
Alzheimer’s Disease Pathogenic Mechanisms: Linking Redox Homeostasis and Mitochondria-Associated Metabolic Pathways Through Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Brain Energy Requirements and the Essential Role of Mitochondria
3. Maintaining Cell Redox Homeostasis
3.1. Generation and Regulation of Reactive Oxygen Species
3.2. Production and Regulation of Reactive Nitrogen Species
4. Mitochondrial and Bioenergetic Alterations in Alzheimer’s Disease
5. Mitochondrial and Bioenergetic Alterations in Transgenic Models
6. Linking ROS Homeostasis and Mitochondria-Associated Metabolic Pathways Through Nrf2
6.1. Structural Characteristics of Nrf2
6.2. Regulation and Activation of Nrf2
6.3. Nrf2 Network Links Cell Metabolic Paths, Redox Homeostasis, and Blood-Brain Barrier Integrity
6.4. Nrf2 in Aging and Alzheimer’s Disease
7. Therapeutic Strategies for Alzheimer’s Disease Targeting Oxidative Stress and Mitochondrial Dysfunction
7.1. Mitochondria-Targeted Antioxidants
7.2. Phytochemicals and Natural Antioxidants
7.3. Compounds Targeting Nrf2 Pathway
7.3.1. Targeting Keap-l Mediated Nrf2 Degradation
7.3.2. Targeting TrCP-Mediated Nrf2 Degradation
8. Conclusions and Future Directions
Author Contributions
Funding
Institutional Board Statement
Data Availability Statement
Conflicts of Interest
References
- Lanctôt, K.L.; Hviid Hahn-Pedersen, J.; Eichinger, C.S.; Freeman, C.; Clark, A.; Tarazona, L.R.S.; Cummings, J. Burden of Illness in People with Alzheimer’s Disease: A Systematic Review of Epidemiology, Comorbidities and Mortality. J. Prev. Alzheimers Dis. 2024, 11, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Rostagno, A.; Holton, J.L.; Lashley, T.; Revesz, T.; Ghiso, J. Cerebral amyloidosis: Amyloid subunits, mutants and phenotypes. Cell. Mol. Life Sci. 2010, 67, 581–600. [Google Scholar] [CrossRef] [PubMed]
- Neuner, S.M.; Tcw, J.; Goate, A.M. Genetic architecture of Alzheimer’s disease. Neurobiol. Dis. 2020, 143, 104976. [Google Scholar] [CrossRef]
- Hrelia, P.; Sita, G.; Ziche, M.; Ristori, E.; Marino, A.; Cordaro, M.; Molteni, R.; Spero, V.; Malaguti, M.; Morroni, F.; et al. Common Protective Strategies in Neurodegenerative Disease: Focusing on Risk Factors to Target the Cellular Redox System. Oxid. Med. Cell. Longev. 2020, 2020, 8363245. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr71. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Selkoe, D.J. A beta oligomers—a decade of discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef]
- Caughey, B.; Lansbury, P.T.J. Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the inocent bystanders. Annu. Rev. Neurosci. 2003, 26, 267–298. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Shea, D.; Daggett, V. Amyloid-β Oligomers: Multiple Moving Targets. Biophysica 2022, 2, 91–110. [Google Scholar] [CrossRef]
- Viola, K.L.; Bicca, M.A.; Bebenek, A.M.; Kranz, D.L.; Nandwana, V.; Waters, E.A.; Haney, C.R.; Lee, M.; Gupta, A.; Brahmbhatt, Z.; et al. The Therapeutic and Diagnostic Potential of Amyloid β Oligomers Selective Antibodies to Treat Alzheimer’s Disease. Front. Neurosci. 2022, 15, 768646. [Google Scholar] [CrossRef] [PubMed]
- Rasool, S.; Martinez-Coria, H.; Wu, J.W.; LaFerla, F.; Glabe, C.G. Systemic vaccination with anti-oligomeric monoclonal antibodies improves cognitive function by reducing Aβ deposition and tau pathology in 3xTg-AD mice. J. Neurochem. 2013, 126, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Kamatham, P.T.; Shukla, R.; Khatri, D.K.; Vora, L.K. Pathogenesis, diagnostics, and therapeutics for Alzheimer’s disease: Breaking the memory barrier. Ageing Res. Rev. 2024, 101, 102481. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid β-based therapy for Alzheimer’s disease: Challenges, successes and future. Signal Transduct. Target. Ther. 2023, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.R.; Bowen, D.M.; Davison, A.N. [14C]acetylcholine synthesis and [14C]carbon dioxide production from [U-14C]glucose by tissue prisms from human neocortex. Biochem. J. 1981, 196, 867–876. [Google Scholar] [CrossRef]
- Sims, N.R.; Bowen, D.M.; Neary, D.; Davison, A.N. Metabolic processes in Alzheimer’s disease: Adenine nucleotide content and production of 14CO2 from [U-14C]glucose in vitro in human neocortex. J. Neurochem. 1983, 41, 1329–1334. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.R.; Finegan, J.M.; Blass, J.P.; Bowen, D.M.; Neary, D. Mitochondrial function in brain tissue in primary degenerative dementia. Brain Res. 1987, 436, 30–38. [Google Scholar] [CrossRef]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 2010, 1802, 2–10. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, M.C.B.; Kanaan, S.; Geller, M.; Praticò, D.; Daher, J.P.L. Mitochondrial dysfunction in Alzheimer’s disease. Ageing Res. Rev. 2025, 107, 102713. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheime’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef]
- Su, B.; Wang, X.; Nunomura, A.; Moreira, P.I.; Lee, H.G.; Perry, G.; Smith, M.A.; Zhu, X. Oxidative stress signaling in Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Nantachai, G.; Vasupanrajit, A.; Tunvirachaisakul, C.; Solmi, M.; Maes, M. Oxidative stress and antioxidant defenses in mild cognitive impairment: A systematic review and meta-analysis. Ageing Res. Rev. 2022, 79, 101639. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Poon, H.F.; St Clair, D.; Keller, J.N.; Pierce, W.M.; Klein, J.B.; Markesbery, W.R. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: Insights into the development of Alzheimer’s disease. Neurobiol. Dis. 2006, 22, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Schmitt, F.A.; Scheff, S.W.; Ding, Q.; Chen, Q.; Butterfield, D.A.; Markesbery, W.R. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 2005, 64, 1152–1156. [Google Scholar] [CrossRef]
- Maes, M.; Galecki, P.; Chang, Y.S.; Berk, M. A review on the oxidative and nitrosative stress (O&NS) pathways in major depression and their possible contribution to the (neuro)degenerative processes in that illness. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 676–692. [Google Scholar] [PubMed]
- Torres, L.L.; Quaglio, N.B.; de Souza, G.T.; Garcia, R.T.; Dati, L.M.; Moreira, W.L.; Loureiro, A.P.; de Souza-Talarico, J.N.; Smid, J.; Porto, C.S.; et al. Peripheral oxidative stress biomarkers in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis. 2011, 26, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Schrag, M.; Mueller, C.; Zabel, M.; Crofton, A.; Kirsch, W.M.; Ghribi, O.; Squitti, R.; Perry, G. Oxidative stress in blood in Alzheimer’s disease and mild cognitive impairment: A meta-analysis. Neurobiol. Dis. 2013, 59, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Fossati, S.; Ghiso, J.; Rostagno, A. Insights into Caspase-Mediated Apoptotic Pathways Induced by Amyloid-β in Cerebral Microvascular Endothelial Cells. Neurodegener. Dis. 2012, 10, 324–328. [Google Scholar] [CrossRef]
- Fossati, S.; Cam, J.; Meyerson, J.; Mezhericher, E.; Romero, I.A.; Couraud, P.-O.; Weksler, B.; Ghiso, J.; Rostagno, A. Differential activation of mitochondrial apoptotic pathways by vasculotropic amyloid-β variants in cells composing the cerebral vessel walls. FASEB J. 2010, 24, 229–241. [Google Scholar] [CrossRef]
- Viana, R.J.; Nunes, A.F.; Castro, R.E.; Ramalho, R.M.; Meyerson, J.; Fossati, S.; Ghiso, J.; Rostagno, A.; Rodrigues, C.M. Tauroursodeoxycholic acid prevents E22Q Alzheimer’s Abeta toxicity in human cerebral endothelial cells. Cell. Mol. Life Sci. 2009, 66, 1094–1104. [Google Scholar] [CrossRef]
- Perez-Cruz, C.; Nolte, M.W.; van Gaalen, M.M.; Rustay, N.R.; Termont, A.; Tanghe, A.; Kirchhoff, F.; Ebert, U. Reduced spine density in specific regions of CA1 pyramidal neurons in two transgenic mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 3926–3934. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.J.; Sheu, J.R.; Lin, C.H.; Shen, M.Y.; Hsu, C.Y. Mitochondrial mechanisms in amyloid beta peptide-induced cerebrovascular degeneration. Biochim. Biophys. Acta 2010, 1800, 290–296. [Google Scholar] [CrossRef]
- Pozueta, J.; Lefort, R.; Ribe, E.M.; Troy, C.M.; Arancio, O.; Shelanski, M. Caspase-2 is required for dendritic spine and behavioural alterations in J20 APP transgenic mice. Nat. commun. 2013, 4, 1939. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.T.; Zhu, S.; Younger, S.; Jan, L.Y.; Jan, Y.N. Identification of E2/E3 ubiquitinating enzymes and caspase activity regulating Drosophila sensory neuron dendrite pruning. Neuron 2006, 51, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.W.; Kondo, S.; Krzyzanowska, A.; Hiromi, Y.; Truman, J.W. Local caspase activity directs engulfment of dendrites during pruning. Nat. Neurosci. 2006, 9, 1234–1236. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.; Tessier-Lavigne, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 457, 981–989. [Google Scholar] [CrossRef]
- Li, Z.; Jo, J.; Jia, J.M.; Lo, S.C.; Whitcomb, D.J.; Jiao, S.; Cho, K.; Sheng, M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 2010, 141, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Li, Z. Nonapoptotic function of BAD and BAX in long-term depression of synaptic transmission. Neuron 2011, 70, 758–772. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Yan, S. Synaptic mitochondrial pathology in Alzheimer’s disease. Antioxid. Redox Signal. 2012, 16, 1467–1475. [Google Scholar] [CrossRef]
- Lee, J.-T.; Xu, J.; Lee, J.-M.; Ku, G.; Han, X.; Yang, D.-I.; Chen, S.; Hsu, C.Y. Amyloid-ß peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J. Cell Biol. 2004, 164, 123–131. [Google Scholar] [CrossRef]
- Xu, J.; Chen, S.; Ahmed, S.H.; Chen, H.; Ku, G.; Goldberg, M.P.; Hsu, C.Y. Amyloid-beta peptides are cytotoxic to oligodendrocytes. J. Neurosci. 2001, 21, RC118. [Google Scholar] [CrossRef] [PubMed]
- Klein, W.L. Synaptic targeting by Aβ oligomers (ADDLS) as a basis for memory loss in early Alzheimer’s disease. Alzheimer’s Dement. 2006, 2, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Brkic, M.; Balusu, S.; Libert, C.; Vandenbroucke, R.E. Friends or Foes: Matrix Metalloproteinases and Their Multifaceted Roles in Neurodegenerative Diseases. Mediators Inflamm. 2015, 2015, 620581. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, S.; Jiang, Q.; Lee, C.Y.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Landreth, G.E. Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J. Neurosci. 2009, 29, 4252–4262. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef]
- Olszewska, A.; Szewczyk, A. Mitochondria as a pharmacological target: Magnum overview. IUBMB Life 2013, 65, 273–281. [Google Scholar] [CrossRef]
- Dumont, M.; Beal, M.F. Neuroprotective strategies involving ROS in Alzheimer disease. Free. Rad. Biol. Med. 2010, 51, 1014–1026. [Google Scholar] [CrossRef]
