
Negative Air Ions Attenuate Nicotine-Induced Vascular Endothelial Dysfunction by Suppressing AP1-Mediated FN1 and SPP1

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Treatments
2.2. Cell Culture and Transfection
2.3. Cell Viability Assay
2.4. Histopathological Analysis
2.5. Oxidative Damage Index, NO, and ET-1 Assays
2.6. RT–PCR
2.7. Western Blotting
2.8. RNA-Seq and Data Processing
2.8.1. RNA Isolation
2.8.2. RNA-Seq Library Construction and Sequencing
2.8.3. RNA-Seq Data Analysis
2.9. ChIP–qPCR
2.10. Statistical Analysis
3. Results
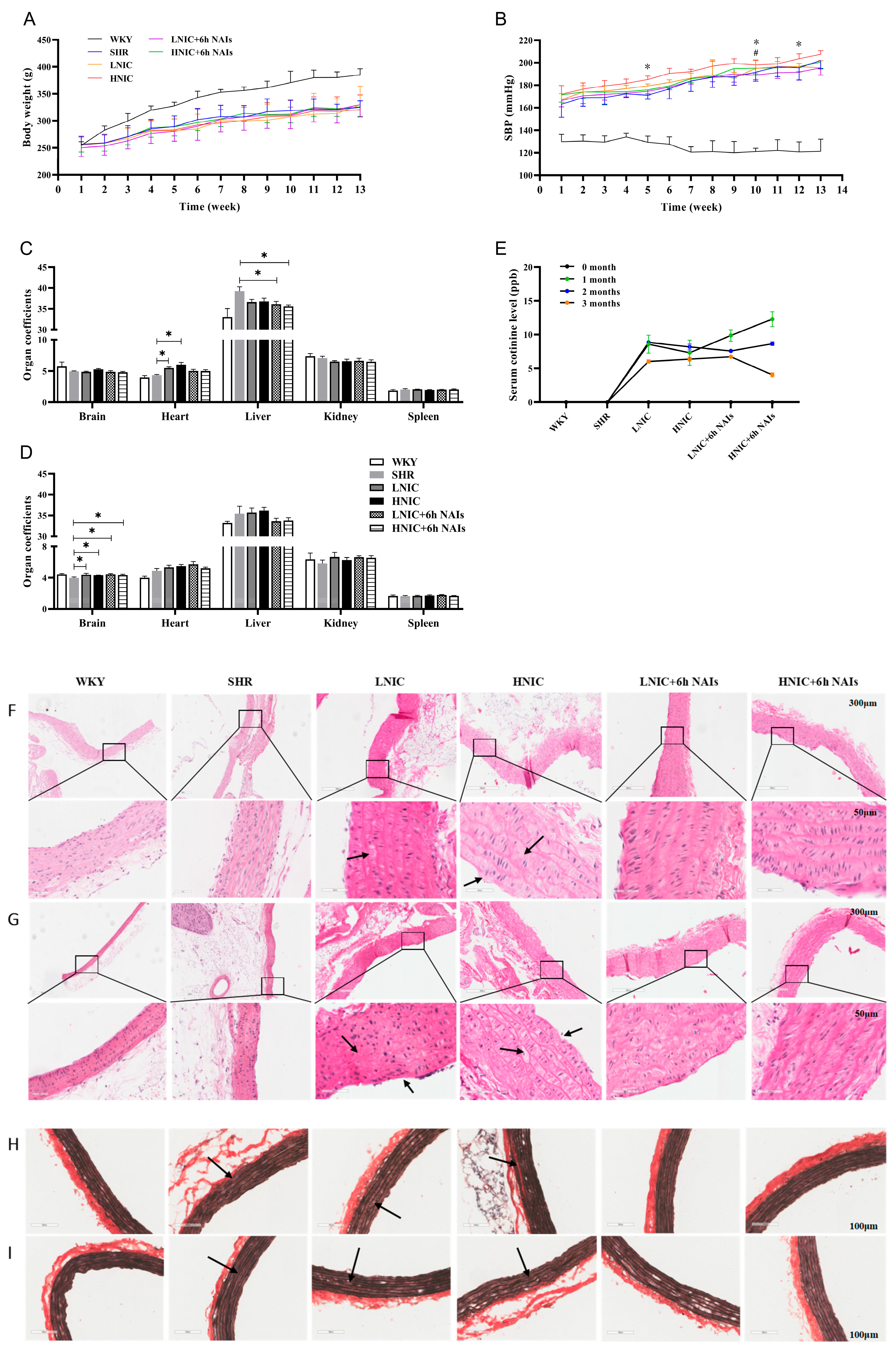
3.1. NAIs Attenuate Nicotine-Aggravated Increases in SBP in SHRs
3.2. NAIs Alleviate Nicotine-Induced Thoracic Aortic Injury
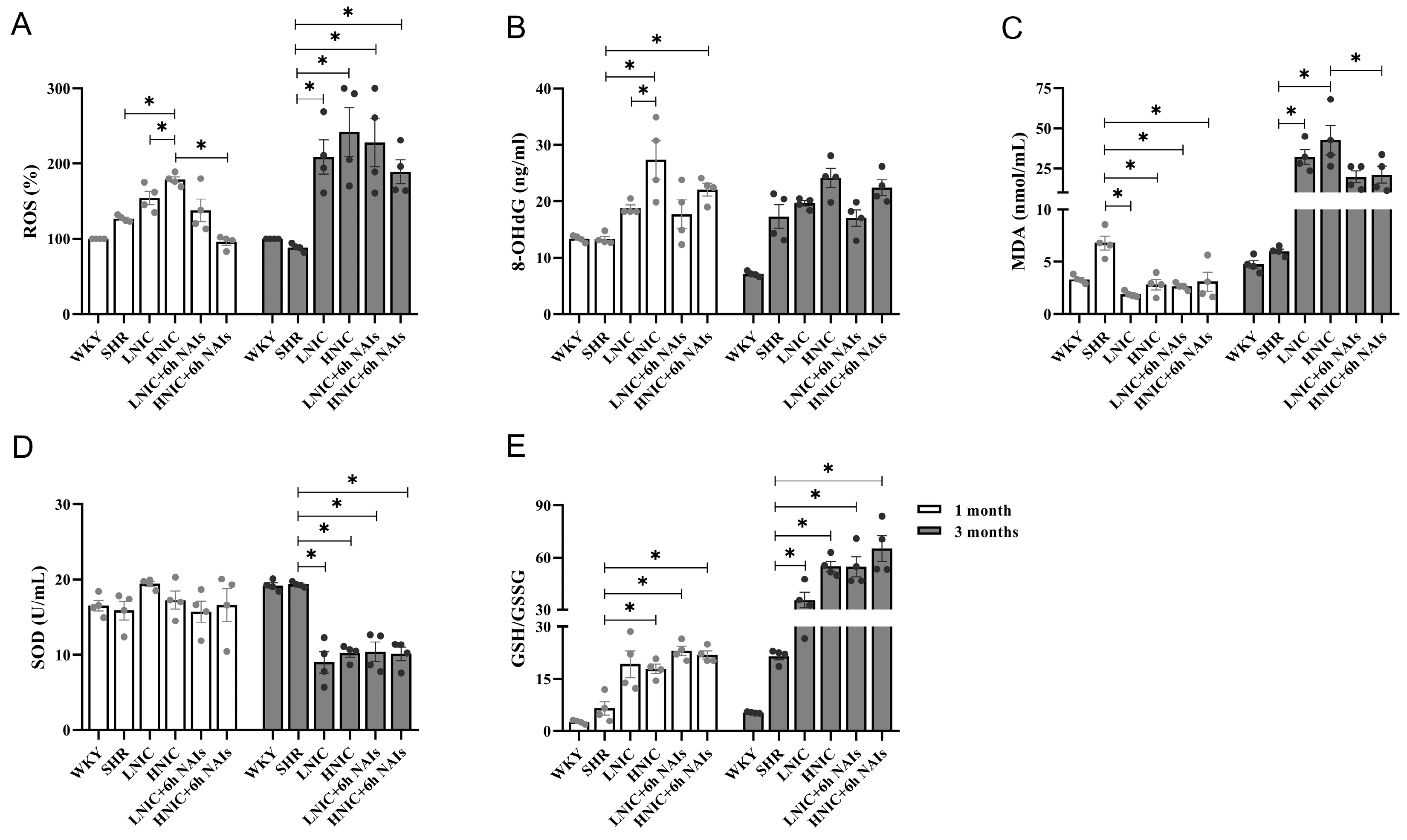
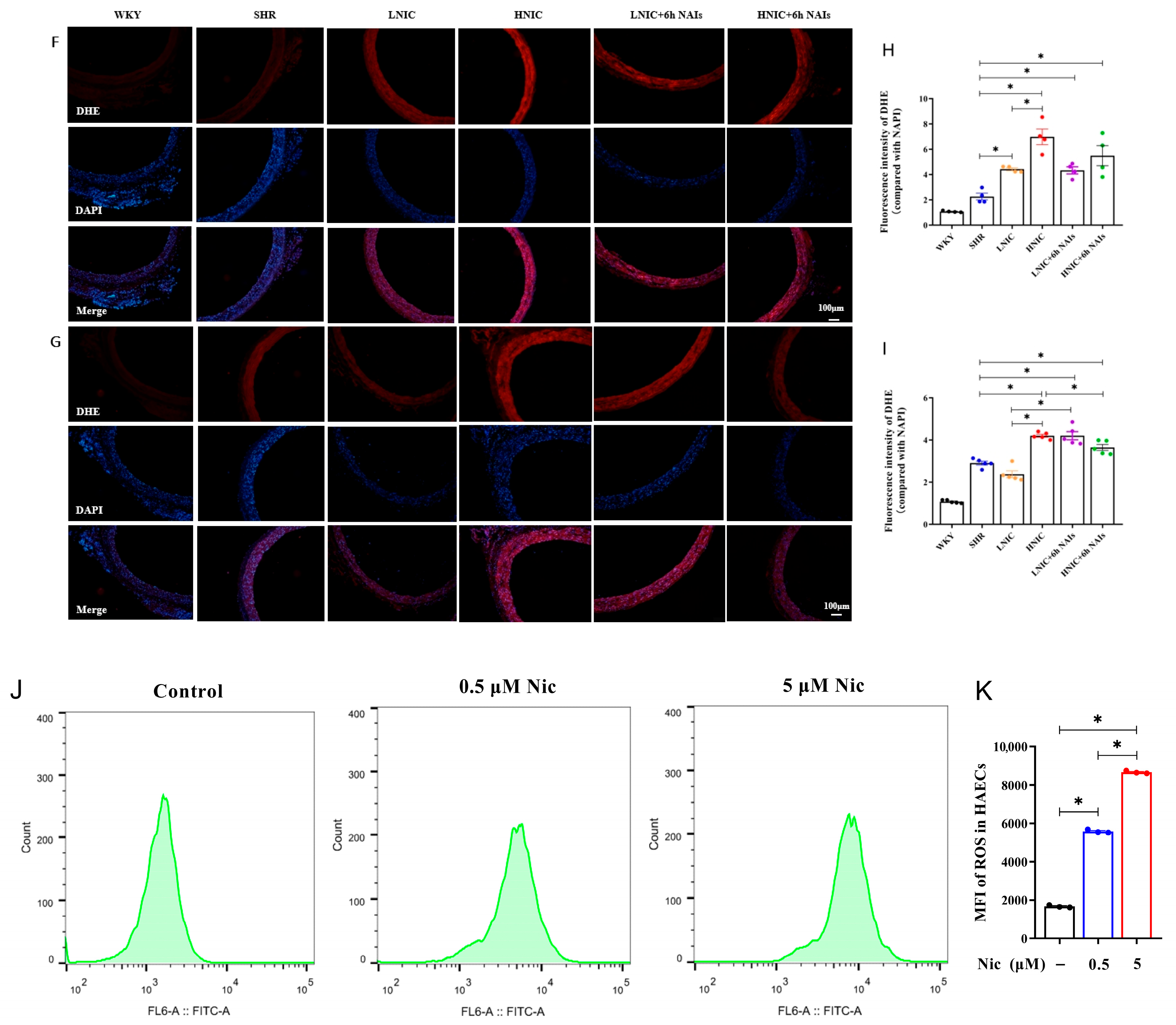
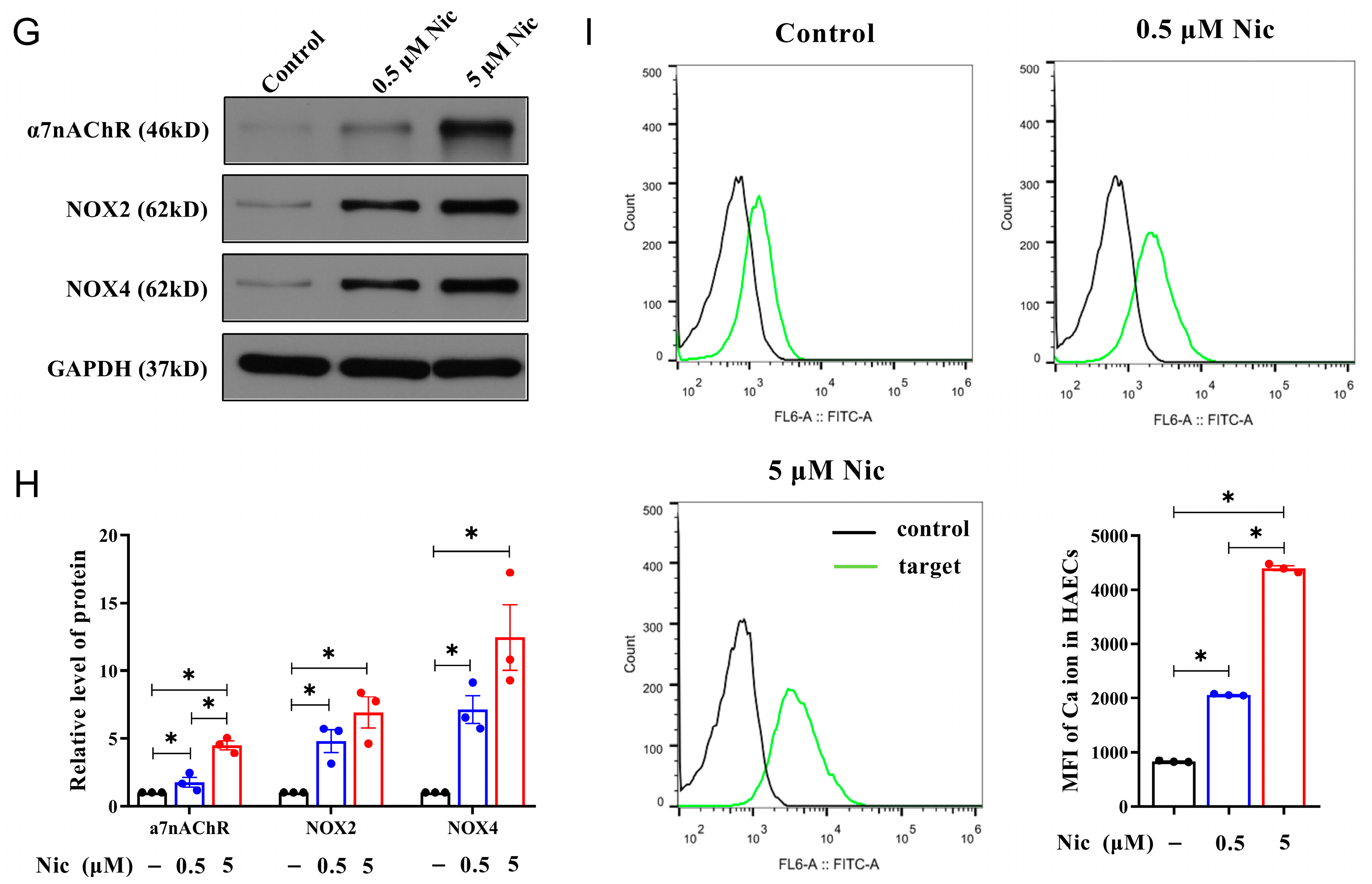
3.3. NAIs Reduced Nicotine-Induced Oxidative Damage Through α7nAChR
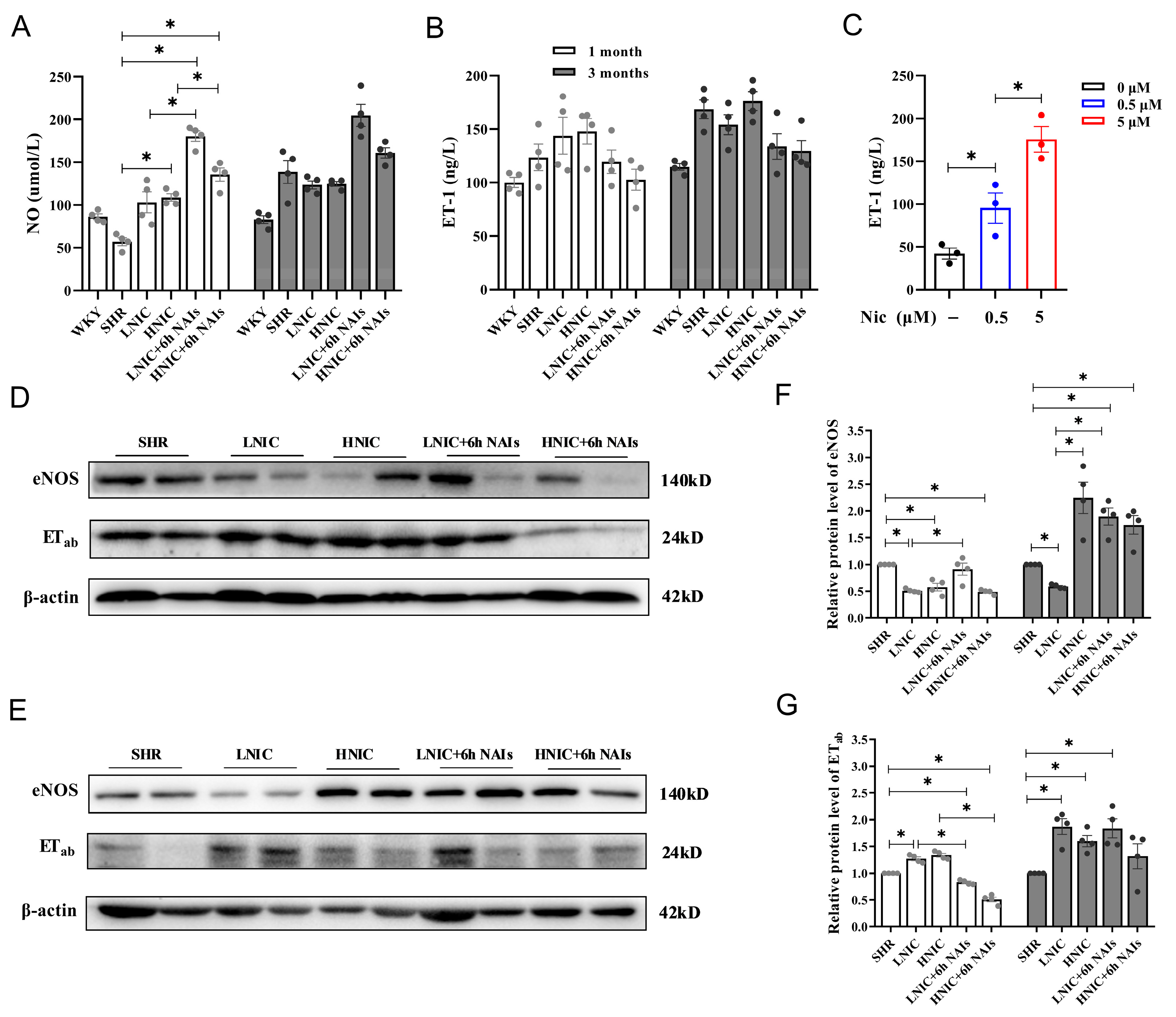
3.4. Nicotine Exposure Reduces NO and Elevates ET-1 Levels in Rat Serum
3.5. Nicotine Exposure Activates the JNK and JUN Signaling Pathways
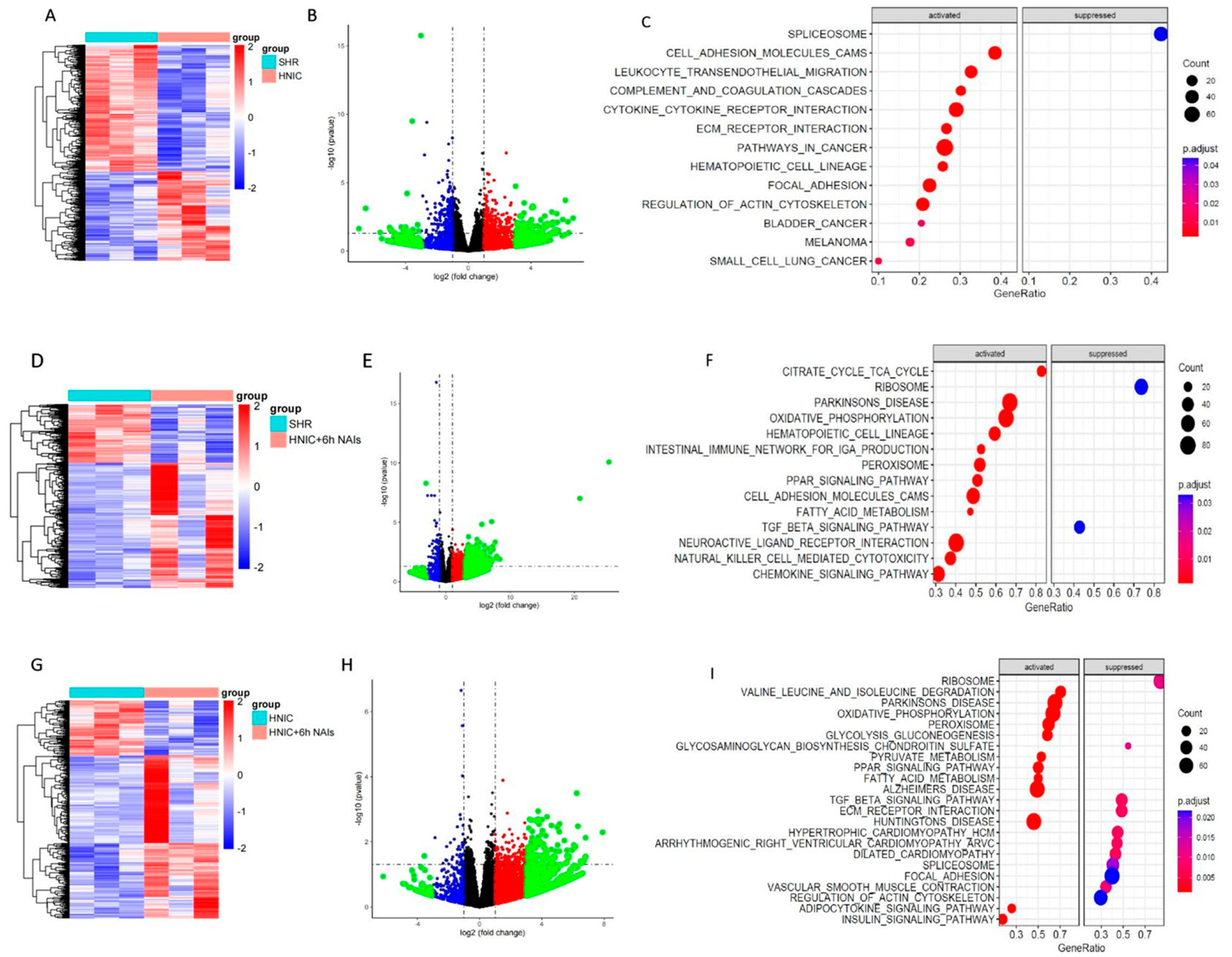
3.6. Exploration of Differentially Expressed Genes in the Thoracic Aortas of Rats Treated with Nicotine and NAIs Using RNA-Seq
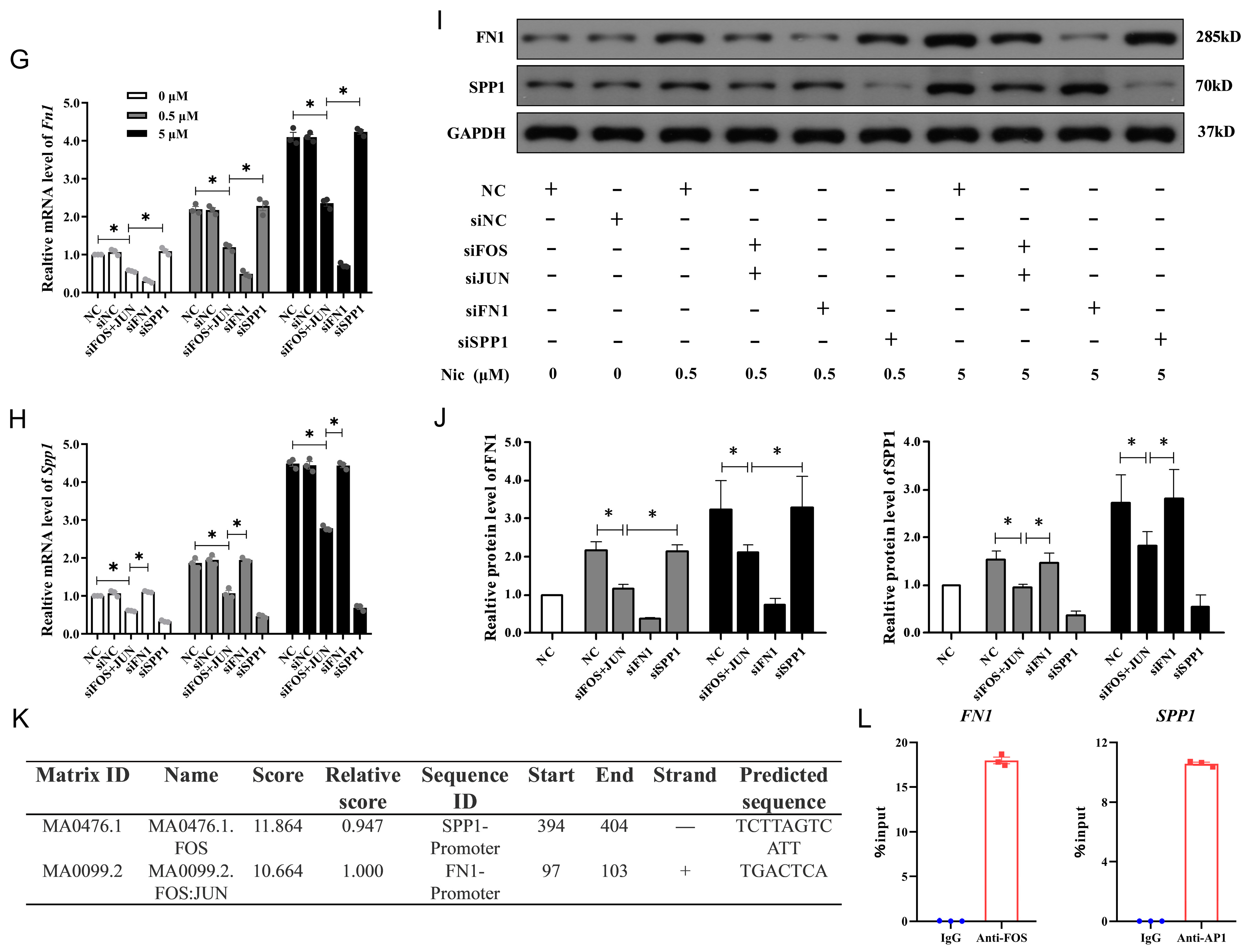
3.7. FN1 and SPP1 Are Regulated by AP1 and Involved in NAIs Alleviating Nicotine-Induced Damage
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| nAChRs | Nicotinic Acetylcholine Receptors |
| ROS | Reactive Oxygen Species |
| NOX | NADPH Oxidases |
| VECs | Vascular Endothelial Cells |
| NO | Nitric Oxide |
| AP1 | Activator Protein-1 |
| MAPK | Mitogen-Activated Protein Kinase |
| ERK | Extracellular Signal-Regulated Kinases |
