
OTUB1-SLC7A11 Axis Mediates 4-Octyl Itaconate Protection Against Acetaminophen-Induced Ferroptotic Liver Injury


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Animals and Treatments
2.3. Cell Culture
2.4. Cell Viability Assay
2.5. Metabolomics Profiling
2.5.1. Metabolites Extraction
2.5.2. UHPLC-QTOF-MS Analysis
2.5.3. UHPLC-QTOF-MS Data Processing
2.5.4. Metabolic Set Enrichment Analysis (MSEA) and Pathway Analysis
2.6. ROS Detection
2.7. Quantitative Real-Time PCR (RT-qPCR)
2.8. Western Blot Analysis
2.9. Immunofluorescence Staining
2.10. Co-Immunoprecipitation (Co-IP) and Ubiquitination Assays
2.11. OTUB1 Knockdown
2.12. Hematoxylin–Eosin (H&E) Staining
2.13. Immunohistochemistry (IHC)
2.14. Measurement of Alanine Aminotransferase (ALT) and Aspartate Aminotransferase (AST) Levels
2.15. Measurement of Hepatic Iron Levels
2.16. GSH Detection
2.17. Measurement of SOD and MDA Levels
2.18. Statistical Analysis
3. Results
3.1. 4-OI Protects Against APAP-Induced Liver Injury by Modulating Inflammatory Response and Oxidative Stress
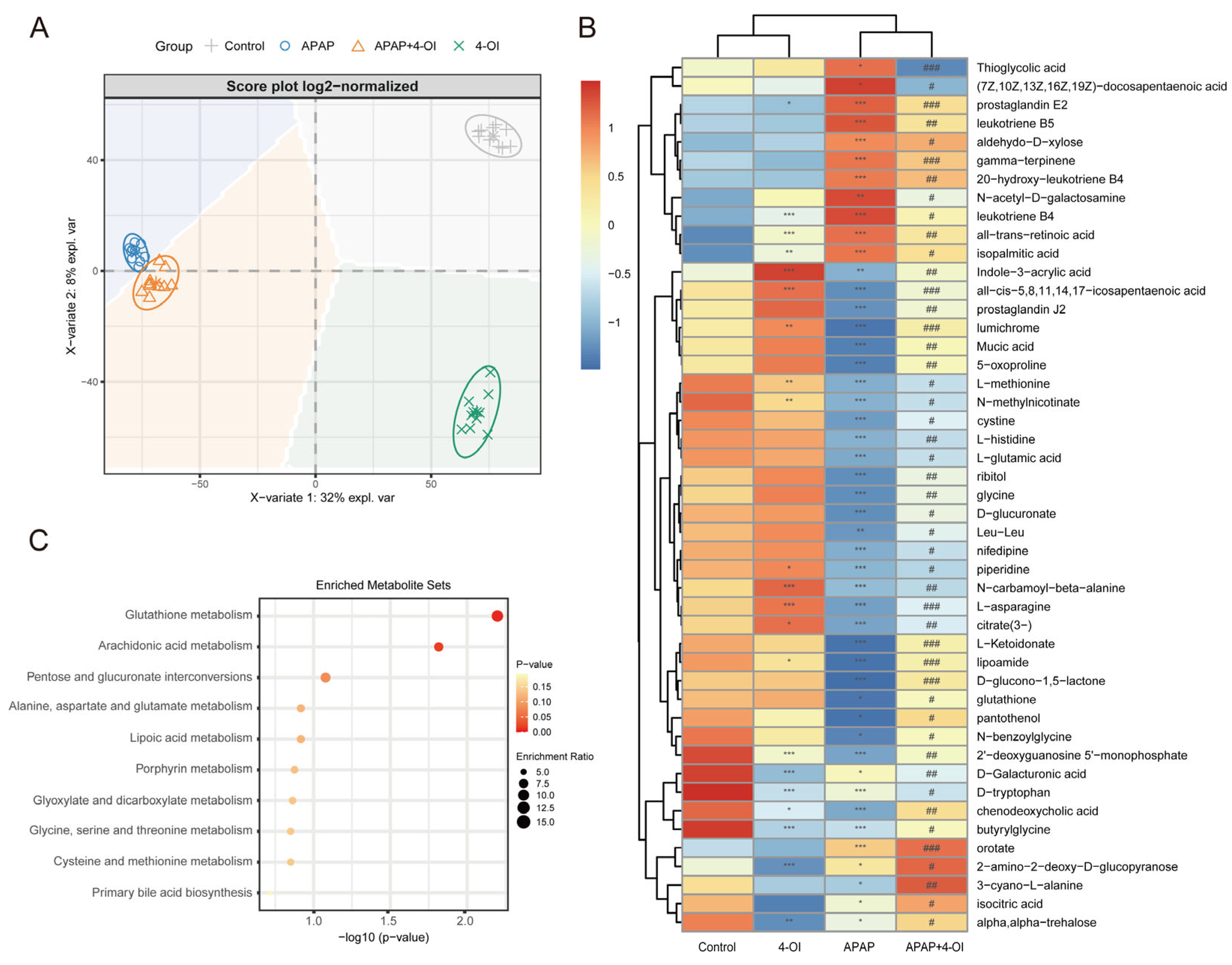
3.2. 4-OI Modulates Both Inflammatory and Antioxidant Metabolic Pathways in APAP-Induced Hepatotoxicity
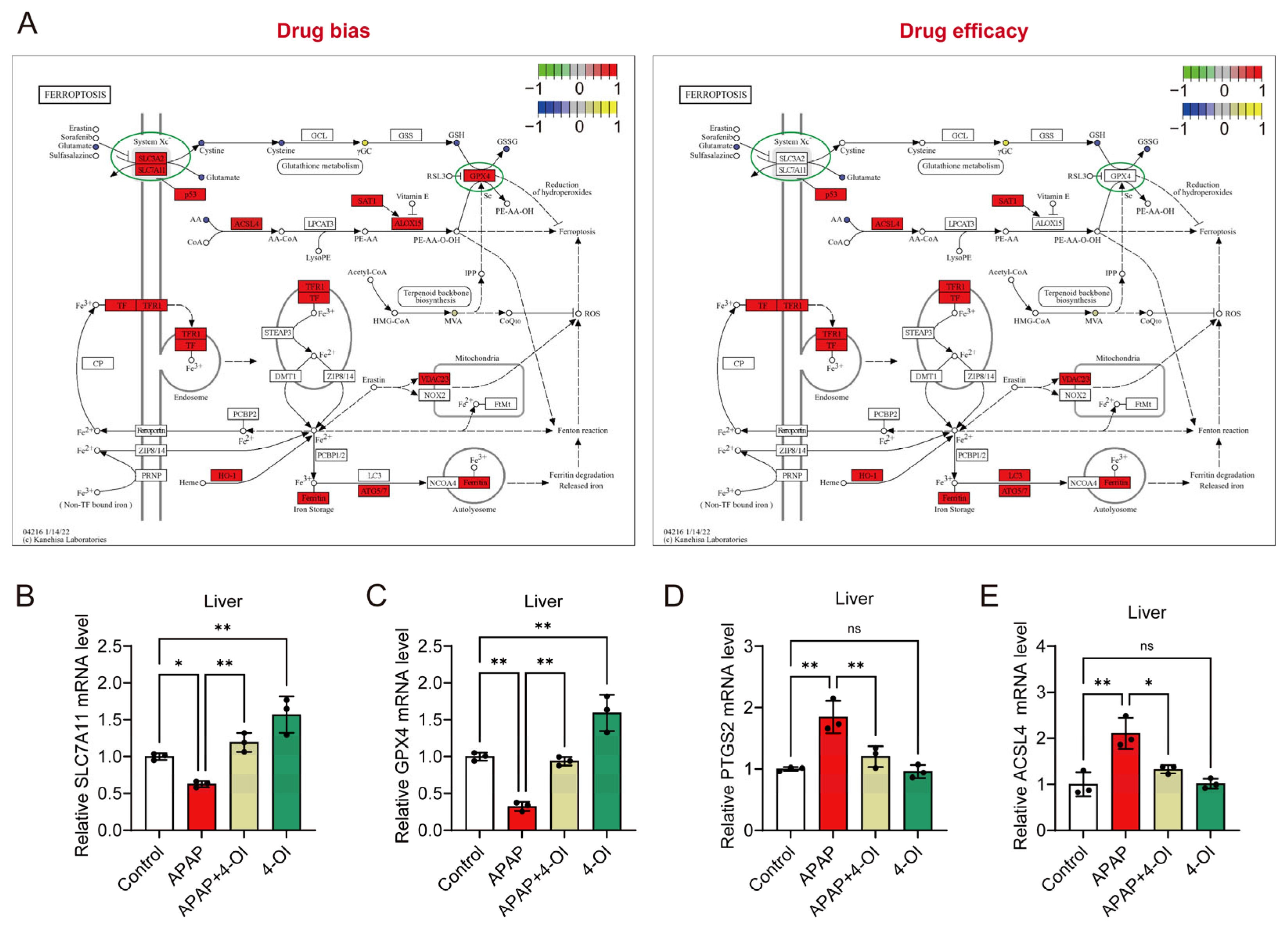
