Abstract
In chronic respiratory diseases (CRDs), oxidative stress and inflammation are closely linked, driving disease onset, progression, and comorbidities. Oxidative stress activates inflammatory pathways, while chronic inflammation promotes further reactive oxygen species (ROS) production, creating a vicious cycle leading to airway remodeling, reduced lung function, and exacerbations. This review highlights the central roles of inflammation and oxidative stress in chronic obstructive pulmonary disease (COPD) and obstructive sleep apnea (OSA). In COPD, chronic hypoxemia associates with emphysema, appearing with disease progression. In OSA, beyond systemic consequences, pulmonary inflammation and oxidative stress contribute to lung injury as well. Although COPD and OSA are distinct conditions, some patients present with “overlap syndrome”, a term used in this review to describe the coexistence of both. This combination poses unique diagnostic and therapeutic challenges. We also examine the role of hypoxia and its transcriptional effects via hypoxia-inducible factors (HIFs) in promoting oxidative stress and inflammation. Finally, we explore how artificial intelligence (AI) offers promising tools to improve diagnosis, monitoring, and management of CRDs and may help elucidate mechanistic links between hypoxia, inflammation, and oxidative stress, contributing to more personalized therapeutic strategies.
1. Introduction
Globally, chronic respiratory diseases (CRDs) are estimated to cause one death every 10 s (equivalent to 3 million annually) and are projected to become the third leading cause of mortality by 2030, according to the World Health Organization (WHO). Data from the Global Burden of Diseases, Injuries, and Risk Factors Study [1] indicates that total deaths and the prevalence of CRDs have increased by 28.5% and 39.8%, respectively [1]. Chronic obstructive pulmonary disease (COPD) and obstructive sleep apnea (OSA) are highly prevalent chronic respiratory conditions associated with significant morbidity. The global prevalence of COPD among adults is estimated at approximately 12% [2,3], whereas the prevalence of OSA may reach up to 38% [4]. Both conditions are characterized by reduced arterial oxygen tension (hypoxemia), increased production of ROS, and an exacerbated inflammatory response, all of which shape the clinical manifestations observed in these patients. Notably, the clinical course of these individuals is highly variable and largely influenced by the development of comorbidities, including cardiovascular, metabolic, and neoplastic diseases, that are often the primary cause of mortality. Despite this, the underlying mechanisms that govern the onset and progression of these comorbidities remain poorly defined, and robust biomarkers to predict comorbidome risk have yet to be identified.
COPD is defined by the presence of respiratory symptoms and persistent airflow limitation resulting from structural abnormalities in the airways and/or alveoli [5]. Although traditionally associated with older adults, COPD can also arise from early-life developmental alterations [6,7]. While smoking remains the primary etiological factor and is strongly associated with adverse clinical outcomes and increased mortality in genetically susceptible individuals worldwide, only approximately 15% of smokers develop an exaggerated local inflammatory response to ROS in cigarette smoke. This leads to irreversible pulmonary damage [8,9] and the emergence of a systemic proinflammatory state [10]. This pathological condition contributes to the progression and prognosis of COPD, promoting the development of comorbidities linked to systemic dysfunction, reduced quality of life, increased hospitalizations, suboptimal therapeutic responses, and elevated mortality rates [11,12].
Frequent comorbidities observed in COPD patients include cardiovascular diseases, metabolic disorders, pulmonary fibrosis, and lung cancer [13,14,15]. It is estimated that 78.6% of individuals with COPD present with at least one clinically relevant comorbidity, 68.8% with two or more, and nearly 47.9% with three or more. Although patients with advanced airflow limitation often succumb to respiratory failure, the majority of deaths among COPD patients are due to non-respiratory causes, chiefly cardiovascular disease and cancer [14,16]. Despite these clinical correlations, the pathophysiological mechanisms underlying the association between COPD and its comorbidities remain poorly understood. Beyond aging, sedentary lifestyle, and tobacco use, both hypoxia and systemic inflammation, hallmarks of COPD, are believed to significantly contribute to the high burden of comorbid conditions in this population [3,17].
OSA is another prevalent respiratory disorder characterized by repeated collapse of the upper airways during sleep, leading to intermittent hypoxia (IH), periodic drops in oxygen saturation, and sleep fragmentation (micro-awakenings). These disturbances result in non-restorative sleep, causing fatigue and daytime sleepiness, and contribute to the development of respiratory, neuropsychiatric, metabolic, cardiovascular, and oncological disorders [18]. OSA is also associated with a high mortality rate, primarily linked to daytime sleepiness and the onset of comorbidities, particularly cardiovascular and metabolic conditions [19,20,21,22]. Severe OSA has been specifically associated with an increased risk of dyslipidemia, non-alcoholic fatty liver disease, and, most notably, diabetes [23]. Additionally, it is also associated with a higher risk of arrhythmias, hypertension, atherosclerosis, ischemic heart disease, and stroke [24,25]. Although the high prevalence of hypertension in OSA patients, exceeding 50% in some cohorts, has been proposed as the main driver of its cardiovascular complications [26], other risk factors, particularly those promoting atherosclerosis, also play a critical role [27,28]. Among these, dyslipidemia and inflammation, both highly prevalent in OSA, stand out as key proatherosclerotic contributors [29,30].
Moreover, several epidemiological studies, patient cohort analyses, and animal models have shown that OSA is associated with an increased incidence, aggressiveness, and mortality of various types of cancer [31,32]. IH has been proposed as a potential mechanism, promoting tumor vascularization, facilitating tumor growth, and modulating immune responses by disrupting immune surveillance, thus allowing the emergence of more aggressive tumors [33,34,35]. In addition, the repetitive cycles of hypoxia and reoxygenation alter redox homeostasis in OSA, promoting ROS production and reducing endogenous antioxidant defenses. This oxidative imbalance correlates with disease severity [36].
Overlap syndrome, as commonly known to refer to the condition of both COPD and OSA, is relatively frequent and is characterized by nocturnal hypoxemia. This condition leads to worse clinical outcomes than either disease on its own [37]. Recent studies have demonstrated that patients with overlap syndrome exhibit elevated levels of systemic inflammatory biomarkers compared to individuals with isolated COPD or OSA, potentially contributing to the increased cardiovascular risk observed in this subgroup [38]. However, inflammation alone may not fully account for the heightened cardiovascular risk, highlighting the need for further investigation into alternative pathogenic mechanisms.
In the context of CRDs, persistent hypoxic conditions trigger a key adaptive response mediated by transcriptional programs governed primarily by hypoxia-inducible factors (HIFs). HIF-1α and HIF-2α are central mediators of cellular adaptation to low oxygen levels, regulating a wide array of genes involved in essential adaptive processes [39,40]. However, in the context of CRDs, this regulation is complex and significantly contributes to disease pathogenesis and progression. While initially protective, chronic activation of HIF pathways can become maladaptive, initiating a cascade of detrimental effects. In this review, we highlight several HIF-mediated pathophysiological responses, with a particular focus on pulmonary vascular adaptation to hypoxia.
Despite advances in understanding the pathophysiological mechanisms underpinning COPD and OSA, substantial gaps remain in early diagnosis, prognosis, and personalized treatment approaches. The complexity and heterogeneity of CRDs, particularly the interplay between hypoxia, oxidative stress, inflammation, and comorbidities, pose significant challenges for conventional clinical methods. In this context, artificial intelligence (AI) has emerged as a promising solution to overcome these limitations. By integrating and analyzing large-scale, multimodal clinical and molecular data, AI tools offer the potential to improve diagnostic accuracy, stratify patient risk, and unveil novel insights into disease mechanisms. These capabilities make AI particularly valuable for refining our understanding and improving diagnosis and prognosis in diseases such as COPD [41] and OSA [42], as well as the underlying molecular pathways and its use in personalized medicine. Advancing AI systems in respiratory medicine require multidisciplinary teams that bring together clinicians, data scientists, engineers, and ethicists to ensure that models are both technically robust and clinically relevant.
2. Transcriptional Response to Sustained and Intermittent Hypoxia in CRDs
As we have mentioned, despite their diverse etiology and clinical manifestations, both OSA and COPD share a common feature: the development of hypoxemia. While in COPD these hypoxic conditions are primarily caused by alveolar damage, known as emphysema, in the case of OSA intermittent hypoxia due to repeated upper airway obstruction during sleep results in temporary drops in oxygen saturation. Although the degree and duration of oxygen desaturation vary widely depending on disease severity and adherence to treatment, some degree of tissue hypoxia is expected in the majority of cases [43,44]. Moreover, tissue hypoxia is increasingly recognized as a potential major contributor to the pathogenesis of chronic respiratory diseases (CRDs) [43,45].
To maintain oxygen homeostasis, the body employs various mechanisms, broadly categorized into immediate physiological adjustments [46] and longer-term transcriptional responses [39]. Acute responses are triggered within seconds of oxygen deprivation, such as the activation of chemoreceptors like the carotid body to modulate respiration, and vascular changes that enhance oxygen delivery. However, these mechanisms are insufficient for adaptation to prolonged hypoxia. Instead, transcriptional responses become central over hours or days, adjusting metabolism and oxygen supply and playing a critical role in adapting to CRDs. Additionally, hypoxia-induced transcriptional reprogramming may help precondition tissues to better tolerate future hypoxic episodes [47,48], providing an essential adaptive mechanism in chronic respiratory conditions where tissues are repeatedly subjected to low oxygen levels.
2.1. HIF Signaling and Gene Expression Dynamics in Sustained and Intermittent Hypoxia
A universal property of nucleated cells is their ability to autonomously sense oxygen levels and regulate members of the HIF family of transcription factors, which orchestrate a broad gene expression response to hypoxia. HIFs, consisting of an oxygen-regulated alpha subunit and a constitutively expressed beta subunit (aryl hydrocarbon receptor nuclear translocator, ARNT), drive a metabolic shift from oxidative phosphorylation to glycolysis, reducing both oxygen consumption and the generation of ROS. They also promote the transcription of genes involved in angiogenesis and erythropoiesis, facilitating oxygen delivery to hypoxic tissues [39]. While these adaptations enhance survival, as mentioned above chronic HIF activation is implicated in pathological processes including tissue remodeling, inflammation, and abnormal vascularization, hallmarks of chronic respiratory and cardiovascular diseases [40].
The oxygen-dependent regulation of HIF and its role in hypoxia adaptation have been extensively characterized, a contribution recognized by the 2019 Nobel Prize in Physiology or Medicine. The HIF pathway depends on a set of oxygen-sensitive enzymes that act as cellular oxygen sensors [49]. Under normoxic conditions, EGLN family prolyl hydroxylases hydroxylate specific proline residues on HIF-α, marking it for recognition by a ubiquitin ligase complex containing the von Hippel–Lindau (VHL) tumor suppressor protein and targeting it for proteasomal degradation [49]. These EGLN enzymes require iron (Fe2+), 2-oxoglutarate, and molecular oxygen for their activity, making them highly sensitive to oxygen levels. Even mild hypoxia inhibits EGLN activity, leading to HIF-α stabilization and activation of its downstream transcriptional targets. HIF transcriptional activity is also regulated by another oxygen-dependent enzyme factor inhibiting HIF (FIH), which hydroxylates an asparagine residue on HIF-α [49]. The central role of HIFs in hypoxia is further reinforced by early studies showing that most hypoxia-induced genes are dependent on HIF activity [50].
Nevertheless, while HIFs are central players, other transcription factors also contribute to the hypoxic gene expression program by integrating diverse cellular signals. NF-κB is a key example, especially relevant for inflammatory responses to hypoxia. Its activation is closely linked to the inhibition of EGLN enzymes, paralleling HIF regulation [51]. Importantly, a recent report suggests a large part of the hypoxia-regulated genes depend, at least in part, on NF-κB [52]. This interplay between NF-κB and HIF is particularly significant in CRDs, where inflammation and hypoxia coexist, with NF-κB acting as a key transcription factor in inflammatory responses and HIF playing a central role in the cellular response to hypoxia. FOXO3 is another transcription factor regulated under hypoxia through prolyl hydroxylation [53]. This factor modulates responses such as oxidative stress resistance and metabolic adaptation [54]. Notch signaling also intersects with hypoxic pathways via direct interactions between HIF-1α and the Notch intracellular domain (NICD), resulting in the hypoxic induction of Notch target genes [55]. Direct hydroxylation of NICD in an oxygen-dependent manner has also been reported. AP-1, in contrast, becomes more prominent following prolonged hypoxic exposure [56], suggesting a role in later stages of adaptation rather than in the immediate response [57,58]. Similarly, the Hippo pathway, which regulates transcriptional enhanced associated domain (TEAD) transcription factors, is modulated by oxygen levels, potentially through the hypoxia-inducible ubiquitin ligase Siah2 [59]. This interaction may contribute to the development of pulmonary hypertension [60,61] and other cardiovascular diseases [62], which are common comorbidities associated with CRDs. Other transcription factors, such as TP53 and Nrf2, may respond to hypoxia in more context-dependent ways. TP53 is generally induced in severe hypoxia or anoxia, which may explain conflicting findings regarding its regulation by oxygen [63]. Nrf2, a master regulator of oxidative stress, is also likely to be indirectly involved in hypoxia adaptation [64], particularly during reoxygenation and in conditions that promote ROS production [65]. Given the critical role of ROS in the progression of CRDs, the involvement of Nrf2 in hypoxia-related responses, albeit indirect, may be especially important in this context. Thus, while HIFs are central, a diverse network of transcription factors contributes to the hypoxic response, integrating cues from inflammation, metabolism, and cellular stress.
Beyond transcription factor regulation, oxygen availability also influences gene expression via epigenetic mechanisms, adding another layer of dynamic control during hypoxic adaptation. Epigenetic modifications are crucial for modulating hypoxia-induced gene expression by altering chromatin accessibility. Seminal studies have demonstrated that hypoxia inhibits histone demethylases such as KDM5A and KDM6A, leading to increased levels of histone marks like H3K4me3 and H3K27me, respectively, modifications that contribute to hypoxic responses independently of HIF [66,67]. These and other findings indicate that DNA and histone methylation states shift dynamically in response to oxygen levels. Many demethylases are 2-oxoglutarate-dependent dioxygenases that require oxygen as a cosubstrate, positioning them as oxygen sensors analogous to HIF prolyl hydroxylases [68,69]. Members of the KDM4, KDM5, and KDM6 families, which have a high affinity for oxygen, are particularly susceptible to inhibition under physiological hypoxia. As a result, hypoxia can profoundly alter chromatin landscapes, modulating gene expression in ways that extend beyond the HIF pathway [68,69]. These insights highlight the importance of oxygen-dependent epigenetic regulation in shaping hypoxic responses and potentially influencing long-term disease progression.
The convergence of these transcriptional and epigenetic regulators underscores the complexity of the cellular response to hypoxia and the importance of considering multiple regulatory pathways. However, caution is warranted when extrapolating findings to CRDs. A significant limitation is that much of the current knowledge comes from in vitro cellular models, often derived from tumor cell lines that are not representative of lung tissue. This raises uncertainty regarding the contribution of these transcriptional mechanisms to hypoxia adaptation in pulmonary cell types in vivo. Moreover, most studies focus on sustained hypoxia, even though oxygen levels fluctuate in CRDs. OSA represents an extreme case, with severe patients experiencing more than 30 apnea episodes per hour, and some exceeding 80 [70]. These cycles of hypoxia and reoxygenation create a physiological context distinct from continuous hypoxia. Whether IH elicits the same transcriptional program as sustained hypoxia remains unclear, especially in cases involving high-frequency, short-duration hypoxic episodes.
2.2. Role of HIFs in Vascular Dysfunction in COPD and OSA
Hypoxic conditions in the vascular endothelium trigger the proliferation of smooth muscle cells, increasing their coverage around pulmonary vessels. This vascular remodeling raises pulmonary arterial pressure, potentially leading to right ventricular hypertrophy and, ultimately, heart failure. Previous studies have demonstrated that hypoxia-induced pulmonary vascular remodeling is mediated by HIFs. Specifically, HIF-induced vascular remodeling involves the emergence of pulmonary artery smooth muscle cells (PASMCs), which results in the muscularization of alveolar wall vessels resulting in increased pulmonary arterial pressure. This vascular remodeling leads to the thickening of the tunica media and adventitia of pulmonary arteries.
In this context, mice with heterozygous germline deletion of HIF-1α or HIF-2α exhibit attenuated development of pulmonary hypertension upon chronic hypoxic exposure [71,72,73]. Further investigations have confirmed the pivotal role of endothelial HIF-2α in hypoxia-driven pulmonary hypertension in which endothelial-cell-specific deletion of HIF-2α protects against the development of hypoxia-induced pulmonary hypertension [49,74,75,76,77,78]. Conversely, sustained activation of HIF-2α in pulmonary artery endothelial cells in mice leads to pulmonary hypertension and right ventricular hypertrophy [74,77,79].
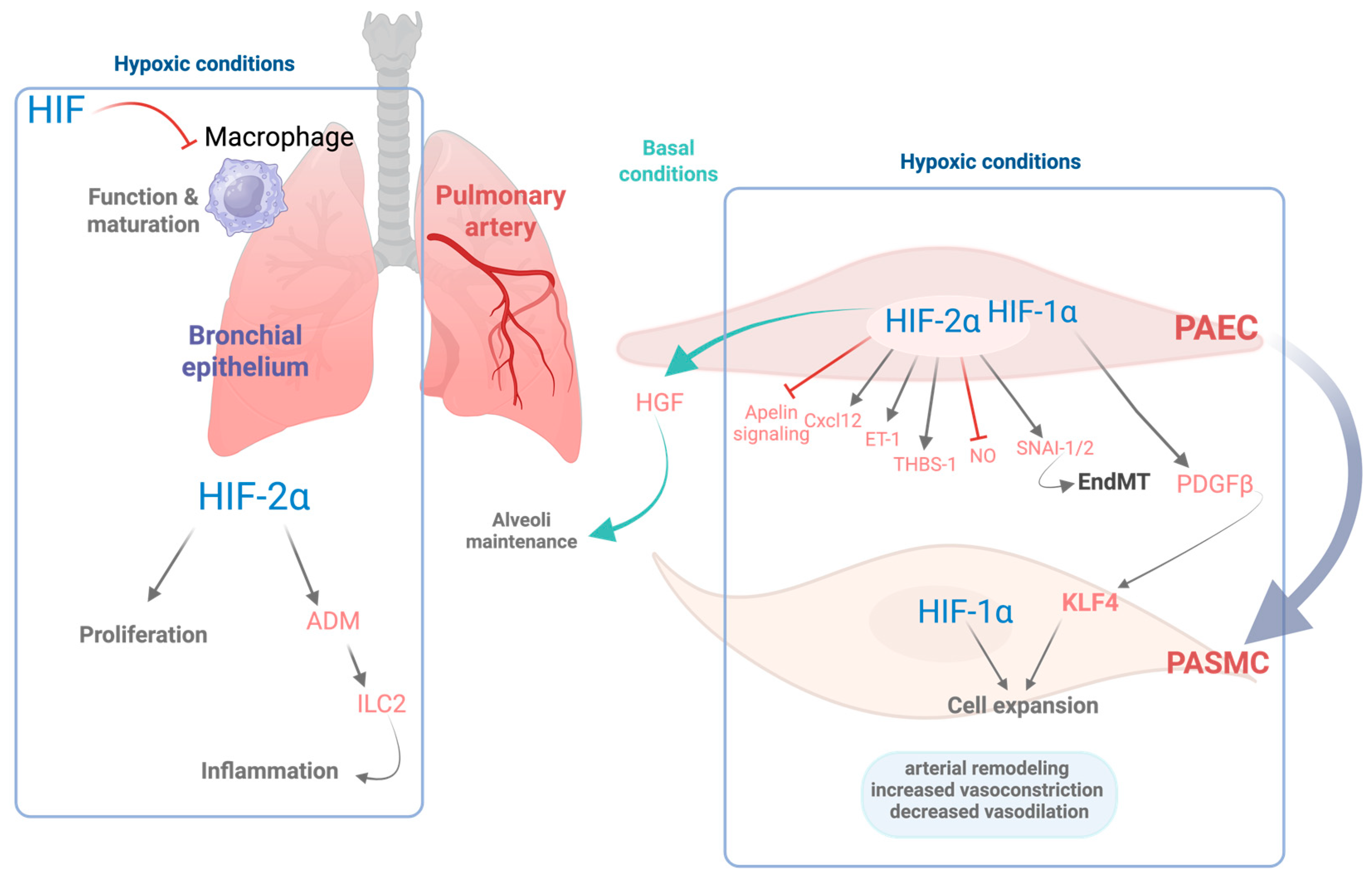
In line with these findings, both human studies and mouse models carrying gain-of-function mutations in HIF-2α have demonstrated severe pulmonary hypertension phenotypes [80,81]. Additionally, HIF-2α regulates the expression of key endothelial genes that enhance vasoconstriction and impair vasodilation, including endothelin-1, arginase activity, as well as suppression of apelin signaling. It also upregulates molecules such as CXC motif chemokine ligand (CXCL12), which promotes the proliferation and migration of pulmonary vascular smooth muscle cells [77,78,82]. HIF-2α has been shown to upregulate the expression of thrombospondin-1 (TSP-1) in hypoxic lungs, contributing to vascular remodeling and vasoconstriction [83]. In addition, HIF-2α has the potential to induce the expression of the transcription factors SNAI-1/2, key regulators of endothelial-to-mesenchymal transition (EndMT), which in turn contributes to pulmonary vascular remodeling [84]. In parallel, HIF-1α activity in both endothelial and smooth muscle cells has been shown to be implicated in the pathogenesis of hypoxia-induced pulmonary hypertension. Specifically, HIF-1α drives the expression of platelet-derived growth factor B (PDGF-B) in endothelial cells, which in turn promotes the expansion of PASMC via activation of the pluripotency factor Krüppel-like factor 4 (KLF4) [73,85] (see Figure 1). Apart from the role of HIF-2α overactivation in pulmonary hypertension, decreased HIF-2α activity in lung endothelial cells also plays an important role in other pathological conditions, including COPD. Notably, these patients exhibit decreased expression of HIF-2α in the lungs [86]. Consistent with these findings, an independent study further demonstrated that lung tissue from COPD patients contains a reduced number of CD31+ endothelial cells, along with lower HIF-2α expression per CD31+ cell, an effect that is particularly pronounced in advanced stages of the disease [87]. Similarly, diminished endothelial HIF-2α expression has been observed in murine models of COPD following chronic exposure to cigarette smoke [86,87].