- Rohn, T.T.; Head, E.; Nesse, W.H.; Cotman, C.W.; Cribbs, D.H. Activation of caspase-8 in the Alzheimer’s disease brain. Neurobiol. Dis. 2001, 8, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Hoeffer, C.A.; Wong, H.; Massaad, C.A.; Zhou, P.; Iadecola, C.; Murphy, M.P.; Pautler, R.G.; Klann, E. Amyloid β-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. J. Neurosci. 2011, 31, 5589–5595. [Google Scholar] [CrossRef]
- Deshpande, A.; Mina, E.; Glabe, C.; Busciglio, J. Different conformations of amyloid β induce neurotoxicity by distinct mechanisms in human cortical neurons. J. Neurosci. 2006, 26, 6011–6018. [Google Scholar] [CrossRef]
- Gray, N.E.; Sampath, H.; Zweig, J.A.; Quinn, J.F.; Soumyanath, A. Centella asiatica Attenuates Amyloid-β-Induced Oxidative Stress and Mitochondrial Dysfunction. J. Alzheimers Dis. 2015, 45, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Folin, M.; Baiguera, S.; Fioravanzo, L.; Conconi, M.T.; Grandi, C.; Nussdorfer, G.G.; Parnigotto, P.P. Caspase-8 activation and oxidative stress are involved in the cytotoxic effect of beta-amyloid on rat brain microvascular endothelial cells. Int. J. Mol. Med. 2006, 17, 431–435. [Google Scholar]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef]
- Devi, L.; Ohno, M. Mitochondrial dysfunction and accumulation of the β-secretase-cleaved C-terminal fragment of APP in Alzheimer’s disease transgenic mice. Neurobiol. Dis. 2012, 45, 417–424. [Google Scholar] [CrossRef]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; McWeeney, S.; Park, B.S.; Manczak, M.; Gutala, R.V.; Partovi, D.; Jung, Y.; Yau, V.; Searles, R.; Mori, M.; et al. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: Up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum. Molec. Genet. 2004, 13, 1225–1240. [Google Scholar] [CrossRef]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Calingasan, N.Y.; Yu, F.; Mauck, W.M.; Toidze, M.; Almeida, C.G.; Takahashi, R.H.; Carlson, G.A.; Beal, M.F.; Lin, M.T.; et al. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J. Neurochem. 2004, 89, 13081312. [Google Scholar] [CrossRef]
- Eckert, A.; Hauptmann, S.; Scherping, I.; Rhein, V.; Müller-Spahn, F.; Götz, J.; Müller, W.E. Soluble beta-amyloid leads to mitochondrial defects in amyloid precursor protein and tau transgenic mice. Neurodegener. Dis. 2008, 5, 157–159. [Google Scholar] [CrossRef]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Dröse, S.; Brandt, U.; et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef]
- Drago, D.; Cavaliere, A.; Mascetra, N.; Ciavardelli, D.; di Ilio, C.; Zatta, P.; Sensi, S.L. Aluminum modulates effects of beta amyloid(1-42) on neuronal calcium homeostasis and mitochondria functioning and is altered in a triple transgenic mouse model of Alzheimer’s disease. Rejuvenation Res. 2008, 11, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Resende, R.; Moreira, P.I.; Proença, T.; Deshpande, A.; Busciglio, J.; Pereira, C.; Oliveira, C.R. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic. Biol. Med. 2008, 44, 2051–2057. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Rapposelli, I.G.; Frazzini, V.; Mascetra, N. Altered oxidant-mediated intraneuronal zinc mobilization in a triple transgenic mouse model of Alzheimer’s disease. Exp. Gerontol. 2008, 43, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef]
- Dragicevic, N.; Mamcarz, M.; Zhu, Y.; Buzzeo, R.; Tan, J.; Arendash, G.W.; Bradshaw, P.C. Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S535–S550. [Google Scholar] [CrossRef]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.J.; Reddy, P.H. Assessment of newly synthesized mitochondrial DNA using BrdU labeling in primary neurons from Alzheimer’s disease mice: Implications for impaired mitochondrial biogenesis and synaptic damage. Biochim. Biophys. Acta 2011, 1812, 1182–1189. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef]
- Bielarczyk, H.; Jankowska-Kulawy, A.; Höfling, C.; Ronowska, A.; Gul-Hinc, S.; Roßner, S.; Schliebs, R.; Pawelczyk, T.; Szutowicz, A. AβPP-Transgenic 2576 Mice Mimic Cell Type-Specific Aspects of Acetyl-CoA-Linked Metabolic Deficits in Alzheimer’s Disease. J. Alzheimers Dis. 2015, 48, 1083–1094. [Google Scholar] [CrossRef]
- Robinson, R.A.; Lange, M.B.; Sultana, R.; Galvan, V.; Fombonne, J.; Gorostiza, O.; Zhang, J.; Warrier, G.; Cai, J.; Pierce, W.M.; et al. Differential expression and redox proteomics analyses of an Alzheimer disease transgenic mouse model: Effects of the amyloid-β peptide of amyloid precursor protein. Neuroscience 2011, 17, 207–222. [Google Scholar] [CrossRef]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; Macvicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Luiten, P.G.M. Cerebral microvascular pathology in aging and Alzheimer’s disease. Progress Neurobiol. 2001, 64, 575–611. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Correia, S.C.; Santos, R.X.; Cardoso, S.; Moreira, P.I.; Clark, T.A.; Zhu, X.; Smith, M.A.; Perry, G. Role of mitochondrial-mediated signaling pathways in Alzheimer disease and hypoxia. J. Bioenerg. Biomembr. 2009, 41, 433–440. [Google Scholar] [CrossRef]
- Dalsgaard, M.K.; Ide, K.; Cai, Y.; Quistorff, B.; Secher, N.H. The intent to exercise influences the cerebral O(2)/carbohydrate uptake ratio in humans. J. Physiol. 2002, 540, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Graham, G.D. Arteriovenous malformations in the brain. Curr. Treat. Options Neurol. 2002, 4, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Fragkouli, A.; Papatheodoropoulos, C.; Georgopoulos, S.; Stamatakis, A.; Stylianopoulou, F.; Tsilibary, E.C.; Tzinia, A.K. Enhanced neuronal plasticity and elevated endogenous sAPPα levels in mice over-expressing MMP9. J. Neurochem. 2012, 121, 239–251. [Google Scholar] [CrossRef]
- D’Amelio, M.; Sheng, M.; Cecconi, F. Caspase-3 in the central nervous system: Beyond apoptosis. Trends Neurosci. 2012, 35, 700–709. [Google Scholar] [CrossRef]
- D’Amelio, M.; Cavallucci, V.; Middei, S.; Marchetti, C.; Pacioni, S.; Ferri, A.; Diamantini, A.; De Zio, D.; Carrara, P.; Battistini, L.; et al. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2011, 14, 69–76. [Google Scholar] [CrossRef]
- D’Amelio, M.; Cavallucci, V.; Cecconi, F. Neuronal caspase-3 signaling: Not only cell death. Cell Death Differ. 2010, 17, 1104–1114. [Google Scholar] [CrossRef]
- Giacomotto, J.; Pertl, C.; Borrel, C.; Walter, M.C.; Bulst, S.; Johnsen, B.; Baillie, D.L.; Lochmüller, H.; Thirion, C.; Ségalat, L. Evaluation of the therapeutic potential of carbonic anhydrase inhibitors in two animal models of dystrophin deficient muscular dystrophy. Hum. Mol. Genet. 2009, 18, 4089–4101. [Google Scholar] [CrossRef]
- Johnson, J.A.; Mercado-Ayon, E.; Mercado-Ayon, Y.; Dong, Y.N.; Halawani, S.; Ngaba, L.; Lynch, D.R. Mitochondrial dysfunction in the development and progression of neurodegenerative diseases. Arch. Biochem. Biophys. 2021, 702, 108698. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, A.; Jetto, C.T.; Manjithaya, R. Dysfunctional mitochondria and mitophagy as drivers of Alzheimer’s disease pathogenesis. Front. Aging Neurosci. 2019, 11, 311. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Zhao, X.; Song, F. Aging-Dependent Mitophagy Dysfunction in Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 2362–2378. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimer’s Dis. 2010, 20, S265–S279. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Voos, W. Mitochondria as potential targets in Alzheimer disease therapy: An update. Front. Pharmacol. 2019, 10, 1–20. [Google Scholar] [CrossRef]
- Park, H.; Kim, J.; Shin, C.; Lee, S. Intersection between Redox Homeostasis and Autophagy: Valuable Insights into Neurodegeneration. Antioxidants 2021, 10, 694. [Google Scholar] [CrossRef]
- Satrustegui, J.; Richter, C. The role of hydroperoxides as calcium release agents in rat brain mitochondria. Arch. Biochem. Biophys. 1984, 233, 736–740. [Google Scholar] [CrossRef]
- Adam-Vizi, V. Production of reactive oxygen species in brain mitochondria: Contribution by electron transport chain and non-electron transport chain sources. Antioxid. Redox Signal. 2005, 7, 1140–1149. [Google Scholar] [CrossRef]
- Halliwell, B. Free radicals and antioxidants: Updating a personal view. Nutr. Rev. 2012, 70, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Formella, I.; Svahn, A.J.; Radford, R.A.W.; Don, E.K.; Cole, N.J.; Hogan, A.; Lee, A.; Chung, R.S.; Morsch, M. Real-time visualization of oxidative stress-mediated neurodegeneration of individual spinal motor neurons in vivo. Redox Biol. 2018, 19, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F.; Boveris, A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem. J. 1980, 191, 421–427. [Google Scholar] [CrossRef]
- Turrens, J.F.; Freeman, B.A.; Levitt, J.G.; Crapo, J.D. The effect of hyperoxia on superoxide production by lung submitochondrial particles. Arch. Biochem. Biophys. 1982, 217, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef]
- Forman, H.J.; Azzi, A. On the virtual existence of superoxide anions in mitochondria: Thoughts regarding its role in pathophysiology. FASEB J. 1997, 11, 374–375. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Loschen, G.; Azzi, A.; Flohe, L. Mitochondrial H2O2 formation: Relationship with energy conservation. FEBS Lett. 1973, 33, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Loschen, G.; Azzi, A.; Richter, C.; Flohe, L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974, 42, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Maiorino, M.; Ursini, F. Signaling functions of reactive oxygen species. Biochemistry 2010, 49, 835–842. [Google Scholar] [CrossRef]
- Alfonso-Pireto, M.; Biarnes, X.; Vidossich, P.; Rovira, C. The molecular mechanism of the catalase reaction. J. Am. Chem. Soc. 2009, 131, 11751–11761. [Google Scholar] [CrossRef] [PubMed]
- Desagher, S.; Glowinski, J.; Premont, J. Astrocytes protect neurons from hydrogen peroxide toxicity. J. Neurosci. 1996, 16, 2553–2562. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Mustacich, D.; Powis, G. Thioredoxin reductase. Biochem. J. 2000, 346, 1–8. [Google Scholar] [CrossRef]
- Minich, W.B. Selenium Metabolism and Biosynthesis of Selenoproteins in the Human Body. Biochemistry (Mosc) 2022, 87, S168–S177. [Google Scholar] [CrossRef]
- Groitl, B.; Jakob, U. Thiol-based redox switches. Biochim. Biophys. Acta 2014, 1844, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Zitka, O.; Skalickova, S.; Gumulec, J.; Masarik, M.; Adam, V.; Hubalek, J.; Trnkova, L.; Kruseova, J.; Eckschlager, T.; Kizek, R. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol. Lett. 2012, 4, 1247–1253. [Google Scholar] [CrossRef]
- Rebrin, I.; Kamzalov, S.; Sohal, R.S. Effects of age and caloric restriction on glutathione redox state in mice. Free Radic. Biol. Med. 2003, 35, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Owen, J.B.; Butterfield, D.A. Measurement of oxidized/reduced glutathione ratio. Methods Mol. Biol. 2010, 648, 269–277. [Google Scholar] [PubMed]
