
Impact of Obesity Caused by a High-Fat Diet on the Heart’s Redox Balance

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Animals and Dietary Model
2.3. Obtaining Blood Samples and Preparation of Cardiac Homogenates After 12 Weeks of Control Diet or High-Fat Diet
2.4. Determination of Biochemical Parameters in Plasma
2.5. Echocardiographic Determinations
2.6. NOX4 Determination
2.7. Lipid Peroxidation
2.8. Protein Carbonylation
2.9. Glutathione Quantification
2.10. Quantification of Pyridine Nucleotides
2.11. RNA Isolation and qRT-PCR
2.12. Enzymatic Activities of Antioxidant Enzymes
2.13. Statistical Analysis
3. Results
3.1. Effect of HFD on Mice’s Physical, Biochemical, and Echocardiographic Values
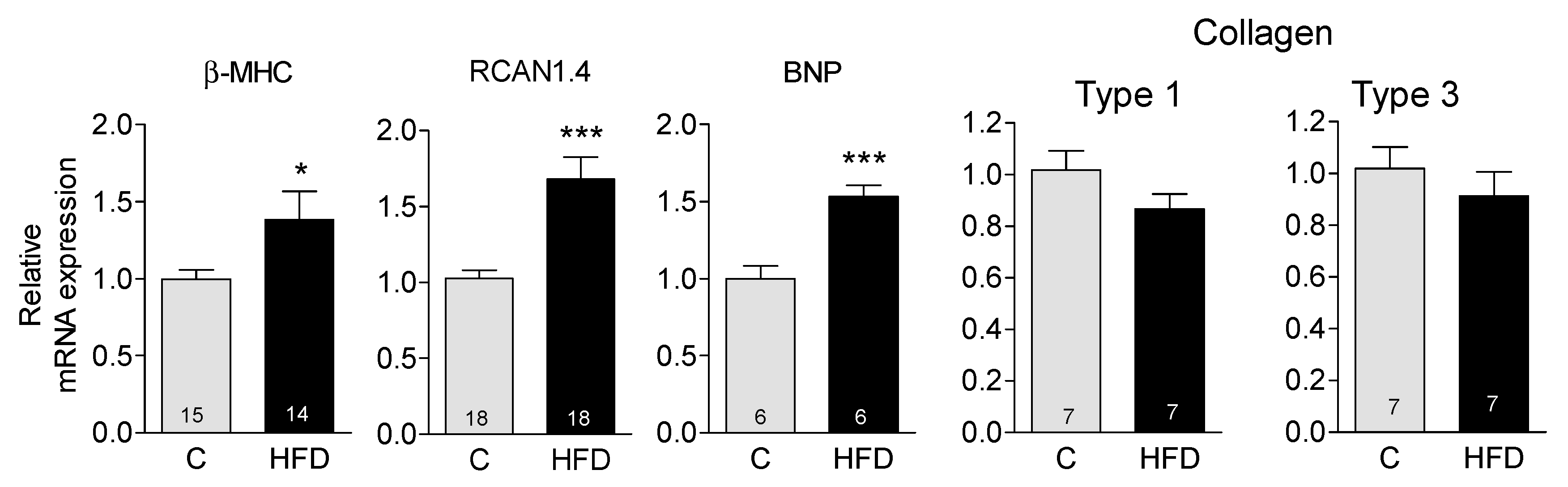
3.2. HFD Induces the Appearance of Markers Indicative of Pathological Hypertrophy
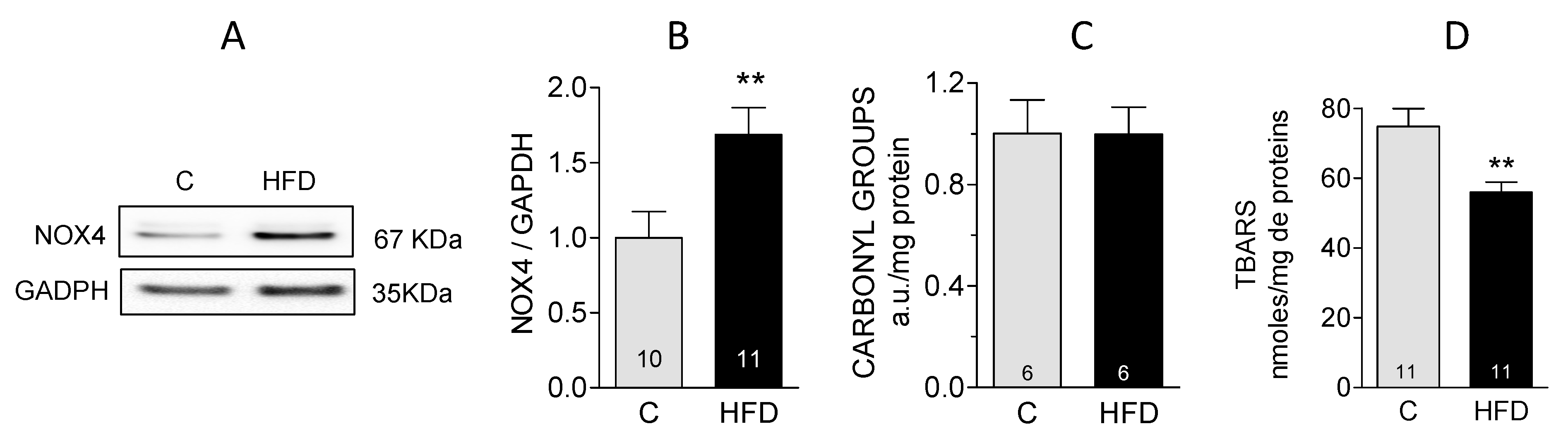
3.3. Evaluation of Oxidative Stress in Cardiac Tissue
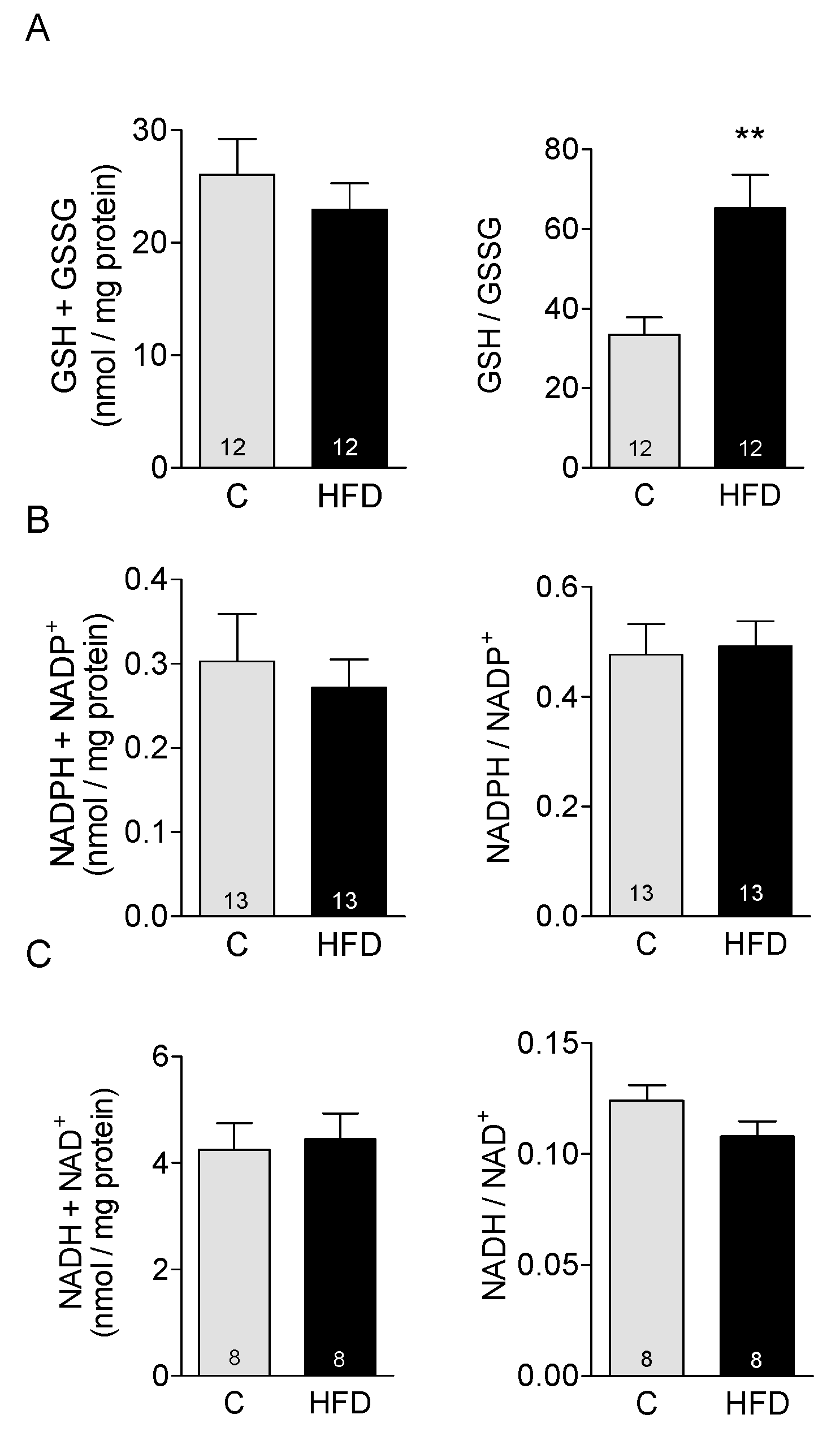
3.4. Effect of HFD on Redox Buffers in Cardiac Tissue
3.5. Effect of HFD on Antioxidant Enzymes in Cardiac Tissue
4. Discussion
5. Conclusions
6. Limitations of This Work
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| βMHC | myosin heavy chain β-isoform |
| BNP | brain natriuretic peptide |
| GSH | reduced glutathione |
| GSSG | oxidized glutathione |
| HFD | high-fat diet |
| NADH/NAD+ | nicotinamide adenine dinucleotide reduced/oxidized form |
| NADPH/NADP+ | nicotinamide adenine dinucleotide phosphate reduced/oxidized form |
| NOX4 | NADPH oxidase 4 |
| RCAN 1.4 | regulator of calcineurin 1 isoform 4 |
| ROS | reactive oxygen species |
| TBARS | thiobarbituric acid reactive substances |
References
- Carbone, S.; Lavie, C.J.; Elagizi, A.; Arena, R.; Ventura, H.O. The Impact of Obesity in Heart Failure. Heart Fail. Clin. 2020, 16, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.D.; Litwin, S.E.; Sweeney, G. Cardiac remodeling in obesity. Physiol. Rev. 2008, 88, 389–419. [Google Scholar] [CrossRef]
- Wang, H.T.; Liu, C.F.; Tsai, T.H.; Chen, Y.L.; Chang, H.W.; Tsai, C.Y.; Leu, S.; Zhen, Y.Y.; Chai, H.T.; Chung, S.Y.; et al. Effect of obesity reduction on preservation of heart function and attenuation of left ventricular remodeling, oxidative stress and inflammation in obese mice. J. Transl. Med. 2012, 10, 145–157. [Google Scholar] [CrossRef]