In agreement with these results, genetic inactivation of HIF-2α specifically in pulmonary endothelial cells results in an emphysematous phenotype characterized by enlarged alveolar spaces and increased apoptosis of alveolar structural cells [87]. These findings suggest that reduced HIF-2α expression in pulmonary endothelial cells may act as a pathogenic driver in the progression of COPD. Moreover, its deficiency in lung endothelial cells leads to a concomitant reduction in both endothelial cells and pericytes. This is associated with diminished expression of hepatocyte growth factor, a key mediator involved in the preservation of the alveolar architecture by sustaining vascular survival [86]. Importantly, this emphysematous phenotype is not observed following endothelial-specific deletion of HIF-1α, underscoring the pivotal role of HIF-2α in preserving lung vascular homeostasis [87]. Collectively, these findings support the idea that endothelial HIF-2α plays a critical protective role in maintaining alveolar architecture and limiting pathological inflammation in the lung. Nevertheless, excessive HIF-2α activation in endothelial cells, such as that observed in pulmonary hypertension, can be detrimental, which highlights the importance of well-balanced HIF-2α levels in the endothelium [86]. A detailed scheme of HIF signaling within the lung is included in Figure 1.
In addition to the role of the HIF pathway in pulmonary hypertension, HIF isoforms have been also involved in systemic hypertension in OSA [88]. Indeed, IH-dependent HIF-1α activity has been proposed to hyperactivate the central and peripheral nervous system leading to hypertension. At a molecular level HIF-1α induces the prooxidant protein NADPH oxidase (NOX)-2, which provokes oxidative stress and subsequent inhibition of heme-oxygenase 2 and generation of hydrogen sulfide [88,89]. In contrast, HIF-2α counteracts this pathological action of HIF-1α by inducing the antioxidant enzyme SOD2, which can counteract HIF-1α-dependent oxidative stress. In this line, IH evoked a degradation of HIF-2α therefore facilitating oxidative-stress-dependent hypertension [90]. In addition to the central and peripheral nervous system, IH induces HIF-1α in heart and aortic tissue, which leads to oxidative stress, vascular smooth muscle cell migration, vascular remodeling, and systemic hypertension [91]. This HIF-1α-dependent vascular remodeling has been associated with the activation of the Hippo-YAP pathway [91].
2.3. Role of HIF Pathway in Airway Inflammation
HIF signaling is also activated in other pulmonary cell types, including epithelial Club cells, where HIF-2α promotes epithelial cell proliferation [92]. Notably, the progression of COPD is associated with a chronic inflammatory state in the airways [93], a process in which HIF activity may play a significant role. Supporting this notion, activation of HIF-2α in Club cells triggers a type 2 helper T cell (Th2)-skewed inflammatory response in the lung, characterized by the activation of group 2 innate lymphoid cells (ILC2s) [94]. At the molecular level, this effect is mediated through HIF2α-induced expression of adrenomedullin (ADM), a gene which contains a hypoxia response element (HRE) in its proximal promoter [94]. ADM enhances ILC2 activation by increasing the expression of surface markers such as Sca-1 and KLRG1 and by amplifying their capacity to secrete IL-5 [94] (see Figure 1).
These findings establish a novel link between HIF-2α activity, ILC2 biology, and Th2-driven airway inflammation. Additionally, recent clinical evidence demonstrates that patients with severe COPD present elevated numbers of ILC2s in sputum samples [95]. In parallel, circulating ADM levels have been identified as a potential biomarker for COPD exacerbation [96], while proadrenomedullin (proADM) has been proposed as a predictor of survival in COPD patients [97]. Collectively, these data suggest that aberrant HIF-2α activity in the bronchial epithelium may exacerbate COPD by promoting airway inflammation through the ADM–ILC2 axis. Conversely, evidence suggests that IH preferentially activates NF-κB over HIFs, particularly in OSA models, where inflammation rather than canonical HIF-driven adaptation appears to dominate [98]. Supporting this, IH has been associated with NF-κB-mediated inflammatory gene expression in adipose tissue, a key contributor to the cardiovascular complications of OSA [99]. These observations highlight the need for further studies to systematically compare the transcriptional effects of sustained versus IH, particularly in CRDs, where oxygenation patterns have critical implications for disease progression and treatment strategies.
In addition to epithelial cells, HIF transcription factors also modulate immune responses in alveolar macrophages, which are essential for maintaining immune homeostasis within the alveolar space. HIF-1α expression in myeloid cells has been shown to be critical for sustaining inflammatory responses [100], with HIF-1α generally associated with M1 macrophage polarization, whereas HIF-2α has been linked to M2 polarization [101]. However, this paradigm appears more complex in the context of alveolar macrophages. Constitutive activation of HIF-1α signaling, through deletion of tumor suppressor gene VHL, leads to impaired Th2 responses, evidenced by reduced eosinophil recruitment and lower IL-5 and IL-13 production [102]. Furthermore, VHL inactivation in alveolar macrophages induces an immature phenotype and impairs their capacity for surfactant clearance [103] (see Figure 1).
These findings indicate that, contrary to its role in promoting proinflammatory M1 phenotypes in other contexts, HIF-1α may act by suppressing specific immune functions of alveolar macrophages. Importantly, alveolar macrophage function is known to be compromised in COPD [104]; however, the potential contribution of HIF signaling to this dysfunction remains unexplored and requires further investigation.
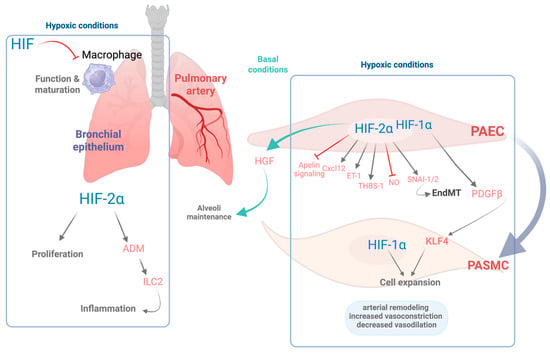
Figure 1.
Role of HIFs in pulmonary cellular responses and vascular remodeling. This schematic illustrates the cellular and molecular effects of hypoxia mediated by HIF-1α and HIF-2α isoforms as well as their role in basal conditions in various pulmonary cell types in baseline and hypoxic conditions. Under hypoxic conditions, HIF signaling is activated, orchestrating the transcription of numerous genes that contribute to pulmonary arterial remodeling and favor vessel contraction. In pulmonary artery endothelial cells (PAECs), HIF-2α regulates both pro- and antiremodeling pathways, including the induction of CXC motif chemokine ligand 12 (CXCL12), endothelin-1 (ET-1), thrombospondin-1 (THBS-1), and the zinc-finger transcription factors SNAI-1/2, which contributes to endothelial-to-mesenchymal transition (EndMT). Conversely, HIF-2α concurrently inhibits apelin and nitric oxide (NO) signaling. In turn, endothelial HIF-1α stimulates the release of platelet-derived growth factor beta (PDGFβ) leading to the activation of the pluripotency factor Krüppel-like factor 4 (KLF4) in both PAECs and PASMCs, which together with HIF-1α activation promotes expansion of PASMCs and vascular remodeling. Collectively, these responses lead to increased vasoconstriction, impaired vasodilation, and structural changes in the pulmonary vasculature. Under basal oxygen conditions, endothelial HIF-2α plays homeostatic roles, supporting alveolar maintenance via hepatocyte growth factor (HGF). Moreover, in the bronchial epithelium, HIF-2α induces proliferation of the bronchial epithelial cells as well as increased expression of adrenomedullin (ADM), which activates the group 2 innate lymphoid cells (ILC2s), contributing to Th2 airway inflammation. In the alveolar macrophages, HIF signaling counteracts macrophage maturation and function. Overall, this figure highlights the complementary and context-dependent roles of HIF-1α and HIF-2α in pulmonary adaptation to hypoxia, with critical implications for inflammation and vascular remodeling. Figure created with Biorender.com.
Figure 1.
Role of HIFs in pulmonary cellular responses and vascular remodeling. This schematic illustrates the cellular and molecular effects of hypoxia mediated by HIF-1α and HIF-2α isoforms as well as their role in basal conditions in various pulmonary cell types in baseline and hypoxic conditions. Under hypoxic conditions, HIF signaling is activated, orchestrating the transcription of numerous genes that contribute to pulmonary arterial remodeling and favor vessel contraction. In pulmonary artery endothelial cells (PAECs), HIF-2α regulates both pro- and antiremodeling pathways, including the induction of CXC motif chemokine ligand 12 (CXCL12), endothelin-1 (ET-1), thrombospondin-1 (THBS-1), and the zinc-finger transcription factors SNAI-1/2, which contributes to endothelial-to-mesenchymal transition (EndMT). Conversely, HIF-2α concurrently inhibits apelin and nitric oxide (NO) signaling. In turn, endothelial HIF-1α stimulates the release of platelet-derived growth factor beta (PDGFβ) leading to the activation of the pluripotency factor Krüppel-like factor 4 (KLF4) in both PAECs and PASMCs, which together with HIF-1α activation promotes expansion of PASMCs and vascular remodeling. Collectively, these responses lead to increased vasoconstriction, impaired vasodilation, and structural changes in the pulmonary vasculature. Under basal oxygen conditions, endothelial HIF-2α plays homeostatic roles, supporting alveolar maintenance via hepatocyte growth factor (HGF). Moreover, in the bronchial epithelium, HIF-2α induces proliferation of the bronchial epithelial cells as well as increased expression of adrenomedullin (ADM), which activates the group 2 innate lymphoid cells (ILC2s), contributing to Th2 airway inflammation. In the alveolar macrophages, HIF signaling counteracts macrophage maturation and function. Overall, this figure highlights the complementary and context-dependent roles of HIF-1α and HIF-2α in pulmonary adaptation to hypoxia, with critical implications for inflammation and vascular remodeling. Figure created with Biorender.com.
3. Role of Oxidative Stress and Inflammation in CRDs
As mentioned above, oxidative stress is a key mechanism in CRD development, making the identification of oxidative-stress-related biomarkers beneficial for improving its diagnosis and treatment. Repeated cycles of hypoxia and reoxygenation, along with exposure to indoor and outdoor air pollutants and other toxic chemicals, are key factors that stimulate the endogenous production of ROS in respiratory diseases such as OSA and COPD [8,105,106]. At low concentrations, ROS act as secondary messengers, playing a crucial role in redox signaling and regulating a variety of essential physiological processes, including bacterial killing during phagocytosis, vasodilation, tissue repair, and regeneration [107,108]. However, when present at elevated levels, ROS can cause irreversible damage to cellular biomolecules, such as lipids, carbohydrates, and nucleic acids, impairing their function, altering cellular metabolism, and ultimately leading to cell and tissue injury [105]. Importantly, oxidative stress and inflammation are not isolated processes but mutually reinforcing: oxidative stress activates inflammatory responses, which in turn further enhance ROS production, creating a self-sustaining pathogenic loop. This dynamic interplay contributes to the onset and progression of chronic respiratory diseases and has been increasingly recognized as a non-linear, bidirectional process [109,110].
Recent findings from multivariate logistic regression models have revealed a negative correlation between oxidative balance scores and the likelihood of COPD prevalence [111]. This imbalance is partly attributable to free radicals in cigarette smoke, which enhance endogenous ROS production and trigger inflammatory responses at the cellular level. Consequently, the respiratory systems of smokers exhibit age-related characteristics, partially due to increased oxidative stress, accumulation of damaged proteins, and elevated inflammation. On this matter, expression of the ROS-generating enzyme NOX4 is enhanced in airway smooth muscle from patients with COPD, which is also correlated with disease severity and lung function decline [112]. Indeed, in end-stage COPD, not only NOX4 but also other isoforms such as NOX1, NOX2, and NOX5 remain active. Among them, NOX1 and NOX4 are notably involved in oxidative stress and inflammation following acute exposure to cigarette smoke [113]. Thus, NOX enzymes are considered potential therapeutic targets for COPD. Supporting this, apocynin, a selective NOX inhibitor, partially improved cigarette-smoke-induced lung neutrophilia and reversed systemic inflammation and oxidative stress [114] in mice, although clinical trials have not been reported yet in COPD. Conversely, others demonstrate protective roles for certain NOX/DUOX family members in COPD models [115,116,117]. These findings suggest that non-specific antioxidant therapies may be ineffective or even counterproductive.
COPD patients exhibit a distinctive inflammatory profile characterized by elevated levels of macrophages, neutrophils, and both T and B lymphocytes in airway secretions [118,119]. This inflammatory response involves both the innate and adaptive immune systems, interconnected through dendritic cell activation. Compared to smokers without airway obstruction, COPD patients show a heightened inflammatory response that persists even after smoking cessation [120]. Although smoking is the major environmental risk factor, only a subset of smokers develop COPD, likely due to genetic predispositions, epigenetic modifications, and oxidative stress, all of which may amplify inflammation [119]. Persistent oxidative stress and inflammation accelerate epithelial cell senescence, weaken barrier integrity, and drive chronic airway inflammation and tissue remodeling [121]. During acute exacerbations, systemic inflammation often worsens [122,123], and population studies link elevated systemic inflammatory markers to higher risks of diabetes, cardiovascular disease, lung cancer, and mortality [120,124].
OSA is another chronic respiratory condition wherein oxidative stress and inflammation play significant roles. The cyclical pattern of oxygen deprivation and reoxygenation intrinsic to OSA leads to excessive ROS production, overwhelming cellular antioxidant defenses and triggering oxidative stress. Such stress contributes to OSA-related complications, including cardiovascular disorders [36,125]. Despite evidence of elevated ROS in OSA, no consensus exists on reliable oxidative stress biomarkers, highlighting the need for further research. Unlike continuous hypoxia, OSA-associated IH includes a posthypoxic reoxygenation phase, marked by frequent, severe oxygen desaturation and fluctuations [126]. This pattern amplifies ROS production, enhancing oxidative stress and inflammation. Oxidative stress also activates NF-κB, promoting the release of proinflammatory cytokines such as IL-1α, IL-1β, IL-6, and TNF-α [127,128]. Recent studies demonstrate that IH in OSA patients induces cardiomyocyte apoptosis, through the calcium-permeable cation channel TRPC5, contributing to increased ROS production, disrupting mitochondrial function, and disturbing calcium balance. These findings shed light on the mechanisms of TRPC5-mediated cardiac damage and highlight its potential as both a diagnostic biomarker and therapeutic target for OSA-related cardiovascular complications [129].
Thus, in both OSA and COPD, hypoxia, oxidative stress, and inflammation form a tightly interconnected network that mutually amplifies each component. Continuous positive airway pressure (CPAP) therapy, the gold standard for OSA treatment, effectively minimizes the frequency and severity of hypoxic episodes and reduces apneas and hypopneas while lowering oxidative stress [130,131]. However, the damage caused by oxidative stress in OSA patients is not limited to the airway; it extends to vital organs, including the cardiovascular system. Chronic oxidative damage is a key factor linking OSA to conditions such as hypertension, atherosclerosis, and other cardiovascular diseases [132]. Despite the beneficial effects of CPAP therapy on reducing hypoxic episodes, it does not directly address the underlying oxidative stress or reverse the oxidative damage already incurred. Indeed, some individuals may continue to exhibit residual oxidative stress despite consistent and effective use of CPAP [133].
Do Antioxidant Therapies Effectively Prevent Symptoms of COPD and OSA?
Despite the well-established role of oxidative stress in COPD and OSA, antioxidant therapies have shown limited clinical success to date. N-acetylcysteine (NAC), a glutathione precursor and mucolytic agent, has demonstrated beneficial effects using in vitro and in vivo COPD models [134]. However, these effects have not been clearly confirmed in clinical studies. Indeed, it has been shown that long-term treatment with high-dose NAC neither significantly reduced the annual rate of total exacerbations nor improved lung function in patients with mild-to-moderate COPD [135]. Other antioxidant compounds, such as vitamin C, have also been reported to increase the levels of antioxidant enzymes in serum and improve lung function [136]. Similarly, other articles suggest that supplementation with vitamins and antioxidants may have a beneficial effect on COPD symptoms, respiratory function, exacerbation, and quality of life [137]. Nevertheless, there is not enough clinical evidence to recommend antioxidant vitamins as standard therapy for COPD, mainly due to the lack of robust and large-scale randomized controlled trials demonstrating long-term efficacy and safety. Overall, the limited clinical success of antioxidant therapy underscores the complexity of redox regulation in COPD and highlights the need for more targeted and disease-specific antioxidant strategies. In some COPD patients, particularly those with overlap syndrome [138] or acute hypercapnic respiratory failure [139], CPAP has demonstrated clinical benefits. In such cases, CPAP may improve gas exchange, enhance ventilatory efficiency, and mitigate nocturnal hypoxemia, which is a key contributor to oxidative stress.
In line with this, the combination of polyphenol-rich antioxidants sourced from Aronia melanocarpa berries with CPAP therapy has shown synergistic benefits in the treatment of OSA by lowering the levels of proinflammatory cytokines, such as TNF-α and IL-6, which are commonly elevated in OSA patients [140]. This anti-inflammatory effect may contribute to improved cardiovascular health and a lower risk of long-term complications. Moreover, the polyphenols in Aronia enhance endothelial function by increasing nitric oxide (NO) availability. This supports vascular tone regulation and helps reduce blood pressure, thereby potentially lowering the risk of atherosclerosis and other cardiovascular issues, supporting improved overall health outcomes in OSA patients [133]. Similar studies show that tempol, a membrane-permeable radical scavenger, known to be a stable nitroxide, effectively reduces inflammation and oxidative stress, significantly alleviating IH-induced lung injury by modulating the miRNA-145-5p/Nrf2 signaling pathway [141]. Additionally, experimental studies in animal models have shown that antioxidant vitamins may help lower oxidative and carbonyl stress, key contributors to cardiovascular complications in OSA, although these studies did not assess cardiovascular remodeling [131]. Thus, the combination of CPAP with antioxidant therapies might be a potential therapeutic approach to reduce the risk of health complications by further reducing oxidative stress in OSA.
4. Oxidative Stress and Inflammation in Comorbidities of Hypoxemic Respiratory Diseases (COPD and OSA)
4.1. Role of Oxidative Stress and Inflammation in Cancer Associated with Respiratory Diseases
A growing body of evidence highlights the pivotal role of inflammation in cancer development, particularly by facilitating key processes involved in tumor progression [142]. Inflammation contributes to tumor initiation and promotes the accumulation of oncogenic mutations. Furthermore, inflammatory conditions elevate oxidative stress, resulting in enhanced DNA damage and increased mutation rates [143]. Inflammatory mediators are also implicated in processes such as epithelial to mesenchymal transition (EMT) and epigenetic alterations that favor oncogenesis. In addition, chronic inflammation leads to immune exhaustion, fostering the development of an immunosuppressive tumor microenvironment and facilitating immune evasion [144]. Patients with CRDs, including COPD and OSA, typically present a persistent low-grade inflammatory state, partly driven by hypoxemia [38,120,145].
In the context of COPD, the combination of chronic hypoxia, oxidative stress, and inflammation promotes a tumor-promoting environment in the airways, especially increasing the risk of lung cancer [146]. Although lung cancer can occur regardless of airflow limitation severity, numerous studies have underscored chronic inflammation as a central factor in promoting cellular transformation and disrupting immune regulation, thereby facilitating tumor initiation and progression [120,147]. In COPD patients, inflammatory processes are thought to support carcinogenesis via several mechanisms, including oxidative damage, immune dysregulation, genomic instability, and tissue remodeling [148,149]. In particular, sustained production of ROS, driven by cigarette smoke and repeated hypoxia–reoxygenation, contributes to oxidative DNA damage, lipid peroxidation, and protein carbonylation, all of which impair cellular homeostasis and promote genomic instability [150]. Moreover, chronic inflammation fosters immune dysregulation, which includes impaired immune surveillance and persistent release of proinflammatory cytokines such as TNF-α, IL-6, and IL-1β [151]. These cytokines activate key transcription factors like NF-κB and STAT3, which promote cell proliferation, angiogenesis, and resistance to apoptosis, contributing to malignant transformation [151]. Additionally, repeated cycles of epithelial injury and repair, driven by both oxidative stress, can lead to tissue remodeling and EMT processes known to support tumor invasion and metastasis [152].
Similarly, evidence indicates that OSA is associated with increased cancer incidence, aggressiveness, and mortality [32]. IH experienced during sleep in OSA patients induces oxidative stress and promotes tumorigenic processes, including angiogenesis and epithelial-to-mesenchymal transition [35,153,154,155]. More importantly, there is solid evidence elucidating how the immune surveillance in OSA patients is disrupted due to chronic inflammation, which modulates immune cell subsets and their immune functionality. For instance, a switch to tumor-associated macrophages and a defect in natural killer (NK) cell maturation have been reported as well as suppression of T cell function mediated by overexpression of immune checkpoint axes [35,156,157,158,159].