| JNK | c-Jun N-terminal Kinases |
| p38 MAPK | Stress-Activated Protein Kinases |
| MMP-9 | Matrix Metalloproteinase-9 |
| NAIs | Negative Air Ions |
| SOD | Superoxide Dismutase |
| SHR | Spontaneously Hypertensive Rat |
| Fn1 | Fibronectin 1 |
| Spp1 | Secreted Phosphoprotein 1 |
| WKY | Wistar-Kyoto |
| SBP | Systolic Blood Pressure |
| HAECs | Human Aortic Endothelial Cells |
| 8-OHdG | 8-Hydroxylated Deoxyguanosine |
| ECM | Extracellular Matrix |
| ITGA5 | Integrin-α5 |
| EMT | Epithelial–Mesenchymal Transition |
| ETS1 | E26 Transcription Factor-1 |
References
- Liu, S.; Li, Y.; Zeng, X.; Wang, H.; Yin, P.; Wang, L.; Liu, Y.; Liu, J.; Qi, J.; Ran, S.; et al. Burden of cardiovascular diseases in China, 1990–2016: Findings from the 2016 global burden of disease study. JAMA Cardiol. 2019, 4, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Münzel, T.; Hahad, O.; Kuntic, M.; Keaney, J.F.; Deanfield, J.E.; Daiber, A. Effects of tobacco cigarettes, e-cigarettes, and waterpipe smoking on endothelial function and clinical outcomes. Eur. Heart J. 2020, 41, 4057–4070. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, A.K.; Erwin, A.P.; Yue, X. Nicotine and vascular dysfunction. Acta Physiol. 2021, 231, e13631. [Google Scholar] [CrossRef]
- Garcia, P.D.; Gornbein, J.A.; Middlekauff, H.R. Cardiovascular autonomic effects of electronic cigarette use: A systematic review. Clin. Auton. Res. 2020, 30, 507–519. [Google Scholar] [CrossRef]
- Wittenberg, R.E.; Wolfman, S.L.; De Biasi, M.; Dani, J.A. Nicotinic acetylcholine receptors and nicotine addiction: A brief introduction. Neuropharmacology 2020, 177, 108256. [Google Scholar] [CrossRef]