- Murdoch, C.E.; Zhang, M.; Cave, A.C.; Shah, A.M. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodeling and failure. Cardiovasc. Res. 2006, 71, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Seddon, M.; Looi, Y.H.; Shah, A.M. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart 2007, 93, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Surwit, R.S.; Kuhn, C.M.; Cochrane, C.; Mc Cubbin, J.A.; Feinglos, M.N. Diet-induced type II diabetes in C57BL/6J mice. Diabetes 1988, 37, 1163–1167. [Google Scholar] [CrossRef]
- Winzell, M.S.; Ahrén, B. The high-fat diet-fed mouse: A model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes 2004, 53 (Suppl. S3), S215–S219. [Google Scholar] [CrossRef]
- Sanchez, G.; Araneda, F.; Peña, J.P.; Finkelstein, J.P.; Riquelme, J.A.; Montecinos, L.; Barrientos, G.; Llanos, P.; Pedrozo, Z.; Said, M.; et al. High-Fat-Diet- Induced Obesity Produces Spontaneous Ventricular Arrhythmias and Increases the Activity of Ryanodine Receptors in Mice. Int. J. Mol. Sci. 2018, 19, 533. [Google Scholar] [CrossRef]
- Ramachandra, C.J.A.; Cong, S.; Chan, X.; Yap, E.P.; Yu, F.; Hausenloy, D.J. Oxidative stress in cardiac hypertrophy: From molecular mechanisms to novel therapeutic targets. Free Radic. Biol. Med. 2021, 166, 297–312. [Google Scholar] [CrossRef]
- Jones, D.P.; Sies, H. The Redox Code. Antioxid. Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef]
- Matuz-Mares, D.; Riveros-Rosas, H.; Vilchis-Landeros, M.M.; Vazquez-Meza, H. Glutathione participation in the prevention of cardiovascular diseases. Antioxidants 2021, 10, 1220. [Google Scholar] [CrossRef]
- Tan, M.; Yin, Y.; Ma, X.; Zhang, J.; Pan, W.; Tan, M.; Zhao, Y.; Yang, T.; Jiang, T.; Li, H. Glutathione system enhancement for cardiac protection: Pharmacological options against oxidative stress and ferroptosis. Cell Death Dis. 2023, 14, 131. [Google Scholar] [CrossRef] [PubMed]
- Francisco, A.; Figueira, T.R.; Castilho, R.F. Mitochondrial NAD(P)+ Transhydrogenase: From Molecular Features to Physiology and Disease. Antioxid. Redox Signal. 2022, 36, 864–884. [Google Scholar] [CrossRef] [PubMed]