4.2. Role of Oxidative Stress and Inflammation in Cardiovascular Complications Associated with CRDs
Chronic inflammation is a central pathological mechanism implicated in a wide range of metabolic and cardiovascular disorders. Defined as a prolonged and persistent immune activation, chronic inflammation results in tissue injury, oxidative stress, mitochondrial dysfunction, and disruption of physiological homeostasis [160,161,162]. Several inflammatory pathways link respiratory diseases with cardiometabolic comorbidities, with particular emphasis on NF-κB signaling, NLRP3 inflammasome activation, and oxidative stress [162,163]. Although both hypoxemia and ROS are known to stimulate NF-κB and NLRP3, current evidence points to NLRP3 as a major driver of systemic inflammation [164,165]. Sterile stimulation activates the inflammasome complex, enhancing the production of inflammatory cytokines such as IL-18 and IL-1β [166]. These cytokines further exacerbate oxidative stress and hypoxemia. Altogether, chronic inflammation contributes to every phase of cardiovascular disease development, from early onset to overt clinical complications [167,168]. Furthermore, in CRDs, inflammation acts as a persistent noxious stimulus that promotes tissue remodeling, exacerbates inflammatory responses, and induces immune dysregulation. COPD is characterized by persistent inflammation of the airways and lung parenchyma, which intensifies during acute exacerbations and is also linked to systemic inflammation. This inflammation occurs in response to inhaled irritant particles, such as cigarette smoke, which tend to deposit in the lungs and may activate surface macrophages and airway epithelial cells. These cells, in turn, release various chemotactic mediators that attract circulating neutrophils, monocytes, and lymphocytes into the lungs. Indeed, inflammation in COPD is marked by an increased presence of alveolar macrophages, neutrophils, and T-lymphocytes recruited from the bloodstream [169,170,171]. As far as is known, cardiovascular disease and COPD share various risk factors, including smoking, and overlapping mechanisms such as accelerated aging, making cardiovascular mortality more common than respiratory failure in COPD patients [172,173]. Many inflammatory mediators have been implicated in COPD as well as in cardiovascular problems, including lipids, free radicals, cytokines, chemokines, growth factors, and air pollution, and all these are able to activate NF-κB and the NLRP3 inflammasome, leading to an inflammatory response. Indeed, the incidence of major adverse cardiovascular events is 25% higher in patients with COPD compared to those without the condition [174]. Although shared risk factors such as age, smoking, and socioeconomic status contribute to this association, they do not fully explain it. Pathophysiological mechanisms such as hypoxemia, oxidative stress, and systemic inflammation are likely key contributors. In line with this, a systematic review by Chen et al. reported that individuals with COPD are at significantly higher risk of developing cardiovascular disease than the general population (OR 2.46, 95% CI 2.02–3.00) [100,175,176].
OSA patients also exhibit a chronic inflammatory phenotype characterized by oxidative stress and elevated levels of proinflammatory cytokines and adhesion molecules [177]. This inflammatory state is known to induce or exacerbate cardiovascular and metabolic comorbidities such as type 2 diabetes mellitus, dyslipidemia, atherosclerosis, and ischemic heart disease [18,178,179,180]. This “sterile” inflammation occurs in response to damage-associated molecular patterns (DAMPs), such as high-mobility group box 1 protein (HMGB1) and oxidized low-density lipoprotein (oxLDL) [181], often as a consequence of IH and oxidative stress [182,183]. These stimuli activate inflammatory pathways, including NF-κB and the NLRP3 inflammasome [128,184]. The inflammasome activation results in increased levels of the inflammatory cytokines IL-1β and IL-18 in combination with elevated release of the major trigger of coagulation cascade, tissue factor, enhancing systemic inflammation and coagulability [185,186]. Additionally, oxLDL potentiates NF-κB signaling and promotes foam cell formation, contributing to the progression of atherosclerosis [187]. Altogether, this network of IH, oxidative stress, and systemic inflammation fosters endothelial dysfunction and promotes cardiovascular complications in OSA patients.
4.3. Role of Oxidative Stress and Inflammation in Pulmonary Hypertension Associated with CRDs
Pulmonary hypertension (PH) is one of the most significant cardiovascular complications associated with CRDs. This condition is defined by an elevation in mean pulmonary arterial pressure exceeding 20 mmHg [188]. Group 3 PH, as classified by the World Symposium on Pulmonary Hypertension, encompasses PH related to CRDs and represents one of the most prevalent subtypes. It includes conditions with diverse etiologies such as COPD, interstitial lung diseases (e.g., pulmonary fibrosis), non-parenchymal restrictive disorders (e.g., OSA), hypoxia without underlying lung disease (e.g., high-altitude exposure), and developmental lung conditions (e.g., bronchopulmonary dysplasia) [189].
The development of PH in the context of CRDs is associated with increased morbidity, reduced quality of life, and poorer prognosis [190]. The underlying pathophysiology is complex and multifactorial, involving disease-specific mechanisms [191]. Hypoxia represents a major common factor in these conditions and much of the insight concerning the molecular mechanisms responsible for pulmonary vasculopathy in CRDs comes from experimental animal models with exposure to hypoxia [192]. Acute hypoxia induces a compensatory response known as hypoxic pulmonary vasoconstriction (HPV), aimed at optimizing ventilation–perfusion matching. However, sustained hypoxia promotes persistent HPV and vascular remodeling, resulting in elevated pulmonary vascular resistance and PH development [192,193]. The involvement of ROS in HPV has been widely documented, although some controversy remains regarding whether ROS levels increase or decrease under hypoxic conditions. Current evidence predominantly supports an increase in ROS, which activates multiple downstream effectors that elevate intracellular calcium and induce contraction of PASMCs [194,195,196,197]. In chronic hypoxia, persistently elevated ROS levels are well-established contributors to pulmonary arterial remodeling and enhanced pulmonary vasoconstriction, as observed in chronic-hypoxia-induced PH [198,199,200]. Similar mechanisms have also been identified in chronic IH, a widely used model for OSA-associated PH [201,202].
Mitochondria-derived ROS are considered pivotal in HPV [196,197,203], although other sources, such as NOX, may amplify the response [194,195]. Mitochondrial respiratory chain [204] and members of the NOX family [205], particularly the isoforms NOX2 and NOX4, are considered key sources of this hypoxic ROS generation. NOX2 is primarily expressed in phagocytes and has been associated with inflammatory responses [206], while NOX4 is constitutively active in vascular endothelial and smooth muscle cells and is implicated in vascular remodeling under oxidative stress conditions [207,208]. Intriguingly, there is also evidence of decreased ROS in PASMCs after chronic hypoxia [197]. Excessive oxidative stress has also been postulated as a key driver of lung fibrosis [209,210]. Indeed, TGF-β, a key mediator of pulmonary fibrosis and associated pulmonary vasculopathy, enhances the production of ROS through mitochondrial pathways and NOX activation [209]. Conversely, ROS can further stimulate the release of TGF-β from alveolar cells, creating a self-amplifying cycle [211]. Moreover, the protective effects of phosphodiesterase type 5A (PDE5A) inhibition in bleomycin-induced pulmonary fibrosis and PH were associated with inhibition of ROS formation [212]. In the perinatal period, ROS-mediated injury to the developing lung is also recognized as a critical factor in the pathogenesis of neonatal pulmonary vascular diseases, including persistent pulmonary hypertension of the newborn and bronchopulmonary dysplasia [195,213,214,215].
As already mentioned, inflammation is increasingly recognized as a key contributor to pulmonary vascular disease and plays a significant role in the pathogenesis of various forms of PH, including those associated with CRDs [191,193,216]. Many immune cells, and multiple cytokines, adhesion molecules, and differentiation factors, have been suggested to participate in the complex inflammatory responses promoting the pulmonary vascular remodeling characteristic of PH [191,193,217]. Thus, elevated levels of inflammatory cytokines and chemokines, including TNF-α, IL-1β, IL-6, Il-8, IL-17, IL-21, CCl2, and CX3CL1, among others, originating from a variety of inflammatory cell types are considered hallmarks of CRD-associated PH [201,218,219,220,221,222]. Extensive evidence highlights monocytes and macrophages as central mediators of lung inflammation and vascular remodeling in hypoxia-induced PH [221,223,224,225]. Upon hypoxic exposure, circulatory monocytes are recruited into the lungs where they differentiate into interstitial macrophages. Both non-classical [221,223] and classical monocytes [222,226] have been proposed as precursors of recruited interstitial macrophages. The expression of TSP-1 in these recruited interstitial macrophages was shown to activate TGF-β, resulting in enhanced pulmonary vasoconstriction and remodeling [222,224,226]. There is also the involvement of lymphoid cells in hypoxia-induced PH. Thus, CD4+ T cells, specifically Th17 cells, have been demonstrated to contribute to the development of hypoxia-induced PH in murine models [220,227] and their inhibition lowers right ventricle systolic pressure in established PH [227]. Similarly, Th17 cells have been suggested to play a detrimental role in PH secondary to lung fibrosis [201].
In addition to hypoxia, other pathogenic environmental factors involved in the development of respiratory diseases such as cigarette smoke, hyperoxia, and others (air pollution, chemicals, and toxins such as asbestos or heavy metals) may produce pulmonary vascular injury through increased ROS or inflammation. Thus, recent evidence suggests that cigarette smoke, a major risk factor in COPD, may directly impact the pulmonary vasculature, independent of low oxygen levels [228]. This is particularly relevant in early-stage COPD and even in smokers without airway obstruction. In advanced COPD, the mean pulmonary artery pressure shows only a weak correlation with hypoxemia and no correlation with airflow limitation [229]. Additionally, vascular changes such as medial thickening are seen in mild COPD and in smokers without airway obstruction [230]. Furthermore, animal studies show that cigarette-smoke-induced PH can occur before emphysema development and without hypoxia [231]. These findings point to early pulmonary artery involvement, likely driven by cigarette smoke itself, even before hypoxia plays a role. This is critical, as CS can compromise epithelial barriers, allowing toxic substances to reach and damage nearby blood vessels [232]. Cigarette smoke also induces pulmonary vascular dysfunction through the secretion of inflammatory molecules and increased ROS production [228]. In particular, cigarette smoke promotes the production of mitochondria-derived ROS, affecting the redox state of the soluble guanylyl cyclase, which severely impairs the NO-soluble guanylyl cyclase signaling pathway in the pulmonary vasculature [8]. Using a murine model of nicotine/hypoxia coexposure to better illustrate the pathological conditions of COPD-PH, Truong et al. have recently identified a major role of Rieske iron–sulfur protein (RISP)-mediated mitochondrial ROS and NF-κB-dependent inflammation in PH development [233]. Other sources of ROS such as xanthine oxidoreductase have also been shown to contribute to the harmful effects of cigarette smoke in pulmonary vascular endothelial cells [234]. In addition, cigarette smoke has been shown to promote dysregulated inflammatory signaling due to increased recruitment of inflammatory cells or ROS-induced inflammation in pulmonary arteries [228,233,235]. An excess of ROS production is also recognized as a critical factor for hyperoxia-induced pulmonary vascular damage in several conditions such as respiratory distress syndrome [236] or pulmonary bronchodysplasia [237].
Many efforts have been made to target oxidative stress and the inflammatory component underlying the pulmonary vascular injury linked to CRDs (Table 1). Thus, targeting ROS with unspecific antioxidants such as NAC [238] or with strategies to specifically reduce mitochondrial ROS [198,199,204] show protective effects in chronic hypoxia-PH. Similarly, antioxidants such as the glutathione peroxidase mimetic ebselen [239] or the NOX inhibitor apocynin [240] show protective effects on vascular dysfunction due to cigarette smoke exposure. Other strategies such as dietary antioxidants, Nrf2 activators, or nitrative stress inhibitors have shown benefit in animal models of COPD [241] and might be useful in ameliorating the associated pulmonary vascular dysfunction. Alternatively, numerous intervention strategies for inflammation have been proven as effective in attenuating hypoxic PH. These include, among others: glucocorticoids [222], blocking IL-6 [218,219,220], NF-κB activation [204], CX3CR1 [221], the SDF-1/CXCR4 axis [242], the CCR2-CCL2 axis [222], inhibiting macrophages [220], switching macrophage polarity toward an anti-inflammatory phenotype [225], or adoptive transfer of M2 regulatory macrophages [243].

Table 1.
Effects of treatment with antioxidants or anti-inflammatory agents on pulmonary vascular damage associated with CRD in preclinical studies.
Despite substantial evidence supporting the pathogenic role of oxidative stress and inflammation in pulmonary-vascular-damage-associated CRDs, and the demonstrated benefits of therapies targeting these pathways in preclinical studies, neither antioxidants nor anti-inflammatory drugs are indicated in PH associated with CRD. So far, clinical trials with antioxidants in COPD have shown disappointing results due to the lack of proven efficacy or occurrence of adverse effects [241]. The development of more effective antioxidants or anti-inflammatory drugs with limited side effects confirmed in clinical trials will determine whether these strategies can be successfully implemented in clinical practice.
5. Relevance of AI in Respiratory Medicine
Machine learning (ML) is a subfield of artificial intelligence that enables computer systems to learn patterns from data and make predictions or decisions without being explicitly programmed. At the core of any ML system is a model, typically represented by a function h, which maps an input x (e.g., patient data, medical images, or sensor readings) to an output (e.g., a diagnosis, risk score, or recommended treatment). This function h is not predefined; rather, it is learned from a dataset during a training phase, where the model adjusts its internal parameters to approximate the underlying relationship between inputs and outputs.
In medicine, the application of ML has grown exponentially, evolving from traditional methods to advanced deep learning (DL) models. Traditional ML models learn patterns from clinical features such as symptoms, spirometry values, or imaging metrics, demonstrating effectiveness in both diagnostic and prognostic predictions for conditions such as COPD and OSA, and more modern DL models can learn from images, audio, or complex tabular data [41,42,247,248]. Also, so-called foundational models have been important to empower AI applications in many fields of medicine. Foundational models are mainly based on the Transformer architecture [249] that are trained with huge amounts of data for general purpose tasks and that can be refined or transferred to specific tasks with a more reasonable amount of data and computing resources. This architecture allows the training of large AI systems integrating data from multiple sources (tabular, text, image, and audio) enhancing the human–machine interaction with specialized chatbots [250,251], generative models capable of synthesizing data with to train and evaluate AI systems [252,253], and a huge potential for analysis and exploration of molecular pathways in order to improve diagnosis and prognosis and also to find comorbidities [254,255,256]. Moreover, the role of AI in personalized and precision medicine is paramount, and its value has been increasing in recent times [257,258,259].
The application of AI in respiratory medicine faces several challenges in the coming years. Table 2 outlines some of these key challenges and highlights their importance for advancing the field.
Table 2.
Main challenges of AI systems applied to respiratory medicine.
5.1. Databases for Respiratory AI Research
ML applications in COPD and OSA research have been made possible by recent publications of extensive clinical datasets. For COPD, some of the most prominent cohorts include COPDGene [267], ECLIPSE [268], SPIROMICS [269], and CanCOLD [270], which offer clinical data, genetic profiles, spirometry results, biomarkers, and chest computed tomography (CT) imaging from patients with and without COPD, covering diverse stages of disease severity. Additionally, intensive care databases like MIMIC-III [260], MIMIC-IV [261], eICU [262], and HiRID [263] provide detailed respiratory data specific to patients with critical respiratory conditions, including those with acute exacerbations of COPD [271]. In sleep apnea research, prominent databases, including Apnea-ECG [272], UCD Sleep Apnea Database [273], and SHHS [274], offer detailed polysomnographic recordings, ECG, EEG, EOG signals, apnea event annotations, and extensive longitudinal clinical data, enabling a wide range of analytical approaches.
5.2. Predictive Machine Learning for Diagnosis and Prognosis
Regarding traditional AI methods, temporal physiological signals from portable sensors have proven effective in diagnosing and predicting outcomes in respiratory diseases. In COPD, models like XGBoost, RF, and SVM have achieved strong performance using data from capnography, air quality monitors, remote spirometry, and electronic stethoscopes [275,276,277]. For OSA, inputs from polysomnography (e.g., ECG, airflow, muscle activity), as well as voice and tracheal sounds [42,248], have been effectively analyzed using SVM, RF, and LR models [219,220,221]. Radiomics features from chest CT scans have shown strong diagnostic and prognostic value in COPD and OSA, particularly when analyzed with ML models like LR, SVM, and RF, outperforming demographic models and basic emphysema measures [278,279,280]. Radiomics has also helped distinguish COPD from asthma and predict disease progression, especially when combined with clinical data [281,282], sometimes surpassing CNN-based methods [283]. Tabular clinical data remains widely used with ML models such as XGBoost, RF, and LightGBM to predict outcomes like respiratory failure, ventilation need, mortality, and exacerbations in COPD, often integrating demographics, comorbidities, and test results [284,285,286]. In OSA, clinical and anthropometric features have enabled accurate prediction of the apnea–hypopnea index and phenotype classification using neural networks, SVMs, RF, and LR models [42,248,287]. In parallel, there is growing interest in enhancing these models by incorporating molecular biomarkers to enrich predictive capabilities. In recent years, various studies have highlighted the role of molecular factors such as HIF1A (the gene encoding HIF-1α), markers of chronic oxidative stress (e.g., TXN, EGR1, CDKN1A), receptors associated with persistent inflammation such as TLR2, and ADM, particularly its mid-regional form, in the pathophysiology of both COPD and OSA. Their relevance in pulmonary diseases has been validated using classical statistical analyses (ROC curves, regression, etc.) as well as ML algorithms, demonstrating their potential as biomarkers for AI-based models [288,289,290,291,292,293]. Currently, emerging studies are exploring the integration of these features into ML and DL models to enhance risk stratification and support clinical decision making [294,295]. Nevertheless, significant limitations persist due to the scarcity of standardized molecular data across cohorts. Recent DL advances have enabled models like CNNs, RNNs, and transformers to achieve expert-level performance in clinical tasks [296,297,298,299,300,301], with some gaining regulatory approval [296]. In COPD and OSA, DL models using temporal signals (e.g., lung sounds, ECG, PSG) have shown high accuracy for classification and event detection [302,303,304,305,306,307,308,309,310,311,312,313], while CNNs and GCNs applied to chest CT and X-rays have outperformed traditional ML in diagnosis, severity staging, and mortality prediction [279,297,314,315,316,317,318,319,320,321]. For OSA, CNN-based methods have accurately detected apnea using various image-based signal representations [307,308,310,311,312,322,323]. Although DL has also been used with clinical tabular data via MLPs and attention-based models like HiTANet [324,325], traditional ML remains competitive, especially in low-dimensional, standardized datasets [42,247,248].
5.3. Foundational Models for Diagnosis and Prognosis
Foundational models are large, Transformer-based neural networks trained with self-supervised learning on massive datasets, enabling adaptation to downstream tasks through fine-tuning [298]. Unlike specialized models tailored to specific tasks, foundational or generalist models are designed to handle a wide range of inputs and tasks, representing a shift toward more versatile AI in medicine. Notable examples include ChatGPT 3.5 (used in clinical text generation and exams), CLIP [326], and BioBERT [327] for biomedical text mining. In respiratory medicine, foundational models offer a promising path for integrating multimodal data such as text, images, and biosignals in unified diagnostic frameworks.
In audio analysis, the OPERA framework [328] introduced three models—OPERA-CT, OPERA-CE, and OPERA-GT—trained on over 136,000 audio samples and benchmarked across 19 respiratory health tasks (e.g., COPD detection, pulmonary function estimation), performing competitively against baselines like OpenSMILE [329], VGGish [330], AudioMAE [331], and CLAP [332]. In medical imaging, M3FM [333] is a general-purpose model trained on over 128,000 3D CT scans and multimodal data using a question–answering framework. It significantly outperformed prior models such as Sybil [334], Tri2D-Net [335], and GPT-4o, especially in tasks like emphysema and lung cancer detection. For clinical text and EHR analysis, BEHRT [250] was trained on data from ~1.6 million patients in the UK CPRD database [336]. It predicted the onset of 301 conditions (including COPD), outperforming previous models such as Deepr [337] and RETAIN (RNN with reverse-time attention) [338], particularly in first-incidence disease prediction.
Foundational AI models, particularly those based on Transformer architectures, have demonstrated transformative potential in the integration and interpretation of high-dimensional molecular data for the diagnosis and prognosis of complex diseases, including respiratory disorders. These models are uniquely suited to handling multiomics inputs—such as gene expression, proteomics, and epigenetic profiles—alongside clinical and imaging data, enabling the discovery of novel molecular patterns associated with disease phenotypes and progression [254,255,256]. In respiratory diseases such as COPD and obstructive sleep apnea (OSA), foundational models facilitate the identification of shared molecular pathways and stratify patients according to their genetic and inflammatory signatures, supporting early diagnosis and risk prediction. By leveraging self-supervised learning and transfer learning, these models can be adapted across datasets and conditions, improving generalizability in heterogeneous patient populations, including the exploration of comorbidities with other diseases. Furthermore, foundational models enhance the potential for precision and personalized medicine by predicting individual responses to therapy based on molecular profiles, thereby optimizing treatment decisions and improving outcomes in chronic respiratory conditions [41].
Together, these models demonstrate strong potential for unifying multimodal AI tools in chronic respiratory care, offering adaptability and improved performance across diverse tasks and data types.
5.4. Ethical, Legal, and Social Aspects of AI Respiratory Medicine
AI, particularly generative AI, is becoming an increasingly powerful tool in medicine, with the potential to function as a comprehensive assistant or even as an autonomous system in clinical environments. These models can handle multiple tasks with minimal adjustment, suggesting a future in which medical AI may play an integral role in therapeutic decision making. However, despite this promise, AI also presents considerable risks and challenges, especially when used in high-stakes contexts such as healthcare. The lack of explainability and biased results of AI-based systems are important concerns for both research community and society. Many AI models, and particularly generative AI models, are trained using mathematical optimization techniques, which can sometimes make the interpretation of their results challenging. AI models learn from data, which may underrepresent specific populations, leading to less accurate predictions for specific demographic groups [339]. This could perpetuate or even exacerbate existing disparities in healthcare delivery and outcomes. Regulators and practitioners increasingly demand interpretability or, at minimum, reliable confidence metrics before integrating AI into clinical workflows [298,340].