- Noviello, C.M.; Gharpure, A.; Mukhtasimova, N.; Cabuco, R.; Baxter, L.; Borek, D.; Sine, S.M.; Hibbs, R.E. Structure and gating mechanism of the α7 nicotinic acetylcholine receptor. Cell 2021, 184, 2121–2134.e13. [Google Scholar] [CrossRef]
- Vang, A.; da Silva Gonçalves Bos, D.; Fernandez-Nicolas, A.; Zhang, P.; Morrison, A.R.; Mancini, T.J.; Clements, R.T.; Polina, I.; Cypress, M.W.; Jhun, B.S.; et al. α7 Nicotinic acetylcholine receptor mediates right ventricular fibrosis and diastolic dysfunction in pulmonary hypertension. JCI Insight 2021, 6, e142945. [Google Scholar] [CrossRef]
- Amponsah-Offeh, M.; Diaba-Nuhoho, P.; Speier, S.; Morawietz, H. Oxidative Stress, Antioxidants and Hypertension. Antioxidants 2023, 12, 281. [Google Scholar] [CrossRef]
- Trimm, E.; Red-Horse, K. Vascular endothelial cell development and diversity. Nat. Rev. Cardiol. 2023, 20, 197–210. [Google Scholar] [CrossRef]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Karakaslar, E.O.; Katiyar, N.; Hasham, M.; Youn, A.; Sharma, S.; Chung, C.H.; Marches, R.; Korstanje, R.; Banchereau, J.; Ucar, D. Transcriptional activation of Jun and Fos members of the AP-1 complex is a conserved signature of immune aging that contributes to inflammaging. Aging Cell 2023, 22, e13792. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Padia, R.; Li, T.; Li, Y.; Li, B.; Jin, L.; Huang, S. Signaling of MK2 sustains robust AP1 activity for triple negative breast cancer tumorigenesis through direct phosphorylation of JAB1. NPJ Breast Cancer 2021, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tan, X.; Zhang, J.; Wu, J.; Shi, H. The emerging roles of MAPK-AMPK in ferroptosis regulatory network. Cell Commun. Signal. 2023, 21, 200. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Pan, H.; Yang, J.; Chen, D.; Wang, Y.; Zhang, H.; Cheng, Y. Xuanfei Baidu formula alleviates impaired mitochondrial dynamics and activated NLRP3 inflammasome by repressing NF-κB and MAPK pathways in LPS-induced ALI and inflammation models. Phytomedicine 2023, 108, 154545. [Google Scholar] [CrossRef]
