Zare-Mehrjardi, A. Protective effect of ghrelin on acetaminophen-induced liver injury in rat. Peptides 2010, 31, 2114–2117. [Google Scholar] [CrossRef]
- Bai, J.; Zhang, Y.; Tang, C.; Hou, Y.; Ai, X.; Chen, X.; Zhang, Y.; Wang, X.; Meng, X. Gallic acid: Pharmacological activities and molecular mechanisms involved in inflammation-related diseases. Biomed. Pharmacother. 2021, 133, 110985. [Google Scholar] [CrossRef] [PubMed]
- Rasool, M.K.; Sabina, E.P.; Ramya, S.R.; Preety, P.; Patel, S.; Mandal, N.; Mishra, P.P.; Samuel, J. Hepatoprotective and antioxidant effects of gallic acid in paracetamol-induced liver damage in mice. J. Pharm. Pharmacol. 2010, 62, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.S.; Zhao, J.; Kim, M.K.; Tae, H.J.; Kim, I.S.; Ahn, D.; Hwang, H.P.; Mao, M.X.; Park, B.Y. Veronica persica ameliorates acetaminophen-induced murine hepatotoxicity via attenuating oxidative stress and inflammation. Biomed. Pharmacother. 2023, 169, 115898. [Google Scholar] [CrossRef]
- Yang, R.; Song, C.; Chen, J.; Zhou, L.; Jiang, X.; Cao, X.; Sun, Y.; Zhang, Q. Limonin ameliorates acetaminophen-induced hepatotoxicity by activating Nrf2 antioxidative pathway and inhibiting NF-kappaB inflammatory response via upregulating Sirt1. Phytomedicine 2020, 69, 153211. [Google Scholar] [CrossRef]
- Xu, J.; Zhao, L.; Zhang, X.; Ying, K.; Zhou, R.; Cai, W.; Wu, X.; Jiang, H.; Xu, Q.; Miao, D.; et al. Salidroside ameliorates acetaminophen-induced acute liver injury through the inhibition of endoplasmic reticulum stress-mediated ferroptosis by activating the AMPK/SIRT1 pathway. Ecotoxicol. Environ. Saf. 2023, 262, 115331. [Google Scholar] [CrossRef]
- Karch, J.; Molkentin, J.D. Regulated necrotic cell death: The passive aggressive side of Bax and Bak. Circ. Res. 2015, 116, 1800–1809. [Google Scholar] [CrossRef]
- Cherian, D.A.; Peter, T.; Narayanan, A.; Madhavan, S.S.; Achammada, S.; Vynat, G.P. Malondialdehyde as a Marker of Oxidative Stress in Periodontitis Patients. J. Pharm. Bioallied Sci. 2019, 11, S297–S300. [Google Scholar] [CrossRef]
- Leng, J.; Wang, Z.; Fu, C.L.; Zhang, J.; Ren, S.; Hu, J.N.; Jiang, S.; Wang, Y.P.; Chen, C.; Li, W. NF-kappaB and AMPK/PI3K/Akt signaling pathways are involved in the protective effects of Platycodon grandiflorum saponins against acetaminophen-induced acute hepatotoxicity in mice. Phytother. Res. 2018, 32, 2235–2246. [Google Scholar] [CrossRef]
- Xiao, K.; Liu, C.; Tu, Z.; Xu, Q.; Chen, S.; Zhang, Y.; Wang, X.; Zhang, J.; Hu, C.A.; Liu, Y. Activation of the NF-kappaB and MAPK Signaling Pathways Contributes to the Inflammatory Responses, but Not Cell Injury, in IPEC-1 Cells Challenged with Hydrogen Peroxide. Oxid. Med. Cell Longev. 2020, 2020, 5803639. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.U.; Stamper, B.D. Polyphenols reported to shift APAP-induced changes in MAPK signaling and toxicity outcomes. Chem. Biol. Interact. 2017, 277, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Sherlock, L.G.; Balasubramaniyan, D.; Zheng, L.; Grayck, M.; McCarthy, W.C.; De Dios, R.C.; Zarate, M.A.; Orlicky, D.J.; De Dios, R.; Wright, C.J. APAP-Induced IkappaBbeta/NFkappaB Signaling Drives Hepatic Il6 Expression and Associated Sinusoidal Dilation. Toxicol. Sci. 2022, 185, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Liu, Y.; Shang, J.; Xie, Q.; Li, J.; Yan, M.; Xu, J.; Niu, J.; Liu, J.; Watkins, P.B.; et al. Incidence and Etiology of Drug-Induced Liver Injury in Mainland China. Gastroenterology 2019, 156, 2230–2241.e11. [Google Scholar] [CrossRef]
- Sun, Y.K.; Zhang, Y.F.; Xie, L.; Rong, F.; Zhu, X.Y.; Xie, J.; Zhou, H.; Xu, T. Progress in the treatment of drug-induced liver injury with natural products. Pharmacol. Res. 2022, 183, 106361. [Google Scholar] [CrossRef]