3.3. 4-OI Exerts Dual Properties of Iron Homeostasis Regulation During APAP-Induced Liver Injury
3.4. 4-OI Modulates Ferroptosis-Related Gene Expression and Exhibits Selective Protection Against APAP-Induced Liver Injury
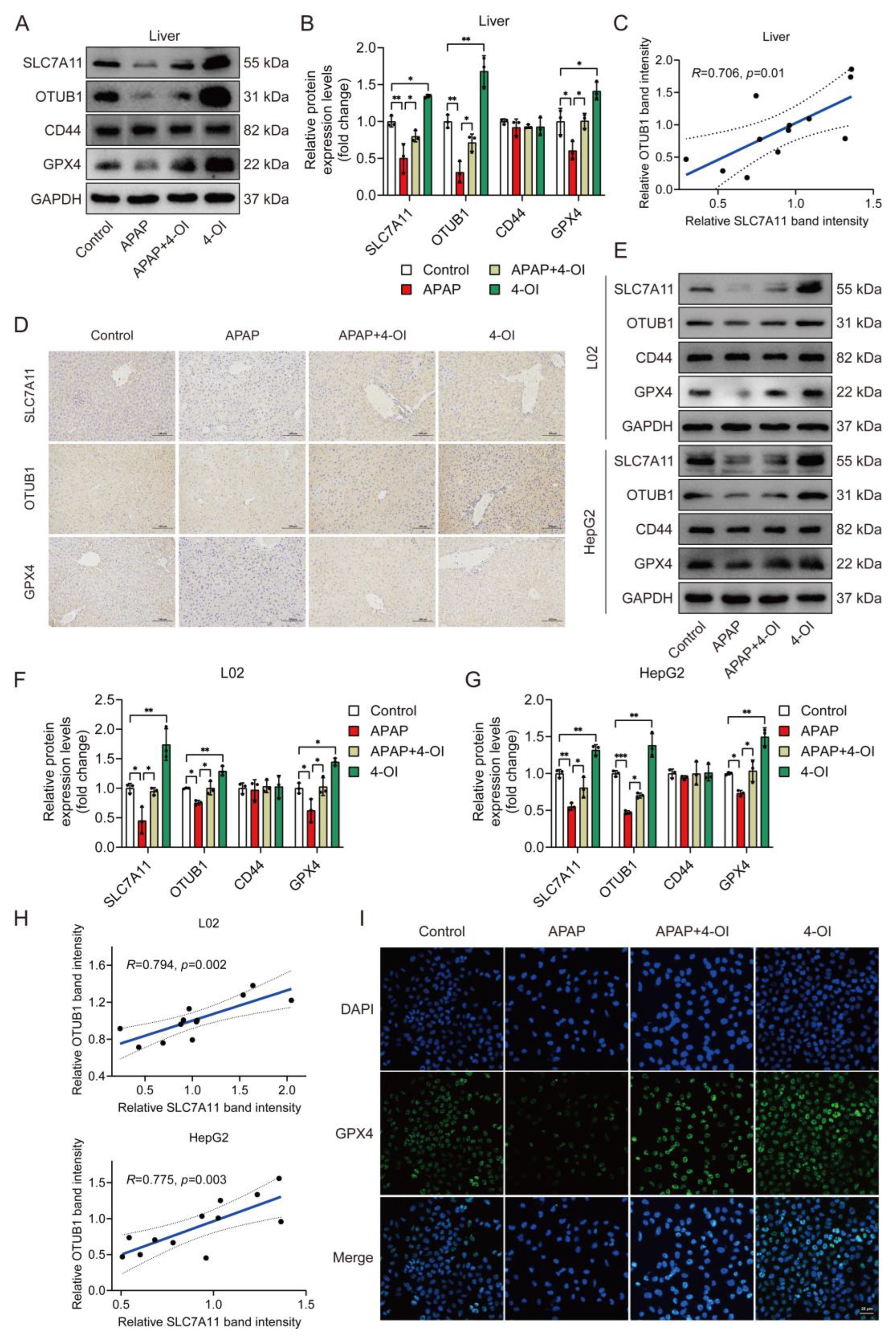
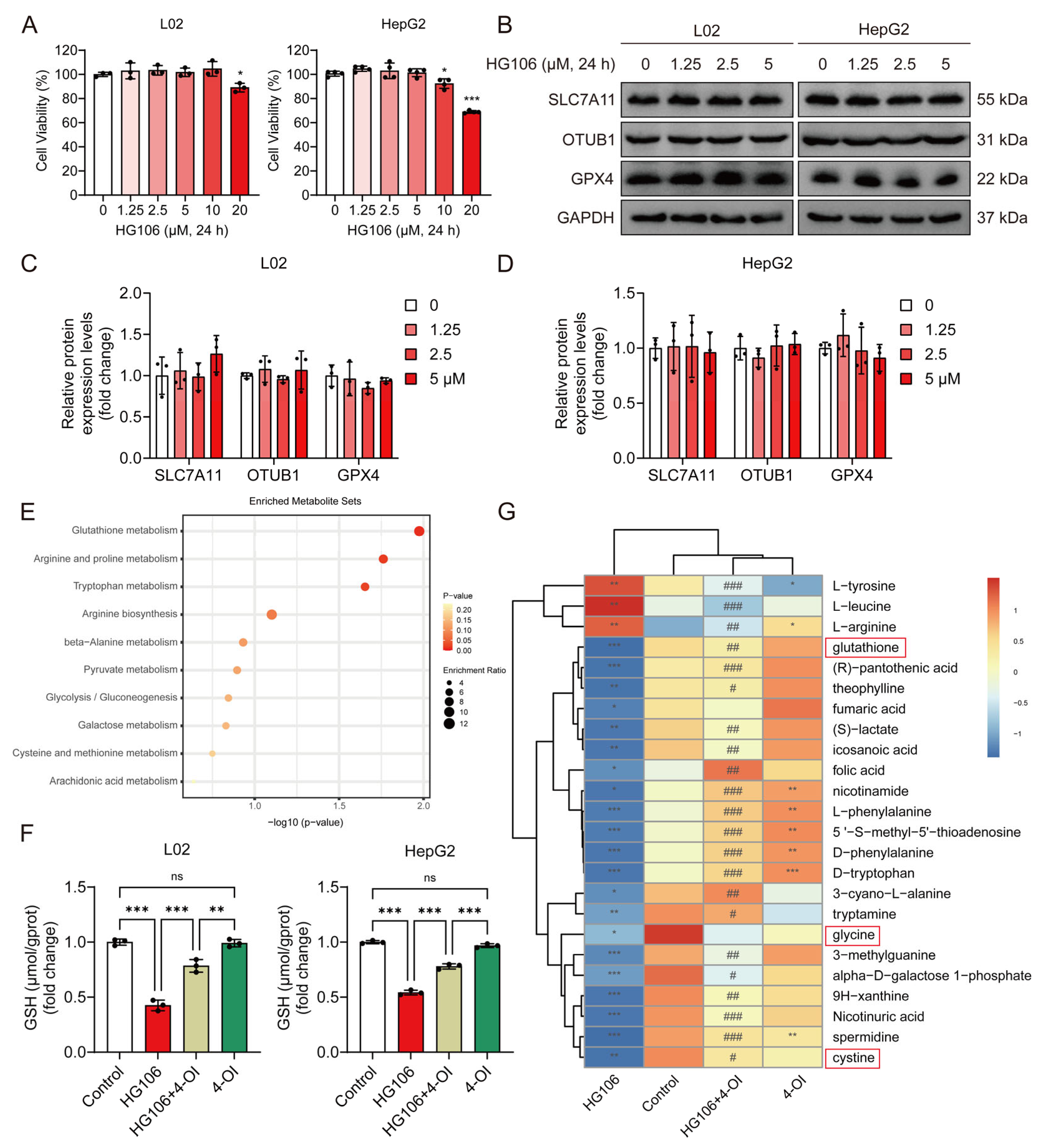
3.5. Coordinated Upregulation of SLC7A11 and OTUB1 Mediates 4-OI’s Hepatoprotective Effects
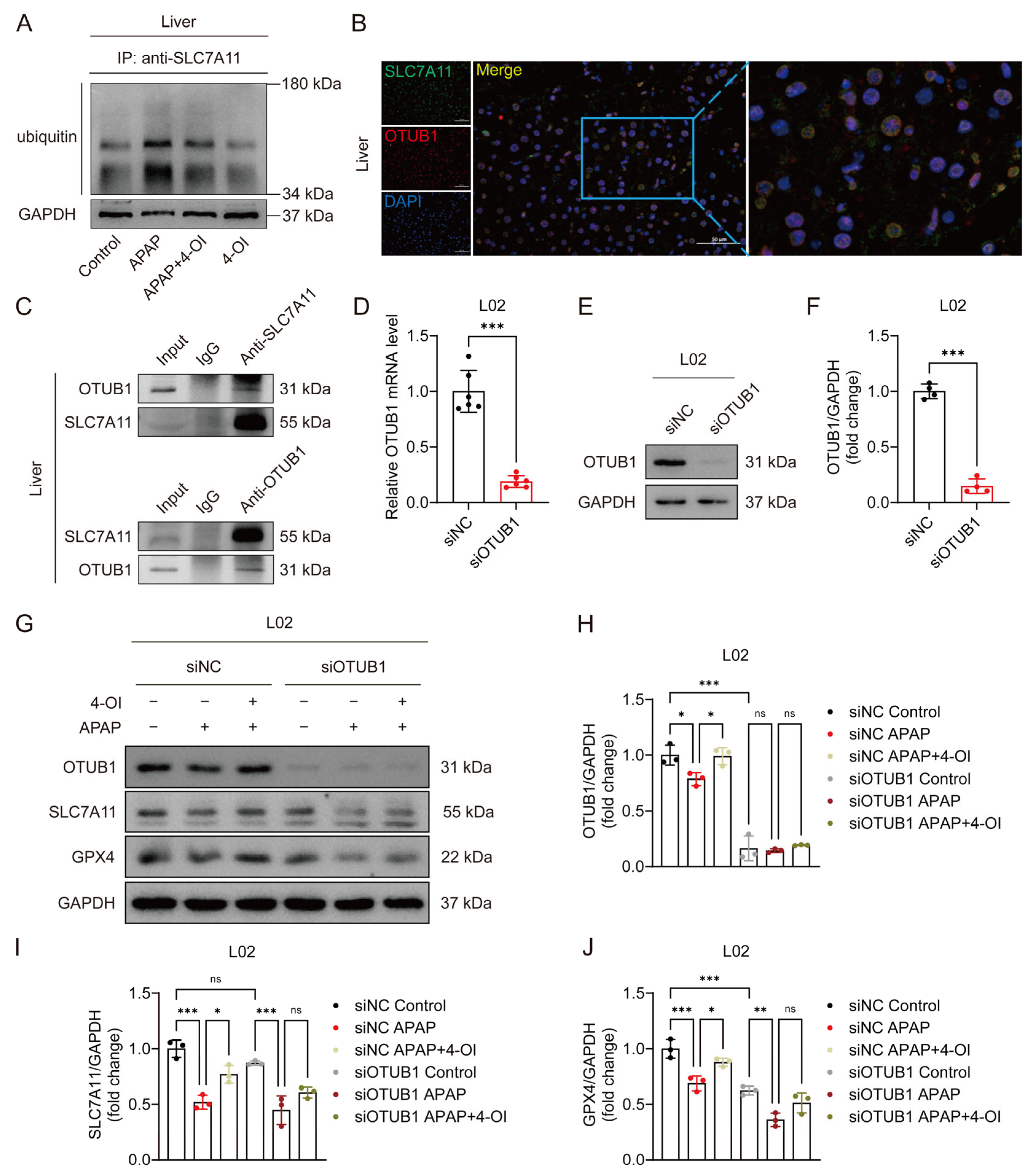
3.6. 4-OI Stabilizes SLC7A11 Through OTUB1-Mediated Deubiquitination in APAP-Induced Liver Injury
3.7. SLC7A11 Function Is Required for Optimal 4-OI-Mediated Enhancement of Cellular Glutathione Synthesis
3.8. Nrf2 Partially Mediates 4-OI’s Anti-Ferroptotic Effects in APAP-Induced Hepatotoxicity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-OI | 4-octyl itaconate |
| ALT | alanine aminotransferase |
| ANOVA | one-way analysis of variance |
| APAP | acetaminophen |
| AST | aspartate aminotransferase |
| BSA | bovine serum albumin |
| CE | collision energy |
| Co-IP | co-immunoprecipitation |
| Cy3 | cyanine 3 |
| DAB | 3,3′-diaminobenzidine |
| ECL | enhanced chemiluminescence |
| ESI | electrospray ionization |
| FBS | fetal bovine serum |
| FITC | fluorescein isothiocyanate |
| FPN1 | ferroportin 1 |
| GSH | glutathione |
| HMDB | Human Metabolome Database |