- Donoso, P.; Finkelstein, J.P.; Montecinos, L.; Said, M.; Sanchez, G.; Vittone, L.; Bull, R. Stimulation of NOX2 in isolated hearts reversibly sensitizes RyR2 channels to activation by cytoplasmic calcium. J. Mol. Cell Cardiol. 2014, 68, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Rottman, J.N.; Ni, G.; Brown, M. Echocardiographic evaluation of ventricular function in mice. Echocardiography 2007, 24, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal. Biochem. 1980, 106, 207–212. [Google Scholar] [CrossRef]
- Murphy, M.P. Mitochondrial thiols in antioxidant protection and redox signaling distinct roles for glutathionylation and other thiol modifications. Antioxid. Redox Signal. 2012, 16, 476–495. [Google Scholar] [CrossRef]
- Márquez-Álvarez, C.M.; Hernández-Cruz, E.Y.; Pedraza-Chaverri, J. Oxidative stress in animal models of obesity caused by hypercaloric diets: A systematic review. Life Sci. 2023, 331, 122019. [Google Scholar] [CrossRef]
- Toye, A.A.; Lippiat, J.D.; Proks, P.; Shimomura, K.; Bentley, L.; Hugill, A.; Mijat, V.; Goldsworthy, M.; Moir, L.; Haynes, A.; et al. A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia 2005, 48, 675–686. [Google Scholar] [CrossRef]
- Ronchi, J.A.; Francisco, A.; Passos, L.A.; Figueira, T.R.; Castilho, R.F. The Contribution of Nicotinamide Nucleotide Transhydrogenase to Peroxide Detoxification Is Dependent on the Respiratory State and Counterbalanced by Other Sources of NADPH in Liver Mitochondria. J. Biol. Chem. 2016, 291, 20173–20187. [Google Scholar] [CrossRef]
- Karthikeyan, S.K.; Nallasamy, P.; Cleveland, J.M.; Arulmani, A.; Raveendran, A.; Karimi, M.; Ansari, M.O.; Challa, A.K.; Ponnusamy, M.P.; Benjamin, I.J.; et al. ProteotoxomiRs: Diagnostic and pathologic miRNA signatures for reductive stress induced proteotoxic heart disease. Redox Biol. 2025, 81, 103525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Min, X.; Li, C.; Benjamin, I.J.; Qian, B.; Zhang, X.; Ding, Z.; Gao, X.; Yao, Y.; Ma, Y.; et al. Involvement of reductive stress in the cardiomyopathy in transgenic mice with cardiac-specific overexpression of heat shock protein 27. Hypertension 2010, 55, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, G.; Narasimhan, M.; Tamowski, S.; Darley-Usmar, V.; Rajasekaran, N.S. Constitutive activation of Nrf2 induces a stable reductive state in the mouse myocardium. Redox Biol. 2017, 12, 937–945. [Google Scholar] [CrossRef]