- Hrycay, E.G.; Bandiera, S.M. Involvement of Cytochrome P450 in Reactive Oxygen Species Formation and Cancer. Adv. Pharmacol. 2015, 74, 35–84. [Google Scholar] [PubMed]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Siegel, S.J.; Bieschke, J.; Powers, E.T.; Kelly, J.W. The oxidative stress metabolite 4-hydroxynonenal promotes Alzheimer protofibril formation. Biochemistry 2007, 46, 1503–1510. [Google Scholar] [CrossRef]
- Nemali, M.R.; Reddy, M.K.; Usuda, N.; Reddy, P.G.; Comeau, L.D.; Rao, M.S.; Reddy, J.K. Differential induction and regulation of peroxisomal enzymes: Predictive value of peroxisome proliferation in identifying certain nonmutagenic carcinogens. Toxicol. Appl. Pharmacol. 1989, 97, 72–87. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Zhu, X.; Lee, H.G.; Petersen, R.B.; Nunomura, A.; Smith, M.A.; Perry, G.; Moreira, P.I. Nuclear and mitochondrial DNA oxidation in Alzheimer’s disease. Free Radic. Res. 2012, 46, 565–576. [Google Scholar] [CrossRef]
- Zarkovic, N. 4-hydroxynonenal as a bioactive marker of pathophysiological processes. Mol. Aspects Med. 2003, 24, 281–291. [Google Scholar] [CrossRef] [PubMed]
- McGrath, L.T.; McGleenon, B.M.; Brennan, S.; McColl, D.; McILroy, S.; Passmore, A.P. Increased oxidative stress in Alzheimer’s disease as assessed with 4-hydroxynonenal but not malondialdehyde. Quarterly J. Med. 2001, 94, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Martínez, M.C.; Andriantsitohaina, R. Reactive nitrogen species: Molecular mechanisms and potential significance in health and disease. Antioxid. Redox Signal. 2009, 11, 669–702. [Google Scholar] [CrossRef]
- Goshtasbi, H.; Pakchin, P.S.; Movafeghi, A.; Barar, J.; Castejon, A.M.; Omidian, H.; Omidi, Y. Impacts of oxidants and antioxidants on the emergence and progression of Alzheimer’s disease. Neurochem. Int. 2022, 153, 105268. [Google Scholar] [CrossRef] [PubMed]
- Poon, C.H.; Tsui, K.C.; Chau, S.C.; Chong, P.S.; Lui, S.W.Y.; Aquili, L.; Wong, K.H.; Lim, L.W. Functional roles of neuronal nitric oxide synthase in neurodegenerative diseases and mood disorders. Curr. Alzheimer Res. 2021, 18, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Oh, C.K.; Zhang, X.; Lipton, S.A. Protein S-nitrosylation and oxidation contribute to protein misfolding in neurodegeneration. Free Radic. Biol. Med. 2021, 172, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. What nitrates tyrosine? Is nitrotyrosine specific as a biomarker of peroxynitrite formation in vivo? FEBS Lett. 1997, 411, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Matsubara, K.; Fujikawa, Y.; Nagahiro, Y.; Shimizu, K.; Umegae, N.; Hayase, N.; Shiono, H.; Kobayashi, S. Nitration of manganese superoxide dismutase in cerebrospinal fluids is a marker for peroxynitrite-mediated oxidative stress in neurodegenerative diseases. Ann. Neurol. 2000, 47, 524–527. [Google Scholar] [CrossRef]
- Gkaliagkousi, E.; Lazaridis, A.; Soner Dogan, S.; Fraenkel Guvenc Tuna, B.; Mozos, I.; Vukicevic, M.; Yalcin, O.; Gopcevic, K. Theories and molecular basis of vascular aging: A review of the literature from VascAgeNet Group on pathophysiological mechanisms of vascular aging. Int. J. Mol. Sci. 2022, 23, 8672. [Google Scholar] [CrossRef]
- Butler, A.R.; Megson, I.L.; Wright, P.G. Diffusion of nitric oxide and scavenging by blood in the vasculature. Biochim. Biophys. Acta 1998, 1425, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.S.; Ferguson, T.B.J.; Han, T.H.; Hyduke, D.R.; Liao, J.C.; Rassaf, T.; Bryan, N.; Feelisch, M.; Lancaster, J.R.J. Nitric oxide is consumed, rather than conserved, by reaction with oxyhemoglobin under physiological conditions. Proc. Natl. Acad. Sci. USA 2002, 99, 10341–10346. [Google Scholar] [CrossRef] [PubMed]
- Radi, R.; Beckman, J.S.; Bush, K.M.; Freeman, B.A. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J. Biol. Chem. 1991, 266, 4244–4250. [Google Scholar] [CrossRef]
- Takakura, K.; Beckman, J.S.; MacMillan-Crow, L.A.; Crow, J.P. Rapid and irreversible inactivation of protein tyrosine phosphatases PTP1B, CD45, LAR by peroxynitrite. Arch. Biochem. Biophys. 1999, 369, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.R.; Cho, E.J.; Eom, J.W.; Oh, S.S.; Nakamura, T.; Oh, C.K.; Lipton, S.A.; Kim, Y.H. S-Nitrosylation of cathepsin B affects autophagic flux and accumulation of protein aggregates in neurodegenerative disorders. Cell Death Differ. 2022, 29, 2137–2150. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.F.; Yu, J.T.; Tan, L. S-Nitrosylation in Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 268–280. [Google Scholar] [CrossRef]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef]
- Onyango, I.G.; Bennett, J.P.; Stokin, G.B. Mitochondrially-Targeted Therapeutic Strategies for Alzheimer’s Disease. Curr. Alzheimer Res. 2021, 18, 753–771. [Google Scholar] [CrossRef]
- Pantiya, P.; Thonusin, C.; Chattipakorn, N.; Chattipakorn, S.C. Mitochondrial abnormalities in neurodegenerative models and possible interventions: Focus on Alzheimer’s disease, Parkinson’s disease, Huntington’s disease. Mitochondrion 2020, 55, 14–47. [Google Scholar] [CrossRef]
- Parker, W.D.J.; Parks, J.; Filley, C.M.; Kleinschmidt-DeMasters, B.K. Electron transport chain defects in Alzheimer’s disease brain. Neurology 1994, 44, 1090–1096. [Google Scholar] [CrossRef]
- Canevari, L.; Abramov, A.Y.; Duchen, M.R. Toxicity of amyloid b peptide: Tales of calcium, mitochondria, and oxidative stress. Neurochem. Res. 2004, 29, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Keil, U.; Bonert, A.; Marques, C.A.; Scherping, I.; Weyermann, J.; Strosznajder, J.B.; Muller-Spahn, F.; Haass, C.; Czech, C.; Pradier, L.; et al. Amyloid β-induced changes in nitric oxide production and mitochondrial activity lead to apoptosis. J. Biol. Chem. 2004, 279, 50310–50320. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Lee, J.H.; Lee, J.P.; Kim, E.M.; Chang, K.A.; Park, C.H.; Jeong, S.J.; Wittendorp, M.C.; Seo, J.H.; Choi, S.H.; et al. Amyloid beta peptide induces cytochrome C release from isolated mitochondria. Neuroreport 2002, 13, 1989–1993. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.-J.; Lee, J.-M.; Chen, H.; Xu, J.; Hsu, C.Y. Ab25–35 alters Akt activity, resulting in Bad translocation and mitochondrial dysfunction in cerebrovascular endothelial cells. J. Cereb. Blood Flow Metab. 2005, 25, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef]
- Fossati, S.; Todd, K.; Sotolongo, K.; Ghiso, J.; Rostagno, A. Differential contribution of isoaspartate post-translational modifications to the fibrillization and toxic properties of amyloid β and the Asn23 Iowa mutation. Biochem. J. 2013, 456, 347–360. [Google Scholar] [CrossRef]
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P.; Calkins, M.J.; Reddy, A.P.; Mao, P.; et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta 2012, 1822, 639–649. [Google Scholar] [CrossRef]
- Smith, M.A.; Perry, G.; Richey, P.L.; Sayre, L.M.; Anderson, V.E.; Beal, M.F.; Kowall, N. Oxidative damage in Alzheimer’s. Nature 1996, 382, 120–121. [Google Scholar] [CrossRef]
- Maurer, I.; Zierz, S.; Möller, H.J. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 2000, 21, 455–462. [Google Scholar] [CrossRef]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef]
- Manczak, M.; Park, B.S.; Jung, Y.; Reddy, P.H. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: Implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 2004, 5, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D.J.; Filley, C.M.; Parks, J.K. Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology 1990, 40, 1302–1303. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid beta-peptide. Trends Mol, Med. 2001, 7, 548–554. [Google Scholar] [CrossRef]
- Mosconi, L.; de Leon, M.; Murray, J.; Lu, J.; Javier, E.; McHugh, P.; Swerdlow, R.H. Reduced mitochondria cytochrome oxidase activity in adult children of mothers with Alzheimer’s disease. J. Alzheimers Dis. 2011, 27, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Sotolongo, K.; Ghiso, J.; Rostagno, A. Nrf2 activation through the PI3K/GSK-3 axis protects neuronal cells from Aβ-mediated oxidative and metabolic damage. Alzheimers Res. Ther. 2020, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef]
- Grasmick, K.A.; Hu, H.; Hone, E.A.; Farooqi, I.; Rellick, S.L.; Simpkins, J.W.; Ren, X. Uncoupling of the Electron Transport Chain Compromises Mitochondrial Oxidative Phosphorylation and Exacerbates Stroke Outcomes. J. Neuroinfect. Dis. 2018, 9, 283. [Google Scholar] [CrossRef]
- Hill, B.G.; Benavides, G.A.; Lancaster, J.R.J.; Ballinger, S.; Dell’Italia, L.; Jianhua, Z.; Darley-Usmar, V.M. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol. Chem. 2012, 393, 1485–1512. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef]
- Harris, R.A.; Tindale, L.; Cumming, R.C. Age-dependent metabolic dysregulation in cancer and Alzheimer’s disease. Biogerontology 2014, 15, 559–577. [Google Scholar] [CrossRef]
- Marcus, C.; Mena, E.; Subramaniam, R.M. Brain PET in the diagnosis of Alzheimer’s disease. Clin. Nucl. Med. 2014, 39, e413–e426. [Google Scholar] [CrossRef]
- Mosconi, L.; Berti, V.; Glodzik, L.; Pupi, A.; De Santi, S.; de Leon, M.J. Pre-clinical detection of Alzheimer’s disease using FDG-PET, with or without amyloid imaging. J. Alzheimers Dis. 2010, 20, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; Sorbi, S.; de Leon, M.J.; Li, Y.; Nacmias, B.; Myoung, P.S.; Tsui, W.; Ginestroni, A.; Bessi, V.; Fayyazz, M.; et al. Hypometabolism exceeds atrophy in presymptomatic early-onset familial Alzheimer’s disease. J. Nucl. Med. 2006, 47, 1778–1786. [Google Scholar] [PubMed]
- de Leon, M.J.; Ferris, S.H.; George, A.E.; Christman, D.R.; Fowler, J.S.; Gentes, C.; Reisberg, B.; Gee, B.; Emmerich, M.; Yonekura, Y.; et al. Positron emission tomographic studies of aging and Alzheimer disease. Am. J. Neuroradiol. 1983, 4, 568–571. [Google Scholar] [PubMed]
- Gibson, G.E.; Shi, Q. A mitocentric view of Alzheimer’s disease suggests multi-faceted treatments. J. Alzheimers Dis. 2010, 20, S591–S607. [Google Scholar] [CrossRef]
- Lu, D.; Popuri, K.; Ding, G.W.; Balachandar, R.; Beg, M.F.; Initiative, A.s.D.N. Multiscale deep neural network based analysis of FDG-PET images for the early diagnosis of Alzheimer’s disease. Med. Image Anal. 2018, 46, 26–34. [Google Scholar] [CrossRef]
- Kapogiannis, D.; Mattson, M.P. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol. 2011, 10, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Frings, L.; Blazhenets, G.; Brumberg, J.; Rau, A.; Urbach, H.; Meyer, P.T. Deformation-based morphometry applied to FDG PET data reveals hippocampal atrophy in Alzheimer’s disease. Sci. Rep. 2024, 14, 20030. [Google Scholar] [CrossRef]
- Ou, Y.N.; Xu, W.; Li, J.Q.; Guo, Y.; Cui, M.; Chen, K.L.; Huang, Y.Y.; Dong, Q.; Tan, L.; Yu, J.T.; et al. FDG-PET as an independent biomarker for Alzheimer’s biological diagnosis: A longitudinal study. Alzheimers Res. Ther. 2019, 11, 57. [Google Scholar] [CrossRef]