| HRP | horseradish peroxidase |
| IDA | information-dependent acquisition |
| IHC | immunohistochemistry |
| IRG1 | immunoresponsive gene 1 |
| MSEA | metabolic set enrichment analysis |
| NAPQI | N-acetyl-p-benzoquinone imine |
| NC | nitrocellulose |
| NEM | N-ethylmaleimide |
| PLS-DA | partial least squares discriminant analysis |
| RT-qPCR | real-time quantitative PCR |
| SD | standard deviation |
| TCA | tricarboxylic acid |
| TFR1 | transferrin receptor 1 |
References
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.-P.; Chen, Y.; Wei, X.; Yu, B.; Xiong, Z.-G.; Lu, C.; Hu, W. Novel insights into ferroptosis: Implications for age-related diseases. Theranostics 2020, 10, 11976–11997. [Google Scholar] [CrossRef] [PubMed]
- Wyart, E.; Hsu, M.Y.; Sartori, R.; Mina, E.; Rausch, V.; Pierobon, E.S.; Mezzanotte, M.; Pezzini, C.; Bindels, L.B.; Lauria, A.; et al. Iron supplementation is sufficient to rescue skeletal muscle mass and function in cancer cachexia. EMBO Rep. 2022, 23, e53746. [Google Scholar] [CrossRef]
- Vogt, A.S.; Arsiwala, T.; Mohsen, M.; Vogel, M.; Manolova, V.; Bachmann, M.F. On Iron Metabolism and Its Regulation. Int. J. Mol. Sci. 2021, 22, 4591. [Google Scholar] [CrossRef]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Worwood, M. Ferritin. Blood Rev. 1990, 4, 259–269. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Jaeschke, H.; McGill, M.R.; Ramachandran, A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: Lessons learned from acetaminophen hepatotoxicity. Drug Metab. Rev. 2012, 44, 88–106. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Jaeschke, H. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J. Hepatol. 2017, 66, 836–848. [Google Scholar] [CrossRef]
- Cai, X.; Hua, S.; Deng, J.; Du, Z.; Zhang, D.; Liu, Z.; Khan, N.U.; Zhou, M.; Chen, Z. Astaxanthin Activated the Nrf2/HO-1 Pathway to Enhance Autophagy and Inhibit Ferroptosis, Ameliorating Acetaminophen-Induced Liver Injury. ACS Appl. Mater. Interfaces 2022, 14, 42887–42903. [Google Scholar] [CrossRef]
- Tak, J.; Kim, Y.S.; Kim, T.H.; Park, G.C.; Hwang, S.; Kim, S.G. Galpha(12) overexpression in hepatocytes by ER stress exacerbates acute liver injury via ROCK1-mediated miR-15a and ALOX12 dysregulation. Theranostics 2022, 12, 1570–1588. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Karasawa, T.; Kimura, H.; Watanabe, S.; Komada, T.; Kamata, R.; Sampilvanjil, A.; Ito, J.; Nakagawa, K.; Kuwata, H.; et al. Ferroptosis driven by radical oxidation of n-6 polyunsaturated fatty acids mediates acetaminophen-induced acute liver failure. Cell Death Dis. 2020, 11, 144. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.T.; Han, C.; Xu, D.Q.; Fu, X.W.; Wang, J.S.; Kong, L.Y. 4-Octyl itaconate inhibits aerobic glycolysis by targeting GAPDH to exert anti-inflammatory effects. Nat. Commun. 2019, 10, 5091. [Google Scholar] [CrossRef]
- Hu, Z.; Xu, D.; Meng, H.; Liu, W.; Zheng, Q.; Wang, J. 4-octyl itaconate protects against oxidative stress-induced liver injury by activating the Nrf2/Sirt3 pathway through AKT and ERK1/2 phosphorylation. Biochem. Pharmacol. 2024, 220, 115992. [Google Scholar] [CrossRef]
- Ishimoto, T.; Nagano, O.; Yae, T.; Tamada, M.; Motohara, T.; Oshima, H.; Oshima, M.; Ikeda, T.; Asaba, R.; Yagi, H.; et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 2011, 19, 387–400. [Google Scholar] [CrossRef]
- Liu, T.; Jiang, L.; Tavana, O.; Gu, W. The Deubiquitylase OTUB1 Mediates Ferroptosis via Stabilization of SLC7A11. Cancer Res. 2019, 79, 1913–1924. [Google Scholar] [CrossRef]
- Sun, S.; Shen, J.; Jiang, J.; Wang, F.; Min, J. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal Transduct. Target. Ther. 2023, 8, 372. [Google Scholar] [CrossRef] [PubMed]