- Xu, Q.; Liu, M.; Chao, X.; Zhang, C.; Yang, H.; Chen, J.; Zhao, C.; Zhou, B. Acidifiers Attenuate Diquat-Induced Oxidative Stress and Inflammatory Responses by Regulating NF-κB/MAPK/COX-2 Pathways in IPEC-J2 Cells. Antioxidants 2022, 11, 2002. [Google Scholar] [CrossRef]
- Li, S.; Khoi, P.N.; Yin, H.; Sah, D.K.; Kim, N.H.; Lian, S.; Jung, Y.D. Sulforaphane Suppresses the Nicotine-Induced Expression of the Matrix Metalloproteinase-9 via Inhibiting ROS-Mediated AP-1 and NF-κB Signaling in Human Gastric Cancer Cells. Int. J. Mol. Sci. 2022, 23, 5172. [Google Scholar] [CrossRef]
- Wölkart, G.; Kollau, A.; Stessel, H.; Russwurm, M.; Koesling, D.; Schrammel, A.; Schmidt, K.; Mayer, B. Effects of flavoring compounds used in electronic cigarette refill liquids on endothelial and vascular function. PLoS ONE 2019, 14, e0222152. [Google Scholar] [CrossRef]
- Antonelli, M.; Donelli, D.; Maggini, V.; Gallo, E.; Mascherini, V.; Firenzuoli, F.; Gavazzi, G.; Zabini, F.; Venturelli, E.; Margheritini, G.; et al. Demographic, Psychosocial, and Lifestyle-Related Characteristics of Forest Therapy Participants in Italy: A Multicenter Cross-Sectional Survey. Healthcare 2023, 11, 1627. [Google Scholar] [CrossRef]
- Li, A.; Li, Q.; Zhou, B.; Ge, X.; Cao, Y. Temporal dynamics of negative air ion concentration and its relationship with environmental factors: Results from long-term on-site monitoring. Sci. Total Environ. 2022, 832, 155057. [Google Scholar] [CrossRef]