- Doering, E.; Antonopoulos, G.; Hoenig, M.; van Eimeren, T.; Daamen, M.; Boecker, H.; Jessen, F.; Düzel, E.; Eickhoff, S.; Patil, K.; et al. MRI or 18F-FDG PET for Brain Age Gap Estimation: Links to Cognition, Pathology, and Alzheimer Disease Progression. J. Nucl. Med. 2024, 65, 147–155. [Google Scholar] [CrossRef]
- Kato, T.; Inui, Y.; Nakamura, A.; Ito, K. Brain fluorodeoxyglucose (FDG) PET in dementia. Ageing Res. Rev. 2016, 30, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.K.; Perry, R.H.; Tomlinson, B.E.; Blessed, G.; Gibson, P.H. Coenzyme A-acetylating enzymes in Alzheimer’s disease: Possible cholinergic ‘compartment’ of pyruvate dehydrogenase. Neurosci. Lett. 1980, 18, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Sorbi, S.; Bird, E.D.; Blass, J.P. Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann. Neurol. 1983, 13, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F.; Besnard, A.M. Thiamine-dependent enzyme changes in temporal cortex of patients with Alzheimer’s disease. Metab. Brain Dis. 1990, 5, 179–184. [Google Scholar] [CrossRef]
- Krugel, U.; Bigl, V.; Eschrich, K.; Bigl, M. Deafferentation of the septo-hippocampal pathway in rats as a model of the metabolic events in Alzheimer’s disease. Int. J. Dev. Neurosci. 2001, 19, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Haroutunian, V.; Zhang, H.; Park, L.C.; Shi, Q.; Lesser, M.; Mohs, R.C.; Sheu, R.K.; Blass, J.P. Mitochondrial damage in Alzheimer’s disease varies with apolipoprotein E genotype. Ann. Neurol. 2000, 48, 297–303. [Google Scholar] [CrossRef]
- Gibson, G.E.; Sheu, K.F.; Blass, J.P. Abnormalities of mitochondrial enzymes in Alzheimer disease. J. Neural Transm. 1998, 105, 855–870. [Google Scholar] [CrossRef]
- Mastrogiacoma, F.; Lindsay, J.G.; Bettendorff, L.; Rice, J.; Kish, S.J. Brain protein and alpha-ketoglutarate dehydrogenase complex activity in Alzheimer’s disease. Ann. Neurol.. 1996, 39, 592–598. [Google Scholar] [CrossRef]
- Gibson, G.E.; Zhang, H.; Sheu, K.F.; Bogdanovich, N.; Lindsay, J.G.; Lannfelt, L.; Vestling, M.; Cowburn, R.F. Alpha-ketoglutarate dehydrogenase in Alzheimer brains bearing the APP670/671 mutation. Ann. Neurol. 1998, 44, 676–681. [Google Scholar] [CrossRef]
- Bubber, P.; Haroutunian, V.; Fisch, G.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar] [CrossRef]
- Banerjee, K.; Munshi, S.; Frank, D.E.; Gibson, G.E. Abnormal Glucose Metabolism in Alzheimer’s Disease: Relation to Autophagy/Mitophagy and Therapeutic Approache. Neurochem. Res. 2015, 40, 2557–2569. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimers Dement. 2018, 14, 318–329. [Google Scholar] [CrossRef]
- Haley, A.; Knight-Scott, J.; Simnad, V.; Manning, C.A. Increased glucose concentration in the hippocampus in early Alzheimer’s disease following oral glucose ingestion. Magn. Reson. Imaging. 2006, 24, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Mookerjee, S.A.; Divakaruni, A.S.; Jastroch, M.; Brand, M.D. Mitochondrial uncoupling and lifespan. Mech. Ageing Dev. 2010, 131, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado-Tejedor, M.; Cabodevilla, J.F.; Zamarbide, M.; Gómez-Isla, T.; Franco, R.; Perez-Mediavilla, A. Age-related mitochondrial alterations without neuronal loss in the hippocampus of a transgenic model of Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Yang, A.; Chen, X.; Xiao, S.; Liu, X.; Lin, J.; Zhao, Y.; Zhang, K.; Li, C.; Ke, J.; et al. Proteomic Profiling of Cerebrum Mitochondria, Myelin Sheath, and Synaptosome Revealed Mitochondrial Damage and Synaptic Impairments in Association with 3 × Tg-AD Mice Model. Cell. Mol. Neurobiol. 2022, 42, 1745–1763. [Google Scholar] [CrossRef]
- Trushina, E.; Nemutlu, E.; Zhang, S.; Christensen, T.; Camp, J.; Mesa, J.; Siddiqui, A.; Tamura, Y.; Sesaki, H.; Wengenack, T.M.; et al. Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer’s disease. PLoS ONE 2012, 7, e32737. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef]
- Nimmrich, V.; Ebert, U. Is Alzheimer’s disease a result of presynaptic failure? Synaptic dysfunctions induced by oligomeric beta-amyloid. Rev. Neurosci. 2009, 20, 1–12. [Google Scholar] [CrossRef]
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Audousset, C.; McGovern, T.; Martin, J. Role of Nrf2 in Disease: Novel Molecular Mechanisms and Therapeutic Approaches— Pulmonary Disease/Asthma. Front. Physiol. 2021, 12, 727806. [Google Scholar] [CrossRef]
- Sandberg, M.; Patil, J.; D’Angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: Implication of cerebral inflammation. Neuropharmacol. 2014, 79, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.I.; Kobayashi, A.; Wakabayashi, N.; Kim, S.G.; Yamamoto, M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2046–2051. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Arozamena, C.; Martí-Marí, O.; Estrada, M.; de la Fuente Revenga, M.; Rodríguez-Franco, M.I. Recent Advances in Neurogenic Small Molecules as Innovative Treatments for Neurodegenerative Diseases. Molecules 2016, 21, pii:E1165. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 activation in the treatment of neurodegenerative diseases: A focus on its role in mitochondrial bioenergetics and function. Biol. Chem. 2016, 397, 383–400. [Google Scholar] [CrossRef]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef]
- Ooi, B.K.; Chan, K.G.; Goh, B.H.; Yap, W.H. The role of natural products in targeting cardiovascular diseases via Nrf2 pathway: Novel molecular mechanisms and therapeutic approaches. Front. Pharmacol. 2018, 9, 1308. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes. Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes. Cells 2001, 6, 857–868. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tang, C.; Ferruzzi, M.G.; Gong, B.; Song, B.J.; Janle, E.M.; Chen, T.Y.; Cooper, B.; Varghese, M.; Cheng, A.; et al. Role of standardized grape polyphenol preparation as a novel treatment to improve synaptic plasticity through attenuation of features of metabolic syndrome in a mouse model. Mol. Nutr. Food Res. 2013, 57, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef]
- Holland, R.; Fishbein, J.C. Chemistry of the cysteine sensors in Kelch-like ECH-associated protein 1. Antioxid. Redox. Signal. 2010, 13, 1749–1761. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobón-Velasco, J.C.; Devijver, H.; García-Mayoral, M.F.; Van Leuven, F.; et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/β-TrCP axis. Mol. Cell Biol. 2012, 32, 3486–3499. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef]
- Wild, A.C.; Moinova, H.R.; Mulcahy, R.T. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J. Biol. Chem. 1999, 274, 33336–33627. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar] [PubMed]
- Lee, J.M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem. 2003, 278, 12029–12038. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Sykiotis, G.P.; Habeos, I.G.; Samuelson, A.V.; Bohmann, D. The role of the antioxidant and longevity-promoting Nrf2 pathway in metabolic regulation. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.; Angelova, P.R.; Zhang, Y.; Abramov, A.Y.; Dinkova-Kostova, A.T. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem. J. 2014, 457, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Yates, M.S.; Tran, Q.T.; Dolan, P.M.; Osburn, W.O.; Shin, S.; McCulloch, C.C.; Silkworth, J.B.; Taguchi, K.; Yamamoto, M.; Williams, C.R.; et al. Genetic versus chemoprotective activation of Nrf2 signaling: Overlapping yet distinct gene expression profiles between Keap1 knockout and triterpenoid-treated mice. Carcinogenesis 2009, 30, 1024–1031. [Google Scholar] [CrossRef]
- Zou, H.; Leah, T.; Huang, Z.; He, X.; Mameli, E.; Caporali, A.; Dando, O.; Qiu, J. Endothelial cell Nrf2 controls neuroinflammation following a systemic insult. iScience 2025, 28, 112630. [Google Scholar] [CrossRef]
- Cazalla, E.; Cuadrado, A.; García-Yagüe, Á.J. Role of the transcription factor NRF2 in maintaining the integrity of the Blood-Brain Barrier. Fluids Barriers CNS 2024, 21, 93. [Google Scholar] [CrossRef]
- Chen, B.; Lu, Y.; Chen, Y.; Cheng, J. The role of Nrf2 in oxidative stress-induced endothelial injuries. J. Endocrinol. 2015, 225, R83–R99. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, F.; Ikonomovic, M.; Yang, T. The Role of NRF2 in Cerebrovascular Protection: Implications for Vascular Cognitive Impairment and Dementia (VCID). Int. J. Mol. Sci. 2024, 25, 3833. [Google Scholar] [CrossRef]
- Chen, X.L.; Dodd, G.; Thomas, S.; Zhang, X.; Wasserman, M.A.; Rovin, B.H.; Kunsch, C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1862–H1870. [Google Scholar] [CrossRef]
- Song, J.; Kang, S.M.; Lee, W.T.; Park, K.A.; Lee, K.M.; Lee, J.E. Glutathione protects brain endothelial cells from hydrogen peroxide-induced oxidative stress by increasing nrf2 expression. Exp. Neurobiol. 2014, 23, 93–103. [Google Scholar] [CrossRef]
- Zhou, H.G.; Liu, L.; Zhang, Y.; Huang, Y.Y.; Tao, Y.H.; Zhang, S.; Su, J.J.; Tang, Y.P.; Guo, Z.L.; Hu, R.M.; et al. Glutathione prevents free fatty acids-induced oxidative stress and apoptosis in human brain vascular endothelial cells through Akt pathway. CNS Neurosci. Ther. 2013, 19, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Moore, A.N.; Redell, J.B.; Dash, P.K. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J. Neurosci. 2007, 27, 10240–10248. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrixmetalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow. Metab. 2007, 27, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Candelario-Jalil, E.; Dijkhuizen, R.M.; Magnus, T. Neuroinflammation, Stroke, Blood-Brain Barrier Dysfunction, and Imaging Modalities. Stroke 2022, 53, 1473–1486. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Lei Jiang, H.E.; Johnson, J.A.; Murphy, T.H. Coordinate Regulation of Glutathione Biosynthesis and Release by Nrf2-Expressing Glia Potently Protects Neurons from Oxidative Stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef] [PubMed]
- van Muiswinkel, F.L.; Kuiperij, H.B. The Nrf2-ARE Signalling pathway: Promising drug target to combat oxidative stress in neurodegenerative disorders. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Silva-Palacios, A.; Ostolga-Chavarría, M.; Zazueta, C.; Königsberg, M. Nrf2: Molecular and epigenetic regulation during aging. Ageing Res. Rev. 2018, 47, 31–40. [Google Scholar] [CrossRef] [PubMed]
- De Plano, L.M.; Calabrese, G.; Rizzo, M.G.; Oddo, S.; Caccamo, A. The Role of the Transcription Factor Nrf2 in Alzheimer’s Disease: Therapeutic Opportunities. Biomolecules 2023, 13, 549. [Google Scholar] [CrossRef] [PubMed]
- George, M.; Tharakan, M.; Culberson, J.; Reddy, A.P.; Reddy, P.H. Role of Nrf2 in aging, Alzheimer’s and other neurodegenerative diseases. Ageing Res. Rev. 2022, 82, 101756. [Google Scholar] [CrossRef] [PubMed]
- von Otter, M.; Landgren, S.; Nilsson, S.; Zetterberg, M.; Celojevic, D.; Bergstrom, P.; Minthon, L.; Bogdanovic, N.; Andreasen, N.; Gustafson, D.R.; et al. Nrf2-encoding NFE2L2 haplotypes influence disease progression but not risk in Alzheimer’s disease and age-related cataract. Mech. Ageing Dev. 2010, 131, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, S.; Chen, X.; Yang, H.; Li, X.; Xu, Y.; Zhu, X. Orientin alleviates cognitive deficits and oxidative stress in Aβ1-42-induced mouse model of Alzheimer’s disease. Life Sci. 2015, 121, 104–109. [Google Scholar] [CrossRef]