- Galy, B.; Conrad, M.; Muckenthaler, M. Mechanisms controlling cellular and systemic iron homeostasis. Nat. Rev. Mol. Cell Biol. 2024, 25, 133–155. [Google Scholar] [CrossRef]
- Zhou, H.; Zhou, Y.L.; Mao, J.A.; Tang, L.F.; Xu, J.; Wang, Z.X.; He, Y.; Li, M. NCOA4-mediated ferritinophagy is involved in ionizing radiation-induced ferroptosis of intestinal epithelial cells. Redox Biol. 2022, 55, 102413. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Silva, I.; Peccerella, T.; Mueller, S.; Rausch, V. IL-1 beta-mediated macrophage-hepatocyte crosstalk upregulates hepcidin under physiological low oxygen levels. Redox Biol. 2019, 24, 101209. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Fan, Z.; Rauh, M.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene 2017, 36, 5593–5608. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, L.; Zhang, X.; Cui, W.; Liu, Y.; Sun, Q.R.; He, Q.; Zhao, S.; Zhang, G.A.; Wang, Y.; et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 2019, 20, e47563. [Google Scholar] [CrossRef]
- Rochette, L.; Dogon, G.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis. Int. J. Mol. Sci. 2022, 24, 449. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Qiang, Z.; Chai, D.; Peng, J.; Xia, Y.; Hu, R.; Jiang, H. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging 2020, 12, 12943–12959. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhai, Y.; Chen, J.; Xu, X.; Wang, H. Kaempferol Ameliorates Oxygen-Glucose Deprivation/Reoxygenation-Induced Neuronal Ferroptosis by Activating Nrf2/SLC7A11/GPX4 Axis. Biomolecules 2021, 11, 923. [Google Scholar] [CrossRef]
- Tang, H.M.; Tang, H.L. Cell recovery by reversal of ferroptosis. Biol. Open 2019, 8, bio043182. [Google Scholar] [CrossRef]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2021, 17, 2054–2081. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar]
- Sies, H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999, 27, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Alcivar, A.L.; Ma, J.; Foo, T.K.; Zywea, S.; Mahdi, A.; Huo, Y.; Kensler, T.W.; Gatza, M.L.; Xia, B. NRF2 Induction Supporting Breast Cancer Cell Survival Is Enabled by Oxidative Stress-Induced DPP3-KEAP1 Interaction. Cancer Res. 2017, 77, 2881–2892. [Google Scholar] [CrossRef]
- Tuo, L.; Xiang, J.; Pan, X.; Gao, Q.; Zhang, G.; Yang, Y.; Liang, L.; Xia, J.; Wang, K.; Tang, N. PCK1 Downregulation Promotes TXNRD1 Expression and Hepatoma Cell Growth via the Nrf2/Keap1 Pathway. Front. Oncol. 2018, 8, 611. [Google Scholar] [CrossRef]