- Rindler, P.M.; Plafker, S.M.; Szweda, L.I.; Kinter, M. High dietary fat selectively increases catalase expression within cardiac mitochondria. J. Biol. Chem. 2013, 288, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, N.; Gao, Z.; Gao, J.; Wang, X.; Xie, H.; Wang, C.Y.; Zhang, S. Reductive stress: The key pathway in metabolic disorders induced by overnutrition. J. Adv. Res. 2025; in press. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Cole, M.A.; Murray, A.J.; Cochlin, L.E.; Heather, L.C.; McAleese, S.; Knight, N.S.; Sutton, E.; Jamil, A.A.; Parassol, N.; Clarke, K. A high fat diet increases mitochondrial fatty acid oxidation and uncoupling to decrease efficiency in rat heart. Basic Res. Cardiol. 2011, 106, 447–457. [Google Scholar] [CrossRef]
- Rosca, M.G.; Vazquez, E.J.; Chen, Q.; Kerner, J.; Kern, T.S.; Hoppel, C.L. Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes 2012, 61, 2074–2083. [Google Scholar] [CrossRef]
- Xiao, W.; Loscalzo, J. Metabolic Responses to Reductive Stress. Antioxid. Redox Signal. 2020, 32, 1330–1347. [Google Scholar] [CrossRef]
- Schonfeld, P.; Wojtczak, L. Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radic. Biol. Med. 2008, 45, 231–241. [Google Scholar] [CrossRef]
- Jezek, P.; Holendova, B.; Garlid, K.D.; Jaburek, M. Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling. Antioxid. Redox Signal. 2018, 29, 667–714. [Google Scholar] [CrossRef]
- Wojtczak, L.; Schonfeld, P. Effect of fatty acids on energy coupling processes in mitochondria. Biochim. Biophys. Acta 1993, 1183, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Tocchetti, C.G.; Caceres, V.; Stanley, B.A.; Xie, C.; Shi, S.; Watson, W.H.; O’Rourke, B.; Spadari-Bratfisch, R.C.; Cortassa, S.; Akar, F.G.; et al. GSH or palmitate preserves mitochondrial energetic/redox balance, preventing mechanical dysfunction in metabolically challenged myocytes/hearts from type 2 diabetic mice. Diabetes 2012, 61, 3094–3105. [Google Scholar] [CrossRef] [PubMed]