- Hong, Y.; An, Z. Hesperidin attenuates learning and memory deficits in APP/PS1 mice through activation of Akt/Nrf2 signaling and inhibition of RAGE/NF-κB signaling. Arch. Pharm. Res. 2018, 41, 655–663. [Google Scholar] [CrossRef]
- Branca, C.; Ferreira, E.; Nguyen, T.-V.; Doyle, K.; Caccamo, A.; Oddo, S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 4823–4835. [Google Scholar] [CrossRef]
- Uruno, A.; Matsumaru, D.; Ryoke, R.; Saito, R.; Kadoguchi, S.; Saigusa, D.; Saito, T.; Saido, T.C.; Kawashima, R.; Yamamoto, M. Nrf2 Suppresses Oxidative Stress and Inflammation in App Knock-in Alzheimer’s Disease Model Mice. Mol. Cell. Biol. 2020, 40, e00467-19. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Park, G.H.; Lee, S.R.; Jang, J.H. Attenuation of β-amyloid-induced oxidative cell death by sulforaphane via activation of NF-E2-related factor 2. Oxid. Med. Cell Longev. 2013, 2013, 313510. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Bai, X.; Yu, S.; Zhao, W.; Qiao, J.; Liu, Y.; Zhao, D.; Wang, J.; Wang, S. Ginsenoside Re Inhibits ROS/ASK-1 Dependent Mitochondrial Apoptosis Pathway and Activation of Nrf2-Antioxidant Response in Beta-Amyloid-Challenged SH-SY5Y Cells. Molecules 2019, 24, pii:E2687. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Miao, Y.; Mir, A.Z.; Cheng, L.; Wang, L.; Zhao, L.; Cui, Q.; Zhao, W.; Wang, H. Inhibition of beta-amyloid-induced neurotoxicity by pinocembrin through Nrf2/HO-1 pathway in SH-SY5Y cells. J. Neurol. Sci. 2016, 368, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Miao, Q.W.; Zhu, C.X.; Zhao, Y.; Liu, L.; Yang, J.; An, L. Sulforaphane ameliorates neurobehavioral deficits and protects the brain from amyloid β deposits and peroxidation in mice with Alzheimer-like lesions. Am. J. Alzheimers Dis. Other Demen. 2015, 30, 183–191. [Google Scholar] [CrossRef]
- Gray, N.E.; Zweig, J.A.; Caruso, M.; Zhu, J.Y.; Wright, K.M.; Quinn, J.F.; Soumyanath, A. Centella asiatica attenuates hippocampal mitochondrial dysfunction and improves memory and executive function in β-amyloid overexpressing mice. Mol. Cell. Neurosci. 2018, 93, 1–9. [Google Scholar] [CrossRef]
- Gray, N.E.; Zweig, J.A.; Matthews, D.G.; Caruso, M.; Quinn, J.F.; Soumyanath, A. Centella asiatica Attenuates Mitochondrial Dysfunction and Oxidative Stress in Aβ-Exposed Hippocampal Neurons. Oxid. Med. Cell. Longev. 2017, 2017, 7023091. [Google Scholar] [CrossRef]
- Kerr, F.; Sofola-Adesakin, O.; Ivanov, D.K.; Gatliff, J.; Gomez Perez-Nievas, B.; Bertrand, H.C.; Martinez, P.; Callard, R.; Snoeren, I.; Cocheme, H.M.; et al. Direct Keap1-Nrf2 disruption as a potential therapeutic target for Alzheimer’s disease. PLoS Genet. 2017, 13, e1006593. [Google Scholar] [CrossRef]
- Llorens-Martín, M.; Jurado, J.; Hernández, F.; Avila, J. GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar]
- Ochalek, A.; Mihalik, B.; Avci, H.X.; Chandrasekaran, A.; Téglási, A.; Bock, I.; Giudice, M.L.; Táncos, Z.; Molnár, K.; László, L.; et al. Neurons derived from sporadic Alzheimer’s disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimers Res. Ther. 2017, 9, 90. [Google Scholar] [CrossRef]
- Talman, V.; Pascale, A.; Jäntti, M.; Amadio, M.; Tuominen, R.K. Protein Kinase C Activation as a Potential Therapeutic Strategy in Alzheimer’s Disease: Is there a Role for Embryonic Lethal Abnormal Vision-like Proteins? Basic. Clin. Pharmacol. Toxicol. 2016, 119, 149–160. [Google Scholar] [CrossRef]
- Zuo, J.; Zhang, Z.; Luo, M.; Zhou, L.; Nice, E.C.; Zhang, W.; Wang, C.; Huang, C. Redox signaling at the crossroads of human health and disease. MedComm 2022, 3, e127. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, X. Antioxidant therapies for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2012, 2012, 472932. [Google Scholar] [CrossRef]
- Plascencia-Villa, G.; Perry, G. Preventive and Therapeutic Strategies in Alzheimer’s Disease: Focus on Oxidative Stress, Redox Metals, and Ferroptosis. Antioxid. Redox Signal. 2021, 34, 591–610. [Google Scholar] [CrossRef] [PubMed]
- Pritam, P.; Deka, R.; Bhardwaj, A.; Srivastava, R.; Kumar, D.; Jha, A.K.; Jha, N.K.; Villa, C.; Jha, S.K. Antioxidants in Alzheimer’s Disease: Current Therapeutic Significance and Future Prospects. Biology 2022, 11, 212. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, J.P.; de Castro, A.A.; Soares, F.V.; da Cunha, E.F.F.; Ramalho, T.C. Future Therapeutic Perspectives into the Alzheimer’s Disease Targeting the Oxidative Stress Hypothesis. Molecules 2019, 24, 4410. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.E.; Saleh, T.M.; Kalisch, B.E. Naturally Occurring Antioxidant Therapy in Alzheimer’s Disease. Antioxidants (Basel) 2022, 11, 213. [Google Scholar] [CrossRef]
- Guo, J.; Zhu, Y.; Zhi, J.; Lou, Q.; Bai, R.; He, Y. Antioxidants in anti-Alzheimer’s disease drug discovery. Ageing Res. Rev. 2025, 107, 102707. [Google Scholar]
- Yao, J.; Brinton, R.D. Targeting mitochondrial bioenergetics for Alzheimer’s prevention and treatment. Curr. Pharm. Des. 2011, 17, 3474–3479. [Google Scholar]
- Packer, L.; Cadenas, E. Lipoic acid: Energy metabolism and redox regulation of transcription and cell signaling. J. Clin. Biochem. Nutr. 2011, 48, 26–32. [Google Scholar] [CrossRef]
- Zhang, C.; Rodriguez, C.; Spaulding, J.; Aw, T.Y.; Feng, J. Age-dependent and tissue-related glutathione redox status in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2012, 28, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.F.; Bussiere, J.R.; Hammond, R.S.; Montine, T.J.; Henson, E.; Jones, R.E.; Stackman, R.W.J. Chronic dietary alpha-lipoic acid reduces deficits in hippocampal memory of aged Tg2576 mice. Neurobiol. Aging 2007, 28, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Bentinger, M.; Tekle, M.; Dallner, G. Coenzyme Q–biosynthesis and functions. Biochem. Biophys. Res. Commun. 2010, 396, 74–79. [Google Scholar] [CrossRef]
- Arenas-Jal, M.; Suñé-Negre, J.M.; García-Montoya, E. Coenzyme Q10 supplementation: Efficacy, safety, and formulation challenges. Compr. Rev. Food Sci. Food Saf. 2020, 19, 574–594. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, K.; Ikeda, A.; Moriyama, Y.; Chei, C.L.; Noda, H.; Umesawa, M.; Cui, R.; Nagao, M.; Kitamura, A.; Yamamoto, Y.; et al. Serum coenzyme Q10 and risk of disabling dementia: The Circulatory Risk in Communities Study (CIRCS). Atherosclerosis 2014, 237, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.S.; Chou, H.H.; Lai, T.J.; Yen, C.H.; Pan, J.C.; Lin, P.T. Investigation of coenzyme Q10 status, serum amyloid-β, and tau protein in patients with dementia. Front. Aging Neurosci. 2022, 14, 910289. [Google Scholar] [CrossRef] [PubMed]
- Shults, C.W. Therapeutic role of coenzyme Q(10) in Parkinson’s disease. Pharmacol. Ther. 2005, 107, 120–130. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, K.L.; Jenkins, B.G.; Hersch, S.M.; Beal, M.F. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington’s disease. J. Neurosci. 2002, 22, 1592–1599. [Google Scholar] [CrossRef]
- Dumont, M.; Kipiani, K.; Yu, F.; Wille, E.; Katz, M.; Calingasan, N.Y.; Gouras, G.K.; Lin, M.T.; Beal, M.F. Coenzyme Q10 decreases amyloid pathology and improves behavior in a transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2011, 27, 211–223. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Gan, R.Y.; Li, S.; Zhou, Y.; Li, A.N.; Xu, D.P.; Li, H.B. Antioxidant Phytochemicals for the Prevention and Treatment of Chronic Diseases. Molecules 2015, 20, 21138–21156. [Google Scholar] [CrossRef]
- Rivas, F.; Poblete-Aro, C.; Pando, M.E.; Allel, M.J.; Fernandez, V.; Soto, A.; Nova, P.; Garcia-Diaz, D. Effects of Polyphenols in Aging and Neurodegeneration Associated with Oxidative Stress. Curr. Med. Chem. 2022, 29, 1045–1060. [Google Scholar] [CrossRef] [PubMed]
- Barchielli, G.; Capperucci, A.; Tanini, D. Role of Selenium in Pathologies: An Updated Review. Antioxidants 2022, 11, 251. [Google Scholar] [CrossRef] [PubMed]
- Hertzog da Silva Leme, A.G.; Cardoso, B.R. Selenium and Alzheimer’s disease. Genet. Neurol. Behav. Diet. Dementia 2020, 2, 739–747. [Google Scholar]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Chambers, I.G.; Ratan, R.R. Selenium abandons selenoproteins to inhibit ferroptosis rapidly. Nat. Metab. 2024, 6, 200–202. [Google Scholar] [CrossRef]
- Akbaraly, N.T.; Arnaud, J.; Hininger-Favier, I.; Gourlet, V.; Roussel, A.M.; Berr, C. Selenium and mortality in the elderly: Results from the EVA study. Clin. Chem. 2005, 51, 2117–2123. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.R.; Kim, Y.; Jeong, H.J.; Kang, J.S.; Lee, S.H.; Kim, Y.; Lee, S.H.; Ho, W.K. Impaired pattern separation in Tg2576 mice is associated with hyperexcitable dentate gyrus caused by Kv4.1 downregulation. Mol. Brain 2021, 14, 62. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alloza, M.; Dodwell, S.A.; Meyer-Luehmann, M.; Hyman, B.T.; Bacskai, B.J. Plaque-derived oxidative stress mediates distorted neurite trajectories in the Alzheimer mouse model. J. Neuropatho.l Exp. Neurol. 2006, 65, 1082–1089. [Google Scholar] [CrossRef]
- Montiel, T.; Quiroz-Baez, R.; Massieu, L.; Arias, C. Role of oxidative stress on beta-amyloid neurotoxicity elicited during impairment of energy metabolism in the hippocampus: Protection by antioxidants. Exp. Neurol. 2006, 200, 496–508. [Google Scholar] [CrossRef]
- Gugliandolo, A.; Bramanti, P.; Mazzon, E. Role of Vitamin E in the Treatment of Alzheimer’s Disease: Evidence from Animal Models. Int. J. Mol. Sci. 2017, 18, 2504. [Google Scholar] [CrossRef] [PubMed]
- Yatin, S.M.; Varadarajan, S.; Butterfield, D.A. Vitamin E Prevents Alzheimer’s Amyloid beta-Peptide (1-42)-Induced Neuronal Protein Oxidation and Reactive Oxygen Species Production. J. Alzheimers Dis. 2000, 2, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Browne, D.; McGuinness, B.; Woodside, J.V.; McKay, G.J. Vitamin E and Alzheimer’s disease: What do we know so far? Clin. Interv. Aging 2019, 14, 1303–1317. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E. A critical review of vitamin C for the prevention of age-related cognitive decline and Alzheimer’s disease. J. Alzheimers Dis. 2012, 29, 711–726. [Google Scholar] [CrossRef] [PubMed]
- Dixit, S.; Bernardo, A.; Walker, J.M.; Kennard, J.A.; Kim, G.Y.; Kessler, E.S.; Harrison, F.E. Vitamin C deficiency in the brain impairs cognition, increases amyloid accumulation and deposition, and oxidative stress in APP/PSEN1 and normally aging mice. ACS Chem. Neurosci. 2015, 6, 570–581. [Google Scholar] [CrossRef]
- Berr, C. Cognitive impairment and oxidative stress in the elderly: Results of epidemiological studies. Biofactors 2000, 13, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Berr, C. Oxidative stress and cognitive impairment in the elderly. J. Nutr. Health Aging 2002, 6, 261–266. [Google Scholar]