- Zhu, L.R.; Li, S.S.; Zheng, W.Q.; Ni, W.J.; Cai, M.; Liu, H.P. Targeted modulation of gut microbiota by traditional Chinese medicine and natural products for liver disease therapy. Front. Immunol. 2023, 14, 1086078. [Google Scholar] [CrossRef]
- Zhuang, X.; Li, L.; Liu, T.; Zhang, R.; Yang, P.; Wang, X.; Dai, L. Mechanisms of isoniazid and rifampicin-induced liver injury and the effects of natural medicinal ingredients: A review. Front. Pharmacol. 2022, 13, 1037814. [Google Scholar] [CrossRef]
- Hadidi, M.; Linan-Atero, R.; Tarahi, M.; Christodoulou, M.C.; Aghababaei, F. The Potential Health Benefits of Gallic Acid: Therapeutic and Food Applications. Antioxidants 2024, 13, 1001. [Google Scholar] [CrossRef]
- Ahmed, H.H.; Galal, A.F.; Shalby, A.B.; Abd-Rabou, A.A.; Mehaya, F.M. Improving Anti-Cancer Potentiality and Bioavailability of Gallic Acid by Designing Polymeric Nanocomposite Formulation. Asian Pac. J. Cancer Prev. 2018, 19, 3137–3146. [Google Scholar] [CrossRef]
- Deng, X.; Li, Y.; Chen, Y.; Hu, Q.; Zhang, W.; Chen, L.; Lu, X.; Zeng, J.; Ma, X.; Efferth, T. Paeoniflorin protects hepatocytes from APAP-induced damage through launching autophagy via the MAPK/mTOR signaling pathway. Cell Mol. Biol. Lett. 2024, 29, 119. [Google Scholar] [CrossRef]
- Jaeschke, H.; Ramachandran, A. Acetaminophen Hepatotoxicity: Paradigm for Understanding Mechanisms of Drug-Induced Liver Injury. Annu. Rev. Pathol. 2024, 19, 453–478. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Adelusi, O.B.; Akakpo, J.Y.; Nguyen, N.T.; Sanchez-Guerrero, G.; Umbaugh, D.S.; Ding, W.X.; Ramachandran, A. Recommendations for the use of the acetaminophen hepatotoxicity model for mechanistic studies and how to avoid common pitfalls. Acta Pharm. Sin. B. 2021, 11, 3740–3755. [Google Scholar] [CrossRef]
- Yang, G.; Zhang, L.; Ma, L.; Jiang, R.; Kuang, G.; Li, K.; Tie, H.; Wang, B.; Chen, X.; Xie, T.; et al. Glycyrrhetinic acid prevents acetaminophen-induced acute liver injury via the inhibition of CYP2E1 expression and HMGB1-TLR4 signal activation in mice. Int. Immunopharmacol. 2017, 50, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.; Bhattacharyya, S.; McCullough, S.; Letzig, L.; Mishra, P.J.; Luo, C.; Dweep, H.; James, L. MicroRNA regulation of CYP 1A2, CYP3A4 and CYP2E1 expression in acetaminophen toxicity. Sci. Rep. 2017, 7, 12331. [Google Scholar] [CrossRef] [PubMed]
- Massart, J.; Begriche, K.; Hartman, J.H.; Fromenty, B. Role of Mitochondrial Cytochrome P450 2E1 in Healthy and Diseased Liver. Cells 2022, 11, 288. [Google Scholar] [CrossRef]
- Guo, H.; Sun, J.; Li, D.; Hu, Y.; Yu, X.; Hua, H.; Jing, X.; Chen, F.; Jia, Z.; Xu, J. Shikonin attenuates acetaminophen-induced acute liver injury via inhibition of oxidative stress and inflammation. Biomed. Pharmacother. 2019, 112, 108704. [Google Scholar] [CrossRef]
- Xun, T.; Zhang, M.; Wei, S.; Zhao, C.; Lin, Z.; Feng, H.; Wang, X.; Zhao, J.; Yang, X. CYP2E1 mediated advanced oxidation protein products exacerbate acetaminophen induced drug-derived liver injury in vitro and in vivo. Eur. J. Pharm. Sci. 2024, 200, 106829. [Google Scholar] [CrossRef]
- McDonald, B.; Kubes, P. Innate Immune Cell Trafficking and Function During Sterile Inflammation of the Liver. Gastroenterology 2016, 151, 1087–1095. [Google Scholar] [CrossRef]
- Yang, T.; Wang, H.; Wang, X.; Li, J.; Jiang, L. The Dual Role of Innate Immune Response in Acetaminophen-Induced Liver Injury. Biology 2022, 11, 1057. [Google Scholar] [CrossRef]
- Bieghs, V.; Trautwein, C. The innate immune response during liver inflammation and metabolic disease. Trends Immunol. 2013, 34, 446–452. [Google Scholar] [CrossRef]