- Norris, K.M.; Okie, W.; Kim, W.K.; Adhikari, R.; Yoo, S.; King, S.; Pazdro, R. A high-fat diet differentially regulates glutathione phenotypes in the obesity-prone mouse strains DBA/2J, C57BL/6J, and AKR/J. Nutr. Res. 2016, 36, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Margaritelis, N.V.; Kyparos, A.; Paschalis, V.; Theodorou, A.A.; Panayiotou, G.; Zafeiridis, A.; Dipla, K.; Nikolaidis, M.G.; Vrabas, I.S. Reductive stress after exercise: The issue of redox individuality. Redox Biol. 2014, 2, 520–528. [Google Scholar] [CrossRef]
- Fulghum, K.; Collins, H.E.; Jones, S.P.; Hill, B.G. Influence of biological sex and exercise on murine cardiac metabolism. J. Sport Health Sci. 2022, 11, 479–494. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Moulin, M.; Piquereau, J.; Lemaire, C.; Mericskay, M.; Veksler, V.; Garnier, A. Mitochondria: A central target for sex differences in pathologies. Clin. Sci. 2017, 131, 803–822. [Google Scholar] [CrossRef]
- Grayson, C.; Faerman, B.; Koufos, O.; Mailloux, R.J. Fatty acid oxidation drives mitochondrial hydrogen peroxide production by α-ketoglutarate dehydrogenase. J. Biol. Chem. 2024, 300, 107159. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gen Bank Accession Number | Primer Sequences (5′-3′) Forward(F) Reverse (R) | Length (bp) | Annealing T (°C) | Fragment Size (bp) |
|---|---|---|---|---|---|
| β-MHC | NM_001425737.1 | F: AAGCAGCAGTTGGATGAGCG | 20 | 55 | 133 |
| R: CCTCGATGCGTGCCTGAAGC | 20 | ||||
| RCAN1.4 | NM_019466.4 | F: CCCGTGAAAAAGCAGAATGC | 20 | 55 | 141 |
| R: TCCTTGTCATATGTTCTGAAGAGGG | 25 | ||||
| BNP | NM_008726.6 | F: CATGGATCTCCTGAAGGTGC | 20 | 55 | 188 |
| R: CCTTCAAGAGCTGTCTCTGG | 20 | ||||
| Collagen Type 1 | NM_007742.4 | F: CTGACGCATGGCCAAGAAGA | 20 | 55 | 94 |
| R: AGCATACCTCGGGTTTCCAC | 20 | ||||
| Collagen Type 3 | NM_009930.2 | F: TCCCCTGGAATCTGTGAATC | 20 | 55 | 63 |
| R: TGAGTCGAATTGGGGAGAAT | 20 | ||||
| GR | NM_010344.4 | F: CAGTTCCTCACGAGAGCCAG | 20 | 55 | 123 |
| R: TCTCCACAGCAATGTACCCG | 20 | ||||
| GPX4 | NM_001037741.4 | F: TTACGAATCCTGGCCTTCCC | 20 | 55 | 82 |