- Martin, A.; Youdim, K.; Szprengiel, A.; Shukitt-Hale, B.; Joseph, J. Roles of vitamins E and C on neurodegenerative diseases and cognitive performance. Nutr. Rev. 2002, 60, 308–326. [Google Scholar] [CrossRef]
- Fillenbaum, G.G.; Kuchibhatla, M.N.; Hanlon, J.T.; Artz, M.B.; Pieper, C.F.; Schmader, K.E.; Dysken, M.W.; Gray, S.L. Dementia and Alzheimer’s disease in community-dwelling elders taking vitamin C and/or vitamin E. Ann. Pharmacother. 2005, 39, 2009–2014. [Google Scholar] [CrossRef]
- Ghezzi, P.; Jaquet, V.; Marcucci, F.; Schmidt, H.H.H.W. The oxidative stress theory of disease: Levels of evidence and epistemological aspects. Br. J. Pharmacol. 2017, 174, 1784–1796. [Google Scholar] [CrossRef]
- Pappolla, M.A.; Martins, R.N.; Poeggeler, B.; Omar, R.A.; Perry, G. Oxidative Stress in Alzheimer’s Disease: The Shortcomings of Antioxidant Therapies. J. Alzheimers Dis. 2024, 101, S155–S178. [Google Scholar] [CrossRef] [PubMed]
- Gomes, B.A.Q.; Silva, J.P.B.; Romeiro, C.F.R.; Dos Santos, S.M.; Rodrigues, C.A.; Gonçalves, P.R.; Sakai, J.T.; Mendes, P.F.S.; Varela, E.L.P.; Monteiro, M.C. Neuroprotective Mechanisms of Resveratrol in Alzheimer’s Disease: Role of SIRT1. Oxid. Med. Cell. Longev. 2018, 2018, 8152373. [Google Scholar] [CrossRef]
- Kim, E.N.; Lim, J.H.; Kim, M.Y.; Ban, T.H.; Jang, I.A.; Yoon, H.E.; Park, C.W.; Chang, Y.S.; Choi, B.S. Resveratrol, an Nrf2 activator, ameliorates aging-related progressive renal injury. Aging (Albany NY) 2018, 10, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Karuppagounder, S.S.; Pinto, J.T.; Xu, H.; Chen, H.L.; Beal, M.F.; Gibson, G.E. Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer’s disease. Neurochem. Int. 2009, 54, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shi, G.W.; Liang, Z.M.; Sheng, S.Y.; Shi, Y.S.; Peng, L.; Wang, Y.P.; Wang, F.; Zhang, X.M. Resveratrol improves cognition and decreases amyloid plaque formation in Tg6799 mice. Mol. Med. Rep. 2019, 19, 3783–3790. [Google Scholar] [CrossRef] [PubMed]
- Pasinetti, G.M.; Wang, J.; Ho, L.; Zhao, W.; Dubner, L. Roles of resveratrol and other grape-derived polyphenols in Alzheimer’s disease prevention and treatment. Biochim. Biophys. Acta 2015, 1852, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Ozpak, L.; Bağca, B.G. Neuroprotective effects of resveratrol through modulation of PI3K/Akt/GSK-3β pathway and metalloproteases. IUBMB Life 2024, 76, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Shahcheraghi, S.H.; Salemi, F.; Small, S.; Syed, S.; Salari, F.; Alam, W.; Cheang, W.S.; Saso, L.; Khan, H. Resveratrol regulates inflammation and improves oxidative stress via Nrf2 signaling pathway: Therapeutic and biotechnological prospects. Phytother. Res. 2023, 37, 1590–1605. [Google Scholar] [CrossRef]
- Wang, J.; Ho, L.; Zhao, Z.; Seror, I.; Humala, N.; Dickstein, D.L.; Thiyagarajan, M.; Percival, S.S.; Talcott, S.T.; Pasinetti, G.M. Moderate consumption of Cabernet Sauvignon attenuates Abeta neuropathology in a mouse model of Alzheimer’s disease. FASEB J. 2006, 20, 2313–2320. [Google Scholar] [CrossRef]
- Kung, H.C.; Lin, K.J.; Kung, C.T.; Lin, T.K. Oxidative Stress, Mitochondrial Dysfunction, and Neuroprotection of Polyphenols with Respect to Resveratrol in Parkinson’s Disease. Biomedicines 2021, 9, 918. [Google Scholar] [CrossRef]
- Squillaro, T.; Cimini, A.; Peluso, G.; Giordano, A.; Melone, M.A.B. Nano-delivery systems for encapsulation of dietary polyphenols: An experimental approach for neurodegenerative diseases and brain tumors. Biochem. Pharmacol. 2018, 154, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Kancheva, V.D.; Dettori, M.A.; Fabbri, D.; Alov, P.; Angelova, S.E.; Slavova-Kazakova, A.K.; Carta, P.; Menshov, V.A.; Yablonskaya, O.I.; Trofimov, A.V.; et al. Natural Chain-Breaking Antioxidants and Their Synthetic Analogs as Modulators of Oxidative Stress. Antioxidants 2021, 10, 624. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Peterson, T.; Fan, Z.; Wang, S. The Commonly Used Stabilizers for Phytochemical-Based Nanoparticles: Stabilization Effects, Mechanisms, and Applications. Nutrients 2023, 15, 3881. [Google Scholar] [CrossRef] [PubMed]
- Lal, R.; Dharavath, R.N.; Chopra, K. Nrf2 Signaling Pathway: A Potential Therapeutic Target in Combating Oxidative Stress and Neurotoxicity in Chemotherapy-Induced Cognitive Impairment. Mol. Neurobiol. 2024, 61, 593–608. [Google Scholar] [CrossRef]
- Halliwell, B. Understanding mechanisms of antioxidant action in health and disease. Nat. Rev. Mol. Cell Biol. 2024, 25, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Cloer, E.W.; Goldfarb, D.; Schrank, T.P.; Weissman, B.E.; Major, M.B. NRF2 Activation in Cancer: From DNA to Protein. Cancer Res. 2019, 79, 889–898. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative stress in cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Hur, W.; Gray, N.S. Small molecule modulators of antioxidant response pathway. Curr. Opin. Chern. Biol. 2011, 15, 162–173. [Google Scholar] [CrossRef]
- Mao, L.; Yang, T.; Li, X.; Lei, X.; Sun, Y.; Zhao, Y.; Zhang, W.; Gao, Y.; Sun, B.; Zhang, F. Protective effects of sulforaphane in experimental vascular cognitive impairment: Contribution of the Nrf2 pathway. J. Cereb. Blood Flow. Metab. 2019, 39, 352–366. [Google Scholar] [CrossRef]
- Soane, L.; Li Dai, W.; Fiskum, G.; Bambrick, L.L. Sulforaphane protects immature hippocampal neurons against death caused by exposure to hemin or to oxygen and glucose deprivation. J. Neurosci. Res. 2010, 88, 1355–1363. [Google Scholar] [CrossRef]
- Liu, X.; Yang, L.; Zhang, G.; Ling, J. Neuroprotective Effects of Phenolic Antioxidant Tert-butylhydroquinone (tBHQ) in Brain Diseases. Mol. Neurobiol. 2023, 60, 4909–4923. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tucker, L.D.; Dong, Y.; Lu, Y.; Yang, L.; Wu, C.; Li, Y.; Zhang, Q. Tert-butylhydroquinone post-treatment attenuates neonatal hypoxic-ischemic brain damage in rats. Neurochem. Int. 2018, 116, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.W.; Liang, J.; Yan, J.X.; Ye, Y.C.; Wang, J.J.; Chen, C.; Sun, H.T.; Chen, F.; Tu, Y.; Li, X.H. TBHQ improved neurological recovery after traumatic brain injury by inhibiting the overactivation of astrocytes. Brain Res. 2020, 1739, 146818. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y.; Li, P.; Murphy, T.H. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J. Neurosci. 2005, 25, 10321–10335. [Google Scholar] [CrossRef]
- Eftekharzadeh, B.; Maghsoudi, N.; Khodagholi, F. Stabilization of transcription factor Nrf2 by tBHQ prevents oxidative stress-induced amyloid beta formation in NT2N neurons. Biochimie 2010, 92, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Akhter, H.; Katre, A.; Li, L.; Liu, X.; Liu, R.M. Therapeutic potential and anti-amyloidosis mechanisms of tert-butylhydroquinone for Alzheimer’s disease. J. Alzheimers Dis. 2011, 26, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Taghibiglou, C. The Mechanisms of Action of Curcumin in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 1003–1016. [Google Scholar] [CrossRef]
- Ak, T.; Gulcin, I. Antioxidant and radical scavenging properties of curcumin. Chem. Biol. Interact. 2008, 174, 27–37. [Google Scholar] [CrossRef]
- Gibellini, L.; Bianchini, E.; De Biasi, S.; Nasi, M.; Cossarizza, A.; Pinti, M. Natural compounds modulating mitochondrial functions. Evid. Based Complement. Alternat. Med. 2015, 2015, 1–13. [Google Scholar] [CrossRef]
- Uğuz, A.C.; Öz, A.; Nazıroğlu, M. Curcumin inhibits apoptosis by regulating intracellular calcium release, reactive oxygen species and mitochondrial depolarization levels in SH-SY5Y neuronal cells. J. Recept. Signal Transduct. Res. 2016, 36, 395–401. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Yin, X.; Grady, M.C.; Mitchell, A.; Tonk, S.; Kuruva, C.S.; Bhatti, J.S.; Kandimalla, R.; Vijayan, M.; et al. Protective Effects of Indian Spice Curcumin Against Amyloid-β in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 61, 843–866. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Su, C.; Li, R.; Wang, H.; Ren, Y.; Sun, H.; Yang, J.; Sun, J.; Shi, J.; Tian, J.; et al. Mechanisms and effects of curcumin on spatial learning and memory improvement in APPswe/PS1dE9 mice. J. Neurosci. Res. 2014, 92, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Li, Z.H.; Liu, L.; Tang, W.X.; Wang, Y.; Dong, M.R.; Xiao, C. Curcumin Attenuates Beta-Amyloid-Induced Neuroinflammation via Activation of Peroxisome Proliferator-Activated Receptor-Gamma Function in a Rat Model of Alzheimer’s Disease. Front. Pharmacol. 2016, 19, 261. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wang, P.; Wei, P.; Feng, H.; Ren, Y.; Yang, J.; Rao, Y.; Shi, J.; Tian, J. Effects of curcumin on synapses in APPswe/PS1dE9 mice. Int. J. Immunopathol. Pharmacol. 2016, 29, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.; Mojumder, D.; Detoledo, J.; Lucius, R.; Wilms, H. Pathophysiological processes in multiple sclerosis: Focus on nuclear factor erythroid-2-related factor 2 and emerging pathways. Clin. Pharmacol. 2014, 6, 35–42. [Google Scholar] [PubMed]
- Uruno, A.; Yamamoto, M. The KEAP1-NRF2 System and Neurodegenerative Diseases. Antioxid. Redox Signal. 2023, 38, 974–988. [Google Scholar] [CrossRef]
- Satoh, T.; Lipton, S. Recent advances in understanding NRF2 as a druggable target: Development of pro-electrophilic and non-covalent NRF2 activators to overcome systemic side effects of electrophilic drugs like dimethyl fumarate. F1000 Res. 2017, 6, 2138. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.Y.; Lee, P.; Adany, P.; Hughes, A.J.; Belliston, S.; Denney, D.R.; Lynch, S.G. In vivo evidence of oxidative stress in brains of patients with progressive multiple sclerosis. Mult. Scler. 2018, 24, 1029–1038. [Google Scholar] [CrossRef]
- Hammer, A.; Waschbisch, A.; Kuhbandner, K.; Bayas, A.; Lee, D.H.; Duscha, A.; Haghikia, A.; Gold, R.; Linker, R.A. The NRF2 pathway as potential biomarker for dimethyl fumarate treatment in multiple sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 668–676. [Google Scholar] [CrossRef]
- Carlström, K.E.; Ewing, E.; Granqvist, M.; Gyllenberg, A.; Aeinehband, S.; Enoksson, S.L.; Checa, A.; Badam, T.V.S.; Huang, J.; Gomez-Cabrero, D.; et al. Therapeutic efficacy of dimethyl fumarate in relapsing-remitting multiple sclerosis associates with ROS pathway in monocytes. Nat. Commun. 2019, 10, 3081. [Google Scholar] [CrossRef]
- Leroy, K.; Yilmaz, Z.; Brion, J.P. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell. Res. 2020, 1867, 118664. [Google Scholar] [CrossRef]
- Freeman, M.P.; Freeman, S.A. Lithium: Clinical considerations in internal medicine. Am. J. Med. 2006, 119, 478–481. [Google Scholar] [CrossRef]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.V.; Andersson, U.; Andersson, S.; Knerr, L.; Bauer, U.; Sundgren-Andersson, A.K. The Conundrum of GSK3 Inhibitors: Is it the Dawn of a New Beginning? J. Alzheimers Dis. 2018, 64, S547–S554. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Begum, A.N.; Jones, M.R.; Oh, M.S.; Beech, W.K.; Beech, B.H.; Yang, F.; Chen, P.; Ubeda, O.J.; Kim, P.C.; et al. GSK3 inhibitors show benefits in an Alzheimer’s disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol. Dis. 2009, 33, 193–206. [Google Scholar]