- Iorga, A.; Donovan, K.; Shojaie, L.; Johnson, H.; Kwok, J.; Suda, J.; Lee, B.T.; Aghajan, M.; Shao, L.; Liu, Z.X.; et al. Interaction of RIPK1 and A20 modulates MAPK signaling in murine acetaminophen toxicity. J. Biol. Chem. 2021, 296, 100300. [Google Scholar] [CrossRef] [PubMed]
- Jansen, T.; Kvandova, M.; Daiber, A.; Stamm, P.; Frenis, K.; Schulz, E.; Munzel, T.; Kroller-Schon, S. The AMP-Activated Protein Kinase Plays a Role in Antioxidant Defense and Regulation of Vascular Inflammation. Antioxidants 2020, 9, 525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Meng, X.; Jiang, H.; Ge, H.; Qian, K.; Zheng, Y.; Park, Y.; Wang, J. Restoration of energy homeostasis under oxidative stress: Duo synergistic AMPK pathways regulating arginine kinases. PLoS Genet. 2023, 19, e1010843. [Google Scholar] [CrossRef] [PubMed]
- Marino, A.; Hausenloy, D.J.; Andreadou, I.; Horman, S.; Bertrand, L.; Beauloye, C. AMP-activated protein kinase: A remarkable contributor to preserve a healthy heart against ROS injury. Free Radic. Biol. Med. 2021, 166, 238–254. [Google Scholar] [CrossRef]
- Qi, J.; Zhou, Z.; Lim, C.W.; Kim, J.W.; Kim, B. Amlexanox ameliorates acetaminophen-induced acute liver injury by reducing oxidative stress in mice. Toxicol. Appl. Pharmacol. 2019, 385, 114767. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Target | Primer | Size | Accession No. | |
|---|---|---|---|---|
| CYP2E1 | Forward | GCAGTATCCCACGCGAATTT | 184 | NM_021282 |
| Reverse | CCATGTGGTTGCTGGGATTT | |||
| Catalase | Forward | AGGTGTTGAACGAGGAGGAG | 196 | NM_009804 |
| Reverse | TGCGTGTAGGTGTGAATTGC | |||
| SOD-1 | Forward | ATGGGTTCCACGTCCATCAG | 132 | NM_011434 |
| Reverse | CATTGCCCAGGTCTCCAACA | |||
| SOD-2 | Forward | GAGAATCTCAGTGCTCACTC | 160 | NM_013671 |
| Reverse | GGAACCCTAAATGCTGCCAG | |||
| Bcl-2 | Forward | CGGGAGAACAGGGTATGA | 149 | NM_009741 |
| Reverse | CAGGCTGGAAGGAGAAGAT | |||
| Bax | Forward | GCAGGGAGGATGGCTGGGGAGA | 352 | NM_001411995 |
| Reverse | TCGAGACAAGCAGCCGCTCACG | |||
| GAPDH | Forward | CACTGAGCATCTCCCTCACA | 111 | NM_008084 |
| Reverse | GTGGGTGCAGCGAACTTTAT | |||
| Parameter | Normal Control | APAP | APAP + GA 50 | APAP + GA 100 | APAP + NAC 150 |
|---|---|---|---|---|---|
| Body weight (g) | 26.15 ± 0.366 | 26.28 ± 0.237 | 26.24 ± 0.196 | 26.22 ± 0.403 | 26.02 ± 0.430 |
| Liver weight (g) | 1.15 ± 0.016 | 1.29 ± 0.019 *** | 1.24 ± 0.014 | 1.214 ± 0.022 * | 1.14 ± 0.022 * |
| Liver/Body × 100 | 4.41 ± 0.025 | 4.92 ± 0.047 **** | 4.72 ± 0.041 | 4.62 ± 0.031 *** | 4.60 ± 0.043 **** |
| Receptor | NF-κB | JNK | ERK | p38 | AMPK |
|---|---|---|---|---|---|
| Binding energy (kcal/mol) | −3.28 | −5.4 | −5.6 | −6.3 | −5.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Zhao, Y.; Song, S.; Zhang, S.; Yang, G.; Qiu, Y.; Tian, W. Gallic Acid Alleviates Acetaminophen-Induced Acute Liver Injury by Regulating Inflammatory and Oxidative Stress Signaling Proteins. Antioxidants 2025, 14, 860. https://doi.org/10.3390/antiox14070860
Zhao J, Zhao Y, Song S, Zhang S, Yang G, Qiu Y, Tian W. Gallic Acid Alleviates Acetaminophen-Induced Acute Liver Injury by Regulating Inflammatory and Oxidative Stress Signaling Proteins. Antioxidants. 2025; 14(7):860. https://doi.org/10.3390/antiox14070860
Chicago/Turabian StyleZhao, Jing, Yuan Zhao, Shuzhe Song, Sai Zhang, Guodong Yang, Yan Qiu, and Weishun Tian. 2025. "Gallic Acid Alleviates Acetaminophen-Induced Acute Liver Injury by Regulating Inflammatory and Oxidative Stress Signaling Proteins" Antioxidants 14, no. 7: 860. https://doi.org/10.3390/antiox14070860
APA StyleZhao, J., Zhao, Y., Song, S., Zhang, S., Yang, G., Qiu, Y., & Tian, W. (2025). Gallic Acid Alleviates Acetaminophen-Induced Acute Liver Injury by Regulating Inflammatory and Oxidative Stress Signaling Proteins. Antioxidants, 14(7), 860. https://doi.org/10.3390/antiox14070860