Beyond these, there are growing concerns about privacy, security, and accountability. AI systems trained on patient data must comply with strict data protection laws such as HIPAA and GDPR. They must also be resilient against adversarial attacks and ensure robust mechanisms for ethical and legal responsibility when outcomes are incorrect or harmful. To mitigate these risks, regulatory frameworks are evolving. The European Union has classified these systems as high-risk technologies, subject to strict compliance requirements under the EU Artificial Intelligence Act [341]. This Act, the world’s first comprehensive AI legislation, adopts a risk-based approach and mandates the following for medical AI applications (e.g., diagnostic tools for COPD or OSA): (1) adequate risk assessment and mitigation plans. (2) High-quality training datasets to minimize bias. (3) Logging capabilities to ensure traceability. (4) Comprehensive documentation for regulatory review. (5) Clear deployment instructions and information. (6) Human oversight mechanisms. (7) High levels of robustness, accuracy, and cybersecurity. In contrast, the United States currently lacks a comprehensive AI law. However, the FDA leads the regulation of AI- and ML-based medical devices. The FDA requires clinical evidence of safety and efficacy, typically through validation studies, before approving or authorizing such systems. Notably, the FDA has approved autonomous AI systems [296], marking an important precedent.
5.5. Integration of the AI in Practitioner’s Decisions
AI can significantly enhance clinical decision making but must remain a support tool, with healthcare professionals retaining ultimate responsibility. Decision theory provides a formal framework for managing uncertainty in high-stakes fields like oncology and respiratory diseases such as COPD and OSA [342]. For AI to aid practitioners effectively, its outputs must be interpretable—something enabled by probabilistic models that offer well-calibrated predictions and support personalized, cost-effective care [343]. However, the utility of these models depends on their calibration, as misaligned probabilities can lead to harmful over- or underestimations that may affect treatment, diagnosis, or prognosis [344,345]. Techniques like Platt scaling, isotonic regression, or Bayesian postprocessing are thus vital for ensuring reliable predictions, especially in imbalanced clinical data [346]. Integrating calibrated models into decision-theoretic frameworks enhances interpretability, trust, and actionable decision making in healthcare and offers practitioners an invaluable decision support tool.
6. Conclusions, Future Research, and Directions
COPD and OSA are both hypoxemic lung diseases that share overlapping pathophysiological mechanisms, including hypoxia-induced oxidative stress and systemic inflammation, which exacerbate disease progression and comorbidities. In OSA, recurrent apneic episodes during sleep result in IH, which in turn promotes excessive ROS production and systemic inflammation. Similarly, in COPD, chronic hypoxia and exposure to environmental insults such as cigarette smoke promote oxidative stress and a persistent inflammatory response. These common pathways contribute to endothelial dysfunction, vascular remodeling, and elevated cardiovascular risk. While antioxidant therapies offer promising strategies for mitigating oxidative-stress-related lung damage, a thorough understanding of the evolving role of ROS in pulmonary inflammation is essential for the development of more effective treatments. In addition, the high prevalence of pulmonary hypertension in both conditions underscores the need to elucidate shared mechanisms and identify new therapeutic strategies.
In this respect, understanding the role of HIFs in CRDs provides opportunities for targeted therapies. While initially adaptive, sustained HIF activation may exacerbate inflammation and tissue remodeling, suggesting a dual role that must be carefully navigated in therapeutic design. Despite the promise of antioxidant and anti-inflammatory treatments, their clinical translation remains limited due to the complexity of redox and inflammatory signaling networks, highlighting the need for integrative and multidisciplinary approaches.
In this context, AI has emerged as a powerful tool to manage the clinical and biological complexity of OSA and COPD. ML and DL models enable accurate phenotyping, risk stratification, and treatment personalization by integrating multimodal data, including clinical, imaging, and physiological signals. AI-based models have improved the classification of disease subtypes, predicted treatment responses, and consistency in diagnosis, particularly in pulmonary function testing and imaging analysis. Foundational models like OPERA and M3FM exemplify AI’s potential to unify heterogeneous data sources and bridge the gap between molecular insights and clinical decision making, including comorbidities with other diseases, and also the use of all the gathered knowledge in personalized medicine. Despite rapid progress, the integration of AI into routine clinical practice faces critical challenges, including limited availability of high-quality data, suboptimal model accuracy for diagnosis and prognosis, regulatory hurdles, and the complex task of embedding AI decision making into existing clinical workflows. In conclusion, the convergence of OSA and COPD pathophysiology reflects shared systemic consequences of hypoxia, oxidative stress, and inflammation. Advances in molecular understanding, particularly of HIF signaling, and the integration of AI into clinical workflows offer promising avenues for precision medicine. Interdisciplinary strategies combining biomedical research and computational innovation are essential to improve outcomes in these prevalent and complex respiratory diseases.
Author Contributions
All authors contributed to the conceptualization, draft writting, review and editing of the review. In addition, M.J.C was involved in image design and supervision. All authors have read and agreed to the published version of the manuscript.
Funding
Supporting grants: Redes en Biomedicina from Comunidad Autonoma of Madrid; INSPIRACM (P2022/BMD7224).
Conflicts of Interest
The authors have no interests related to the content of the manuscript to declare.
References
- GBD. 2019 Chronic Respiratory Diseases Collaborators. Global burden of chronic respiratory diseases and risk factors, 1990–2019: An update from the Global Burden of Disease Study 2019. eClinicalMedicine 2023, 59, 101936. [Google Scholar] [CrossRef]
- Soriano, J.B.; Alfageme, I.; Miravitlles, M.; de Lucas, P.; Soler-Cataluña, J.J.; García-Río, F.; Casanova, C.; Gonzalez-Moro, J.M.R.; Cosío, B.G.; Sanchez, G.; et al. Prevalence and Determinants of COPD in Spain: EPISCAN II. Arch. Bronconeumol. 2021, 57, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Safiri, S.; Carson-Chahhoud, K.; Noori, M.; Nejadghaderi, S.A.; Sullman, M.J.M.; Ahmadian Heris, J.; Ansarin, K.; Mansournia, M.A.; Collins, G.S.; Kolahi, A.A.; et al. Burden of chronic obstructive pulmonary disease and its attributable risk factors in 204 countries and territories, 1990-2019: Results from the Global Burden of Disease Study 2019. BMJ 2022, 378, e069679. [Google Scholar] [CrossRef] [PubMed]
- Benjafield, A.V.; Ayas, N.T.; Eastwood, P.R.; Heinzer, R.; Ip, M.S.M.; Morrell, M.J.; Nunez, C.M.; Patel, S.R.; Penzel, T.; Pépin, J.-L.; et al. Estimation of the global prevalence and burden of obstructive sleep apnoea: A literature-based analysis. Lancet Respir. Med. 2019, 7, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Bagdonas, E.; Raudoniute, J.; Bruzauskaite, I.; Aldonyte, R. Novel aspects of pathogenesis and regeneration mechanisms in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2015, 10, 995–1013. [Google Scholar]
- Duijts, L.; Reiss, I.K.; Brusselle, G.; de Jongste, J.C. Early origins of chronic obstructive lung diseases across the life course. Eur. J. Epidemiol. 2014, 29, 871–885. [Google Scholar] [CrossRef]
- Fonseca, W.; Lukacs, N.W.; Elesela, S.; Malinczak, C.-A. Role of ILC2 in Viral-Induced Lung Pathogenesis. Front. Immunol. 2021, 12, 675169. [Google Scholar] [CrossRef]
- Sevilla-Montero, J.; Munar-Rubert, O.; Pino-Fadón, J.; Aguilar-Latorre, C.; Villegas-Esguevillas, M.; Climent, B.; Agrò, M.; Choya-Foces, C.; Martínez-Ruiz, A.; Balsa, E.; et al. Cigarette smoke induces pulmonary arterial dysfunction through an imbalance in the redox status of the soluble guanylyl cyclase. Free Radic. Biol. Med. 2022, 193, 9–22. [Google Scholar] [CrossRef]
- MacNee, W. Pulmonary and systemic oxidant/antioxidant imbalance in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2005, 2, 50–60. [Google Scholar] [CrossRef]
- Garcia-Rio, F.; Miravitlles, M.; Soriano, J.B.; Muñoz, L.; Duran-Tauleria, E.; Sánchez, G.; Sobradillo, V.; Ancochea, J.; EPI-SCAN Steering Committee. Systemic inflammation in chronic obstructive pulmonary disease: A population-based study. Respir. Res. 2010, 11, 63. [Google Scholar] [CrossRef]
- Divo, M.J.; Casanova, C.; Marin, J.M.; Pinto-Plata, V.M.; de-Torres, J.P.; Zulueta, J.J.; Cabrera, C.; Zagaceta, J.; Sanchez-Salcedo, P.; Berto, J.; et al. COPD comorbidities network. Eur. Respir. J. 2015, 46, 640–650. [Google Scholar] [CrossRef]
- Heffernan, M.; Rutherford, S. The Intersection of Chronic Obstructive Pulmonary Disease and Cardiovascular Disease: Recent Insights in a Challenging Area. CJC Open 2025, 7, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.; Wrobel, J.P. Epidemiology and clinical impact of major comorbidities in patients with COPD. Int. J. Chronic Obstr. Pulm. Dis. 2014, 9, 871–888. [Google Scholar] [CrossRef]
- Santiago Díaz, C.; Medrano, F.J.; Muñoz-Rivas, N.; Castilla Guerra, L.; Alonso Ortiz, M.B.; En Representación de los Grupos de Trabajo de EPOC y de Riesgo Vascular de la Sociedad Española de Medicina Interna. COPD and cardiovascular risk. Clin. Investig. Arterioscler. 2025, 500757. [Google Scholar] [CrossRef] [PubMed]
- Polman, R.; Hurst, J.R.; Uysal, O.F.; Mandal, S.; Linz, D.; Simons, S. Cardiovascular disease and risk in COPD: A state of the art review. Expert Rev. Cardiovasc. Ther. 2024, 22, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Chen, P.; Chen, S.; Kong, X.; Ma, H.; Cao, C. Relationship between advanced lung cancer inflammation index and all-cause and cause-specific mortality among chronic inflammatory airway diseases patients: A population-based study. Front. Immunol. 2025, 16, 1585927. [Google Scholar] [CrossRef]
- Camiciottoli, G.; Bigazzi, F.; Magni, C.; Bonti, V.; Diciotti, S.; Bartolucci, M.; Mascalchi, M.; Pistolesi, M. Prevalence of comorbidities according to predominant phenotype and severity of chronic obstructive pulmonary disease. Int. J. Chronic Obstr. Pulm. Dis. 2016, 11, 2229–2236. [Google Scholar] [CrossRef]
- Gottlieb, D.J.; Punjabi, N.M. Diagnosis and Management of Obstructive Sleep Apnea: A Review. JAMA 2020, 323, 1389–1400. [Google Scholar] [CrossRef]
- Parra, O.; Arboix, A.; Montserrat, J.M.; Quintó, L.; Bechich, S.; García-Eroles, L. Sleep-related breathing disorders: Impact on mortality of cerebrovascular disease. Eur. Respir. J. 2004, 24, 267–272. [Google Scholar] [CrossRef]
- Martínez-García, M.A.; Soler-Cataluña, J.J.; Ejarque-Martínez, L.; Soriano, Y.; Román-Sánchez, P.; Illa, F.B.; Canal, J.M.M.; Durán-Cantolla, J. Continuous positive airway pressure treatment reduces mortality in patients with ischemic stroke and obstructive sleep apnea: A 5-year follow-up study. Am. J. Respir. Crit. Care Med. 2009, 180, 36–41. [Google Scholar] [CrossRef]
- Marin, J.M.; Carrizo, S.J.; Vicente, E.; Agusti, A.G.N. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: An observational study. Lancet 2005, 365, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Labarca, G.; Vena, D.; Hu, W.-H.; Esmaeili, N.; Gell, L.; Yang, H.C.; Wang, T.-Y.; Messineo, L.; Taranto-Montemurro, L.; Sofer, T.; et al. Sleep Apnea Physiological Burdens and Cardiovascular Morbidity and Mortality. Am. J. Respir. Crit. Care Med. 2023, 208, 802–813. [Google Scholar] [CrossRef]
- Martínez Cerón, E.; Casitas Mateos, R.; García-Río, F. Sleep apnea-hypopnea syndrome and type 2 diabetes. A reciprocal relationship? Arch. Bronconeumol. 2015, 51, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Nieto, F.J.; Young, T.B.; Lind, B.K.; Shahar, E.; Samet, J.M.; Redline, S.; D’Agostino, R.B.; Newman, A.B.; Lebowitz, M.D.; Pickering, T.G. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA 2000, 283, 1829–1836. [Google Scholar] [CrossRef]
- Loke, Y.K.; Brown, J.W.L.; Kwok, C.S.; Niruban, A.; Myint, P.K. Association of obstructive sleep apnea with risk of serious cardiovascular events: A systematic review and meta-analysis. Circ. Cardiovasc. Qual. Outcomes 2012, 5, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Cai, A.; Wang, L.; Zhou, Y. Hypertension and obstructive sleep apnea. Hypertens. Res. 2016, 39, 391–395. [Google Scholar] [CrossRef]
- Nakanishi, R.; Baskaran, L.; Gransar, H.; Budoff, M.J.; Achenbach, S.; Al-Mallah, M.; Cademartiri, F.; Callister, T.Q.; Chang, H.; Chinnaiyan, K.; et al. Relationship of Hypertension to Coronary Atherosclerosis and Cardiac Events in Patients With Coronary Computed Tomographic Angiography. Hypertension 2017, 70, 293–299. [Google Scholar] [CrossRef]
- Al-Mashhadi, R.H.; Al-Mashhadi, A.L.; Nasr, Z.P.; Mortensen, M.B.; Lewis, E.A.; Camafeita, E.; Ravlo, K.; Al-Mashhadi, Z.; Kjær, D.W.; Palmfeldt, J.; et al. Local Pressure Drives Low-Density Lipoprotein Accumulation and Coronary Atherosclerosis in Hypertensive Minipigs. J. Am. Coll. Cardiol. 2021, 77, 575–589. [Google Scholar] [CrossRef]
- Baguet, J.-P.; Hammer, L.; Lévy, P.; Pierre, H.; Launois, S.; Mallion, J.-M.; Peípin, J.-L. The severity of oxygen desaturation is predictive of carotid wall thickening and plaque occurrence. Chest 2005, 128, 3407–3412. [Google Scholar] [CrossRef]
- Savransky, V.; Nanayakkara, A.; Li, J.; Bevans, S.; Smith, P.L.; Rodriguez, A.; Polotsky, V.Y. Chronic intermittent hypoxia induces atherosclerosis. Am. J. Respir. Crit. Care Med. 2007, 175, 1290–1297. [Google Scholar] [CrossRef]
- Campos-Rodriguez, F.; Martinez-Garcia, M.A.; Martinez, M.; Duran-Cantolla, J.; Peña Mde, L.; Masdeu, M.J.; Gonzalez, M.; Campo Fd Gallego, I.; Marin, J.M.; Barbe, F.; et al. Association between obstructive sleep apnea and cancer incidence in a large multicenter Spanish cohort. Am. J. Respir. Crit. Care Med. 2013, 187, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garcia, M.A.; Campos-Rodriguez, F.; Almendros, I.; Garcia-Rio, F.; Sanchez-de-la-Torre, M.; Farre, R.; Gozal, D. Cancer and Sleep Apnea: Cutaneous Melanoma as a Case Study. Am. J. Respir. Crit. Care Med. 2019, 200, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Zapata, C.; Martínez-García, M.Á.; Campos-Rodríguez, F.; Sánchez de la Torre, M.; Nagore, E.; Martorell-Calatayud, A.; Blasco, L.H.; Vives, E.C.; Abad-Capa, J.; Montserrat, J.M.; et al. Soluble PD-L1 is a potential biomarker of cutaneous melanoma aggressiveness and metastasis in obstructive sleep apnoea patients. Eur. Respir. J. 2019, 53, 1801298. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Zapata, C.; Martínez-García, M.Á.; Díaz-García, E.; Jaureguizar, A.; Campos-Rodríguez, F.; Sánchez-de-la-Torre, M.; Nagore, E.; Martorell-Calatayud, A.; Blasco, L.H.; Pastor, E.; et al. Obesity attenuates the effect of sleep apnea on active TGF-ß1 levels and tumor aggressiveness in patients with melanoma. Sci. Rep. 2020, 10, 15528. [Google Scholar] [CrossRef]
- Cubillos-Zapata, C.; Martínez-García, M.Á.; Díaz-García, E.; Toledano, V.; Campos-Rodríguez, F.; Sánchez-de-la-Torre, M.; Nagore, E.; Martorell-Calatayud, A.; Hernández Blasco, L.; Pastor, E.; et al. Proangiogenic factor midkine is increased in melanoma patients with sleep apnea and induces tumor cell proliferation. FASEB J. 2020, 34, 16179–16190. [Google Scholar] [CrossRef]
- Olszewska, E.; Rogalska, J.; Brzóska, M.M. The Association of Oxidative Stress in the Uvular Mucosa with Obstructive Sleep Apnea Syndrome: A Clinical Study. J. Clin. Med. 2021, 10, 1132. [Google Scholar] [CrossRef]
- Sunwoo, B.Y.; Raphelson, J.R.; Malhotra, A. Chronic obstructive pulmonary disease and obstructive sleep apnea overlap: Who to treat and how? Expert Rev. Respir. Med. 2024, 18, 527–537. [Google Scholar] [CrossRef]
- Sanchez-Azofra, A.; Gu, W.; Masso-Silva, J.A.; Sanz-Rubio, D.; Marin-Oto, M.; Cubero, P.; Gil, A.V.; Moya, E.A.; Barnes, L.A.; Mesarwi, O.A.; et al. Inflammation biomarkers in OSA, chronic obstructive pulmonary disease, and chronic obstructive pulmonary disease/OSA overlap syndrome. J. Clin. Sleep Med. 2023, 19, 1447–1456. [Google Scholar] [CrossRef]
- Ratcliffe, P.J. Oxygen sensing and hypoxia signalling pathways in animals: The implications of physiology for cancer. J. Physiol. 2013, 591, 2027–2042. [Google Scholar] [CrossRef]
- Luo, Z.; Tian, M.; Yang, G.; Tan, Q.; Chen, Y.; Li, G.; Zhang, Q.; Li, Y.; Wan, P.; Wu, J. Hypoxia signaling in human health and diseases: Implications and prospects for therapeutics. Signal Transduct. Target. Ther. 2022, 7, 218. [Google Scholar] [CrossRef]
- Chen, Z.; Hao, J.; Sun, H.; Li, M.; Zhang, Y.; Qian, Q. Applications of digital health technologies and artificial intelligence algorithms in COPD: Systematic review. BMC Med. Inform. Decis. Mak. 2025, 25, 77. [Google Scholar] [CrossRef] [PubMed]
- Bazoukis, G.; Bollepalli, S.C.; Chung, C.T.; Li, X.; Tse, G.; Bartley, B.L.; Batool-Anwar, S.; Quan, S.F.; Armoundas, A.A. Application of artificial intelligence in the diagnosis of sleep apnea. J. Clin. Sleep Med. 2023, 19, 1337–1363. [Google Scholar] [CrossRef]