- Xiao, S.; Wei, T.; Petersen, J.D.; Zhou, J.; Lu, X. Biological effects of negative air ions on human health and integrated multiomics to identify biomarkers: A literature review. Environ. Sci. Pollut. Res. Int. 2023, 30, 69824–69836. [Google Scholar] [CrossRef]
- Iwama, H. Negative air ions created by water shearing improve erythrocyte deformability and aerobic metabolism. Indoor Air 2004, 14, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Jeong, G.J.; Hong, J.Y.; Park, K.Y.; Lee, M.K.; Seo, S.J. Negative Air Ions Alleviate Particulate Matter-Induced Inflammation and Oxidative Stress in the Human Keratinocyte Cell Line HaCaT. Ann. Dermatol. 2021, 33, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Wei, T.; Xiao, M.; An, Z.; Shan, M.; Luo, Z.; Zhou, J.; Li, N.; Lu, X. Negative air ions alleviate nicotine-induced renal damage of spontaneously hypertensive rats via inhibiting oxidative stress and TGF-β/Smad pathway. Ecotoxicol. Environ. Saf. 2025, 291, 117882. [Google Scholar] [CrossRef]
- Chen, J.; Xiao, H.; Xue, R.; Kumar, V.; Aslam, R.; Mehdi, S.F.; Luo, H.; Malhotra, A.; Lan, X.; Singhal, P. Nicotine exacerbates diabetic nephropathy through upregulation of Grem1 expression. Mol. Med. 2023, 29, 92. [Google Scholar] [CrossRef]
- Le Foll, B.; Piper, M.E.; Fowler, C.D.; Tonstad, S.; Bierut, L.; Lu, L.; Jha, P.; Hall, W.D. Tobacco and Nicotine Use. Nat. Rev. Dis. Primers 2022, 8, 19. [Google Scholar] [CrossRef]
- Tidey, J.W.; Snell, L.M.; Colby, S.M.; Cassidy, R.N.; Denlinger-Apte, R.L. Effects of very low nicotine content cigarettes on smoking across vulnerable populations. Prev. Med. 2022, 165 Pt B, 107099. [Google Scholar] [CrossRef]
- Gould, T.J. Epigenetic and Long-Term Effects of Nicotine on Biology, Behavior, and Health. Pharmacol. Res. 2023, 192, 106741. [Google Scholar] [CrossRef]