| R: CGGCTGCAAACTCCTTGATT | 20 | ||||
| SOD1 | NM_011434.2 | F: CCAGTGCAGGACCTCATTTT | 20 | 55 | 216 |
| R: CACCTTTGCCCAAGTCATCT | 20 | ||||
| SOD2 | NM_013671.3 | F: GGCCAAGGGAGATGTTACAA | 20 | 55 | 216 |
| R: GAACCTTGGACTCCCACA | 18 | ||||
| Catalase | NM_009804.2 | F: TGCAGCTCCGCAATCCTAC | 19 | 55 | 113 |
| R: CTCCGGTGGTCAGGACATCA | 20 |
| Control | HFD | p | |
|---|---|---|---|
| n | 34 | 34 | |
| Physical Parameters | |||
| Body Weight (g) | 28.2 ± 2.5 | 43.5 ± 4.7 | <0.001 |
| Heart Weight (mg) | 139 ± 15 | 153 ± 16 | <0.001 |
| Tibia Length (mm) | 16.98 ± 0.30 | 16.97 ± 0.40 | ns |
| HW/TL (mg/mm) | 8.22 ± 0.88 | 9.02 ± 0.95 | <0.001 |
| n | 11 | 12 | |
| Biochemical Parameters | |||
| Glucose (mg/dL) | 156 ± 48 | 250 ± 20 | <0.001 |
| Insulin (mIU/L) | 3.6 ± 1.9 | 35.9 ± 11.9 | <0.001 |
| Triglycerides (mg/dL) | 68 ± 14 | 86 ± 9 | <0.001 |
| Cholesterol (mg/dL) | 82 ± 12 | 164 ± 24 | <0.001 |
| Control | HFD | p | |
|---|---|---|---|
| n | 8 | 8 | |
| Heart rate (BPM) | 712 ± 24 | 756 ± 31 | ns |
| LVID diastole, mm | 0.260 ± 0.020 | 0.311 ± 0.025 | ns |
| LVID systole, mm | 0.151 ± 0.017 | 0.150 ± 0.021 | ns |
| FS% | 42.80 ± 2.22 | 49.78 ± 4.90 | ns |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Utreras-Mendoza, Y.; Mujica Valenzuela, I.; Montecinos, L.; Donoso, P.; Sánchez, G. Impact of Obesity Caused by a High-Fat Diet on the Heart’s Redox Balance. Antioxidants 2025, 14, 708. https://doi.org/10.3390/antiox14060708
Utreras-Mendoza Y, Mujica Valenzuela I, Montecinos L, Donoso P, Sánchez G. Impact of Obesity Caused by a High-Fat Diet on the Heart’s Redox Balance. Antioxidants. 2025; 14(6):708. https://doi.org/10.3390/antiox14060708
Chicago/Turabian StyleUtreras-Mendoza, Yildy, Isidora Mujica Valenzuela, Luis Montecinos, Paulina Donoso, and Gina Sánchez. 2025. "Impact of Obesity Caused by a High-Fat Diet on the Heart’s Redox Balance" Antioxidants 14, no. 6: 708. https://doi.org/10.3390/antiox14060708
APA StyleUtreras-Mendoza, Y., Mujica Valenzuela, I., Montecinos, L., Donoso, P., & Sánchez, G. (2025). Impact of Obesity Caused by a High-Fat Diet on the Heart’s Redox Balance. Antioxidants, 14(6), 708. https://doi.org/10.3390/antiox14060708