- Kent, B.D.; Mitchell, P.D.; McNicholas, W.T. Hypoxemia in patients with COPD: Cause, effects, and disease progression. Int. J. Chronic Obstr. Pulm. Dis. 2011, 6, 199–208. [Google Scholar]
- Martinez-Garcia, M.A.; Sánchez-de-la-Torre, M.; White, D.P.; Azarbarzin, A. Hypoxic Burden in Obstructive Sleep Apnea: Present and Future. Arch. Bronconeumol. 2023, 59, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Dewan, N.A.; Nieto, F.J.; Somers, V.K. Intermittent hypoxemia and OSA: Implications for comorbidities. Chest 2015, 147, 266–274. [Google Scholar] [CrossRef]
- López-Barneo, J.; González-Rodríguez, P.; Gao, L.; Fernández-Agüera, M.C.; Pardal, R.; Ortega-Sáenz, P. Oxygen sensing by the carotid body: Mechanisms and role in adaptation to hypoxia. Am. J. Physiol. Cell Physiol. 2016, 310, C629–C642. [Google Scholar] [CrossRef]
- Pescador, N.; Villar, D.; Cifuentes, D.; Garcia-Rocha, M.; Ortiz-Barahona, A.; Vazquez, S.; Ordoñez, A.; Cuevas, Y.; Saez-Morales, D.; Garcia-Bermejo, M.L.; et al. Hypoxia promotes glycogen accumulation through hypoxia inducible factor (HIF)-mediated induction of glycogen synthase 1. PLoS ONE 2010, 5, e9644. [Google Scholar] [CrossRef]
- Arias, C.F.; Acosta, F.J.; Bertocchini, F.; Fernández-Arias, C. Redefining the role of hypoxia-inducible factors (HIFs) in oxygen homeostasis. Commun. Biol. 2025, 8, 446. [Google Scholar] [CrossRef]
- Bishop, T.; Ratcliffe, P.J. Signaling hypoxia by hypoxia-inducible factor protein hydroxylases: A historical overview and future perspectives. Hypoxia 2014, 2, 197–213. [Google Scholar]
- Elvidge, G.P.; Glenny, L.; Appelhoff, R.J.; Ratcliffe, P.J.; Ragoussis, J.; Gleadle, J.M. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: The role of HIF-1alpha, HIF-2alpha, and other pathways. J. Biol. Chem. 2006, 281, 15215–15226. [Google Scholar] [CrossRef]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Shakir, D.; Batie, M.; Kwok, C.-S.; Kenneth, N.S.; Rocha, S. NF-κB Is a Central Regulator of Hypoxia-Induced Gene Expression. bioRxiv 2025. [Google Scholar] [CrossRef]
- Li, L.; Kang, H.; Zhang, Q.; D’Agati, V.D.; Al-Awqati, Q.; Lin, F. FoxO3 activation in hypoxic tubules prevents chronic kidney disease. J. Clin. Investig. 2019, 129, 2374–2389. [Google Scholar] [CrossRef]
- Jensen, K.S.; Binderup, T.; Jensen, K.T.; Therkelsen, I.; Borup, R.; Nilsson, E.; Multhaupt, H.; Bouchard, C.; Quistorff, B.; Kjaer, A.; et al. FoxO3A promotes metabolic adaptation to hypoxia by antagonizing Myc function. EMBO J. 2011, 30, 4554–4570. [Google Scholar] [CrossRef]
- Gustafsson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.; Ruas, J.L.; Poellinger, L.; Lendahl, U.; Bondesson, M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell 2005, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Laderoute, K.R.; Calaoagan, J.M.; Gustafson-Brown, C.; Knapp, A.M.; Li, G.-C.; Mendonca, H.L.; Ryan, H.E.; Wang, Z.; Johnson, R.S. The response of c-jun/AP-1 to chronic hypoxia is hypoxia-inducible factor 1 alpha dependent. Mol. Cell Biol. 2002, 22, 2515–2523. [Google Scholar] [CrossRef]
- Michiels, C.; Minet, E.; Michel, G.; Mottet, D.; Piret, J.P.; Raes, M. HIF-1 and AP-1 cooperate to increase gene expression in hypoxia: Role of MAP kinases. IUBMB Life 2001, 52, 49–53. [Google Scholar] [CrossRef]
- Villar, D.; Ortiz-Barahona, A.; Gómez-Maldonado, L.; Pescador, N.; Sánchez-Cabo, F.; Hackl, H.; Rodriguez, B.A.T.; Trajanoski, Z.; Dopazo, A.; Huang, T.H.M.; et al. Cooperativity of stress-responsive transcription factors in core hypoxia-inducible factor binding regions. PLoS ONE 2012, 7, e45708. [Google Scholar] [CrossRef]
- Ma, B.; Chen, Y.; Chen, L.; Cheng, H.; Mu, C.; Li, J.; Gao, R.; Zhou, C.; Cao, L.; Liu, J.; et al. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nat. Cell Biol. 2015, 17, 95–103. [Google Scholar] [CrossRef]
- Wu, Z.; Zhu, L.; Nie, X.; Wei, L.; Qi, Y. USP15 promotes pulmonary vascular remodeling in pulmonary hypertension in a YAP1/TAZ-dependent manner. Exp. Mol. Med. 2023, 55, 183–195. [Google Scholar] [CrossRef]
- Chen, J.; Lockett, A.; Zhao, S.; Huang, L.S.; Wang, Y.; Wu, W.; Tang, M.; Haider, S.; Rendon, D.V.; Khan, R.; et al. Sphingosine Kinase 1 Deficiency in Smooth Muscle Cells Protects against Hypoxia-Mediated Pulmonary Hypertension via YAP1 Signaling. Int. J. Mol. Sci. 2022, 23, 14516. [Google Scholar] [CrossRef]
- Islam, R.; Hong, Z. YAP/TAZ as mechanobiological signaling pathway in cardiovascular physiological regulation and pathogenesis. Mechanobiol. Med. 2024, 2, 100085. [Google Scholar] [CrossRef] [PubMed]
- Sermeus, A.; Michiels, C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011, 2, e164. [Google Scholar] [CrossRef] [PubMed]
- Potteti, H.R.; Noone, P.M.; Tamatam, C.R.; Ankireddy, A.; Noel, S.; Rabb, H.; Reddy, S.P. Nrf2 mediates hypoxia-inducible HIF1α activation in kidney tubular epithelial cells. Am. J. Physiol. Ren. Physiol. 2021, 320, F464–F474. [Google Scholar] [CrossRef]
- Bae, T.; Hallis, S.P.; Kwak, M.-K. Hypoxia, oxidative stress, and the interplay of HIFs and NRF2 signaling in cancer. Exp. Mol. Med. 2024, 56, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Batie, M.; Frost, J.; Frost, M.; Wilson, J.W.; Schofield, P.; Rocha, S. Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science 2019, 363, 1222–1226. [Google Scholar] [CrossRef]
- Chakraborty, A.A.; Laukka, T.; Myllykoski, M.; Ringel, A.E.; Booker, M.A.; Tolstorukov, M.Y.; Meng, Y.J.; Meier, S.R.; Jennings, R.B.; Creech, A.L.; et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science 2019, 363, 1217–1222. [Google Scholar] [CrossRef]
- Kindrick, J.D.; Mole, D.R. Hypoxic Regulation of Gene Transcription and Chromatin: Cause and Effect. Int. J. Mol. Sci. 2020, 21, 8320. [Google Scholar] [CrossRef]
- Batie, M.; Rocha, S. Gene transcription and chromatin regulation in hypoxia. Biochem. Soc. Trans. 2020, 48, 1121–1128. [Google Scholar] [CrossRef]
- Romero-Peralta, S.; García-Rio, F.; Resano Barrio, P.; Viejo-Ayuso, E.; Izquierdo, J.L.; Sabroso, R.; Castelao, J.; Francés, J.F.; Mediano, O. Defining the Heterogeneity of Sleep Apnea Syndrome: A Cluster Analysis With Implications for Patient Management. Arch. Bronconeumol. 2022, 58, 125–134. [Google Scholar] [CrossRef]
- Yu, A.Y.; Shimoda, L.A.; Iyer, N.V.; Huso, D.L.; Sun, X.; McWilliams, R.; Beaty, T.; Sham, J.S.; Wiener, C.M.; Sylvester, J.T.; et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J. Clin. Investig. 1999, 103, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Brusselmans, K.; Compernolle, V.; Tjwa, M.; Wiesener, M.S.; Maxwell, P.H.; Collen, D.; Carmeliet, P. Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J. Clin. Investig. 2003, 111, 1519–1527. [Google Scholar] [CrossRef]
- Sheikh, A.Q.; Saddouk, F.Z.; Ntokou, A.; Mazurek, R.; Greif, D.M. Cell Autonomous and Non-cell Autonomous Regulation of SMC Progenitors in Pulmonary Hypertension. Cell Rep. 2018, 23, 1152–1165. [Google Scholar] [CrossRef] [PubMed]
- Kapitsinou, P.P.; Rajendran, G.; Astleford, L.; Michael, M.; Schonfeld, M.P.; Fields, T.; Shay, S.; French, J.L.; West, J.; Haase, V.H. The Endothelial Prolyl-4-Hydroxylase Domain 2/Hypoxia-Inducible Factor 2 Axis Regulates Pulmonary Artery Pressure in Mice. Mol. Cell Biol. 2016, 36, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Pullamsetti, S.S.; Mamazhakypov, A.; Weissmann, N.; Seeger, W.; Savai, R. Hypoxia-inducible factor signaling in pulmonary hypertension. J. Clin. Investig. 2020, 130, 5638–5651. [Google Scholar] [CrossRef]
- Hu, C.-J.; Poth, J.M.; Zhang, H.; Flockton, A.; Laux, A.; Kumar, S.; McKeon, B.; Mouradian, G.; Li, M.; Riddle, S.; et al. Suppression of HIF2 signalling attenuates the initiation of hypoxia-induced pulmonary hypertension. Eur. Respir. J. 2019, 54, 1900378. [Google Scholar] [CrossRef]
- Dai, Z.; Li, M.; Wharton, J.; Zhu, M.M.; Zhao, Y.-Y. Prolyl-4 Hydroxylase 2 (PHD2) Deficiency in Endothelial Cells and Hematopoietic Cells Induces Obliterative Vascular Remodeling and Severe Pulmonary Arterial Hypertension in Mice and Humans Through Hypoxia-Inducible Factor-2α. Circulation 2016, 133, 2447–2458. [Google Scholar] [CrossRef]
- Cowburn, A.S.; Crosby, A.; Macias, D.; Branco, C.; Colaço, R.D.D.R.; Southwood, M.; Toshner, M.; Crotty Alexander, L.E.; Morrell, N.W.; Chilvers, E.R.; et al. HIF2α-arginase axis is essential for the development of pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2016, 113, 8801–8806. [Google Scholar] [CrossRef]
- Wang, S.; Zeng, H.; Xie, X.-J.; Tao, Y.-K.; He, X.; Roman, R.J.; Aschner, J.L.; Chen, J.-X. Loss of prolyl hydroxylase domain protein 2 in vascular endothelium increases pericyte coverage and promotes pulmonary arterial remodeling. Oncotarget 2016, 7, 58848–58861. [Google Scholar] [CrossRef]
- Gale, D.P.; Harten, S.K.; Reid, C.D.; Tuddenham, E.G.; Maxwell, P.H. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 alpha mutation. Blood 2008, 112, 919–921. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Kerestes, H.; Percy, M.J.; Pietrofesa, R.; Chen, L.; Khurana, T.S.; Christofidou-Solomidou, M.; Lappin, T.R.; Lee, F.S. Erythrocytosis and pulmonary hypertension in a mouse model of human HIF2A gain of function mutation. J. Biol. Chem. 2013, 288, 17134–17144. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Urrutia, A.A.; Aragonés, J. HIF Oxygen Sensing Pathways in Lung Biology. Biomedicines 2018, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Labrousse-Arias, D.; Castillo-González, R.; Rogers, N.M.; Torres-Capelli, M.; Barreira, B.; Aragonés, J.; Cogolludo, A.; Isenberg, J.S.; Calzada, M.J. HIF-2α-mediated induction of pulmonary thrombospondin-1 contributes to hypoxia-driven vascular remodelling and vasoconstriction. Cardiovasc. Res. 2016, 109, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Babicheva, A.; McDermott, K.M.; Gu, Y.; Ayon, R.J.; Song, S.; Wang, Z.; Gupta, A.; Zhou, T.; Sun, X.; et al. Endothelial HIF-2α contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L256–L275. [Google Scholar]
- Sheikh, A.Q.; Misra, A.; Rosas, I.O.; Adams, R.H.; Greif, D.M. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci. Transl. Med. 2015, 7, 308ra159. [Google Scholar] [CrossRef] [PubMed]
- Pasupneti, S.; Tian, W.; Tu, A.B.; Dahms, P.; Granucci, E.; Gandjeva, A.; Xiang, M.; Butcher, E.C.; Semenza, G.L.; Tuder, R.M.; et al. Endothelial HIF-2α as a Key Endogenous Mediator Preventing Emphysema. Am. J. Respir. Crit. Care Med. 2020, 202, 983–995. [Google Scholar] [CrossRef]
- Yoo, S.; Takikawa, S.; Geraghty, P.; Argmann, C.; Campbell, J.; Lin, L.; Huang, T.; Tu, Z.; Feronjy, R.; Spira, A.; et al. Integrative analysis of DNA methylation and gene expression data identifies EPAS1 as a key regulator of COPD. PLoS Genet. 2015, 11, e1004898. [Google Scholar] [CrossRef]
- Prabhakar, N.R.; Peng, Y.-J.; Nanduri, J. Hypoxia-inducible factors and obstructive sleep apnea. J. Clin. Investig. 2020, 130, 5042–5051. [Google Scholar] [CrossRef]
- Yuan, G.; Khan, S.A.; Luo, W.; Nanduri, J.; Semenza, G.L.; Prabhakar, N.R. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J. Cell. Physiol. 2011, 226, 2925–2933. [Google Scholar] [CrossRef]
- Nanduri, J.; Wang, N.; Yuan, G.; Khan, S.A.; Souvannakitti, D.; Peng, Y.-J.; Kumar, G.K.; Garcia, J.A.; Prabhakar, N.R. Intermittent hypoxia degrades HIF-2alpha via calpains resulting in oxidative stress: Implications for recurrent apnea-induced morbidities. Proc. Natl. Acad. Sci. USA 2009, 106, 1199–1204. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, Y.; Dong, Z.; Jin, M.; Lu, Y.; Xu, M.; Pan, H.; Zhou, G.; Xiao, M. HIF-1α mediates hypertension and vascular remodeling in sleep apnea via hippo-YAP pathway activation. Mol. Med. 2024, 30, 281. [Google Scholar] [CrossRef] [PubMed]
- Torres-Capelli, M.; Marsboom, G.; Li, Q.O.Y.; Tello, D.; Rodriguez, F.M.; Alonso, T.; Sanchez-Madrid, F.; García-Rio, F.; Ancochea, J.; Aragonés, J. Role Of Hif2α Oxygen Sensing Pathway In Bronchial Epithelial Club Cell Proliferation. Sci. Rep. 2016, 6, 25357. [Google Scholar] [CrossRef]
- Woodruff, P.G.; Agusti, A.; Roche, N.; Singh, D.; Martinez, F.J. Current concepts in targeting chronic obstructive pulmonary disease pharmacotherapy: Making progress towards personalised management. Lancet 2015, 385, 1789–1798. [Google Scholar] [CrossRef]
- Han, J.; Wan, Q.; Seo, G.-Y.; Kim, K.; El Baghdady, S.; Lee, J.H.; Kronenberg, M.; Liu, Y.-C. Hypoxia induces adrenomedullin from lung epithelia, stimulating ILC2 inflammation and immunity. J. Exp. Med. 2022, 219, e20211985. [Google Scholar] [CrossRef] [PubMed]
- Flayer, C.H.; Linderholm, A.L.; Ge, M.Q.; Juarez, M.; Franzi, L.; Tham, T.; Teuber, M.; Liao, S.Y.; Schivo, M.; Kuhn, B.; et al. COPD with elevated sputum group 2 innate lymphoid cells is characterized by severe disease. medRxiv 2024. [Google Scholar] [CrossRef]
- Meng, D.-Q.; Li, X.-J.; Song, X.-Y.; Xin, J.-B.; Yang, W.-B. Diagnostic and prognostic value of plasma adrenomedullin in COPD exacerbation. Respir. Care 2014, 59, 1542–1549. [Google Scholar] [CrossRef]
- Stolz, D.; Christ-Crain, M.; Morgenthaler, N.G.; Miedinger, D.; Leuppi, J.; Müller, C.; Bingisser, R.; Struck, J.; Müller, B.; Tamm, M. Plasma pro-adrenomedullin but not plasma pro-endothelin predicts survival in exacerbations of COPD. Chest 2008, 134, 263–272. [Google Scholar] [CrossRef]
- Ryan, S.; Taylor, C.T.; McNicholas, W.T. Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation 2005, 112, 2660–2667. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Kent, B.D.; Crinion, S.J.; McNicholas, W.T.; Ryan, S. Human adipocytes are highly sensitive to intermittent hypoxia induced NF-kappaB activity and subsequent inflammatory gene expression. Biochem. Biophys. Res. Commun. 2014, 447, 660–665. [Google Scholar] [CrossRef]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Förster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 2003, 112, 645–657. [Google Scholar] [CrossRef]
- Takeda, N.; O’Dea, E.L.; Doedens, A.; Kim, J.; Weidemann, A.; Stockmann, C.; Asagiri, M.; Simon, M.C.; Hoffmann, A.; Johnson, R.S. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010, 24, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, Q.; Li, D.; Li, J.; Aki, D.; Liu, Y.-C. The E3 ligase VHL controls alveolar macrophage function via metabolic-epigenetic regulation. J. Exp. Med. 2018, 215, 3180–3193. [Google Scholar] [CrossRef]
- Izquierdo, H.M.; Brandi, P.; Gómez, M.-J.; Conde-Garrosa, R.; Priego, E.; Enamorado, M.; Martínez-Cano, S.; Sánchez, I.; Conejero, L.; Jimenez-Carretero, D.; et al. Von Hippel-Lindau Protein Is Required for Optimal Alveolar Macrophage Terminal Differentiation, Self-Renewal, and Function. Cell Rep. 2018, 24, 1738–1746. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhao, X.; Xiao, C.; Xiong, G.; Ye, X.; Li, L.; Fang, Y.; Chen, H.; Yang, W.; Du, X. The role of lung macrophages in chronic obstructive pulmonary disease. Respir. Med. 2022, 205, 107035. [Google Scholar] [CrossRef] [PubMed]
- Meliante, P.G.; Zoccali, F.; Cascone, F.; Di Stefano, V.; Greco, A.; de Vincentiis, M.; Petrella, C.; Fiore, M.; Minni, A.; Barbato, C. Molecular Pathology, Oxidative Stress, and Biomarkers in Obstructive Sleep Apnea. Int. J. Mol. Sci. 2023, 24, 5478. [Google Scholar] [CrossRef]
- Lavalle, S.; Masiello, E.; Iannella, G.; Magliulo, G.; Pace, A.; Lechien, J.R.; Calvo-Henriquez, C.; Cocuzza, S.; Parisi, F.M.; Favier, V.; et al. Unraveling the Complexities of Oxidative Stress and Inflammation Biomarkers in Obstructive Sleep Apnea Syndrome: A Comprehensive Review. Life 2024, 14, 425. [Google Scholar] [CrossRef]
- Antosova, M.; Mokra, D.; Pepucha, L.; Plevkova, J.; Buday, T.; Sterusky, M.; Bencova, A. Physiology of nitric oxide in the respiratory system. Physiol. Res. 2017, 66, S159–S172. [Google Scholar] [CrossRef]
- Bernard, K.; Hecker, L.; Luckhardt, T.R.; Cheng, G.; Thannickal, V.J. NADPH oxidases in lung health and disease. Antioxid. Redox Signal. 2014, 20, 2838–2853. [Google Scholar] [CrossRef]
- Al Ghouleh, I.; Khoo, N.K.H.; Knaus, U.G.; Griendling, K.K.; Touyz, R.M.; Thannickal, V.J.; Barchowsky, A.; Nauseef, W.M.; Kelley, E.E.; Bauer, P.M.; et al. Oxidases and peroxidases in cardiovascular and lung disease: New concepts in reactive oxygen species signaling. Free Radic. Biol. Med. 2011, 51, 1271–1288. [Google Scholar] [CrossRef]
- Chan, S.M.H.; Selemidis, S.; Bozinovski, S.; Vlahos, R. Pathobiological mechanisms underlying metabolic syndrome (MetS) in chronic obstructive pulmonary disease (COPD): Clinical significance and therapeutic strategies. Pharmacol. Ther. 2019, 198, 160–188. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, W. Association between oxidative balance score and chronic obstructive pulmonary disease: A cross-sectional study. Medicine 2024, 103, e39883. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hao, B.; Ma, A.; He, J.; Liu, X.; Chen, J. The Expression of NOX4 in Smooth Muscles of Small Airway Correlates with the Disease Severity of COPD. Biomed. Res. Int. 2016, 2016, 2891810. [Google Scholar] [CrossRef]
- Wang, X.; Murugesan, P.; Zhang, P.; Xu, S.; Peng, L.; Wang, C.; Cai, H. NADPH Oxidase Isoforms in COPD Patients and Acute Cigarette Smoke-Exposed Mice: Induction of Oxidative Stress and Lung Inflammation. Antioxidants 2022, 11, 1539. [Google Scholar] [CrossRef]
- Alateeq, R.; Akhtar, A.; De Luca, S.N.; Chan, S.M.H.; Vlahos, R. Apocynin Prevents Cigarette Smoke-Induced Anxiety-Like Behavior and Preserves Microglial Profiles in Male Mice. Antioxidants 2024, 13, 855. [Google Scholar] [CrossRef]
- Yao, H.; Edirisinghe, I.; Yang, S.-R.; Rajendrasozhan, S.; Kode, A.; Caito, S.; Adenuga, D.; Rahman, I. Genetic Ablation of NADPH Oxidase Enhances Susceptibility to Cigarette Smoke-Induced Lung Inflammation and Emphysema in Mice. Am. J. Pathol. 2008, 172, 1222–1237. [Google Scholar] [CrossRef] [PubMed]