- Snitow, M.E.; Bhansali, R.S.; Klein, P.S. Lithium and Therapeutic Targeting of GSK-3. Cells 2021, 10, 255. [Google Scholar] [CrossRef]
- Serenó, L.; Coma, M.; Rodríguez, M.; Sánchez-Ferrer, P.; Sánchez, M.B.; Gich, I.; Agulló, J.M.; Pérez, M.; Avila, J.; Guardia-Laguarta, C.; et al. A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 2009, 35, 359–367. [Google Scholar] [CrossRef]
- Wang, H.; Huang, S.; Yan, K.; Fang, X.; Abussaud, A.; Martinez, A.; Sun, H.S.; Feng, Z.P. Tideglusib, a chemical inhibitor of GSK3β, attenuates hypoxic-ischemic brain injury in neonatal mice. Biochim. Biophys. Acta 2016, 1860, 2076–2085. [Google Scholar] [CrossRef]
- Frame, S.; Cohen, P. GSK3 takes center stage more than 20 years after its discovery. Biochem. J. 2001, 359, 1–16. [Google Scholar] [CrossRef]
- Razani, E.; Pourbagheri-Sigaroodi, A.; Safaroghli-Azar, A.; Zoghi, A.; Shanaki-Bavarsad, M.; Bashash, D. The PI3K/Akt signaling axis in Alzheimer’s disease: A valuable target to stimulate or suppress? Cell Stress Chaperones 2021, 26, 871–887. [Google Scholar] [CrossRef] [PubMed]
- Long, H.Z.; Cheng, Y.; Zhou, Z.W.; Luo, H.Y.; Wen, D.D.; Gao, L.C. PI3K/AKT Signal Pathway: A Target of Natural Products in the Prevention and Treatment of Alzheimer’s Disease and Parkinson’s Disease. Front. Pharmacol. 2021, 12, 648636. [Google Scholar] [CrossRef]
- Lei, L.; Luo, Y.; Kang, D.; Yang, F.; Meng, D.; Wang, J.Z.; Liu, R.; Wang, X.; Li, H.L. Gypenoside IX restores Akt/GSK-3β pathway and alleviates Alzheimer’s disease-like neuropathology and cognitive deficits. Aging (Albany NY) 2023, 15, 14172–14191. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.F.; Zu, M.L.; Wang, Y.R.; Cui, W.Y.; Duan, Y.; Yang, C.; Piao, X.L. Protective effects of four new saponins from Gynostemma pentaphyllum against hydrogen peroxide-induced neurotoxicity in SH-SY5Y cells. Bioorg. Chem. 2021, 106, 104470. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Luo, B.; Zhong, B.R.; Li, K.Y.; Wen, Q.X.; Song, L.; Xiang, X.J.; Zhou, G.F.; Hu, L.T.; Deng, X.J.; et al. Sulfuretin exerts diversified functions in the processing of amyloid precursor protein. Genes Dis. 2020, 8, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Lisk, C.; McCord, J.; Bose, S.; Sullivan, T.; Loomis, Z.; Nozik-Grayck, E.; Schroeder, T.; Hamilton, K.; Irwin, D.C. Nrf2 activation: A potential strategy for the prevention of acute mountain sickness. Free Radic. Biol. Med. 2013, 63, 264–273. [Google Scholar] [CrossRef]
- Ding, Z.; Wu, X.; Wang, Y.; Ji, S.; Zhang, W.; Kang, J.; Li, J.; Fei, G. Melatonin prevents LPS-induced epithelial-mesenchymal transition in human alveolar epithelial cells via the GSK-3β/Nrf2 pathway. Biomed. Pharmacother. 2020, 132, 110827. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zhao, Q.; Lu, C.; Yong, W.; Xu, M.; Wang, Z.; Leng, X. Melatonin suppresses atherosclerosis by ferroptosis inhibition via activating NRF2 pathway. FASEB J. 2024, 38, e23678. [Google Scholar] [CrossRef]
- Gou, Z.; Su, X.; Hu, X.; Zhou, Y.; Huang, L.; Fan, Y.; Li, J.; Lu, L. Melatonin improves hypoxic-ischemic brain damage through the Akt/Nrf2/Gpx4 signaling pathway. Brain Res. Bull. 2020, 163, 40–48. [Google Scholar] [CrossRef]
- Qin, T.; Feng, D.; Zhou, B.; Bai, L.; Yin, Y. Melatonin Suppresses LPS-Induced Oxidative Stress in Dendritic Cells for Inflammatory Regulation via the Nrf2/HO-1 Axis. Antioxidants 2022, 11, 2012. [Google Scholar] [CrossRef]
- Supuran, C.T.; Scozzafava, A. Carbonic anhydrase inhibitors and their therapeutic potential. Expert. Opin. Ther. Pat. 2000, 10, 575–600. [Google Scholar] [CrossRef]
- Ciccone, L.; Cerri, C.; Nencetti, S.; Orlandini, E. Carbonic Anhydrase Inhibitors and Epilepsy: State of the Art and Future Perspectives. Molecules 2021, 26, 6380. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhang, H.; Jiang, Y. Methazolamide in high-altitude illnesses. Eur. J. Pharm. Sci. 2020, 148, 105326. [Google Scholar] [CrossRef]
- Belete, T.M. Recent Progress in the Development of New Antiepileptic Drugs with Novel Targets. Ann. Neurosci. 2023, 30, 262–276. [Google Scholar] [CrossRef]
- Aggarwal, M.; Kondeti, B.; McKenna, R. Anticonvulsant/antiepileptic carbonic anhydrase inhibitors: A patent review. Expert. Opin. Ther. Pat. 2013, 23, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Chesler, M. Regulation and modulation of pH in the brain. Physiol. Rev. 2003, 83, 1183–1221. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and Regulators of Intracellular pH. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef]
- Wilkins, M.E.; Hosie, A.M.; Smart, T.G. Proton Modulation of Recombinant GABAAreceptors: Influence of GABA Concentration and the β Subunit TM2-TM3 Domain. J. Physiol. 2005, 567, 365–377. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.; Vance, K.M.; Ogden, K.; Hansen, K.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar]
- Makani, S.; Chen, H.-Y.; Esquenazi, S.; Shah, G.N.; Waheed, A.; Sly, W.S.; Chesler, M. NMDA Receptor-Dependent Afterdepolar-izations Are Curtailed by Carbonic Anhydrase 14: Regulation of a Short-Term Postsynaptic Potentiation. J. Neurosci. 2012, 32, 16754–16762. [Google Scholar] [CrossRef]
- Waldmann, R.; Champigny, G.; Bassilana, F.; Heurteaux, C.; Lazdunski, M. A Proton-Gated Cation Channel Involved in Acid-Sensing. Nature 1997, 386, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Hassel, B. Carboxylation and anaplerosis in neurons and glia. Mol. Neurobiol. 2000, 22, 21–40. [Google Scholar] [CrossRef]
- Sarang, S.S.; Yoshida, T.; Cadet, R.; Valeras, A.S.; Jensen, R.V.; Gullans, S.R. Discovery of molecular mechanisms of neuroprotection using cell-based bioassays and oligonucleotide arrays. Physiol. Genomics 2002, 11, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, W.; Mai, H.; Zhang, X.; Wang, J.; Gao, Y.; Wang, Y.; Deng, G.; Gao, L.; Zhou, S.; et al. Methazolamide improves neurological behavior by inhibition of neuron apoptosis in subarachnoid hemorrhage mice. Sci. Rep. 2016, 6, 35055. [Google Scholar] [CrossRef] [PubMed]
- Anzovino, A.; Canepa, E.; Alves, M.; Lemon, N.L.; Carare, R.O.; Fossati, S. Amyloid Beta Oligomers Activate Death Receptors and Mitochondria-Mediated Apoptotic Pathways in Cerebral Vascular Smooth Muscle Cells; Protective Effects of Carbonic Anhydrase Inhibitors. Cells 2023, 12, 2840. [Google Scholar] [CrossRef]
- Canepa, E.; Parodi-Rullan, R.; Vazquez-Torres, R.; Gamallo-Lana, B.; Guzman-Hernandez, R.; Lemon, N.L.; Angiulli, F.; Debure, L.; Ilies, M.A.; Østergaard, L.; et al. FDA-approved carbonic anhydrase inhibitors reduce amyloid β pathology and improve cognition, by ameliorating cerebrovascular health and glial fitness. Alzheimers Dement. 2023, 19, 5048–5073. [Google Scholar] [CrossRef]
- Solesio, M.E.; Peixoto, P.M.; Debure, L.; Madamba, S.M.; de Leon, M.J.; Wisniewski, T.; Pavlov, E.V.; Fossati, S. Carbonic anhydrase inhibition selectively prevents amyloid β neurovascular mitochondrial toxicity. Aging Cell 2018, 17, e12787. [Google Scholar] [CrossRef]
- Zisapel, N. New perspectives on the role of melatonin in human sleep, circadian rhythms and their regulation. Br. J. Pharmacol. 2018, 175, 3190–3199. [Google Scholar] [CrossRef]
- Jessen, N.A.; Munk, A.S.; Lundgaard, I.; Nedergaard, M. The glymphatic system: A beginner’s guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef]
- Mayer, G.; Frohnhofen, H.; Jokisch, M.; Hermann, D.M.; Gronewold, J. Associations of sleep disorders with all-cause MCI/dementia and different types of dementia—clinical evidence, potential pathomechanisms and treatment options: A narrative review. Front. Neurosci. 2024, 18, 1372326. [Google Scholar] [CrossRef]
- Millán-Plano, S.; Piedrafita, E.; Miana-Mena, F.J.; Fuentes-Broto, L.; Martínez-Ballarín, E.; López-Pingarrón, L.; Sáenz, M.A.; García, J.J. Melatonin and structurally-related compounds protect synaptosomal membranes from free radical damage. Int. J. Mol. Sci. 2010, 11, 312–328. [Google Scholar] [CrossRef] [PubMed]
- Ionov, M.; Burchell, V.; Klajnert, B.; Bryszewska, M.; Abramov, A.Y. Mechanism of neuroprotection of melatonin against beta-amyloid neurotoxicity. Neuroscience 2011, 180, 229–237. [Google Scholar] [CrossRef]
- Steinbach, M.J.; Denburg, N.L. Melatonin in Alzheimer’s Disease: Literature Review and Therapeutic Trials. J. Alzheimers Dis. 2024, 101, S193–S204. [Google Scholar] [CrossRef] [PubMed]
- Poeggeler, B.; Reiter, R.J.; Tan, D.X.; Chen, L.D.; Manchester, L.C. Melatonin, hydroxyl radical-mediated oxidative damage, and aging: A hypothesis. J. Pineal Res. 1993, 14, 151–168. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Plummer, B.F.; Limson, J.; Wintraub, S.T.; Qi, W. Melatonin directly scavenges hydrogen peroxide: A potentially new metabolic pathway of melatonin biotransformation. Free Radic. Biol. Med. 2000, 29, 1177–1185. [Google Scholar] [CrossRef]
- Ximenez, V.F.; Silva, S.O.; Rodrigues, M.R.; Catalani, L.H.; Maghzal, G.J.; Kettle, A.J.; Campa, A. Superoxide dependent oxidation of melatonin by myeloperoxidase. J. Biol. Chem. 2005, 280, 38160–38169. [Google Scholar] [CrossRef] [PubMed]
- O’Neal-Moffitt, G.; Delic, V.; Bradshaw, P.C.; Olcese, J. Prophylactic melatonin significantly reduces Alzheimer’s neuropathology and associated cognitive deficits independent of antioxidant pathways in AβPP(swe)/PS1 mice. Mol. Neurodegener. 2015, 10, 27. [Google Scholar] [CrossRef]
- Pappolla, M.A.; Chyan, Y.J.; Poeggeler, B.; Frangione, B.; Wilson, G.; Ghiso, J.; Reiter, R.J. An assessment of the antioxidant and the antiamyloidogenic properties of melatonin: Implications for Alzheimer’s disease. J. Neural Transm. 2000, 107, 203–231. [Google Scholar] [CrossRef]
- Srinivasan, V.; Pandi-Perumal, S.R.; Cardinali, D.P.; Poeggeler, B.; Hardeland, R. Melatonin in Alzheimer’s disease and other neurodegenerative disorders. Behav. Brain Funct. 2006, 2, 15. [Google Scholar] [CrossRef]
- Chyan, Y.J.; Poeggeler, B.; Omar, R.A.; Chain, D.G.; Frangione, B.; Ghiso, J.; Pappolla, M.A. Potent neuroprotective properties against the Alzheimer beta-amyloid by an endogenous melatonin-related indole structure, indole-3-propionic acid. J. Biol. Chem. 1999, 274, 21937–21942. [Google Scholar] [CrossRef]