- Kuan, V.; Warwick, A.; Hingorani, A.; Tufail, A.; Cipriani, V.; Burgess, S.; Sofat, R. International AMD Genomics Consortium (IAMDGC). Association of Smoking, Alcohol Consumption, Blood Pressure, Body Mass Index, and Glycemic Risk Factors With Age-Related Macular Degeneration: A Mendelian Randomization Study. JAMA Ophthalmol. 2021, 139, 1299–1306. [Google Scholar] [CrossRef]
- Suzuki, S.; Yanagita, S.; Amemiya, S.; Kato, Y.; Kubota, N.; Ryushi, T.; Kita, I. Effects of negative air ions on activity of neural substrates involved in autonomic regulation in rats. Int. J. Biometeorol. 2008, 52, 481–489. [Google Scholar] [CrossRef]
- Kato, T.; Inoue, T.; Morooka, T.; Yoshimoto, N.; Node, K. Short-term passive smoking causes endothelial dysfunction via oxidative stress in nonsmokers. Can. J. Physiol. Pharmacol. 2006, 84, 523–529. [Google Scholar] [CrossRef]
- Kankanamage, R.N.T.; Ghosh, A.B.; Jiang, D.; Gkika, K.; Keyes, T.; Achola, L.A.; Suib, S.; Rusling, J.F. Metabolites of Tobacco- and E-Cigarette-Related Nitrosamines Can Drive Cu2+-Mediated DNA Oxidation. Chem. Res. Toxicol. 2020, 33, 2072–2086. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Hu, X.; Wang, H.; Wang, R.; Sun, Z.; Tan, X.; Liu, S.; Wang, H. Effects of a dammarane-type saponin, ginsenoside Rd, in nicotine-induced vascular endothelial injury. Phytomedicine 2020, 79, 153325. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Huang, Q.; Wu, Y.; Song, Y.; Dong, W.; Chu, M.; Yang, D.; Zhang, X.; Zhang, J.; Chen, C.; et al. Metabolic linkages between indoor negative air ions, particulate matter and cardiorespiratory function: A randomized, double-blind crossover study among children. Environ. Int. 2020, 138, 105663. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, H.; Zhu, W.; Xu, Y.; Liu, M.; Zhu, L.; Yang, F.; Zhang, L.; Liu, X.; Zhong, Z.; et al. Nicotine Accelerates Atherosclerosis in Apolipoprotein E-Deficient Mice by Activating α7 Nicotinic Acetylcholine Receptor on Mast Cells. Arter. Thromb. Vasc. Biol. 2017, 37, 53–65. [Google Scholar] [CrossRef]
- Sowka, A.; Dobrzyn, P. Role of Perivascular Adipose Tissue-Derived Adiponectin in Vascular Homeostasis. Cells 2021, 10, 1485. [Google Scholar] [CrossRef]
- Liu, S.; Li, C.; Chu, M.; Zhang, W.; Wang, W.; Wang, Y.; Guo, X.; Deng, F. Associations of forest negative air ions exposure with cardiac autonomic nervous function and the related metabolic linkages: A repeated-measure panel study. Sci. Total Environ. 2022, 850, 158019. [Google Scholar] [CrossRef]
- Riad, A.; Narasimhulu, C.A.; Deme, P.; Parthasarathy, S. A Novel Mechanism for Atherosclerotic Calcification: Potential Resolution of the Oxidation Paradox. Antioxid. Redox Signal. 2018, 29, 471–483. [Google Scholar] [CrossRef]
- Zollinger, A.J.; Smith, M.L. Fibronectin, the extracellular glue. Matrix. Biol. 2017, 60–61, 27–37. [Google Scholar] [CrossRef]
- Schumacher, J.A.; Wright, Z.A.; Owen, M.L.; Bredemeier, N.O.; Sumanas, S. Integrin α5 and Integrin α4 cooperate to promote endocardial differentiation and heart morphogenesis. Dev. Biol. 2020, 465, 46–57. [Google Scholar] [CrossRef]
- Wernig, G.; Chen, S.Y.; Cui, L.; Van Neste, C.; Tsai, J.M.; Kambham, N.; Vogel, H.; Natkunam, Y.; Gilliland, D.G.; Nolan, G.; et al. Unifying mechanism for different fibrotic diseases. Proc. Natl. Acad. Sci. USA 2017, 114, 4757–4762. [Google Scholar] [CrossRef]