- Schiffers, C.; van de Wetering, C.; Bauer, R.A.; Habibovic, A.; Hristova, M.; Dustin, C.M.; Lambrichts, S.; Vacek, P.M.; Wouters, E.F.; Reynaert, N.L.; et al. Downregulation of epithelial DUOX1 in chronic obstructive pulmonary disease. JCI Insight 2021, 6, e142189. [Google Scholar] [CrossRef] [PubMed]
- Schiffers, C.; Lundblad, L.K.A.; Hristova, M.; Habibovic, A.; Dustin, C.M.; Daphtary, N.; Aliyeva, M.; Seward, D.J.; Janssen-Heininger, Y.M.W.; Wouters, E.F.M.; et al. Downregulation of DUOX1 function contributes to aging-related impairment of innate airway injury responses and accelerated senile emphysema. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L144–L158. [Google Scholar] [CrossRef]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The Nature of Small-Airway Obstruction in Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef]
- Brusselle, G.G.; Joos, G.F.; Bracke, K.R. New insights into the immunology of chronic obstructive pulmonary disease. Lancet 2011, 378, 1015–1026. [Google Scholar] [CrossRef]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef]
- Zuo, L.; He, F.; Sergakis, G.G.; Koozehchian, M.S.; Stimpfl, J.N.; Rong, Y.; Diaz, P.T.; Best, T.M. Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L205–L218. [Google Scholar] [CrossRef]
- Gan, W.Q. Association between chronic obstructive pulmonary disease and systemic inflammation: A systematic review and a meta-analysis. Thorax 2004, 59, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Agustí, A.; Edwards, L.D.; Rennard, S.I.; MacNee, W.; Tal-Singer, R.; Miller, B.E.; Vestbo, J.; Lomas, D.A.; Calverley, P.M.A.; Wouters, E.; et al. Persistent Systemic Inflammation is Associated with Poor Clinical Outcomes in COPD: A Novel Phenotype. PLoS ONE 2012, 7, e37483. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.; Dahl, M.; Lange, P.; Vestbo, J.; Nordestgaard, B.G. Inflammatory Biomarkers and Comorbidities in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2012, 186, 982–988. [Google Scholar] [CrossRef] [PubMed]
- Mastino, P.; Rosati, D.; de Soccio, G.; Romeo, M.; Pentangelo, D.; Venarubea, S.; Fiore, M.; Meliante, P.G.; Petrella, C.; Barbato, C.; et al. Oxidative Stress in Obstructive Sleep Apnea Syndrome: Putative Pathways to Hearing System Impairment. Antioxidants 2023, 12, 1430. [Google Scholar] [CrossRef]
- Abourjeili, J.; Salameh, E.; Noureddine, M.; Bou Khalil, P.; Eid, A.A. Obstructive sleep apnea: Beyond the dogma of obesity! Respir. Med. 2024, 222, 107512. [Google Scholar] [CrossRef]
- Alharbi, K.S.; Fuloria, N.K.; Fuloria, S.; Rahman, S.B.; Al-Malki, W.H.; Javed Shaikh, M.A.; Thangavelu, L.; Singh, S.K.; Allam, V.S.R.R.; Jha, N.K.; et al. Nuclear factor-kappa B and its role in inflammatory lung disease. Chem. Biol. Interact. 2021, 345, 109568. [Google Scholar] [CrossRef]
- Israel, L.P.; Benharoch, D.; Gopas, J.; Goldbart, A.D. A Pro-Inflammatory Role for Nuclear Factor Kappa B in Childhood Obstructive Sleep Apnea Syndrome. Sleep 2013, 36, 1947–1955. [Google Scholar] [CrossRef]
- Qiu, X.; Yao, Y.; Chen, Y.; Li, Y.; Sun, X.; Zhu, X. TRPC5 Promotes Intermittent Hypoxia-Induced Cardiomyocyte Injury Through Oxidative Stress. Nat. Sci. Sleep 2024, 16, 2125–2141. [Google Scholar] [CrossRef]
- Karamanlı, H.; Özol, D.; Ugur, K.S.; Yıldırım, Z.; Armutçu, F.; Bozkurt, B.; Yigitoglu, R. Influence of CPAP treatment on airway and systemic inflammation in OSAS patients. Sleep Breath. 2014, 18, 251–256. [Google Scholar] [CrossRef]
- Celec, P.; Jurkovičová, I.; Buchta, R.; Bartík, I.; Gardlík, R.; Pálffy, R.; Mucska, I.; Hodosy, J. Antioxidant vitamins prevent oxidative and carbonyl stress in an animal model of obstructive sleep apnea. Sleep Breath. 2013, 17, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Thareja, S.; Mandapalli, R.; Shaik, F.; Rajeev Pillai, A.; Palaniswamy, G.; Sahu, S.; Cherukuri, S.P.; Younas, S.; Palaniswamy, G.P., Jr. Impact of Obstructive Sleep Apnea on Cardiovascular Health: A Systematic Review. Cureus 2024, 16, e71940. [Google Scholar] [CrossRef]
- Jelska, A.; Polecka, A.; Zahorodnii, A.; Olszewska, E. The Role of Oxidative Stress and the Potential Therapeutic Benefits of Aronia melanocarpa Supplementation in Obstructive Sleep Apnea Syndrome: A Comprehensive Literature Review. Antioxidants 2024, 13, 1300. [Google Scholar] [CrossRef] [PubMed]
- Mokra, D.; Mokry, J.; Barosova, R.; Hanusrichterova, J. Advances in the Use of N-Acetylcysteine in Chronic Respiratory Diseases. Antioxidants 2023, 12, 1713. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wu, F.; Shi, Z.; Cao, J.; Tian, J.; Yao, W.; Wei, L.; Li, F.; Cai, S.; Shen, Y.; et al. Effect of high-dose N-acetylcysteine on exacerbations and lung function in patients with mild-to-moderate COPD: A double-blind, parallel group, multicentre randomised clinical trial. Nat. Commun. 2024, 15, 8468. [Google Scholar] [CrossRef]
- Lei, T.; Lu, T.; Yu, H.; Su, X.; Zhang, C.; Zhu, L.; Yang, K.; Liu, J. Efficacy of Vitamin C Supplementation on Chronic Obstructive Pulmonary Disease (COPD): A Systematic Review and Meta-Analysis. Int. J. Chronic Obstr. Pulm. Dis. 2022, 17, 2201–2216. [Google Scholar] [CrossRef]
- Fekete, M.; Csípő, T.; Fazekas-Pongor, V.; Fehér, Á.; Szarvas, Z.; Kaposvári, C.; Horváth, K.; Lehoczki, A.; Tarantini, S.; Varga, J.T. The Effectiveness of Supplementation with Key Vitamins, Minerals, Antioxidants and Specific Nutritional Supplements in COPD—A Review. Nutrients 2023, 15, 2741. [Google Scholar] [CrossRef]
- Srivali, N.; Thongprayoon, C.; Tangpanithandee, S.; Cheungpasitporn, W.; Won, C. The use of continuous positive airway pressure in COPD-OSA overlap syndrome: A systematic review. Sleep Med. 2023, 108, 55–60. [Google Scholar] [CrossRef]
- Miro, A.M.; Shivaram, U.; Hertig, I. Continuous Positive Airway Pressure in COPD Patients in Acute Hypercapnic Respiratory Failure. Chest 1993, 103, 266–268. [Google Scholar] [CrossRef]
- Jang, B.-K.; Lee, J.-W.; Choi, H.; Yim, S.-V. Aronia melanocarpa Fruit Bioactive Fraction Attenuates LPS-Induced Inflammatory Response in Human Bronchial Epithelial Cells. Antioxidants 2020, 9, 816. [Google Scholar] [CrossRef]
- Ai, L.; Li, Y.; Liu, Z.; Li, R.; Wang, X.; Hai, B. Tempol attenuates chronic intermittent hypoxia-induced lung injury through the miR-145-5p/Nrf2 signaling pathway. Cell. Mol. Biol. 2023, 69, 225–234. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef]
- Fang, L.; Liu, K.; Liu, C.; Wang, X.; Ma, W.; Xu, W.; Wu, J.; Sun, C. Tumor accomplice: T cell exhaustion induced by chronic inflammation. Front. Immunol. 2022, 13, 979116. [Google Scholar] [CrossRef]
- Vicente, E.; Marin, J.M.; Carrizo, S.J.; Osuna, C.S.; González, R.; Marin-Oto, M.; Forner, M.; Vicente, P.; Cubero, P.; Gil, A.V.; et al. Upper airway and systemic inflammation in obstructive sleep apnoea. Eur. Respir. J. 2016, 48, 1108–1117. [Google Scholar] [CrossRef]
- Houghton, A.M. Mechanistic links between COPD and lung cancer. Nat. Rev. Cancer 2013, 13, 233–245. [Google Scholar] [CrossRef]
- Dubinett, S.M.; Spira, A.E. Impact of Chronic Obstructive Pulmonary Disease on Immune-based Treatment for Lung Cancer. Moving toward Disease Interception. Am. J. Respir. Crit. Care Med. 2018, 197, 278–280. [Google Scholar] [CrossRef] [PubMed]
- Zaynagetdinov, R.; Sherrill, T.P.; Gleaves, L.A.; Hunt, P.; Han, W.; McLoed, A.G.; Saxon, J.A.; Tanjore, H.; Gulleman, P.M.; Young, L.R.; et al. Chronic NF-κB activation links COPD and lung cancer through generation of an immunosuppressive microenvironment in the lungs. Oncotarget 2016, 7, 5470–5482. [Google Scholar] [CrossRef] [PubMed]
- Forder, A.; Zhuang, R.; Souza, V.G.P.; Brockley, L.J.; Pewarchuk, M.E.; Telkar, N.; Stewart, G.L.; Benard, K.; Marshall, E.A.; Reis, P.P.; et al. Mechanisms Contributing to the Comorbidity of COPD and Lung Cancer. Int. J. Mol. Sci. 2023, 24, 2859. [Google Scholar] [CrossRef]
- Kirkham, P.A.; Barnes, P.J. Oxidative Stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef]
- Chung, K.F. Cytokines in chronic obstructive pulmonary disease. Eur. Respir. J. 2001, 18, 50s–59s. [Google Scholar] [CrossRef]
- Sohal, S.; Walters, E. Role of epithelial mesenchymal transition (EMT) in chronic obstructive pulmonary disease (COPD). Respir. Res. 2013, 14, 120. [Google Scholar] [CrossRef]
- Díaz-García, E.; García-Tovar, S.; Casitas, R.; Jaureguizar, A.; Zamarrón, E.; Sánchez-Sánchez, B.; Sastre-Perona, A.; López-Collazo, E.; Garcia-Rio, F.; Cubillos-Zapata, C. Intermittent Hypoxia Mediates Paraspeckle Protein-1 Upregulation in Sleep Apnea. Cancers 2021, 13, 3888. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Zapata, C.; Martínez-García, M.Á.; Díaz-García, E.; García-Tovar, S.; Campos-Rodríguez, F.; Sánchez-de-la-Torre, M.; Nagore, E.; Martorell-Calatayud, A.; Blasco, L.H.; Pastor, E.; et al. Obstructive sleep apnoea is related to melanoma aggressiveness through paraspeckle protein-1 upregulation. Eur. Respir. J. 2023, 61, 2200707. [Google Scholar] [CrossRef] [PubMed]
- Gozal, D.; Lipton, A.J.; Jones, K.L. Circulating Vascular Endothelial Growth Factor Levels in Patients with Obstructive Sleep Apnea. Sleep 2002, 25, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Díaz-García, E.; García-Sánchez, A.; Alfaro, E.; López-Fernández, C.; Mañas, E.; Cano-Pumarega, I.; López-Collazo, E.; García-Río, F.; Cubillos-Zapata, C. PSGL-1: A novel immune checkpoint driving T-cell dysfunction in obstructive sleep apnea. Front. Immunol. 2023, 14, 1277551. [Google Scholar] [CrossRef]
- Hernández-Jiménez, E.; Cubillos-Zapata, C.; Toledano, V.; Pérez de Diego, R.; Fernández-Navarro, I.; Casitas, R.; Carpio, C.; Casas-Martín, J.; Valentin, J.; Varela-Serrano, A.; et al. Monocytes inhibit NK activity via TGF-β in patients with obstructive sleep apnoea. Eur. Respir. J. 2017, 49, 1602456. [Google Scholar] [CrossRef]
- Verduzco, D.; Lloyd, M.; Xu, L.; Ibrahim-Hashim, A.; Balagurunathan, Y.; Gatenby, R.A.; Gillies, R.J. Intermittent Hypoxia Selects for Genotypes and Phenotypes That Increase Survival, Invasion, and Therapy Resistance. PLoS ONE 2015, 10, e0120958. [Google Scholar] [CrossRef]
- Almendros, I.; Wang, Y.; Becker, L.; Lennon, F.E.; Zheng, J.; Coats, B.R.; Schoenfelt, K.S.; Carreras, A.; Hakim, F.; Zhang, S.X.; et al. Intermittent Hypoxia-induced Changes in Tumor-associated Macrophages and Tumor Malignancy in a Mouse Model of Sleep Apnea. Am. J. Respir. Crit. Care Med. 2014, 189, 593–601. [Google Scholar] [CrossRef]
- Pahwa, R.; Goyal, A.; Jialal, I. Chronic Inflammation; National Library of Medicine: Bethesda, MD, USA, 2025. [Google Scholar]
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015, 4, 296–307. [Google Scholar] [CrossRef]
- Kim, J.; Kim, H.-S.; Chung, J.H. Molecular mechanisms of mitochondrial DNA release and activation of the cGAS-STING pathway. Exp. Mol. Med. 2023, 55, 510–519. [Google Scholar] [CrossRef]
- Sharma, B.R.; Kanneganti, T.-D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and Functions of Inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef]
- Gupta, N.; Sahu, A.; Prabhakar, A.; Chatterjee, T.; Tyagi, T.; Kumari, B.; Khan, N.; Nair, V.; Bajaj, N.; Sharma, M.; et al. Activation of NLRP3 inflammasome complex potentiates venous thrombosis in response to hypoxia. Proc. Natl. Acad. Sci. USA 2017, 114, 4763–4768. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, H.; Liu, B.; Zhang, Y.; Pan, X.; Yu, X.-Y.; Shen, Z.; Song, Y.-H. Inflammasomes as therapeutic targets in human diseases. Signal Transduct. Target. Ther. 2021, 6, 247. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.-H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 Inflammasome Links Systemic Low-Grade Inflammation to Functional Decline in Aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef]
- Sutterwala, F.S.; Ogura, Y.; Flavell, R.A. The inflammasome in pathogen recognition and inflammation. J. Leukoc. Biol. 2007, 82, 259–264. [Google Scholar] [CrossRef]
- Barnes, P.J. Cellular and Molecular Mechanisms of Chronic Obstructive Pulmonary Disease. Clin. Chest Med. 2014, 35, 71–86. [Google Scholar] [CrossRef]
- Barnes, P.J. COPD: Inflammatory mechanisms and systemic consequences. In COPD and Comorbidity; European Respiratory Society: Lausanne, Switzerland, 2013. [Google Scholar]
- Eisner, M.D.; Anthonisen, N.; Coultas, D.; Kuenzli, N.; Perez-Padilla, R.; Postma, D.; Romieu, I.; Silverman, E.K.; Balmes, J.R. An Official American Thoracic Society Public Policy Statement: Novel Risk Factors and the Global Burden of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2010, 182, 693–718. [Google Scholar] [CrossRef]
- Campo, G.; Pavasini, R.; Malagù, M.; Mascetti, S.; Biscaglia, S.; Ceconi, C.; Papi, A.; Contoli, M. Chronic Obstructive Pulmonary Disease and Ischemic Heart Disease Comorbidity: Overview of Mechanisms and Clinical Management. Cardiovasc. Drugs Ther. 2015, 29, 147–157. [Google Scholar] [CrossRef]
- Divo, M.; Cote, C.; de Torres, J.P.; Casanova, C.; Marin, J.M.; Pinto-Plata, V.; Zulueta, J.; Cabrera, C.; Zagaceta, J.; Hunninghake, G.; et al. Comorbidities and Risk of Mortality in Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2012, 186, 155–161. [Google Scholar] [CrossRef]
- Onishi, K. Total management of chronic obstructive pulmonary disease (COPD) as an independent risk factor for cardiovascular disease. J. Cardiol. 2017, 70, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.D.; Zakeri, R.; Quint, J.K. Defining the relationship between COPD and CVD: What are the implications for clinical practice? Ther. Adv. Respir. Dis. 2018, 12, 1753465817750524. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Thomas, J.; Sadatsafavi, M.; FitzGerald, J.M. Risk of cardiovascular comorbidity in patients with chronic obstructive pulmonary disease: A systematic review and meta-analysis. Lancet Respir. Med. 2015, 3, 631–639. [Google Scholar] [CrossRef]
- Kheirandish-Gozal, L.; Gozal, D. Obstructive Sleep Apnea and Inflammation: Proof of Concept Based on Two Illustrative Cytokines. Int. J. Mol. Sci. 2019, 20, 459. [Google Scholar] [CrossRef]
- Yeghiazarians, Y.; Jneid, H.; Tietjens, J.R.; Redline, S.; Brown, D.L.; El-Sherif, N.; Mehra, R.; Bozkurt, B.; Ndumele, C.E.; Somers, V.K.; et al. Obstructive Sleep Apnea and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 2021, 144, e56–e67. [Google Scholar] [CrossRef] [PubMed]
- Tietjens, J.R.; Claman, D.; Kezirian, E.J.; De Marco, T.; Mirzayan, A.; Sadroonri, B.; Goldberg, A.N.; Long, C.; Gerstenfeld, E.P.; Yeghiazarians, Y. Obstructive Sleep Apnea in Cardiovascular Disease: A Review of the Literature and Proposed Multidisciplinary Clinical Management Strategy. J. Am. Heart Assoc. 2019, 8, e010440. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, H.; Cai, M.; Zou, Y.; Jiang, X.; Song, L.; Liang, E.; Bian, J.; Wu, H.; Hui, R. Effect of spironolactone on patients with resistant hypertension and obstructive sleep apnea. Clin. Exp. Hypertens. 2016, 38, 464–468. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Lavie, L.; Lavie, P. Molecular mechanisms of cardiovascular disease in OSAHS: The oxidative stress link. Eur. Respir. J. 2009, 33, 1467–1484. [Google Scholar] [CrossRef]
- Feres, M.C.; Fonseca, F.A.H.; Cintra, F.D.; Mello-Fujita, L.; de Souza, A.L.; De Martino, M.C.; Tufik, S.; Poyares, D. An assessment of oxidized LDL in the lipid profiles of patients with obstructive sleep apnea and its association with both hypertension and dyslipidemia, and the impact of treatment with CPAP. Atherosclerosis 2015, 241, 342–349. [Google Scholar] [CrossRef]
- Imamura, T.; Poulsen, O.; Haddad, G.G. Intermittent hypoxia induces murine macrophage foam cell formation by IKK-β-dependent NF-κB pathway activation. J. Appl. Physiol. 2016, 121, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Díaz-García, E.; García-Tovar, S.; Alfaro, E.; Jaureguizar, A.; Casitas, R.; Sánchez-Sánchez, B.; Zamarrón, E.; Fernández-Lahera, J.; López-Collazo, E.; Cubillos-Zapata, C.; et al. Inflammasome Activation: A Keystone of Proinflammatory Response in Obstructive Sleep Apnea. Am. J. Respir. Crit. Care Med. 2022, 205, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Díaz-García, E.; Sanz-Rubio, D.; García-Tovar, S.; Alfaro, E.; Cubero, P.; Gil, A.V.; Marin, J.M.; Cubillos-Zapata, C.; García-Río, F. Inflammasome activation mediated by oxidised low-density lipoprotein in patients with sleep apnoea and early subclinical atherosclerosis. Eur. Respir. J. 2023, 61, 2201401. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.; Bartolome, S.; Denton, C.P.; Gatzoulis, M.A.; Gu, S.; Khanna, D.; Badesch, D.; Montani, D. Definition, classification and diagnosis of pulmonary hypertension. Eur. Respir. J. 2024, 64, 2401324. [Google Scholar] [CrossRef]
- Krompa, A.; Marino, P. Diagnosis and management of pulmonary hypertension related to chronic respiratory disease. Breathe 2022, 18, 220205. [Google Scholar] [CrossRef]
- McNicholas, W.T. Chronic obstructive pulmonary disease and obstructive sleep apnea: Overlaps in pathophysiology, systemic inflammation, and cardiovascular disease. Am. J. Respir. Crit. Care Med. 2009, 180, 692–700. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Meyrick, B.; Galie, N.; Mooi, W.J.; McMurtry, I.F. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L1013–L1032. [Google Scholar] [CrossRef]
- Pugliese, S.C.; Poth, J.M.; Fini, M.A.; Olschewski, A.; El Kasmi, K.C.; Stenmark, K.R. The role of inflammation in hypoxic pulmonary hypertension: From cellular mechanisms to clinical phenotypes. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L229–L252. [Google Scholar] [CrossRef] [PubMed]
- Frazziano, G.; Moreno, L.; Moral-Sanz, J.; Menendez, C.; Escolano, L.; Gonzalez, C.; Villamor, E.; Alvarez-Sala, J.L.; Cogolludo, A.L.; Perez-Vizcaino, F. Neutral sphingomyelinase, NADPH oxidase and reactive oxygen species. Role in acute hypoxic pulmonary vasoconstriction. J. Cell Physiol. 2011, 226, 2633–2640. [Google Scholar] [CrossRef]