- Dkhar, P.; Sharma, R. Attenuation of age-related increase of protein carbonylation in the liver of mice by melatonin and curcumin. Mol. Cell. Biochem. 2013, 380, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Zubero, E.; López-Pingarrón, L.; Ramírez, J.M.; Reyes-Gonzales, M.C.; Azúa-Romeo, F.J.; Soria-Aznar, M.; Agil, A.; García, J.J. Melatonin Preserves Fluidity in Cell and Mitochondrial Membranes against Hepatic Ischemia-Reperfusion. Biomedicines 2023, 11, 1940. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Balmik, A.A.; Chinnathambi, S. Melatonin Reduces GSK3β-Mediated Tau Phosphorylation, Enhances Nrf2 Nuclear Translocation and Anti-Inflammation. ASN Neuro 2020, 12, 1759091420981204. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Kim, M.O. Melatonin ameliorates amyloid beta-induced memory deficits, tau hyperphosphorylation and neurodegeneration via PI 3/Akt/GSk3b pathway in the mouse hippocampus. J. Pineal Res. 2005, 59, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.C.; Liu, X.C.; Yang, S.H.; Song, L.L.; Zhou, S.J.; Deng, S.L.; Tian, L.; Cheng, L.Y. Melatonin inhibits oxidative stress and apoptosis in cryopreserved ovarian tissues via Nrf2/HO-1 signaling pathway. Front. Mol. Biosci. 2020, 7, 163. [Google Scholar] [CrossRef]
- Dhapola, R.; Beura, S.K.; Sharma, P.; Singh, S.K.; HariKrishnaReddy, D. Oxidative stress in Alzheimer’s disease: Current knowledge of signaling pathways and therapeutics. Mol. Biol. Rep. 2024, 51, 48. [Google Scholar] [CrossRef] [PubMed]
- Waseem, M.; Tabassum, H.; Parvez, S. Melatonin modulates permeability transition pore and 5-hydroxydecanoate induced KATP channel inhibition in isolated brain mitochondria. Mitochondrion 2016, 31, 1–8. [Google Scholar] [CrossRef]
- Baydas, G.; Reiter, R.J.; Akbulut, M.; Tuzcu, M.; Tamer, S. Melatonin inhibits neural apoptosis induced by homocysteine in hippocampus of rats via inhibition of cytochrome c translocation and caspase-3 activation and by regulating pro- and anti-apoptotic protein levels. Neuroscience 2005, 135, 879–886. [Google Scholar] [CrossRef]
- Kleszczyński, K.; Zillikens, D.; Fischer, T.W. Melatonin enhances mitochondrial ATP synthesis, reduces reactive oxygen species formation, and mediates translocation of the nuclear erythroid 2-related factor 2 resulting in activation of phase-2 antioxidant enzymes (γ-GCS, HO-1, NQO1) in ultraviolet radiation-treated normal human epidermal keratinocytes (NHEK). J. Pineal Res. 2016, 61, 187–197. [Google Scholar]
- Cheng, Y.; Feng, Z.; Zhang, Q.Z.; Zhang, J.T. Beneficial effects of melatonin in experimental models of Alzheimer disease. Acta Pharmacol. Sin. 2006, 27, 129–139. [Google Scholar] [CrossRef]
- Matsubara, E.; Bryant-Thomas, T.; Pacheco Quinto, J.; Henry, T.L.; Poeggeler, B.; Herbert, D.; Cruz-Sanchez, F.; Chyan, Y.J.; Smith, M.A.; Perry, G.; et al. Melatonin increases survival and inhibits oxidative and amyloid pathology in a transgenic model of Alzheimer’s disease. J. Neurochem. 2003, 85, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Labban, S.; Alshehri, F.S.; Kurdi, M.; Alatawi, Y.; Alghamdi, B.S. Melatonin Improves Short-Term Spatial Memory in a Mouse Model of Alzheimer’s Disease. Degener. Neurol. Neuromuscul. Dis. 2021, 11, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Vincent, B. Protective roles of melatonin against the amyloid-dependent development of Alzheimer’s disease: A critical review. Pharmacol. Res. 2018, 134, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Pappolla, M.; Bozner, P.; Soto, C.; Shao, H.; Robakis, N.K.; Zagorski, M.; Frangione, B.; Ghiso, J. Inhibition of Alzheimer beta-fibrillogenesis by melatonin. J. Biol. Chem. 1998, 273, 7185–7188. [Google Scholar] [CrossRef]

 ) (red star symbol). Notably, many of these integral components, including the transcription factor Nrf2 itself with its concomitant downstream antioxidant response, have been reported downregulated in AD (
) (red star symbol). Notably, many of these integral components, including the transcription factor Nrf2 itself with its concomitant downstream antioxidant response, have been reported downregulated in AD ( ) (yellow star symbol). Thus, enhancing Nrf2 expression and/or upregulation may constitute an attractive translational approach in the field of neurodegeneration and AD, with potential to exert a positive impact not only in the metabolic aspects of the disease but also in boosting the antioxidant response and reducing the detrimental effects of oxidative stress on proteins, lipids and nucleic acids. Complex V of the respiratory chain, also known as ATP synthase, is represented in light blue while Complexes I-IV are illustrated in color beige to differentiate their function. Complex V catalyzes the synthesis of ATP using the proton gradient generated by the complexes I-IV. Abbreviations: G6P, Glucose 6 phosphate; PEP, phosphoenolpyruvate; PK, pyruvate kinase; ME1, malic enzyme 1; PDH, pyruvate dehydrogenase; ICDH, isocitrate dehydrogenase; αKG, alpha ketoglutarate; KGDH, ketoglutarate dehydrogenase; SDH, succinate dehydrogenase; OxalAc, oxaloacetate; CS, citrate synthase; FAs, fatty acids; FATP, fatty acid transport proteins; FAO, fatty acid oxidation; GScx, glutathione synthase complex; MMP, mitochondrial membrane potential; TCA; tricarboxylic acid; CoQ; coenzyme Q; AcCoA; acetyl-coenzyme A; MDH; malate dehydrogenase 6PGD; 6-phosphogluconate dehydrogenase; G6PDH; glucose-6-phosphate dehydrogenase; R5P; ribose 5-phosphate; CPT1, carnitine palmitoyltransferase 1.
) (red star symbol). Notably, many of these integral components, including the transcription factor Nrf2 itself with its concomitant downstream antioxidant response, have been reported downregulated in AD () (yellow star symbol). Thus, enhancing Nrf2 expression and/or upregulation may constitute an attractive translational approach in the field of neurodegeneration and AD, with potential to exert a positive impact not only in the metabolic aspects of the disease but also in boosting the antioxidant response and reducing the detrimental effects of oxidative stress on proteins, lipids and nucleic acids. Complex V of the respiratory chain, also known as ATP synthase, is represented in light blue while Complexes I-IV are illustrated in color beige to differentiate their function. Complex V catalyzes the synthesis of ATP using the proton gradient generated by the complexes I-IV. Abbreviations: G6P, Glucose 6 phosphate; PEP, phosphoenolpyruvate; PK, pyruvate kinase; ME1, malic enzyme 1; PDH, pyruvate dehydrogenase; ICDH, isocitrate dehydrogenase; αKG, alpha ketoglutarate; KGDH, ketoglutarate dehydrogenase; SDH, succinate dehydrogenase; OxalAc, oxaloacetate; CS, citrate synthase; FAs, fatty acids; FATP, fatty acid transport proteins; FAO, fatty acid oxidation; GScx, glutathione synthase complex; MMP, mitochondrial membrane potential; TCA; tricarboxylic acid; CoQ; coenzyme Q; AcCoA; acetyl-coenzyme A; MDH; malate dehydrogenase 6PGD; 6-phosphogluconate dehydrogenase; G6PDH; glucose-6-phosphate dehydrogenase; R5P; ribose 5-phosphate; CPT1, carnitine palmitoyltransferase 1.
) (yellow star symbol). Thus, enhancing Nrf2 expression and/or upregulation may constitute an attractive translational approach in the field of neurodegeneration and AD, with potential to exert a positive impact not only in the metabolic aspects of the disease but also in boosting the antioxidant response and reducing the detrimental effects of oxidative stress on proteins, lipids and nucleic acids. Complex V of the respiratory chain, also known as ATP synthase, is represented in light blue while Complexes I-IV are illustrated in color beige to differentiate their function. Complex V catalyzes the synthesis of ATP using the proton gradient generated by the complexes I-IV. Abbreviations: G6P, Glucose 6 phosphate; PEP, phosphoenolpyruvate; PK, pyruvate kinase; ME1, malic enzyme 1; PDH, pyruvate dehydrogenase; ICDH, isocitrate dehydrogenase; αKG, alpha ketoglutarate; KGDH, ketoglutarate dehydrogenase; SDH, succinate dehydrogenase; OxalAc, oxaloacetate; CS, citrate synthase; FAs, fatty acids; FATP, fatty acid transport proteins; FAO, fatty acid oxidation; GScx, glutathione synthase complex; MMP, mitochondrial membrane potential; TCA; tricarboxylic acid; CoQ; coenzyme Q; AcCoA; acetyl-coenzyme A; MDH; malate dehydrogenase 6PGD; 6-phosphogluconate dehydrogenase; G6PDH; glucose-6-phosphate dehydrogenase; R5P; ribose 5-phosphate; CPT1, carnitine palmitoyltransferase 1.
) (red star symbol). Notably, many of these integral components, including the transcription factor Nrf2 itself with its concomitant downstream antioxidant response, have been reported downregulated in AD () (yellow star symbol). Thus, enhancing Nrf2 expression and/or upregulation may constitute an attractive translational approach in the field of neurodegeneration and AD, with potential to exert a positive impact not only in the metabolic aspects of the disease but also in boosting the antioxidant response and reducing the detrimental effects of oxidative stress on proteins, lipids and nucleic acids. Complex V of the respiratory chain, also known as ATP synthase, is represented in light blue while Complexes I-IV are illustrated in color beige to differentiate their function. Complex V catalyzes the synthesis of ATP using the proton gradient generated by the complexes I-IV. Abbreviations: G6P, Glucose 6 phosphate; PEP, phosphoenolpyruvate; PK, pyruvate kinase; ME1, malic enzyme 1; PDH, pyruvate dehydrogenase; ICDH, isocitrate dehydrogenase; αKG, alpha ketoglutarate; KGDH, ketoglutarate dehydrogenase; SDH, succinate dehydrogenase; OxalAc, oxaloacetate; CS, citrate synthase; FAs, fatty acids; FATP, fatty acid transport proteins; FAO, fatty acid oxidation; GScx, glutathione synthase complex; MMP, mitochondrial membrane potential; TCA; tricarboxylic acid; CoQ; coenzyme Q; AcCoA; acetyl-coenzyme A; MDH; malate dehydrogenase 6PGD; 6-phosphogluconate dehydrogenase; G6PDH; glucose-6-phosphate dehydrogenase; R5P; ribose 5-phosphate; CPT1, carnitine palmitoyltransferase 1.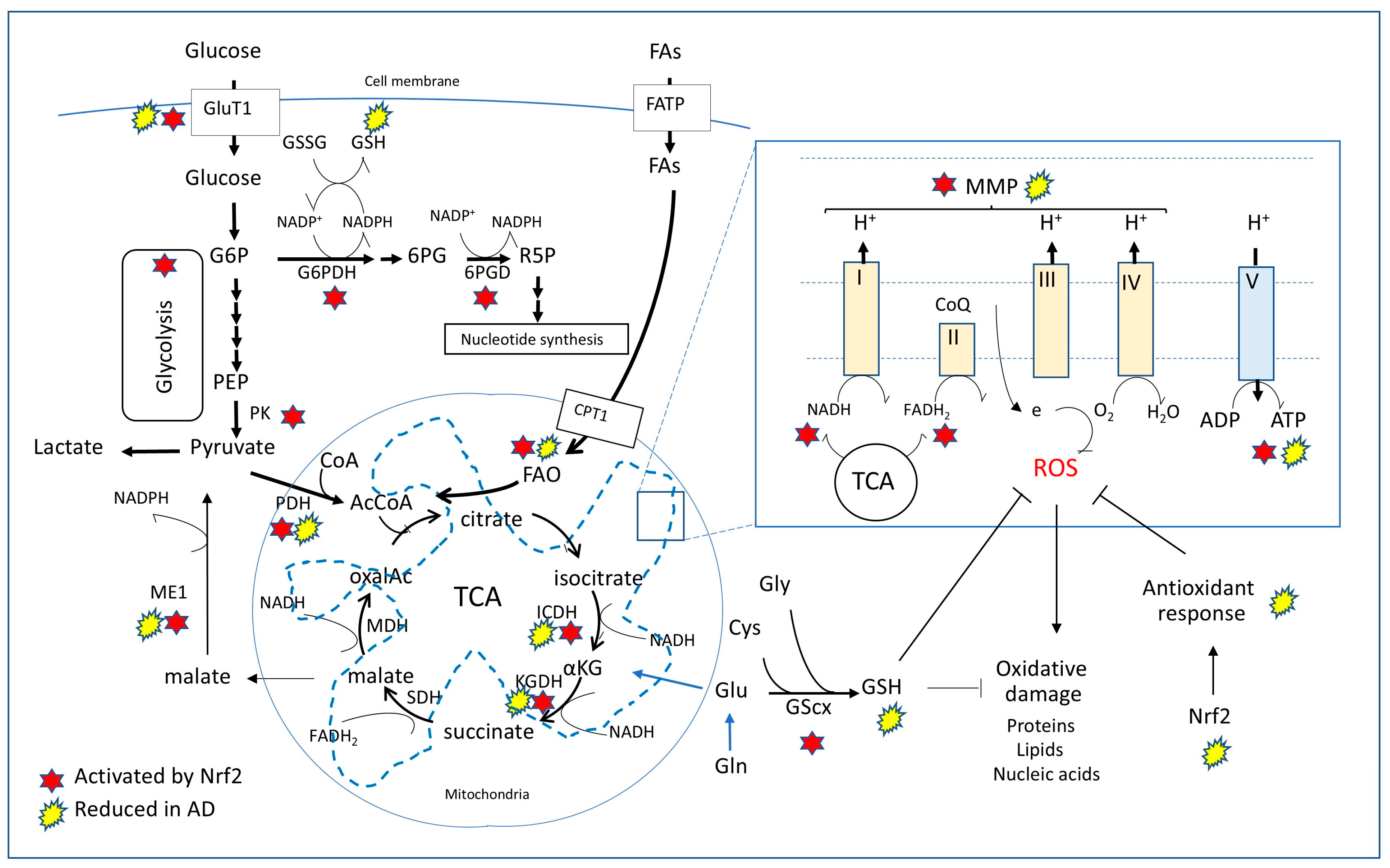
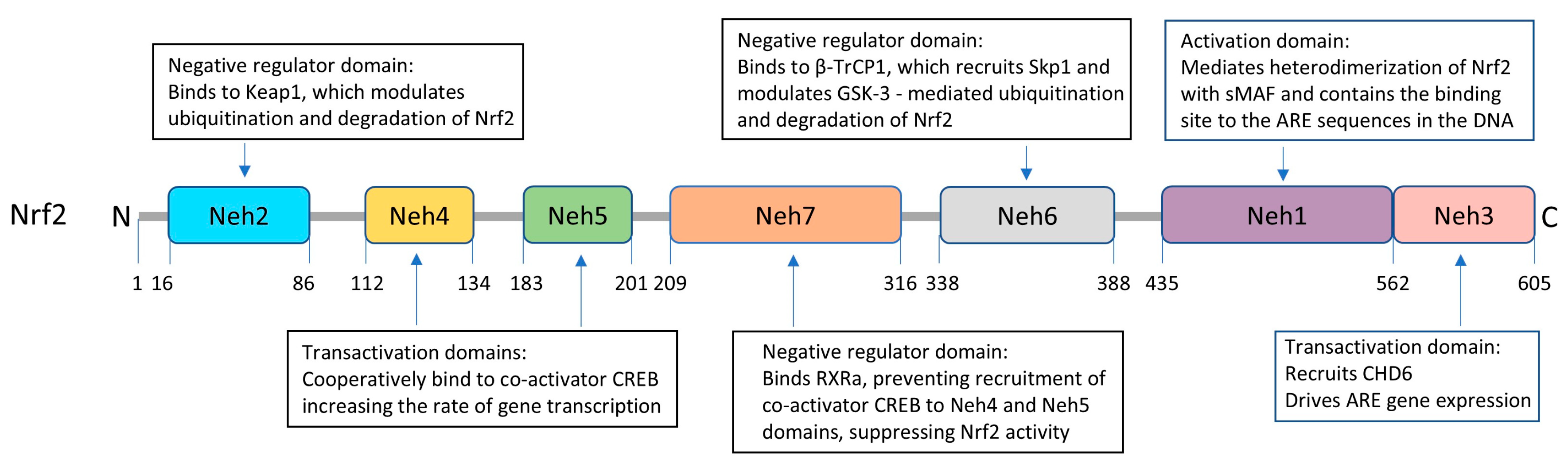
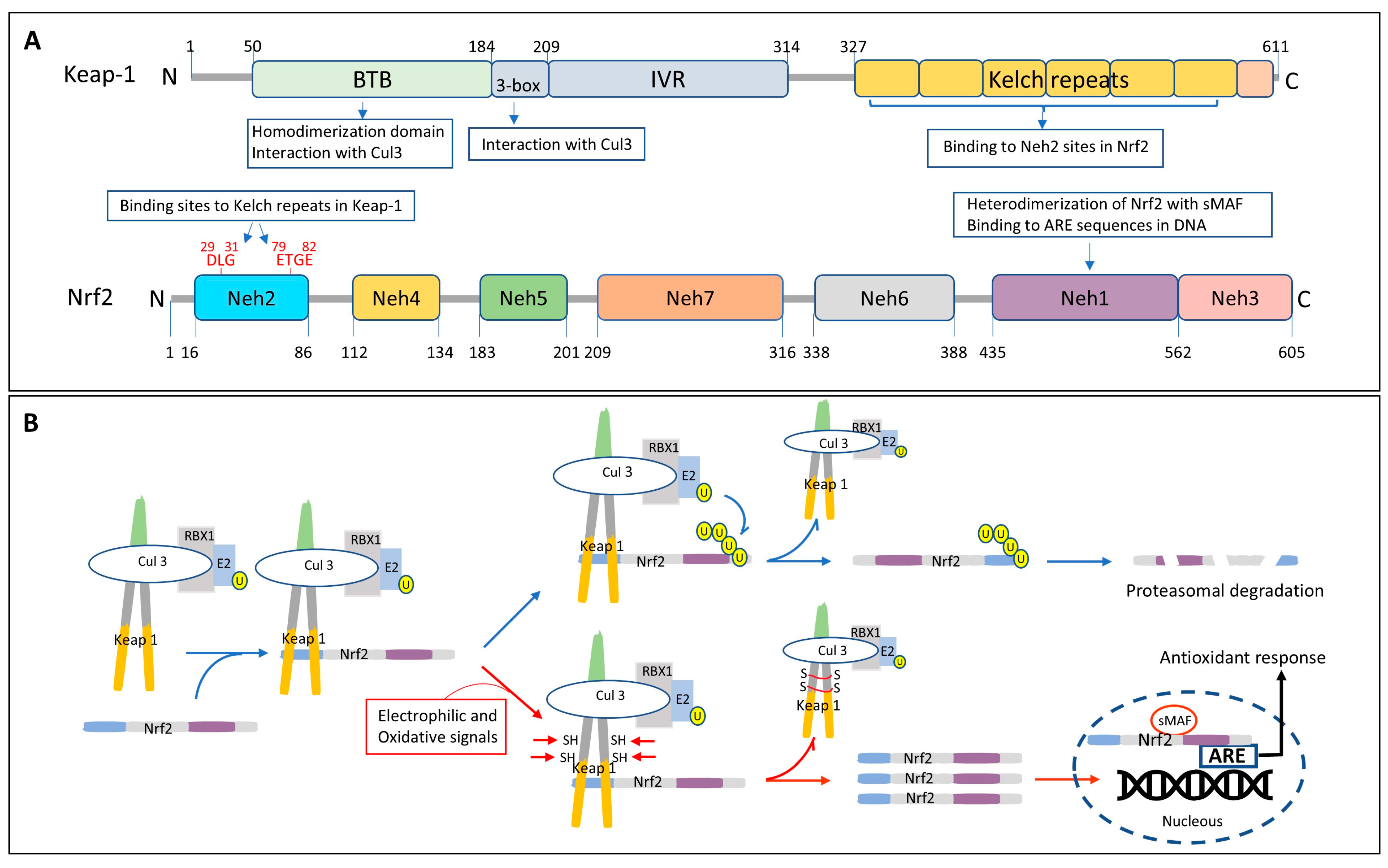
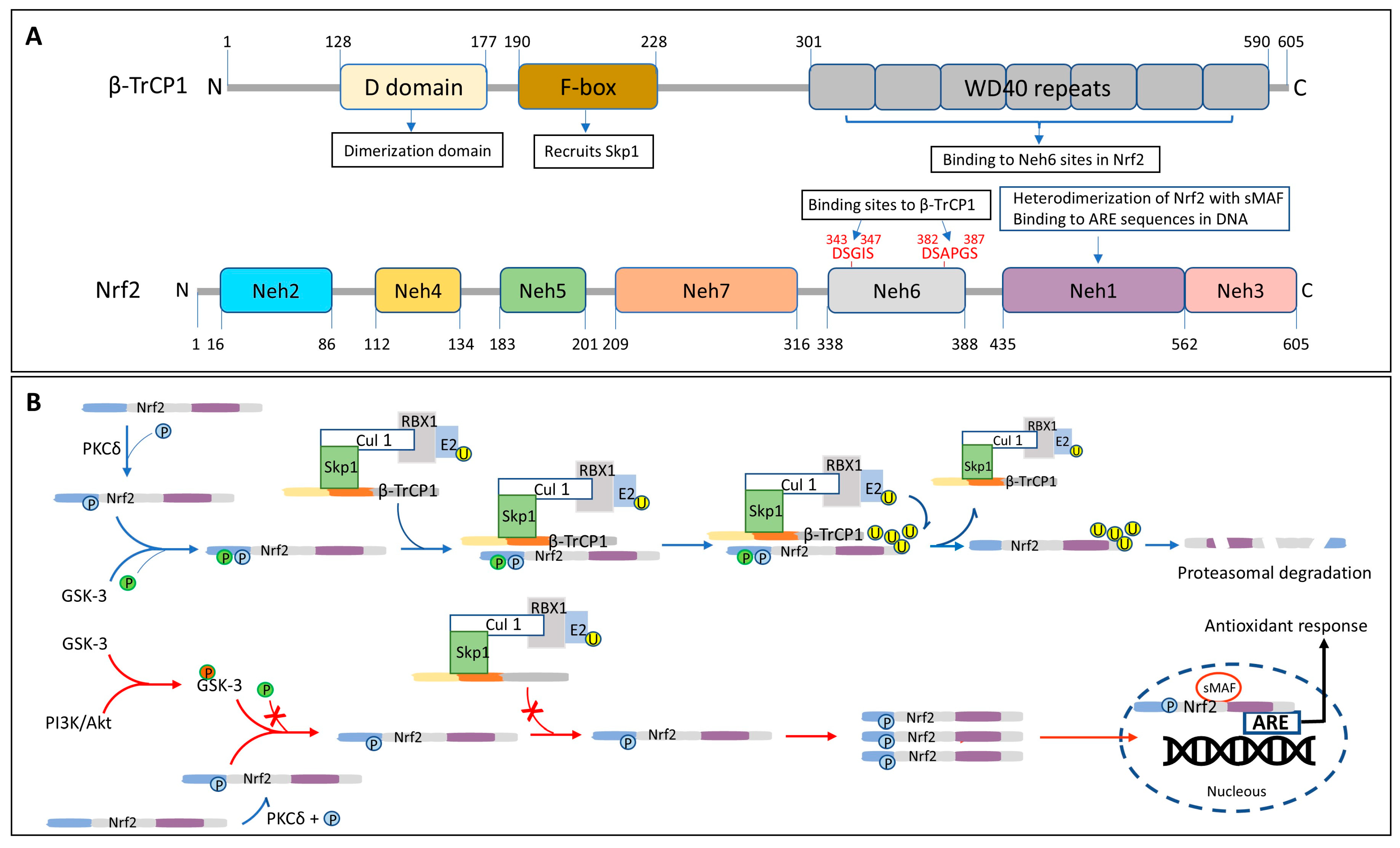
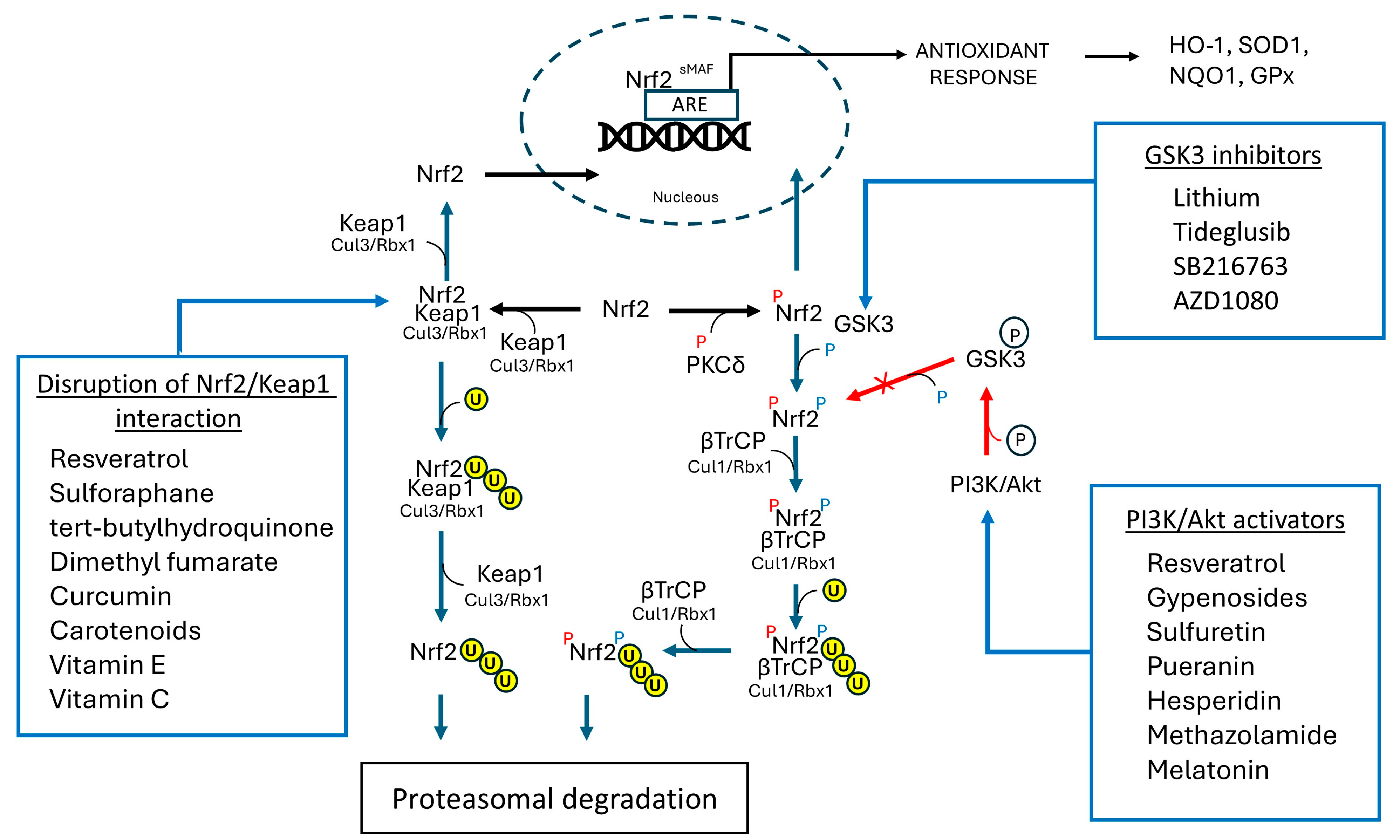
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rostagno, A.; Ghiso, J. Alzheimer’s Disease Pathogenic Mechanisms: Linking Redox Homeostasis and Mitochondria-Associated Metabolic Pathways Through Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2). Antioxidants 2025, 14, 812. https://doi.org/10.3390/antiox14070812
Rostagno A, Ghiso J. Alzheimer’s Disease Pathogenic Mechanisms: Linking Redox Homeostasis and Mitochondria-Associated Metabolic Pathways Through Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2). Antioxidants. 2025; 14(7):812. https://doi.org/10.3390/antiox14070812
Chicago/Turabian StyleRostagno, Agueda, and Jorge Ghiso. 2025. "Alzheimer’s Disease Pathogenic Mechanisms: Linking Redox Homeostasis and Mitochondria-Associated Metabolic Pathways Through Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2)" Antioxidants 14, no. 7: 812. https://doi.org/10.3390/antiox14070812
APA StyleRostagno, A., & Ghiso, J. (2025). Alzheimer’s Disease Pathogenic Mechanisms: Linking Redox Homeostasis and Mitochondria-Associated Metabolic Pathways Through Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2). Antioxidants, 14(7), 812. https://doi.org/10.3390/antiox14070812