- Bill, R.; Wirapati, P.; Messemaker, M.; Roh, W.; Zitti, B.; Duval, F.; Kiss, M.; Park, J.C.; Saal, T.M.; Hoelzl, J.; et al. CXCL9: SPP1 macrophage polarity identifies a network of cellular programs that control human cancers. Science 2023, 381, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Freiholtz, D.; Bergman, O.; Pradhananga, S.; Lång, K.; Poujade, F.A.; Granath, C.; Olsson, C.; Franco-Cereceda, A.; Sahlén, P.; Eriksson, P.; et al. SPP1/osteopontin: A driver of fibrosis and inflammation in degenerative ascending aortic aneurysm? J. Mol. Med. 2023, 101, 1323–1333. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, S.; Wei, T.; Xiao, M.; Shan, M.; An, Z.; Li, N.; Zhou, J.; Zhao, S.; Lu, X. Negative Air Ions Attenuate Nicotine-Induced Vascular Endothelial Dysfunction by Suppressing AP1-Mediated FN1 and SPP1. Antioxidants 2025, 14, 859. https://doi.org/10.3390/antiox14070859
Xiao S, Wei T, Xiao M, Shan M, An Z, Li N, Zhou J, Zhao S, Lu X. Negative Air Ions Attenuate Nicotine-Induced Vascular Endothelial Dysfunction by Suppressing AP1-Mediated FN1 and SPP1. Antioxidants. 2025; 14(7):859. https://doi.org/10.3390/antiox14070859
Chicago/Turabian StyleXiao, Sha, Tianjing Wei, Mingyang Xiao, Mingming Shan, Ziqi An, Na Li, Jing Zhou, Shuang Zhao, and Xiaobo Lu. 2025. "Negative Air Ions Attenuate Nicotine-Induced Vascular Endothelial Dysfunction by Suppressing AP1-Mediated FN1 and SPP1" Antioxidants 14, no. 7: 859. https://doi.org/10.3390/antiox14070859
APA StyleXiao, S., Wei, T., Xiao, M., Shan, M., An, Z., Li, N., Zhou, J., Zhao, S., & Lu, X. (2025). Negative Air Ions Attenuate Nicotine-Induced Vascular Endothelial Dysfunction by Suppressing AP1-Mediated FN1 and SPP1. Antioxidants, 14(7), 859. https://doi.org/10.3390/antiox14070859