- Villamor, E.; Moreno, L.; Mohammed, R.; Pérez-Vizcaíno, F.; Cogolludo, A. Reactive oxygen species as mediators of oxygen signaling during fetal-to-neonatal circulatory transition. Free Radic. Biol. Med. 2019, 142, 82–96. [Google Scholar] [CrossRef]
- Hernansanz-Agustín, P.; Choya-Foces, C.; Carregal-Romero, S.; Ramos, E.; Oliva, T.; Villa-Piña, T.; Moreno, L.; Izquierdo-Álvarez, A.; Cabrera-García, J.D.; Cortés, A.; et al. Na+ controls hypoxic signalling by the mitochondrial respiratory chain. Nature 2020, 586, 287–291. [Google Scholar] [CrossRef]
- Pak, O.; Scheibe, S.; Esfandiary, A.; Gierhardt, M.; Sydykov, A.; Logan, A.; Fysikopoulos, A.; Veit, F.; Hecker, M.; Kroschel, F.; et al. Impact of the mitochondria-targeted antioxidant MitoQ on hypoxia-induced pulmonary hypertension. Eur. Respir. J. 2018, 51, 1701024. [Google Scholar] [CrossRef] [PubMed]
- Adesina, S.E.; Kang, B.-Y.; Bijli, K.M.; Ma, J.; Cheng, J.; Murphy, T.C.; Hart, C.M.; Sutliff, R.L. Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free Radic. Biol. Med. 2015, 87, 36–47. [Google Scholar] [CrossRef]
- Yan, S.; Sheak, J.R.; Walker, B.R.; Jernigan, N.L.; Resta, T.C. Contribution of Mitochondrial Reactive Oxygen Species to Chronic Hypoxia-Induced Pulmonary Hypertension. Antioxidants 2023, 12, 2060. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Q.; Erbynn, E.M.; Folz, R.J. Chronic hypoxia-enhanced murine pulmonary vasoconstriction: Role of superoxide and gp91phox. Chest 2005, 128, 594S–596S. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.; Hou, X.; Bing, X.; Zhu, F.; Wu, X.; Guo, N.; Zhao, H.; Xu, F.; Xia, M. Obstructive sleep apnea-increased DEC1 regulates systemic inflammation and oxidative stress that promotes development of pulmonary arterial hypertension. Apoptosis 2023, 28, 432–446. [Google Scholar] [CrossRef]
- Nisbet, R.E.; Graves, A.S.; Kleinhenz, D.J.; Rupnow, H.L.; Reed, A.L.; Fan, T.-H.M.; Mitchell, P.O.; Sutliff, R.L.; Hart, C.M. The role of NADPH oxidase in chronic intermittent hypoxia-induced pulmonary hypertension in mice. Am. J. Respir. Cell Mol. Biol. 2009, 40, 601–609. [Google Scholar] [CrossRef]
- Smith, K.A.; Schumacker, P.T. Sensors and signals: The role of reactive oxygen species in hypoxic pulmonary vasoconstriction. J. Physiol. 2019, 597, 1033–1043. [Google Scholar] [CrossRef]
- Mei, L.; Zheng, Y.-M.; Song, T.; Yadav, V.R.; Joseph, L.C.; Truong, L.; Kandhi, S.; Barroso, M.M.; Takeshima, H.; Judson, M.A.; et al. Rieske iron-sulfur protein induces FKBP12.6/RyR2 complex remodeling and subsequent pulmonary hypertension through NF-κB/cyclin D1 pathway. Nat. Commun. 2020, 11, 3527. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, C.; Tabeling, C.; Nagy, B.M.; Jain, P.P.; Marsh, L.M.; Papp, R.; Pienn, M.; Witzenrath, M.; Ghanim, B.; Klepetko, W.; et al. Hypoxic vascular response and ventilation/perfusion matching in end-stage COPD may depend on p22phox. Eur. Respir. J. 2017, 50, 1601651. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Zhao, Q.D.; Viswanadhapalli, S.; Williams, P.; Shi, Q.; Tan, C.; Yi, X.; Bhandari, B.; Abboud, H.E. NADPH Oxidase 4 Induces Cardiac Fibrosis and Hypertrophy Through Activating Akt/mTOR and NFκB Signaling Pathways. Circulation 2015, 131, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Schröder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 Is a Protective Reactive Oxygen Species Generating Vascular NADPH Oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gonzalez, F.J.; Chandel, N.S.; Jain, M.; Budinger, G.R.S. Reactive oxygen species as signaling molecules in the development of lung fibrosis. Transl. Res. 2017, 190, 61–68. [Google Scholar] [CrossRef]
- Kinnula, V.L.; Fattman, C.L.; Tan, R.J.; Oury, T.D. Oxidative stress in pulmonary fibrosis: A possible role for redox modulatory therapy. Am. J. Respir. Crit. Care Med. 2005, 172, 417–422. [Google Scholar] [CrossRef]
- Bellocq, A.; Azoulay, E.; Marullo, S.; Flahault, A.; Fouqueray, B.; Philippe, C.; Cadranel, J.; Baud, L. Reactive oxygen and nitrogen intermediates increase transforming growth factor-beta1 release from human epithelial alveolar cells through two different mechanisms. Am. J. Respir. Cell Mol. Biol. 1999, 21, 128–136. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Zaiman, A.; Champion, H.C. PDE5A inhibition attenuates bleomycin-induced pulmonary fibrosis and pulmonary hypertension through inhibition of ROS generation and RhoA/Rho kinase activation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L24–L33. [Google Scholar] [CrossRef]
- Behnke, J.; Dippel, C.M.; Choi, Y.; Rekers, L.; Schmidt, A.; Lauer, T.; Dong, Y.; Behnke, J.; Zimmer, K.-P.; Bellusci, S.; et al. Oxygen Toxicity to the Immature Lung-Part II: The Unmet Clinical Need for Causal Therapy. Int. J. Mol. Sci. 2021, 22, 10694. [Google Scholar] [CrossRef] [PubMed]
- Wedgwood, S.; Steinhorn, R.H. Role of reactive oxygen species in neonatal pulmonary vascular disease. Antioxid. Redox Signal. 2014, 21, 1926–1942. [Google Scholar] [CrossRef]
- Huizing, M.J.; Cavallaro, G.; Moonen, R.M.; González-Luis, G.E.; Mosca, F.; Vento, M.; Villamor, E. Is the C242T Polymorphism of the CYBA Gene Linked with Oxidative Stress-Associated Complications of Prematurity? Antioxid. Redox Signal. 2017, 27, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-R.; Yuan, T.-Y.; Wang, J.-M.; Chen, Y.-C.; Zhao, J.-L.; Li, M.-T.; Fang, L.-H.; Du, G.-H. Immunity and inflammation in pulmonary arterial hypertension: From pathophysiology mechanisms to treatment perspective. Pharmacol. Res. 2022, 180, 106238. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Xu, Q.; Wuren, T. Inflammation and immunity in the pathogenesis of hypoxic pulmonary hypertension. Front. Immunol. 2023, 14, 1162556. [Google Scholar] [CrossRef]
- Lin, D.; Hu, L.; Wei, D.; Li, Y.; Yu, Y.; Wang, Q.; Sun, X.; Shen, Y.; Yu, Y.; Li, K.; et al. Peli1 Deficiency in Macrophages Attenuates Pulmonary Hypertension by Enhancing Foxp1-Mediated Transcriptional Inhibition of IL-6. Hypertension 2025, 82, 445–459. [Google Scholar] [CrossRef]
- Savale, L.; Tu, L.; Rideau, D.; Izziki, M.; Maitre, B.; Adnot, S.; Eddahibi, S. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir. Res. 2009, 10, 6. [Google Scholar] [CrossRef]
- Hashimoto-Kataoka, T.; Hosen, N.; Sonobe, T.; Arita, Y.; Yasui, T.; Masaki, T.; Minami, M.; Inagaki, T.; Miyagawa, S.; Sawa, Y.; et al. Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proc. Natl. Acad. Sci. USA 2015, 112, E2677–E2686. [Google Scholar] [CrossRef]
- Amsellem, V.; Abid, S.; Poupel, L.; Parpaleix, A.; Rodero, M.; Gary-Bobo, G.; Latiri, M.; Dubois-Rande, J.-L.; Lipskaia, L.; Combadiere, C.; et al. Roles for the CX3CL1/CX3CR1 and CCL2/CCR2 Chemokine Systems in Hypoxic Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2017, 56, 597–608. [Google Scholar] [CrossRef]
- Kumar, R.; Nolan, K.; Kassa, B.; Chanana, N.; Palmo, T.; Sharma, K.; Singh, K.; Mickael, C.; Balladares, D.F.; Nilsson, J.; et al. Monocytes and interstitial macrophages contribute to hypoxic pulmonary hypertension. J. Clin. Investig. 2025, 135, e176865. [Google Scholar] [CrossRef]
- Yu, Y.-R.A.; Malakhau, Y.; Yu, C.-H.A.; Phelan, S.-L.J.; Cumming, R.I.; Kan, M.J.; Mao, L.; Rajagopal, S.; Piantadosi, C.A.; Gunn, M.D. Nonclassical Monocytes Sense Hypoxia, Regulate Pulmonary Vascular Remodeling, and Promote Pulmonary Hypertension. J. Immunol. 2020, 204, 1474–1485. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Mickael, C.; Kassa, B.; Gebreab, L.; Robinson, J.C.; Koyanagi, D.E.; Sanders, L.; Barthel, L.; Meadows, C.; Fox, D.; et al. TGF-β activation by bone marrow-derived thrombospondin-1 causes Schistosoma- and hypoxia-induced pulmonary hypertension. Nat. Commun. 2017, 8, 15494. [Google Scholar] [CrossRef] [PubMed]
- Vergadi, E.; Chang, M.S.; Lee, C.; Liang, O.D.; Liu, X.; Fernandez-Gonzalez, A.; Mitsialis, S.A.; Kourembanas, S. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation 2011, 123, 1986–1995. [Google Scholar] [CrossRef]
- Kumar, R.; Mickael, C.; Kassa, B.; Sanders, L.; Hernandez-Saavedra, D.; Koyanagi, D.E.; Kumar, S.; Pugliese, S.C.; Thomas, S.; McClendon, J.; et al. Interstitial macrophage-derived thrombospondin-1 contributes to hypoxia-induced pulmonary hypertension. Cardiovasc. Res. 2020, 116, 2021–2030. [Google Scholar] [CrossRef]
- Maston, L.D.; Jones, D.T.; Giermakowska, W.; Howard, T.A.; Cannon, J.L.; Wang, W.; Wei, Y.; Xuan, W.; Resta, T.C.; Bosc, L.V.G. Central role of T helper 17 cells in chronic hypoxia-induced pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L609–L624. [Google Scholar] [CrossRef] [PubMed]
- Sevilla-Montero, J.; Labrousse-Arias, D.; Fernández-Pérez, C.; Fernández-Blanco, L.; Barreira, B.; Mondéjar-Parreño, G.; Alfaro-Arnedo, E.; López, I.P.; Pérez-Rial, S.; Peces-Barba, G.; et al. Cigarette Smoke Directly Promotes Pulmonary Arterial Remodeling and Kv7.4 Channel Dysfunction. Am. J. Respir. Crit. Care Med. 2021, 203, 1290–1305. [Google Scholar] [CrossRef]
- Chatterjee, K.; Tarawneh, A.R.; Alam, S. Out of proportion pulmonary hypertension in obstructive lung diseases. Curr. Opin. Pulm. Med. 2018, 24, 161–172. [Google Scholar] [CrossRef]
- Santos, S.; Peinado, V.I.; Ramírez, J.; Melgosa, T.; Roca, J.; Rodriguez-Roisin, R.; Barberà, J. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur. Respir. J. 2002, 19, 632–638. [Google Scholar] [CrossRef]
- Seimetz, M.; Parajuli, N.; Pichl, A.; Veit, F.; Kwapiszewska, G.; Weisel, F.C.; Milger, K.; Egemnazarov, B.; Turowska, A.; Fuchs, B.; et al. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell 2011, 147, 293–305. [Google Scholar] [CrossRef]
- Borgas, D.; Chambers, E.; Newton, J.; Ko, J.; Rivera, S.; Rounds, S.; Lu, Q. Cigarette Smoke Disrupted Lung Endothelial Barrier Integrity and Increased Susceptibility to Acute Lung Injury via Histone Deacetylase 6. Am. J. Respir. Cell Mol. Biol. 2016, 54, 683–696. [Google Scholar] [CrossRef]
- Truong, L.N.; Wilson Santos, E.; Zheng, Y.-M.; Wang, Y.-X. Rieske Iron-Sulfur Protein Mediates Pulmonary Hypertension Following Nicotine/Hypoxia Coexposure. Am. J. Respir. Cell Mol. Biol. 2024, 70, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Serebreni, L.; Hamdan, O.; Wang, L.; Parniani, A.; Sussan, T.; Stephens, R.S.; Boyer, L.; Damarla, M.; Hassoun, P.M.; et al. Xanthine oxidoreductase is a critical mediator of cigarette smoke-induced endothelial cell DNA damage and apoptosis. Free Radic. Biol. Med. 2013, 60, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Peinado, V.I.; Barberá, J.A.; Abate, P.; Ramírez, J.; Roca, J.; Santos, S.; Rodriguez-Roisin, R. Inflammatory reaction in pulmonary muscular arteries of patients with mild chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999, 159, 1605–1611. [Google Scholar] [CrossRef]
- Lim, E.Y.; Lee, S.-Y.; Shin, H.S.; Kim, G.-D. Reactive Oxygen Species and Strategies for Antioxidant Intervention in Acute Respiratory Distress Syndrome. Antioxidants 2023, 12, 2016. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dong, W. Oxidative stress and bronchopulmonary dysplasia. Gene 2018, 678, 177–183. [Google Scholar] [CrossRef]
- Lachmanová, V.; Hnilicková, O.; Povýsilová, V.; Hampl, V.; Herget, J. N-acetylcysteine inhibits hypoxic pulmonary hypertension most effectively in the initial phase of chronic hypoxia. Life Sci. 2005, 77, 175–182. [Google Scholar] [CrossRef]
- Brassington, K.; Chan, S.M.H.; De Luca, S.N.; Dobric, A.; Almerdasi, S.A.; Mou, K.; Seow, H.J.; Oseghale, O.; Bozinovski, S.; Selemidis, S.; et al. Ebselen abolishes vascular dysfunction in influenza A virus-induced exacerbations of cigarette smoke-induced lung inflammation in mice. Clin. Sci. 2022, 136, 537–555. [Google Scholar] [CrossRef]
- Chan, S.M.H.; Brassington, K.; Almerdasi, S.A.; Dobric, A.; De Luca, S.N.; Coward-Smith, M.; Wang, H.; Mou, K.; Akhtar, A.; Alateeq, R.A.; et al. Inhibition of oxidative stress by apocynin attenuated chronic obstructive pulmonary disease progression and vascular injury by cigarette smoke exposure. Br. J. Pharmacol. 2023, 180, 2018–2034. [Google Scholar] [CrossRef]
- Barnes, P.J. Oxidative Stress in Chronic Obstructive Pulmonary Disease. Antioxidants 2022, 11, 965. [Google Scholar] [CrossRef]
- Young, K.C.; Torres, E.; Hatzistergos, K.E.; Hehre, D.; Suguihara, C.; Hare, J.M. Inhibition of the SDF-1/CXCR4 axis attenuates neonatal hypoxia-induced pulmonary hypertension. Circ. Res. 2009, 104, 1293–1301. [Google Scholar] [CrossRef]
- Fernandez-Gonzalez, A.; Mukhia, A.; Nadkarni, J.; Willis, G.R.; Reis, M.; Zhumka, K.; Vitali, S.; Liu, X.; Galls, A.; Mitsialis, S.A.; et al. Immunoregulatory Macrophages Modify Local Pulmonary Immunity and Ameliorate Hypoxic Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2024, 44, e288–e303. [Google Scholar] [CrossRef] [PubMed]
- Jankov, R.P.; Kantores, C.; Pan, J.; Belik, J. Contribution of xanthine oxidase-derived superoxide to chronic hypoxic pulmonary hypertension in neonatal rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L233–L245. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhao, J.; Jia, X.; Pan, J.; Xu, S.; Wang, D.; Li, J.; Ji, Y.; Zhu, Z.; Hasnain, M.; et al. Fucoxanthin Ameliorates Vascular Remodeling via Attenuating Oxidative Stress in Hypoxic Pulmonary Hypertension Rats. J. Nutr. Biochem. 2025, 110002. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Liu, M.; Zhang, X.; Pan, J.; Han, J.; Wang, Y.; Lei, H.; Ding, Y.; Yuan, Y. Grape seed procyanidin extract attenuates hypoxic pulmonary hypertension by inhibiting oxidative stress and pulmonary arterial smooth muscle cells proliferation. J. Nutr. Biochem. 2016, 36, 81–88. [Google Scholar] [CrossRef]
- Smith, L.A.; Oakden-Rayner, L.; Bird, A.; Zeng, M.; To, M.-S.; Mukherjee, S.; Palmer, L.J. Machine learning and deep learning predictive models for long-term prognosis in patients with chronic obstructive pulmonary disease: A systematic review and meta-analysis. Lancet Digit. Health 2023, 5, e872–e881. [Google Scholar] [CrossRef]
- Vigil, L.; Zapata, T.; Grau, A.; Bonet, M.; Montaña, M.; Piñar, M. Innovación en sueño. Open Respir. Arch. 2024, 6, 100402. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention Is All You Need. arXiv 2017. [Google Scholar] [CrossRef]
- Li, Y.; Rao, S.; Solares, J.R.A.; Hassaine, A.; Ramakrishnan, R.; Canoy, D.; Zhu, Y.; Rahimi, K.; Salimi-Khorshidi, G. BEHRT: Transformer for Electronic Health Records. Sci. Rep. 2020, 10, 7155. [Google Scholar] [CrossRef]
- Ma, R.; Cheng, Q.; Yao, J.; Peng, Z.; Yan, M.; Lu, J.; Liao, J.; Tian, L.; Shu, W.; Zhang, Y.; et al. Multimodal machine learning enables AI chatbot to diagnose ophthalmic diseases and provide high-quality medical responses. NPJ Digit. Med. 2025, 8, 64. [Google Scholar] [CrossRef]
- Loni, M.; Poursalim, F.; Asadi, M.; Gharehbaghi, A. A review on generative AI models for synthetic medical text, time series, and longitudinal data. NPJ Digit. Med. 2025, 8, 281. [Google Scholar] [CrossRef]
- Giuffrè, M.; Shung, D.L. Harnessing the power of synthetic data in healthcare: Innovation, application, and privacy. NPJ Digit. Med. 2023, 6, 186. [Google Scholar] [CrossRef]
- Liu, X.; Tao, Y.; Cai, Z.; Bao, P.; Ma, H.; Li, K.; Li, M.; Zhu, Y.; Lu, Z.J.; Wren, J. Pathformer: A biological pathway informed transformer for disease diagnosis and prognosis using multi-omics data. Bioinformatics 2024, 40, btae316. [Google Scholar] [CrossRef]
- Xie, J.; Chen, Y.; Luo, S.; Yang, W.; Lin, Y.; Wang, L.; Ding, X.; Tong, M.; Yu, R. Tracing unknown tumor origins with a biological-pathway-based transformer model. Cell Rep. Methods 2024, 4, 100797. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z. Multi-omics/genomics in predictive and personalized medicine. In Artificial Intelligence for Drug Product Lifecycle Applications; Elsevier: Amsterdam, The Netherlands, 2025; pp. 109–120. [Google Scholar]
- Chen, Y.-M.; Hsiao, T.-H.; Lin, C.-H.; Fann, Y.C. Unlocking precision medicine: Clinical applications of integrating health records, genetics, and immunology through artificial intelligence. J. Biomed. Sci. 2025, 32, 16. [Google Scholar] [CrossRef]
- Chang, L.; Liu, J.; Zhu, J.; Guo, S.; Wang, Y.; Zhou, Z.; Wei, X. Advancing precision medicine: The transformative role of artificial intelligence in immunogenomics, radiomics, and pathomics for biomarker discovery and immunotherapy optimization. Cancer Biol. Med. 2025, 22, 33–47. [Google Scholar] [CrossRef]
- Johnson, A.E.W.; Pollard, T.J.; Shen, L.; Lehman, L.H.; Feng, M.; Ghassemi, M.; Moody, B.; Szolovits, P.; Celi, L.A.; Mark, R.G. MIMIC-III, a freely accessible critical care database. Sci. Data 2016, 3, 160035. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.E.W.; Bulgarelli, L.; Shen, L.; Gayles, A.; Shammout, A.; Horng, S.; Pollard, T.J.; Hao, S.; Moody, B.; Gow, B.; et al. Author Correction: MIMIC-IV, a freely accessible electronic health record dataset. Sci. Data 2023, 10, 219. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.J.; Johnson, A.E.W.; Raffa, J.D.; Celi, L.A.; Mark, R.G.; Badawi, O. The eICU Collaborative Research Database, a freely available multi-center database for critical care research. Sci. Data 2018, 5, 180178. [Google Scholar] [CrossRef]
- Faltys, M.; Zimmermann, M.; Lyu, X.; Hüser, M.; Hyland, S.; Rätsch, G.; Merz, T. HiRID, a High Time-Resolution ICU Dataset; PhysioNet: Cambridge, MA, USA, 2021. [Google Scholar]
- ISO/IEC 27001:2022; Information Security, Cybersecurity and Privacy Protection—Information Security Management Systems—Requirements. ISO: Geneva, Switzerland, 2022.
- ISO/TS 82304-2:2021; Health Software. Part 2: Health and Wellness Apps—Quality and Reliability. ISO: Geneva, Switzerland, 2021.