- Liu, J.; Huang, C.; Liu, J.; Meng, C.; Gu, Q.; Du, X.; Yan, M.; Yu, Y.; Liu, F.; Xia, C. Nrf2 and its dependent autophagy activation cooperatively counteract ferroptosis to alleviate acute liver injury. Pharmacol. Res. 2023, 187, 106563. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Z.; Li, Y.; Xu, D.; Meng, H.; Liu, W.; Xu, Q.; Yao, B.; Wang, J. OTUB1-SLC7A11 Axis Mediates 4-Octyl Itaconate Protection Against Acetaminophen-Induced Ferroptotic Liver Injury. Antioxidants 2025, 14, 698. https://doi.org/10.3390/antiox14060698
Hu Z, Li Y, Xu D, Meng H, Liu W, Xu Q, Yao B, Wang J. OTUB1-SLC7A11 Axis Mediates 4-Octyl Itaconate Protection Against Acetaminophen-Induced Ferroptotic Liver Injury. Antioxidants. 2025; 14(6):698. https://doi.org/10.3390/antiox14060698
Chicago/Turabian StyleHu, Ziyun, Yuxin Li, Di Xu, Huihui Meng, Wenya Liu, Qian Xu, Benxing Yao, and Junsong Wang. 2025. "OTUB1-SLC7A11 Axis Mediates 4-Octyl Itaconate Protection Against Acetaminophen-Induced Ferroptotic Liver Injury" Antioxidants 14, no. 6: 698. https://doi.org/10.3390/antiox14060698
APA StyleHu, Z., Li, Y., Xu, D., Meng, H., Liu, W., Xu, Q., Yao, B., & Wang, J. (2025). OTUB1-SLC7A11 Axis Mediates 4-Octyl Itaconate Protection Against Acetaminophen-Induced Ferroptotic Liver Injury. Antioxidants, 14(6), 698. https://doi.org/10.3390/antiox14060698