- Robert, G.C.; Philip Dawid, A.; Lauritzen, S.L.; David, J.S. Probabilistic Networks and Expert Systems; Springer: New York, NY, USA, 1999. [Google Scholar]
- Regan, E.A.; Hokanson, J.E.; Murphy, J.R.; Make, B.; Lynch, D.A.; Beaty, T.H.; Curran-Everett, D.; Silverman, E.K.; Crapo, J.D. Genetic Epidemiology of COPD (COPDGene) Study Design. COPD: J. Chronic Obstr. Pulm. Dis. 2011, 7, 32–43. [Google Scholar] [CrossRef]
- Vestbo, J.; Anderson, W.; Coxson, H.O.; Crim, C.; Dawber, F.; Edwards, L.; Hagan, G.; Knobil, K.; Lomas, D.A.; MacNee, W.; et al. Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points (ECLIPSE). Eur. Respir. J. 2008, 31, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Couper, D.; LaVange, L.M.; Han, M.; Barr, R.G.; Bleecker, E.; Hoffman, E.A.; Kanner, R.; Kleerup, E.; Martinez, F.J.; Woodruff, P.G.; et al. Design of the Subpopulations and Intermediate Outcomes in COPD Study (SPIROMICS): Table 1. Thorax 2014, 69, 492–495. [Google Scholar] [CrossRef]
- Bourbeau, J.; Tan, W.C.; Benedetti, A.; Aaron, S.D.; Chapman, K.R.; Coxson, H.O.; Cowie, R.; Fitzgerald, M.; Goldstein, R.; Hernandez, P.; et al. Canadian Cohort Obstructive Lung Disease (CanCOLD): Fulfilling the Need for Longitudinal Observational Studies in COPD. COPD: J. Chronic Obstr. Pulm. Dis. 2014, 11, 125–132. [Google Scholar] [CrossRef]
- Wu, Z.; Jiang, Y.; Li, S.; Li, A. Enhanced machine learning predictive modeling for delirium in elderly ICU patients with COPD and respiratory failure: A retrospective study based on MIMIC-IV. PLoS ONE 2025, 20, e0319297. [Google Scholar] [CrossRef]
- Penzel, T.; Moody, G.B.; Mark, R.G.; Goldberger, A.L.; Peter, J.H. The apnea-ECG database. In Computers in Cardiology 2000; Cat 00CH37163; IEEE: Cambridge, MA, USA, 2002; Volume 27, pp. 255–258. [Google Scholar]
- Goldberger, A.; Amaral, L.; Glass, L.; Hausdorff, J.; Ivanov, P.C.; Mark, R.; Mietus, J.E.; Moody, G.B.; Peng, C.K.; Stanley, H.E. St. Vincent’s University Hospital/University College Dublin Sleep Apnea Database; PhysioNet: Cambridge, MA, USA, 2007. [Google Scholar]
- Quan, S.F.; Howard, B.V.; Iber, C.; Kiley, J.P.; Nieto, F.J.; O’Connor, G.T.; Rapoport, D.M.; Redline, S.; Robbins, J.; Samet, J.M.; et al. The Sleep Heart Health Study: Design, rationale, and methods. Sleep 1997, 20, 1077–1085. [Google Scholar] [PubMed]
- Talker, L.; Dogan, C.; Neville, D.; Lim, R.H.; Broomfield, H.; Lambert, G.; Selim, A.; Brown, T.; Wiffen, L.; Carter, J.; et al. Diagnosis and Severity Assessment of COPD Using a Novel Fast-Response Capnometer and Interpretable Machine Learning. COPD: J. Chronic Obstr. Pulm. Dis. 2024, 21, 2321379. [Google Scholar] [CrossRef] [PubMed]
- Atzeni, M.; Cappon, G.; Quint, J.K.; Kelly, F.; Barratt, B.; Vettoretti, M. A machine learning framework for short-term prediction of chronic obstructive pulmonary disease exacerbations using personal air quality monitors and lifestyle data. Sci. Rep. 2025, 15, 2385. [Google Scholar] [CrossRef]
- Yin, H.; Wang, K.; Yang, R.; Tan, Y.; Li, Q.; Zhu, W.; Sung, S. A machine learning model for predicting acute exacerbation of in-home chronic obstructive pulmonary disease patients. Comput. Methods Programs Biomed. 2024, 246, 108005. [Google Scholar] [CrossRef]
- Cheplygina, V.; Pena, I.P.; Pedersen, J.H.; Lynch, D.A.; Sorensen, L.; de Bruijne, M. Transfer Learning for Multicenter Classification of Chronic Obstructive Pulmonary Disease. IEEE J. Biomed. Health Inform. 2018, 22, 1486–1496. [Google Scholar] [CrossRef]
- Li, Z.; Liu, L.; Zhang, Z.; Yang, X.; Li, X.; Gao, Y.; Huang, K. A Novel CT-Based Radiomics Features Analysis for Identification and Severity Staging of COPD. Acad. Radiol. 2022, 29, 663–673. [Google Scholar] [CrossRef]
- Amudala Puchakayala, P.R.; Sthanam, V.L.; Nakhmani, A.; Chaudhary, M.F.A.; Kizhakke Puliyakote, A.; Reinhardt, J.M.; Zhang, C.; Bhatt, S.P.; Bodduluri, S. Radiomics for Improved Detection of Chronic Obstructive Pulmonary Disease in Low-Dose and Standard-Dose Chest CT Scans. Radiology 2023, 307, e222998. [Google Scholar] [CrossRef] [PubMed]
- Moslemi, A.; Kontogianni, K.; Brock, J.; Wood, S.; Herth, F.; Kirby, M. Differentiating COPD and asthma using quantitative CT imaging and machine learning. Eur. Respir. J. 2022, 60, 2103078. [Google Scholar] [CrossRef]
- Makimoto, K.; Hogg, J.C.; Bourbeau, J.; Tan, W.C.; Kirby, M. CT Imaging With Machine Learning for Predicting Progression to COPD in Individuals at Risk. Chest 2023, 164, 1139–1149. [Google Scholar] [CrossRef]
- Guan, Y.; Zhang, D.; Zhou, X.; Xia, Y.; Lu, Y.; Zheng, X.; He, C.; Liu, S.; Fan, L. Comparison of deep-learning and radiomics-based machine-learning methods for the identification of chronic obstructive pulmonary disease on low-dose computed tomography images. Quant. Imaging Med. Surg. 2024, 14, 2485–2498. [Google Scholar] [CrossRef]
- Liao, K.-M.; Liu, C.-F.; Chen, C.-J.; Shen, Y.-T. Machine Learning Approaches for Predicting Acute Respiratory Failure, Ventilator Dependence, and Mortality in Chronic Obstructive Pulmonary Disease. Diagnostics 2021, 11, 2396. [Google Scholar] [CrossRef]
- Moll, M.; Qiao, D.; Regan, E.A.; Hunninghake, G.M.; Make, B.J.; Tal-Singer, R.; McGeachie, M.; Castaldi, P.J.; Estepar, R.S.J.; Washko, G.R.; et al. Machine Learning and Prediction of All-Cause Mortality in COPD. Chest 2020, 158, 952–964. [Google Scholar] [CrossRef]
- Zeng, S.; Arjomandi, M.; Tong, Y.; Liao, Z.C.; Luo, G. Developing a Machine Learning Model to Predict Severe Chronic Obstructive Pulmonary Disease Exacerbations: Retrospective Cohort Study. J. Med. Internet Res. 2022, 24, e28953. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Emami, E.; Kauffmann, C.; Rompré, P.; Almeida, F.; Schmittbuhl, M.; van der Stelt, P.; Ge, S.; Lavigne, G.; Huynh, N. Airway Phenotypes and Nocturnal Wearing of Dentures in Elders with Sleep Apnea. J. Dent. Res. 2023, 102, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Atalay, N.A.; Yormaz, B.; Şahin, A.; Sonmez, G.; Colkesen, S.S.; Vatansev, H.; Ozer, H.; Korez, M.K. Elevated HIF-1α as a Diagnostic Biomarker in COPD: Correlations with EPO, Emphysema Index, and Pulmonary Function. Respir. Med. 2025, 108184. [Google Scholar] [CrossRef]
- Liu, C.; Wang, H.; Zhu, C.; Wang, S. Plasma expression of HIF-1α as novel biomarker for the diagnosis of obstructive sleep apnea-hypopnea syndrome. J. Clin. Lab. Anal. 2020, 34, e23545. [Google Scholar] [CrossRef]
- Luo, H.; Yan, J.; Gong, R.; Zhang, D.; Zhou, X.; Wang, X. Identification of biomarkers and pathways for the SARS-CoV-2 infections in obstructive sleep apnea patients based on machine learning and proteomic analysis. BMC Pulm. Med. 2024, 24, 112. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Jiang, C.; Dai, Z.; Xie, B.; Chen, Q.; Lin, J. Identification and verification of mitochondria-related genes biomarkers associated with immune infiltration for COPD using WGCNA and machine learning algorithms. Sci. Rep. 2025, 15, 14347. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Dong, L.; Chi, W.; Song, D. Machine learning identifies potential diagnostic biomarkers associated with ferroptosis in obstructive sleep apnea. Exp. Ther. Med. 2025, 29, 95. [Google Scholar] [CrossRef]
- Zuur-Telgen, M.C.; Citgez, E.; Zuur, A.T.; VanderValk, P.; van der Palen, J.; Kerstjens, H.A.M.; Brusse-Keizer, M. Predicting Mortality in COPD with Validated and Sensitive Biomarkers; Fibrinogen and Mid-Range-Proadrenomedullin (MR-proADM). COPD J. Chronic Obstr. Pulm. Dis. 2021, 18, 643–649. [Google Scholar] [CrossRef]
- Luo, X.; Zeng, W.; Tang, J.; Liu, W.; Yang, J.; Chen, H.; Jiang, L.; Zhou, X.; Huang, J.; Zhang, S.; et al. Multi-modal transcriptomic analysis reveals metabolic dysregulation and immune responses in chronic obstructive pulmonary disease. Sci. Rep. 2024, 14, 22699. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Xing, F.; Ghosh, D.; Hobbs, B.D.; Hersh, C.P.; Banaei-Kashani, F.; Bowler, R.P.; Kechris, K.; Liu, G. Deep learning on graphs for multi-omics classification of COPD. PLoS ONE 2023, 18, e0284563. [Google Scholar] [CrossRef]
- Abràmoff, M.D.; Lavin, P.T.; Birch, M.; Shah, N.; Folk, J.C. Pivotal trial of an autonomous AI-based diagnostic system for detection of diabetic retinopathy in primary care offices. NPJ Digit. Med. 2018, 1, 39. [Google Scholar] [CrossRef]
- Wu, Y.; Xia, S.; Liang, Z.; Chen, R.; Qi, S. Artificial intelligence in COPD CT images: Identification, staging, and quantitation. Respir. Res. 2024, 25, 319. [Google Scholar] [CrossRef]
- Quer, G.; Topol, E.J. The potential for large language models to transform cardiovascular medicine. Lancet Digit. Health 2024, 6, e767–e771. [Google Scholar] [CrossRef]
- Moor, M.; Banerjee, O.; Abad, Z.S.H.; Krumholz, H.M.; Leskovec, J.; Topol, E.J.; Rajpurkar, P. Foundation models for generalist medical artificial intelligence. Nature 2023, 616, 259–265. [Google Scholar] [CrossRef]
- Rasmy, L.; Xiang, Y.; Xie, Z.; Tao, C.; Zhi, D. Med-BERT: Pretrained contextualized embeddings on large-scale structured electronic health records for disease prediction. NPJ Digit. Med. 2021, 4, 86. [Google Scholar] [CrossRef]
- Yang, S.; Yang, X.; Lyu, T.; Huang, J.L.; Chen, A.; He, X.; Braithwaite, D.; Mehta, H.J.; Wu, Y.; Guo, Y.; et al. Extracting Pulmonary Nodules and Nodule Characteristics from Radiology Reports of Lung Cancer Screening Patients Using Transformer Models. J. Healthc. Inform. Res. 2024, 8, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Gyanchandani, B.; Oza, A.; Singh, A. TriSpectraKAN: A novel approach for COPD detection via lung sound analysis. Sci. Rep. 2025, 15, 6296. [Google Scholar] [CrossRef] [PubMed]
- Vodnala, N.; Yarlagadda, P.S.; Bhuvana, K.S.; Ch, M.; Sailaja, K. Novel Deep Learning Approaches to Differentiate Asthma and COPD Based on Cough Sounds. In Proceedings of the 2024 Parul International Conference on Engineering and Technology (PICET), Vadodara, India, 3–4 May 2024; IEEE: Piscataway, NJ, USA, 2004; pp. 1–4. [Google Scholar]
- Bodduluri, S.; Nakhmani, A.; Reinhardt, J.M.; Wilson, C.G.; McDonald, M.-L.; Rudraraju, R.; Jaeger, B.C.; Bhakta, N.R.; Castaldi, P.J.; Sciurba, F.C.; et al. Deep neural network analyses of spirometry for structural phenotyping of chronic obstructive pulmonary disease. JCI Insight 2020, 5, e132781. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Li, X.; Zhou, Y.; Xu, J.; Zhang, Y.; Wan, Y.; Cao, S.; Zhao, Q.; Geng, S.; Xie, J.; et al. Deep learning for detecting and early predicting chronic obstructive pulmonary disease from spirogram time series. NPJ Syst. Biol. Appl. 2025, 11, 18. [Google Scholar] [CrossRef]
- Gadgil, S.; Galanter, J.; Negahdar, M. Transformer-based Time-Series Biomarker Discovery for COPD Diagnosis. arXiv 2024. [Google Scholar] [CrossRef]
- Taghizadegan, Y.; Jafarnia Dabanloo, N.; Maghooli, K.; Sheikhani, A. Prediction of obstructive sleep apnea using ensemble of recurrence plot convolutional neural networks (RPCNNs) from polysomnography signals. Med. Hypotheses 2021, 154, 110659. [Google Scholar] [CrossRef]
- Niroshana, S.M.I.; Zhu, X.; Nakamura, K.; Chen, W. A fused-image-based approach to detect obstructive sleep apnea using a single-lead ECG and a 2D convolutional neural network. PLoS ONE 2021, 16, e0250618. [Google Scholar] [CrossRef]
- Mukherjee, D.; Dhar, K.; Schwenker, F.; Sarkar, R. Ensemble of Deep Learning Models for Sleep Apnea Detection: An Experimental Study. Sensors 2021, 21, 5425. [Google Scholar] [CrossRef]
- Mashrur, F.R.; Islam, M.d.S.; Saha, D.K.; Islam, S.M.R.; Moni, M.A. SCNN: Scalogram-based convolutional neural network to detect obstructive sleep apnea using single-lead electrocardiogram signals. Comput. Biol. Med. 2021, 134, 104532. [Google Scholar] [CrossRef]
- Wang, T.; Lu, C.; Shen, G.; Hong, F. Sleep apnea detection from a single-lead ECG signal with automatic feature-extraction through a modified LeNet-5 convolutional neural network. PeerJ 2019, 7, e7731. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-Y.; Yeh, C.-Y.; Lee, C.-T.; Lin, C.-C. A Sleep Apnea Detection System Based on a One-Dimensional Deep Convolution Neural Network Model Using Single-Lead Electrocardiogram. Sensors 2020, 20, 4157. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, L.; Zhu, S.; Zhou, Y.; Wang, Z.; Ma, L.; Yuan, Y.; Xie, Y.; Niu, X.; Su, Y.; et al. Deep Learning for Obstructive Sleep Apnea Detection and Severity Assessment: A Multimodal Signals Fusion Multiscale Transformer Model. Nat. Sci. Sleep 2025, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.Y.W.; Coxson, H.O.; Lam, S.; Leipsic, J.; Tam, R.C.; Sin, D.D. Towards large-scale case-finding: Training and validation of residual networks for detection of chronic obstructive pulmonary disease using low-dose CT. Lancet Digit. Health 2020, 2, e259–e267. [Google Scholar] [CrossRef]
- Wu, Y.; Qi, S.; Feng, J.; Chang, R.; Pang, H.; Hou, J.; Li, M.; Wang, Y.; Xia, S.; Qian, W. Attention-guided multiple instance learning for COPD identification: To combine the intensity and morphology. Biocybern. Biomed. Eng. 2023, 43, 568–585. [Google Scholar] [CrossRef]
- Xu, C.; Qi, S.; Feng, J.; Xia, S.; Kang, Y.; Yao, Y.; Qian, W. DCT-MIL: Deep CNN transferred multiple instance learning for COPD identification using CT images. Phys. Med. Biol. 2020, 65, 145011. [Google Scholar] [CrossRef]
- Yu, K.; Sun, L.; Chen, J.; Reynolds, M.; Chaudhary, T.; Batmanghelich, K. DrasCLR: A self-supervised framework of learning disease-related and anatomy-specific representation for 3D lung CT images. Med. Image Anal. 2024, 92, 103062. [Google Scholar] [CrossRef]
- González, G.; Ash, S.Y.; Vegas-Sánchez-Ferrero, G.; Onieva Onieva, J.; Rahaghi, F.N.; Ross, J.C.; Díaz, A.; Estépar, R.S.J.; Washko, G.R.; Copdgene, F.T. Disease Staging and Prognosis in Smokers Using Deep Learning in Chest Computed Tomography. Am. J. Respir. Crit. Care Med. 2018, 197, 193–203. [Google Scholar] [CrossRef]
- Zou, X.; Ren, Y.; Yang, H.; Zou, M.; Meng, P.; Zhang, L.; Gong, M.; Ding, W.; Han, L.; Zhang, T. Screening and staging of chronic obstructive pulmonary disease with deep learning based on chest X-ray images and clinical parameters. BMC Pulm. Med. 2024, 24, 153. [Google Scholar] [CrossRef]
- Schroeder, J.D.; Bigolin Lanfredi, R.; Li, T.; Chan, J.; Vachet, C.; Paine, R.; Srikumar, V.; Tasdizen, T. Prediction of Obstructive Lung Disease from Chest Radiographs via Deep Learning Trained on Pulmonary Function Data. Int. J. Chronic Obstr. Pulm. Dis. 2021, 15, 3455–3466. [Google Scholar] [CrossRef]
- Weiss, J.; Raghu, V.K.; Bontempi, D.; Christiani, D.C.; Mak, R.H.; Lu, M.T.; Aerts, H.J. Deep learning to estimate lung disease mortality from chest radiographs. Nat. Commun. 2023, 14, 2797. [Google Scholar] [CrossRef]
- Ryu, S.; Kim, J.H.; Yu, H.; Jung, H.-D.; Chang, S.W.; Park, J.J.; Hong, S.; Cho, H.-J.; Choi, Y.J.; Choi, J.; et al. Diagnosis of obstructive sleep apnea with prediction of flow characteristics according to airway morphology automatically extracted from medical images: Computational fluid dynamics and artificial intelligence approach. Comput. Methods Programs Biomed. 2021, 208, 106243. [Google Scholar] [CrossRef] [PubMed]
- Tsuiki, S.; Nagaoka, T.; Fukuda, T.; Sakamoto, Y.; Almeida, F.R.; Nakayama, H.; Inoue, Y.; Enno, H. Machine learning for image-based detection of patients with obstructive sleep apnea: An exploratory study. Sleep Breath. 2021, 25, 2297–2305. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Ye, M.; Xiao, C.; Ma, F. HiTANet: Hierarchical Time-Aware Attention Networks for Risk Prediction on Electronic Health Records. In Proceedings of the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, San Diego, CA, USA, 23–27 August 2020; pp. 647–656. [Google Scholar]
- Lopez, K.; Li, H.; Lipkin-Moore, Z.; Kay, S.; Rajeevan, H.; Davis, J.L.; Wilson, F.P.; Rochester, C.L.; Gomez, J.L. Deep learning prediction of hospital readmissions for asthma and COPD. Respir. Res. 2023, 24, 311. [Google Scholar] [CrossRef] [PubMed]
- Radford, A.; Kim, J.W.; Hallacy, C.; Ramesh, A.; Goh, G.; Agarwal, S.; Sastry, G.; Askell, A.; Mishkin, P.; Clark, J.; et al. Learning Transferable Visual Models From Natural Language Supervision. arXiv 2021. [Google Scholar] [CrossRef]
- Lee, J.; Yoon, W.; Kim, S.; Kim, D.; Kim, S.; So, C.H.; Kang, J.; Wren, J. BioBERT: A pre-trained biomedical language representation model for biomedical text mining. Bioinformatics 2020, 36, 1234–1240. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, T.; Han, J.; Wu, Y.; Rizos, G.; Liu, Y.; Mosuily, M.; Ch, J.; Mascolo, C. Towards Open Respiratory Acoustic Foundation Models: Pretraining and Benchmarking. arXiv 2024. [Google Scholar] [CrossRef]
- Eyben, F.; Wöllmer, M.; Schuller, B. Opensmile. In Proceedings of the 18th ACM International Conference on Multimedia, Firenze, Italy, 25–29 October 2010; pp. 1459–1462. [Google Scholar]
- Hershey, S.; Chaudhuri, S.; Ellis, D.P.W.; Gemmeke, J.F.; Jansen, A.; Moore, R.C.; Plakal, M.; Platt, D.; Saurous, R.A.; Seybold, B.; et al. CNN architectures for large-scale audio classification. In Proceedings of the 2017 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP), New Orleans, LA, USA, 5–9 March 2017; IEEE: Piscataway, NJ, USA, 2017; pp. 131–135. [Google Scholar]
- Huang, P.-Y.; Xu, H.; Li, J.; Baevski, A.; Auli, M.; Galuba, W.; Metze, F.; Feichtenhofer, C. Masked Autoencoders that Listen. In Proceedings of the NeurIPS Proceedings, New Orleans, LA, USA, 28–29 December 2022. [Google Scholar]
- Elizalde, B.; Deshmukh, S.; Ismail MAl Wang, H. CLAP Learning Audio Concepts from Natural Language Supervision. In Proceedings of the ICASSP 2023—2023 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP), Rhodes Island, Greece, 4–10 June 2023; IEEE: Piscataway, NJ, USA, 2023; pp. 1–5. [Google Scholar]
- Niu, C.; Lyu, Q.; Carothers, C.D.; Kaviani, P.; Tan, J.; Yan, P.; Kalra, M.K.; Whitlow, C.T.; Wang, G. Medical multimodal multitask foundation model for lung cancer screening. Nat. Commun. 2025, 16, 1523. [Google Scholar] [CrossRef]
- Mikhael, P.G.; Wohlwend, J.; Yala, A.; Karstens, L.; Xiang, J.; Takigami, A.K.; Bourgouin, P.P.; Chan, P.; Mrah, S.; Amayri, W.; et al. Sybil: A Validated Deep Learning Model to Predict Future Lung Cancer Risk From a Single Low-Dose Chest Computed Tomography. J. Clin. Oncol. 2023, 41, 2191–2200. [Google Scholar] [CrossRef]
- Chao, H.; Shan, H.; Homayounieh, F.; Singh, R.; Khera, R.D.; Guo, H.; Su, T.; Wang, G.; Kalra, M.K.; Yan, P. Deep learning predicts cardiovascular disease risks from lung cancer screening low dose computed tomography. Nat. Commun. 2021, 12, 2963. [Google Scholar] [CrossRef]
- Herrett, E.; Gallagher, A.M.; Bhaskaran, K.; Forbes, H.; Mathur, R.; van Staa, T.; Smeeth, L. Data Resource Profile: Clinical Practice Research Datalink (CPRD). Int. J. Epidemiol. 2015, 44, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Miotto, R.; Li, L.; Kidd, B.A.; Dudley, J.T. Deep Patient: An Unsupervised Representation to Predict the Future of Patients from the Electronic Health Records. Sci. Rep. 2016, 6, 26094. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.; Bahadori, M.T.; Kulas, J.A.; Schuetz, A.; Stewart, W.F.; Sun, J. RETAIN: An Interpretable Predictive Model for Healthcare Using Reverse Time Attention Mechanism.; Curran Associates Inc.: Red Hook, NY, USA, 2016. [Google Scholar]
- Science & Tech Spotlight: Generative AIin Health Care; U.S. Government Accountability Office: Washington, DC, USA, 2024.
- Topol, E.J. High-performance medicine: The convergence of human and artificial intelligence. Nat. Med. 2019, 25, 44–56. [Google Scholar] [CrossRef] [PubMed]
- EU 2024/1689; Regulation (EU) 2024/1689 of the European Parliament and of the Council of 13 June 2024 Laying down Harmonised Rules on Artificial Intelligence and Amending Regulations (EC) No 300/2008, (EU) No 167/2013, (EU) No 168/2013, (EU) 2018/858, (EU) 2018/1139 and (EU) 2019/2144 and Directives 2014/90/EU, (EU) 2016/797 and (EU) 2020/1828 (Artificial Intelligence Act) (Text with EEA Relevance). European Union: Brussels, Belgium, 2024.
- DeGroot, M.H. Optimal Statistical Decisions; Wiley: Hoboken, NJ, USA, 2004. [Google Scholar]
- Kevin, P.M. Probabilistic Machine Learning: An introduction; MIT Press: Cambridge, MA, USA, 2022. [Google Scholar]
- Dawid, A.P. The Well-Calibrated Bayesian. J. Am. Stat. Assoc. 1982, 77, 605–610. [Google Scholar] [CrossRef]
- Ferrer, L.; Ramos, D. Evaluating Posterior Probabilities: Decision Theory, Proper Scoring Rules, and Calibration. Trans. Mach. Learn. Res. 2025. [Google Scholar] [CrossRef]
- Guo, C.; Pleiss, G.; Sun, Y.; Weinberger, K.Q. On calibration of modern neural networks. In Proceedings of the 34th International Conference on Machine Learning, Sydney, Australia, 6–11 August 2017; pp. 1321–1330. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).